

In this study, Barrales et al. identify Lem2 as a novel factor contributing to heterochromatic gene silencing in S. pombe. The results show that Lem2 mediates distinct silencing and localization of antagonistic factors at heterochromatin, providing novel insight into heterochromatin regulation.
Keywords: heterochromatin, perinuclear silencing, nuclear periphery, lamin-associated proteins, LEM, tethering
Abstract
Transcriptionally silent chromatin localizes to the nuclear periphery, which provides a special microenvironment for gene repression. A variety of nuclear membrane proteins interact with repressed chromatin, yet the functional role of these interactions remains poorly understood. Here, we show that, in Schizosaccharomyces pombe, the nuclear membrane protein Lem2 associates with chromatin and mediates silencing and heterochromatin localization. Unexpectedly, we found that these functions can be separated and assigned to different structural domains within Lem2, excluding a simple tethering mechanism. Chromatin association and tethering of centromeres to the periphery are mediated by the N-terminal LEM (LAP2–Emerin–MAN1) domain of Lem2, whereas telomere anchoring and heterochromatin silencing require exclusively its conserved C-terminal MSC (MAN1–Src1 C-terminal) domain. Particularly, silencing by Lem2 is epistatic with the Snf2/HDAC (histone deacetylase) repressor complex SHREC at telomeres, while its necessity can be bypassed by deleting Epe1, a JmjC protein with anti-silencing activity. Furthermore, we found that loss of Lem2 reduces heterochromatin association of SHREC, which is accompanied by increased binding of Epe1. This reveals a critical function of Lem2 in coordinating these antagonistic factors at heterochromatin. The distinct silencing and localization functions mediated by Lem2 suggest that these conserved LEM-containing proteins go beyond simple tethering to play active roles in perinuclear silencing.
Chromatin is distributed in a nonrandom manner within the eukaryotic nucleus. Chromatin domains with different functions reside in separate subcompartments, and transcriptionally silent heterochromatin is often found at the nuclear periphery or close to the nucleolus. It has been proposed that these nuclear subcompartments establish a special microenvironment enriched for factors required for silencing and to which heterochromatin is recruited, thereby facilitating the establishment, propagation, and maintenance of the repressed state (Taddei and Gasser 2012). This notion is supported by artificial tethering experiments in yeast and mammals, in which a transcriptionally active locus can become repressed when sequestered to the nuclear periphery (for review, see Towbin et al. 2013). However, the endogenous mechanisms that lead to the peripheral sequestration of repressed chromatin remain poorly understood.
In metazoans, beneath the inner nuclear membrane (INM) lies the nuclear lamina that interacts with well-defined chromosomal regions. These lamina-associated domains are enriched for repressed chromatin, and their peripheral association is linked to the methylation of Lys9 of histone H3 (H3K9), a conserved mark of repressive chromatin (Amendola and van Steensel 2014). The nuclear lamina itself is a meshwork of intermediate filaments consisting of A- and B-type lamins that interact with integral proteins of the INM. Several studies demonstrate that lamins affect the perinuclear positioning and repression of heterochromatin (for review, see Towbin et al. 2013; Amendola and van Steensel 2014). A striking example is the inverted nuclear architecture of rod photoreceptor cells in nocturnal animals. These cells express neither lamin A/C nor the lamin B receptor (LBR), which results in the central localization of heterochromatin, yet this inverted architecture can be completely reverted when LBR is ectopically expressed (Solovei et al. 2013). However, unicellular organisms entirely lack lamins despite the conserved presence of peripheral heterochromatin. Furthermore, various peripheral proteins other than lamins interact with repressed chromatin, suggesting that additional tethering pathways do exist (Hirano et al. 2012; Poleshko et al. 2013; Towbin et al. 2013; Zuleger et al. 2013; Amendola and van Steensel 2014). Among lamin-associated proteins (LAPs), several contain a LEM (LAP2–Emerin–MAN1) domain, a 40-amino-acid helix–extension–helix (HEH) motif that is conserved from yeast to humans (Brachner and Foisner 2011). The LEM domain binds to the metazoan-specific barrier to autointegration factors (BAFs), which are sequence-independent DNA-binding proteins. It has been proposed that their interaction with LEM domain proteins might contribute to gene repression (Margalit et al. 2007; Barton et al. 2015), but whether BAF proteins bind specifically to heterochromatin is unknown. LAPs have also been reported to interact directly with histone deacetylases (HDACs) and mediate their recruitment to the nuclear periphery (Somech et al. 2005; Demmerle et al. 2012). Thus, LAPs might use different mechanisms by which they could contribute to heterochromatin silencing through either chromatin tethering or the recruitment of repressive factors. However, the large number of INM proteins and their potential redundancy in metazoans (Brachner and Foisner 2011) make it challenging to study their functional requirement in gene repression.
In the fission yeast Schizosaccharomyces pombe, only three INM proteins with homology with LAPs are present: Lem2, Man1, and Ima1 (Mans et al. 2004). Whereas Ima1 is homologous to Samp1 (NET5) (Gudise et al. 2011; Zuleger et al. 2013), Lem2 and Man1 belong to the group II of LEM proteins (Brachner and Foisner 2011). Members of this group contain (1) a HEH motif homologous to the metazoan LEM domain at the N terminus and (2) a DNA-binding winged helix MSC (MAN1–Src1 C-terminal) domain in the C-terminal region, which are separated by two transmembrane domains. These highly conserved domains project into the nucleoplasm, where they might interact with chromosomal regulatory factors. A recent study demonstrated that Lem2 and Man1 contribute to the integrity of the nuclear structure and to telomere anchoring in fission yeast (Gonzalez et al. 2012). Furthermore, homologs of Lem2 associate with telomeric regions in Saccharomyces cerevisiae (Heh1/Src1) and Caenorhabditis elegans (LEM-2) (Grund et al. 2008; Ikegami et al. 2010), and loss of Heh1 causes defects in the perinuclear positioning of telomeres and the silent rDNA loci in budding yeast (Mekhail et al. 2008; Chan et al. 2011). However, the role of Lem2 in heterochromatin silencing remains elusive. Thus, although yeast does not have either a nuclear lamina or BAF homologs, the essential functions of LAPs appear to be conserved, making S. pombe an ideal model to study the relationship between heterochromatin localization and silencing in a less complex system.
S. pombe has large perinuclear heterochromatic domains spanning up to 40 kb at the pericentromeres, the telomeres, and the silent mating type locus (mat). While the three centromeres colocalize with the mat locus and cluster next to the spindle pole body (SPB), which is equivalent to the mammalian centrosome, the telomeres are positioned opposite at the nuclear envelope—an orientation known as Rabl configuration. Heterochromatin domains are marked by dimethylated H3K9 that is deposited by Clr4, the sole histone H3K9 methyltransferase in S. pombe. This repressive histone modification is recognized by HP1 proteins, which serve as a platform to recruit other silencing factors such as SHREC, a repressor complex comprising a Snf2-like nucleosome remodeler and a HDAC subunit similar to the mammalian NuRD complex (Grewal 2010; Allshire and Ekwall 2015). Binding and distribution of these factors within the HP1 platform is subject to a complex regulation. Recruitment of SHREC is positively regulated through phosphorylation of HP1 proteins, which counteracts the binding of the JmjC protein Epe1, an anti-silencing factor with similarity to histone demethylases (Shimada et al. 2009). This pathway works in parallel to the ubiquitin-dependent degradation of Epe1 by the conserved ubiquitin ligase Cul4-Ddb1Cdt2, which removes Epe1 from the body of heterochromatin (Braun et al. 2011). In fission yeast, the RNAi machinery also contributes to the establishment and maintenance of heterochromatin (Grewal 2010; Allshire and Ekwall 2015). siRNAs are generated from transcripts of heterochromatic repeats by the endonuclease Dicer (Dcr1), the Argonaute-containing complex RITS (RNA-induced transcriptional silencing), and an RNA-dependent RNA polymerase complex (RDRC). Importantly, the assembly of Dcr1 and RDRC at the nuclear envelope through interactions with the putative transmembrane protein Dsh1 is critical for RNAi-mediated silencing (Kawakami et al. 2012).
Here, we show that the nuclear envelope protein Lem2 controls heterochromatin silencing and positioning in S. pombe. Lem2 regulates silencing of all heterochromatic regions, but its contribution is partially masked by redundancy with other pathways. By developing a highly sensitive SGA (synthetic genetic array) approach that uncovers redundancy based on a silencing reporter assay, we identified multiple factors that collaborate with Lem2 in silencing, such as the RNAi machinery and the centromere clustering factor Csi1. In addition to silencing, Lem2 cooperates with these factors to ensure the proper positioning of heterochromatin at the nuclear periphery. To obtain insights into the molecular mechanisms, we separated the structural domains of Lem2. We found that the N-terminal part of Lem2, which contains the conserved LEM domain, mediates binding to centromeres and their correct positioning. Unexpectedly, both telomere localization and silencing of all heterochromatic domains are exclusively dependent on the MSC domain. We further demonstrated that silencing by Lem2 is epistatic with the role of SHREC at telomeres, while it opposes the anti-silencing function of the JmjC protein Epe1. We conclude that the main role of Lem2 in heterochromatin silencing is to maintain a proper balance between SHREC and Epe1. Thus, our study uncovers and functionally dissects principal pathways controlling heterochromatin localization and silencing by Lem2 in S. pombe. This unveils a new perspective on these highly conserved LEM proteins and suggests that the metazoan homologs also play active roles in heterochromatin silencing.
Results
The INM protein Lem2 mediates repression of silent chromatin
To identify novel factors controlling heterochromatin, we performed a genome-wide screen for mutants with defects in pericentromeric silencing. We crossed an S. pombe haploid deletion library with a silencing reporter strain that harbors the ura4+ gene at the left innermost repeat region of centromere 1 (imr1L::ura4+) (Fig. 1A; Ekwall et al. 1999). The repressed state of this silent locus can be monitored by assessing growth on medium containing the drug 5-FOA (5′-fluoroorotic acid) that is converted into a toxic metabolite by the gene product of ura4+. Among various mutants with disturbed silencing, which will be described elsewhere, we identified Lem2 (also known as Heh1), a LEM domain protein of the INM.
Figure 1.

Lem2 contributes globally to transcriptional gene silencing. (A) S. pombe heterochromatic domains (shaded in orange) with insertion sites of the ura4+ reporter and primer positions for RT-qPCR and chromatin immunoprecipitation (ChIP) analysis (red bars). (B) Silencing reporter assay. Fivefold serial dilutions of wild-type (WT) cells and two independent knockout isolates (ko-1 and ko-2) of lem2+; the clr4Δ strain was used as a positive control. (N/S) Nonselective. (C,E) RT-qPCR analysis. Shown are transcript levels relative to wild type after normalization to act1+. tlh1+ and tlh2+ are located on the left and right arms of chromosomes 1 and 2, respectively, but share 100% identity. (D) Silencing reporter assay, as in B. For all quantitative experiments, data are represented as mean ± SEM from n independent experiments; asterisks denote P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****) from two-tailed Student's t-test analysis.
We confirmed the silencing defect of lem2Δ from the genome-wide screen by disrupting the lem2+ locus in the imr1L::ura4+ reporter strain. Compared with cells lacking the sole H3K9 methyltransferase Clr4, the absence of Lem2 causes a moderate growth defect in the presence of 5-FOA (Fig. 1B) that depends on the expression of the reporter gene and is rescued by ectopic expression of lem2+ (Supplemental Fig. S1A,B). Growth of lem2Δ cells is also impaired in the presence of thiabendazole (Supplemental Fig. S1C), consistent with the fact that defects in pericentromeric heterochromatin result in chromosome instability and hypersensitivity toward this microtubule-destabilizing drug. Quantitative analysis by RT-qPCR revealed that lem2Δ cells display a twofold increase in reporter gene expression (Fig. 1C). Increased transcript levels (fivefold to 25-fold) were also found for the mat3::ura4+ reporter, endogenous heterochromatic loci (cen-dg, tlh1/2+), the central core region of centromere 1 (cnt1), and subtelomeric long terminal repeats (LTRs; up to 100-fold) (Fig. 1C; Supplemental Fig. S1D). In contrast, we detected no change in expression for various euchromatic loci (including housekeeping and low-expressed genes) (Supplemental Fig. S1E), suggesting that the repressive function of Lem2 is specific to silent chromatin.
In S. cerevisiae, Lem2 and the nuclear membrane protein Nur1 form a physical complex called CLIP that is involved in the perinuclear localization of the rDNA locus (Mekhail et al. 2008). We deleted the corresponding S. pombe gene, mug154+ (which we refer to here as nur1+) and studied its requirement for silencing. Independent nur1Δ isolates display a subtle but reproducible silencing defect (Supplemental Fig. S2A). Notably, the nur1Δ lem2Δ double mutant exhibits an epistatic phenotype, which resembles the phenotype of the single nur1Δ (Supplemental Fig. S2A,B). Together, these findings indicate that both Lem2 and Nur1 are involved in heterochromatic silencing and act together in the same pathway.
A possible reason for the moderate silencing defect in lem2Δ cells might be partial redundancy with other factors in heterochromatin formation. Two INM proteins, Man1 and Ima1, have been reported to have overlapping functions with Lem2 in ensuring nuclear membrane integrity (Hiraoka et al. 2011). In addition, Man1 was shown to associate with subtelomeric regions (Steglich et al. 2012), which may suggest a role in telomeric silencing. However, in contrast to Lem2, we found that loss of either Man1 or Ima1 does not affect repression of heterochromatin (Fig. 1D,E). Furthermore, we did not detect synthetic defects in heterochromatin silencing for these mutants in combination with lem2Δ. Instead silencing is partially restored in the double mutants (Fig. 1E), and we noticed a subtle but reproducible transcriptional increase (1.6-fold to 1.7-fold) of lnp1+, a homolog of human LUNAPARK that is involved in membrane organization (Supplemental Fig. S2C). Duplication of the genomic locus of lnp1+ has been reported to suppress lem2-associated phenotypes (Y Hiraoka, pers. comm.), which could explain the partial suppression of the silencing defect in these double mutants. In conclusion, whereas Lem2, Man1, and Ima1 may have overlapping roles in other pathways (Hiraoka et al. 2011), heterochromatic silencing is exclusively mediated by Lem2.
Genome-wide isolation of synthetic genetic interactors of lem2+
To test whether the role of Lem2 in silencing is masked by redundancy with other factors, we determined genome-wide genetic interactions by SGA analysis. To make this approach more sensitive and specific for heterochromatin function, we advanced the method to monitor silencing defects instead of pleiotropic phenotypes. To this end, we crossed the pericentromeric imr1L::ura4+ reporter strain deleted for lem2+ with the haploid deletion library. We then measured the silencing defects in the double mutants by determining the ratio of colony sizes on 5-FOA to those on nonselective medium and calculated a genetic interaction score, ε, for each mutant pair (Fig. 2A; Supplemental Fig. S3A). In order to distinguish genetic interactions that are specific for lem2+, we examined two additional mutants of known heterochromatin factors as query strains: Clr2, a subunit of the repressor complex SHREC (Sugiyama et al. 2007), and Ckb1, the catalytic subunit of casein kinase II that phosphorylates HP1 proteins (Shimada et al. 2009). In addition, we included nur1+ due to its epistatic function with lem2+, which predicts that genetic interactions related to silencing should be shared between these two genes.
Figure 2.

lem2+ interacts genetically with multiple factors in transcriptional silencing. (A) Scheme illustrating the SGA approach with subsequent silencing reporter assays and determination of genetic interaction scores (ε values). For more information, see the text and Supplemental Figure S3. (B) Selected genetic interaction data after filtering (211 out of 2918) (see also Supplemental Fig. S3). (Top panel) Correlation matrix of the genetic interaction profiles. (Bottom panels) Interaction scores (ε values) analyzed by hierarchical clustering. (Blue) Negative ε values (synthetic interactions); (yellow) positive ε values (epistatic or suppressive interactions). (C) Gene ontology (GO) analysis for biological processes of genes identified in individual clusters. (D) Pairwise comparison of genetic interaction scores between averaged replicates of different query strains. Symbols in gray and black indicate all (2918) and selected (211) genetic interactions, respectively. Symbols in color show genetic interaction pairs for double mutants enriched for the GO terms depicted in C. The inset graphs at the top and at the right of each graph show the normal distribution of all genetic interactions (2918), with red lines demarcating ±1 standard deviation (SD) from the mean.
From a total of 22 screens and 2918 mutants, we determined ∼64,000 genetic interactions based on the functional readout of the reporter assay. We applied a cutoff to select for robust genetic interactions of the query strains, resulting in 211 mutants (Fig. 2B; Supplemental Fig. S3B). Correlation coefficient analysis of the interaction profiles revealed that lem2Δ is fairly similar to nur1Δ (R2 of 0.39) but differs from clr2Δ (R2 of 0.09) (Fig. 2B [top], D). We then performed hierarchical clustering of the ε values to identify groups with similar genetic interaction profiles (Fig. 2B, bottom). Two clusters (I and II) contain many synthetic interactions that are shared between lem2Δ and nur1Δ but absent in clr2Δ and ckb1Δ. These clusters are enriched for the gene ontology (GO) biology process terms “chromosome organization” and “RNA metabolism” (Fig. 2C). Interestingly, many factors of this group have in common that they associate with the nuclear periphery (e.g., with the SPB), which is highly reminiscent of Lem2. Among those factors are the microtubule-associated proteins Alp14 and Mto1, which we previously identified in an independent screen for silencing factors (Braun et al. 2011), and Csi1, a nuclear factor involved in centromere clustering (Hou et al. 2012). Remarkably, we also found lnp1+ as a synthetic interactor, which matches our reciprocal finding of the partial suppression of lem2Δ when lnp1+ transcript levels are increased (Supplemental Fig. S2C). We confirmed the synthetic silencing defect for lnp1Δ and other selected candidates by reporter assays and RT-qPCR experiments (Supplemental Fig. S4). Another cluster (IV) with synthetic interaction for lem2Δ and nur1Δ shows a GO enrichment for “chromatin silencing.” Consistently, many mutants in this group are epistatic with clr2Δ and ckb1Δ; for example, members of the SHREC complex and the RNAi machinery. In conclusion, this heterochromatin-specific SGA approach unveils the functional redundancy of Lem2 in silencing and identifies several key factors such as SPB-interacting proteins and heterochromatin complexes that coordinate pericentromeric silencing together with Lem2. Furthermore, the substantial overlap between Lem2 and Nur1 reinforces the idea that both factors are part of the same network controlling silencing.
Lem2 cooperates with multiple pathways in heterochromatic silencing at the nuclear periphery
The SGA analysis revealed several redundant factors that localize to the nuclear envelope and cooperate with Lem2 in pericentromeric silencing. To better understand this cooperative role of Lem2, we decided to focus on two peripheral factors that have previously been linked to heterochromatin positioning: Csi1 and Dsh1 (as a representative of the RNAi machinery). Csi1 binds to the SPB and mediates centromere clustering (Hou et al. 2012). Dsh1 is a putative INM protein and mediates the peripheral assembly of the RNAi machinery (Kawakami et al. 2012), which contributes to telomere clustering (Hall et al. 2003). We deleted the corresponding ORF of dsh1+ in wild-type and lem2Δ cells, as the deletion mutant was not included in our large-scale SGA study. We then examined endogenous heterochromatic transcripts from the pericentromeric dg repeats and the subtelomeric tlh1/2+ loci by RT-qPCR in the single and double mutants (Fig. 3A). While the single deletion of csi1+ displays normal transcript levels at centromeres and telomeres, we found synthetic silencing defects in the csiΔ lem2Δ double mutant, particularly for tlh1/2+. In the single dsh1Δ mutant, we observed that cen-dg transcripts are already strongly derepressed, consistent with the requirement for Dsh1 in pericentromeric silencing (Kawakami et al. 2012). However, we found that cen-dg transcripts are further up-regulated in the dsh1Δ lem2Δ double mutant to levels similar to those seen in cells lacking the sole histone H3K9 methyltransferase, Clr4. The synthetic phenotype suggests that Lem2 acts in a pathway different from RNAi, and we found consistently that chromatin binding of Chp1, a subunit of the Argonaute-containing RITS complex, is not affected by lem2Δ (Supplemental Fig. S5A). We further observed a synthetic yet not complete derepression in the dsh1Δ lem2Δ mutant for the tlh1/2+ transcripts, implying that additional pathways are involved in telomeric silencing (Fig. 3A). Since RNAi acts redundantly with the telomere-associated protein Taz1 (Grewal 2010), we tested for a cooperative function with Lem2. We found that disrupting taz1+ in lem2Δ cells indeed causes a synthetic telomeric silencing defect. (Fig. 3B). Strikingly, deleting all three factors results in a further derepression of tlh1/2+. Collectively, these findings indicate that Lem2 coordinates heterochromatic silencing with multiple parallel pathways at the nuclear periphery.
Figure 3.

Lem2 cooperates with various pathways in heterochromatin silencing. (A,B) RT-qPCR of heterochromatic transcript levels, analyzed as in Figure 1C. (C,D) ChIP-qPCR analysis of H3K9me2 levels. ChIP data have been normalized to act1+ and are shown relative to the maximal enrichment in wild-type cells for each heterochromatic domain (cen1, tel1L, and tel2L). Data are represented as mean ± SEM from n independent experiments; asterisks denote P < 0.05 (*) and P < 0.01 (**) from two-tailed Student's t-test analysis.
In contrast to the synergistic defect at telomeres seen in the taz1Δ dsh1Δ lem2Δ triple mutant, we observed a partial suppression at pericentromeres (Supplemental Fig. S5B). We made a similar observation for the cen-dg transcripts in lem2Δ cells in the absence of the transcription factor Atf1 (Supplemental Fig. S5C), which is specific for silencing of the mating type locus (Grewal 2010). Thus, blocking the telomere-specific or mating type-specific pathway restores silencing in a reciprocal manner at pericentromeres. This finding makes it less likely that the silencing defects in lem2Δ mutants are nonspecifically caused by broadly altered transcription, although we noticed that double and triple mutants lacking Lem2 often display pleiotropic phenotypes like slow growth and small colonies (data not shown). Furthermore, as we did not see altered expression of various euchromatic genes in these double mutants (Supplemental Fig. S5D), we conclude that the cooperative role of Lem2 in gene repression is specifically restricted to silent chromatin.
H3K9me is an integral part of the heterochromatin structure and crucial for transcriptional silencing. In S. pombe, the prevalent mark of silent chromatin is dimethylated H3K9 (Supplemental Fig. S6A; Zofall and Grewal 2006; Al-Sady et al. 2013). Cells lacking Lem2 display only a modest reduction of pericentromeric H3K9me2 (Fig. 3C, top) but no further decrease in combination with csi1Δ (Supplemental Fig. S6B). In contrast, maintenance of pericentromeric H3K9me2 requires RNAi (Grewal 2010) and is strongly reduced to 20%–30% in the dsh1Δ mutant (Fig. 3C, bottom). Nonetheless, dsh1Δ cells are not completely devoid of this mark, and additional deletion of lem2+ causes a further reduction of H3K9me2 to only 5%–10% of wild-type levels (Fig. 3C, bottom). This finding implies that Lem2 and RNAi also act synthetically on pericentromeric H3K9me2. However, for most parts of the subtelomeres, H3K9me2 is not decreased below wild-type levels in single or combinatorial mutants of lem2Δ, dsh1Δ, and taz1Δ (Fig. 3D; Supplemental Fig. S6C). In conclusion, pericentromeres and telomeres appear to have different requirements for the maintenance of H3K9me with respect to Lem2. These findings further imply that sustaining high levels of H3K9me is generally not sufficient to maintain repressed transcription at telomeres.
Lem2 cooperates with Csi1 and Dsh1 in heterochromatin positioning
In yeast, interphase heterochromatin adopts the so-called Rabl configuration in which the centromeres cluster next to the SPB at the periphery, while the telomeres are positioned opposite to the SPB. Given the partial redundancy of Lem2 and the centromere clustering factor Csi1 in silencing, we asked whether both also have overlapping functions in centromere localization. To determine the relative localization of centromeres and the SPB, we used GFP-tagged Mis6 (an inner centromere protein) and mCherry-tagged Sad1 (a SUN domain protein associated with the SPB). Live-cell imaging confirmed the proper expression of the fusion proteins, which, in wild-type cells, form a single cluster that represents the three centromeres colocalizing with the SBP (Fig. 4A). Cells lacking Csi1 often display two Mis6-GFP foci (Hou et al. 2012), but we found that the number of cells with three foci is dramatically increased in the csi1Δ lem2Δ double mutant (32% vs. 8% in csi1Δ cells) (Fig. 4A,B; Supplemental Fig. S7A). Additionally, the colocalization of centromeres with the SPB is significantly perturbed in the double mutant, resulting in a substantial increase of cells in which the most SPB-proximal Mis6-GFP signal is fully separated from the mCherry-Sad1 focus (18% and 10% in csi1Δ lem2Δ compared with 0.7% and 1.5% in csi1Δ, respectively) (Fig. 4A; Supplemental Fig. S4B). This colocalization phenotype is not seen for lem2Δ dsh1Δ cells (Supplemental Fig. S7B). Notably, by expressing a nuclear envelope marker (mCherry-Cut11), we confirmed that the centromeres in csi1Δ lem2Δ cells are indeed delocalized from the nuclear periphery (Supplemental Fig. S7C).
Figure 4.

Lem2 cooperates with Csi1 and Dsh1 in centromere and telomere positioning. (A) Representative pictures of two-color live-cell imaging of Mis6-GFP (centromere) and Sad1-mCherry (SPB). Dotted lines delineate cell boundaries. (B) Quantification of cells with distinct numbers of Mis6 foci. Shown is the percentage for a population of n cells of one representative experiment. (C) Quantification of cells displaying distinct degrees of colocalization of Mis6-GFP and Sad1-mCherry. For each cell, the most proximal Mis6-GFP focus next to the SPB is classified as overlapping, proximal, or completely separated from the SPB. Shown is the percentage of n cells of a representative experiment, denoting each type of localization. (D) Zone designation (I–III) and distribution of Taz1-GFP (telomere) within confocal planes with representative pictures (shown at right). The nuclear envelope is visualized by mCherry-Cut11. (E) Quantification of Taz1-GFP distribution relative to the nuclear periphery. Shown is the percentage of telomeres for each nuclear zone for a population of n cells combined from two independent experiments. The asterisks denote P < 0.05 (*) and P < 0.0001 (****) from χ2 test analysis.
Lem2 also contributes to telomere anchoring (Gonzalez et al. 2012). We therefore tested whether Lem2 cooperates with the RNAi assembly factor Dsh1 in localization because of their redundancy in silencing. To determine the frequency of perinuclear anchoring, we analyzed the position of the telomere-associated protein Taz1 (GFP-tagged) relative to the nuclear envelope protein Cut11 (mCherry-tagged) by confocal microscopy. For the quantification, we used a previously described method (Hediger et al. 2004) by which each optical section is divided into three zones with equal areas that correspond to the nuclear periphery (zone I), the neighboring area (zone II), and the nuclear interior (zone III) (Fig. 4D). While random distribution of telomeres would result in a uniform localization between all three zones, Taz1-GFP is found predominantly in zone I (80%) but rarely in zone II (15%) or zone III (6%) in wild-type cells (Fig. 4E). The absence of Dsh1 hardly affects the distribution of Taz1-GFP, whereas loss of Lem2 causes a more frequent delocalization, in agreement with a recent report (Gonzalez et al. 2012). Intriguingly, cells lacking both Dsh1 and Lem2 display an exacerbated phenotype (27% vs. 16% in lem2Δ for zone III; P < 0.05), resulting in a nearly random distribution of telomeres (Fig. 4E). This finding demonstrates that Lem2 and Dsh1 cooperate in not only telomere silencing but also anchoring.
Lem2 binds to chromatin via its LEM-containing N-terminal domain
The striking correlation between heterochromatin silencing and localization prompted us to study whether Lem2 mediates these functions through peripheral tethering of heterochromatin. To investigate the functions of the different Lem2 domains (see above; Fig. 5A), we generated membrane-bound versions lacking one of the nucleoplasmic domains (Lem2ΔN, without LEM domain; Lem2ΔC, without MSC domain) as well as fragments comprising solely the soluble parts (N terminus and C terminus) (Fig. 5A). These fragments are expressed from a plasmid under conditions that complement the lem2Δ phenotype for full-length Lem2 (Supplemental Fig. S1B). Using C-terminal GFP fusions and live-cell imaging, we first verified the correct expression of the fragments. The fragments that contain the transmembrane domains localize predominantly to the nuclear envelope, whereas those without transmembrane domains are mainly nucleoplasmic (Fig. 5B). Furthermore, we confirmed that the C-terminal GFP tag does not interfere with the function of Lem2 in silencing (Supplemental Fig. S8A). We then performed chromatin immunoprecipitation (ChIP) with full-length Lem2-GFP and probed centromere 1 as well as the subtelomeric regions of tel1L and tel2L. In particular, we found that Lem2-GFP is enriched over 10 kb at centromere 1, suggesting that it directly interacts with chromatin (Fig. 5C; Supplemental Fig. S8B). The distribution of Lem2-GFP is strictly confined to the central core cnt1 domain (involved in kinetochore attachment) but is excluded from the pericentromeres (associated with H3K9me). This chromatin enrichment is also seen for the fragment that lacks the C terminus (Lem2ΔC-GFP) but not for the fragments that lack the N terminus (Lem2ΔN-GFP) or contain only the soluble N-terminal or C-terminal domains of Lem2 (Fig. 5C; Supplemental Fig. S8C). Furthermore, none of the constructs display a significant enrichment within the subtelomeric regions. Together, these findings indicate that Lem2 interacts with centromeric chromatin and that the N-terminal part that contains the conserved LEM domain is required and sufficient for binding.
Figure 5.

Lem2 binds to centromeres through its N-terminal domain. (A) Scheme illustrating the topology of Lem2 and its individual domains. Shown below are the truncated versions of Lem2 (GFP-tagged) used in this study. (NE) Nuclear envelope; (TMD) transmembrane domain. (B) Live-cell imaging of the Lem2-GFP fragments. (Green) Lem2-GFP fragments; (red) Sad1-mCherry; (control) empty vector. Sad1-mCherry was used as an SPB marker to indicate the relative positions of the Lem2-GFP fragments. An individual image of one of the z-slices is shown (for some cells, the SPB is out of plane and therefore not visible). (C) ChIP-qPCR experiments showing the binding profiles of Lem2-GFP fragments. The left Y-axes represent Lem2-GFP binding relative to act1+, and the right Y-axes represent H3K9me2 levels relative to the maximum of each domain in wild-type cells (from Fig. 3C,D). Data are shown as mean ± SEM from three independent experiments.
Centromere localization and telomere anchoring are mediated by different Lem2 domains
The specific binding of Lem2 to centromeric chromatin via its N terminus led us to test whether this part is also crucial for tethering centromeres to the SPB. We therefore performed complementation studies with full-length Lem2 or N-terminally and C-terminally truncated versions that are expressed in csi1Δ lem2Δ cells together with Mis6-GFP and Sad1-mCherry. Expression of full-length Lem2 complements the colocalization defect of Mis6-GFP to the level of the csi1Δ single mutant (Fig. 6A). Remarkably, expression of Lem2ΔC but not Lem2ΔN results in a similar rescue of this phenotype, indicating that the N-terminal domain is indeed essential for centromere attachment to the SPB. We also tested the requirement for centromere clustering. In contrast to full-length Lem2, neither of the truncated versions is able to complement the phenotype (Fig. 6B), implying that both domains contribute to centromere clustering.
Figure 6.

Lem2 mediates heterochromatin positioning and silencing through its different domains. (A) Complementation of the centromere SPB colocalization defect in csi1Δ lem2Δ cells expressing different fragments of Lem2. (e/v) Empty vector. MSC refers to the C-terminal nucleoplasmic domain. Shown are pooled data from two independent experiments. (B) Complementation of centromere clustering defects in csi1Δ lem2Δ cells. Shown are pooled data from two independent experiments. (C) Complementation of the Taz1-GFP localization defect in lem2Δ cells. One representative experiment is shown. In A–C, asterisks denote P < 0.01 (**) and P < 0.0001 (****) (χ2 test). Quantitative analysis as described in Figure 4. Note that for the complementation experiments, cells were grown in EMM medium to maintain the expression plasmids, which partially suppresses the clustering and delocalization defects. (D) Complementation of the silencing defect in lem2Δ cells by silencing reporter assays on 5-FOA. Fivefold serial dilutions of indicated strains grown on EMM-Leu plates with (uninduced) or without thiamine (to induce expression of Lem2 fragments). (E) Complementation of the silencing defect in lem2Δ dsh1Δ cells by RT-qPCR. Asterisks denote P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****) from two-tailed Student's t-test analysis of three independent experiments.
We further studied the requirement for telomere recruitment and performed an analogous rescue experiment with lem2Δ cells coexpressing Taz1-GFP and mCherry-Cut11. Unexpectedly, we found that telomere anchoring does not depend on the N-terminal part but instead requires the C-terminal MSC domain (Fig. 6C). Thus, while the N-terminal part is necessary for correct positioning and binding to centromeric chromatin, the C-terminal part is required and sufficient to mediate the proper localization of telomeres.
Lem2 controls heterochromatin silencing through its C-terminal MSC domain
We next investigated which domain of Lem2 is required for transcriptional silencing. Using the pericentromeric imr1L::ura4+ reporter silencing assay, we found that, analogous to telomere anchoring, expression of Lem2ΔN still fully complements the mutant phenotype of lem2Δ on 5-FOA (Fig. 6D). Conversely, no complementation was seen for fragments lacking the MSC domain or the transmembrane domains. These findings imply that the MSC domain is necessary and sufficient to promote Lem2's role in heterochromatic silencing but only when present at the nuclear envelope.
To confirm the complementation of the silencing defect in a more quantitative manner, we examined the levels of heterochromatic transcripts by RT-qPCR in the dsh1Δ lem2Δ double mutant, which is more suitable for quantitative suppression analysis due to the exacerbated phenotype (Fig. 3A). In agreement with the silencing reporter assay, we found that expression of full-length Lem2 and Lem2ΔN represses transcription of the ura4+ reporter and various endogenous heterochromatic loci, whereas no complementation was observed for Lem2ΔC that lacks the MSC domain and for the soluble fragments (Fig. 6E). It is noteworthy that, although the expression level of Lem2ΔC is lower compared with Lem2ΔN, the Lem2ΔC fragment is expressed at the same level as full-length Lem2 (Supplemental Fig. S9A,B). Furthermore, Lem2ΔC and full-length Lem2 localize to the nuclear envelope and associate with chromatin at similar levels (Fig. 5B,C). This excludes the possibility that the expression level of Lem2ΔC is limiting for suppression of the lem2Δ silencing defect. Based on these findings, we conclude that silencing by Lem2 is exclusively mediated by the MSC domain.
Silencing by Lem2 involves the Snf2/HDAC repressor complex SHREC
The MSC domain of human MAN1 binds to DNA in vitro through a conserved stretch of positively charged residues (Caputo et al. 2006). In fission yeast, this positively charged motif is not present in Lem2 (Supplemental Fig. S8D), and binding to chromatin in vivo does not require its MSC domain (Fig. 5C). Therefore, we investigated whether silencing by the MSC domain might alternatively involve the recruitment of factors to heterochromatin. Prominent candidates are HDACs that have been reported to be recruited to the periphery by mammalian LAPs (Amendola and van Steensel 2014). Using a candidate approach, we focused on the Snf2/HDAC repressor complex SHREC that contains the class II HDAC Clr3 (Sugiyama et al. 2007). SHREC acts antagonistically to the anti-silencing protein Epe1 and competes with it for binding to HP1, resulting in an increased chromatin recruitment of Epe1 when binding of SHREC is impaired (Fig. 7A; Zofall and Grewal 2006; Shimada et al. 2009). Using previously described epitope-tagged versions of Clr1 (a subunit of SHREC) and Epe1 (Braun et al. 2011), we performed ChIP to assess whether their association with heterochromatin is affected by Lem2. Remarkably, we found that the ratio between Clr1 and Epe1 is profoundly perturbed in lem2Δ cells, resulting in a nearly 50% decrease of Clr1-Flag and a concomitant 15%–25% increase of Epe1-Flag at the cen-dg repeats and the tlh1/2+ loci (Fig. 7B). These data suggest that Lem2 promotes the association of SHREC with HP1, which in turn prevents the binding of Epe1.
Figure 7.
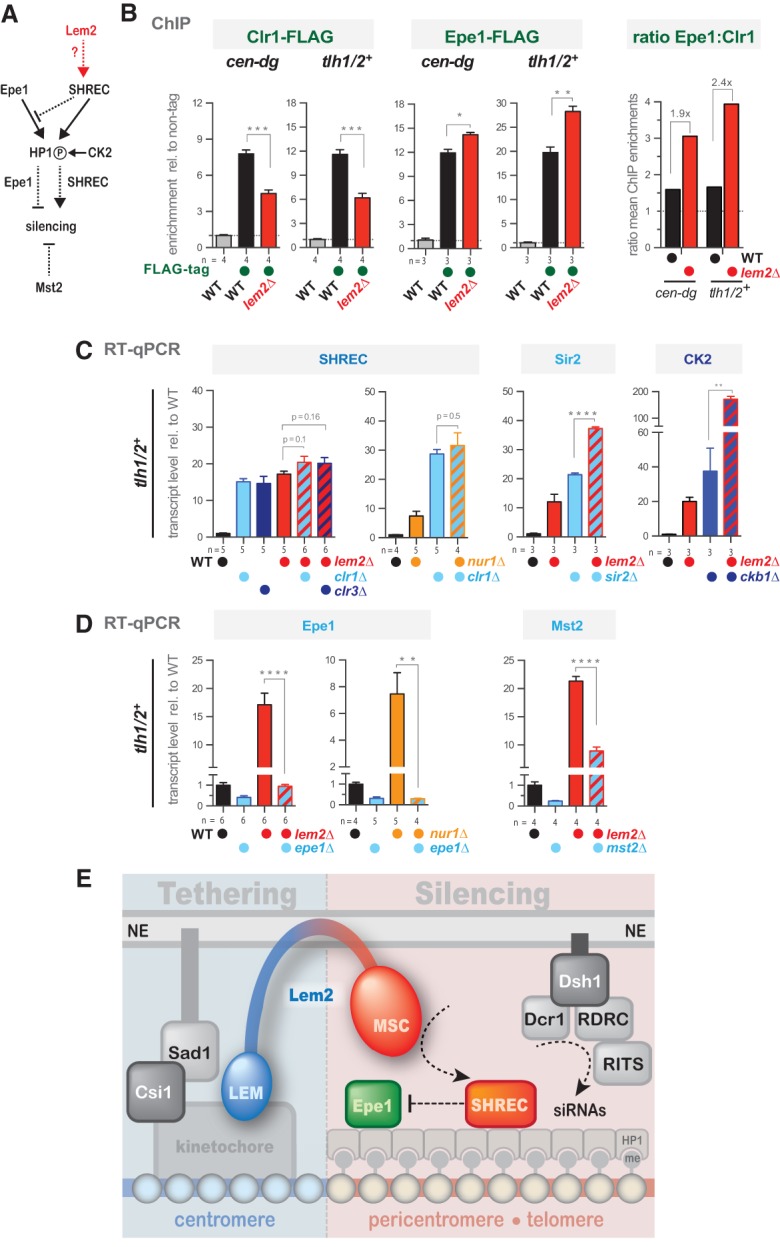
Lem2 contributes to telomeric silencing by acting through the SHREC pathway and opposing the anti-silencing function of Epe1. (A) Scheme illustrating the functional relationship between Epe1, SHREC, CK2, and Mst2. Arrows with solid and dotted lines indicate direct and functional interactions, respectively. The circled “P” indicates phosphorylation of HP1 by CK2. (B) ChIP-qPCR analysis of Clr1-Flag (left) and Epe1-Flag (middle) at the cen-dg and tlh1/2+ loci in wild-type and lem2Δ cells. Shown are enrichments relative to the untagged control. (Right) Ratio of Epe1:Clr1 based on the mean enrichments of Epe1-Flag and Clr1-Flag. (C,D) RT-qPCR analysis of tlh1/2+ mRNA in the indicated mutants. Shown are transcript levels relative to wild type after normalization to act1+. Data are represented as mean ± SEM from n independent experiments; asterisks denote P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****) from two-tailed Student t-test analysis. (E) Model of Lem2's role in centromere tethering and heterochromatin silencing.
If Lem2's role in SHREC binding to heterochromatin is critical for its function in silencing, then both factors should act epistatically. We focused first on telomere silencing, where Lem2 plays a predominant role. Indeed, we found that single mutants of lem2+ and clr1+ display transcript levels of tlh1/2+ nearly equal to that of the double mutant (Fig. 7C). This epistatic interaction is also seen for Clr3, the catalytic HDAC subunit of SHREC, but not for the class III HDAC Sir2 (Fig. 7C). Strikingly, deleting epe1+ completely suppresses the telomeric silencing defect of lem2Δ (Fig. 7D). Partial suppression of the telomeric silencing defect is also seen in the absence of the MYST histone acetyltransferase Mst2 (Fig. 7D), suggesting that it also counteracts the functions of Lem2 and SHREC. We observed, in an analogous manner, an epistatic behavior with clr1Δ and a suppressive phenotype with epe1Δ for cells lacking Nur1, the putative partner of Lem2 (Fig. 7C,D). The bypass of the requirements for Lem2 and Nur1 in silencing when Epe1 is absent resembles the suppression of heterochromatin defects in clr3Δ epe1Δ cells (Zofall and Grewal 2006). However, while efficient binding of SHREC requires the phosphorylation of HP1 proteins by CK2 (Shimada et al. 2009), we did not observe an epistatic interaction between lem2Δ and ckb1Δ (Fig. 7C), implying that Lem2 is not involved in the CK2-dependent recruitment of SHREC. Collectively, our findings demonstrate that Lem2 acts with SHREC in the same pathway at telomeres and preserves a correct balance between SHREC and Epe1.
At the pericentromeres, we found a more complex situation: While clr1Δ lem2Δ (and clr1Δ nur1Δ) cells behave epistatically, the clr3Δ lem2Δ mutant displays instead a synthetic increase of the cen-dg transcripts (Supplemental Fig. S10A). These findings are consistent with the results from our SGA screen for pericentromeric silencing, in which we also observed synthetic defects for lem2Δ and clr3Δ (and other members of the SHREC complex) but not clr1Δ (Fig. 2B). This suggests that Clr3 can act independently of Lem2 and that possibly different SHREC subcomplexes may exist, depending on the chromatin context. Furthermore, we found that the pericentromeric silencing defect of lem2Δ is not suppressed by deleting epe1+ or mst2+ (Supplemental Fig. S10B). Nonetheless, deletion of epe1+ itself causes a silencing defect at pericentromeres (Braun et al. 2011) and interferes with the robust generation of siRNAs (Trewick et al. 2007), which likely explains the lack of suppression. Thus, these nonlinear pathways make it more challenging to assign Lem2 to discrete functions, and further work is needed to dissect the silencing mechanisms at this heterochromatin domain.
Discussion
Perinuclear chromatin physically interacts with a variety of proteins at the nuclear envelope, but its relevance for gene expression remains poorly understood. Here, we demonstrate that the LEM domain protein Lem2 mediates both heterochromatin silencing and localization in S. pombe by cooperating with redundant pathways. Multiple deficiencies in these pathways cause synergistic silencing defects that correlate with aggravated delocalization of centromeres and telomeres, suggesting a link between repression and nuclear positioning. Furthermore, we show that Lem2 interacts with centromeric chromatin via its N terminus that contains the LEM domain. Since LEM proteins have been broadly thought to be involved in heterochromatin tethering (Brachner and Foisner 2011; Barton et al. 2015), a plausible model would be that silencing by Lem2 is mediated through tethering these chromatin domains to the nuclear periphery via its LEM domain. However, we provide several lines of experimental evidence that argue against such a simple mechanism. The key to solving this question was to separate the structural domains of Lem2 and test their functional requirements in vivo. This unveiled a far more complex role of Lem2, which possesses multiple discrete functions that can be attributed to its structural domains. Here we discuss the implications of these functions for chromatin localization and silencing, particularly with respect to the different heterochromatin domains.
Lem2 mediates distinct tethering and silencing functions through its LEM and MSC domains
Lem2 binds specifically to the central centromeric region, and its N-terminal part containing the LEM domains is sufficient for this association (Fig. 5C). This chromatin region is involved in kinetochore formation, and we consistently found that the Lem2 N terminus is also vital for the colocalization of centromeres with the SPB (Fig. 6A). Hence, we propose that the LEM domain of Lem2 mediates chromatin binding and, at least for centromeric chromatin, also tethering to the periphery. Although BAF homologs have not been identified in yeast, Lem2 may bind via its LEM domain to an analogous chromatin-bound protein or directly to DNA.
In stark contrast, the flanking pericentromeric regions (which contain H3K9me2) and subtelomeres are excluded from the binding profile of Lem2; this was not anticipated given its role in silencing and telomere anchoring. Furthermore, none of the repressed regions that we tested require the LEM domain for silencing (Fig. 6E). Instead, we found that the MSC domain is necessary and sufficient for silencing, which evidently is uncoupled from LEM-mediated tethering (see the model in Fig. 7E). Nonetheless, the function of the MSC domain is strictly dependent on its presence at the nuclear membrane, reinforcing the notion that the nuclear periphery is crucial for silencing. Since the MSC domain is apparently not involved in chromatin binding, we suspect that it promotes the perinuclear recruitment of factors that are critical for silencing. Such a scenario would be reminiscent of the situation described in budding yeast and worms: In S. cerevisiae, the perinuclear attachment of telomeres mediates the clustering and local sequestration of SIR proteins, which facilitates the establishment of silent chromatin (Taddei et al. 2009), whereas in C. elegans, the peripheral association of the methyltransferase SET-25 ensures the establishment of H3K9me3, which is required for full repression of perinuclear chromatin (Towbin et al. 2012).
Although tethering has been mechanistically separated from silencing (Taddei et al. 2004; Gonzalez-Sandoval et al. 2015), both take place at the periphery and may still reinforce each other. Indeed, the signals exploited by the recruitment mechanisms are often epigenetic marks that are established during heterochromatin formation (deacetylated histones in budding yeast and H3K9me in worms) (Towbin et al. 2013). However, centromere recruitment by the LEM domain of Lem2 differs from those tethering mechanisms, as it occurs independently of heterochromatin: It does not require either silencing by the MSC domain and RNAi/Dsh1 (Fig. 6; Supplemental Fig. S7B) or the presence of H3K9me, which is mostly absent in the central centromeric region (Fig. 3C). These findings are consistent with the previous observation that loss of H3K9me or the RNAi pathway does not result in a detectable dissociation of centromeres and telomeres from the periphery (Ekwall et al. 1996; Hall et al. 2003). However, it is noteworthy that the penetrance of centromere localization defects in S. pombe is <50% even in the lem2Δ csi1Δ double mutant (Fig. 4B), implying that additional tethering pathways exist. Interestingly, the RITS complex associates with chromatin through binding to H3K9me via Chp1, whereas it is recruited to the nuclear envelope via Dsh1. Thus, it is conceivable that this physical association contributes to centromere recruitment, although such a redundant function would be masked by Lem2 and Csi1. In conclusion, while other tethering mechanisms in S. pombe could be functionally coupled to heterochromatin, the LEM-mediated centromere recruitment and the MSC-dependent silencing are independent mechanisms, although they are mediated by the same protein. The only exception where we found that both functions functionally cooperate is centromere clustering, which requires both domains (Fig. 6B).
Telomere recruitment differs substantially from centromere localization, as it does not depend on the LEM domain and cannot be separated from silencing, since both functions require the MSC domain. Other pathways that cooperate with Lem2 in telomere recruitment—i.e., the RNAi machinery (Fig. 4E) and the Taz1–Rap1–Bqt3/Bqt4 anchoring pathway (Chikashige et al. 2009)—are also linked to telomere silencing. We speculate that anchoring and silencing by Lem2 may involve the same mechanism. Since Lem2 promotes the binding of SHREC to heterochromatin, its components (e.g., Clr3) could also be candidates for mediating telomere anchoring either directly or through deacetylation of histones or other factors. Interestingly, Clr3 affects the expression and peripheral localization of two clusters of stress-induced genes, one of which is in close proximity to telomeres (Alfredsson-Timmins et al. 2009). Moreover, the SIR complex plays a dual role in histone deacetylation and telomere anchoring in budding yeast (Taddei et al. 2004). Together, these examples suggest that one strategy to couple gene repression and telomere anchoring might be the recruitment of multifunctional HDAC complexes to the nuclear periphery.
Redundant and competitive silencing pathways
Loss of Lem2 alone modestly affects transcriptional silencing of all major heterochromatin domains that are marked with H3K9me; however, it also contributes to the repression of the centromeric cnt1 domain and several LTRs (Fig. 1). These loci are mostly devoid of H3K9me2, and we consistently found that silencing at these regions is more dependent on Lem2 than on Clr4. Hence, silencing by Lem2 at the major heterochromatic regions appears to be masked by redundant pathways (see below), while its impact is more evident where those pathways are less active or inactive (e.g., at LTRs). Collectively, these data show that Lem2 is a global regulator of heterochromatin silencing in S. pombe that can act independently of H3K9me2.
Homologs of Lem2 in S. cerevisiae and C. elegans have also been linked to gene repression, although a global role in heterochromatin silencing has not yet been described (Grund et al. 2008; Ikegami et al. 2010; Chan et al. 2011; Mattout et al. 2011). This role might have been missed due to further redundancy with other factors. Using a highly sensitive SGA approach, we identified a large number of mutants with synthetic silencing defects in combination with lem2Δ (Fig. 2). Various genes that we found encode proteins that are associated with the nuclear envelope/ER membrane or the cytoskeleton (e.g., Csi1, Lnp1, Alp14, and Mto1). How they contribute to heterochromatin silencing is not yet understood. Although several of them localize to the SPB (i.e., in close vicinity to the centromere), they also affect other heterochromatin domains (e.g., telomeres). This may suggest that these mutants also cause secondary silencing defects (for example, through an abnormal morphology of the nuclear envelope), which then could affect the activity or stability of other INM proteins involved in chromatin binding.
Besides these membrane/cytoskeleton factors, we identified several known factors with well-characterized functions in heterochromatin silencing; for example, members of the RNAi machinery. Using precise quantitative approaches, we determined how Lem2, the RNAi factor Dsh1, and the telomere-associated protein Taz1 cooperate in pericentromeric and telomeric silencing (Supplemental Fig. S5E). At pericentromeres, Lem2 and Dsh1 are responsible for the maximal repression seen for H3K9me-dependent silencing even though the RNAi pathway prevails. In contrast, full repression of telomeres requires at least one additional pathway, Taz1, yet here we found that Lem2 plays a predominant role among the redundant pathways. Remarkably, inhibiting the telomere-specific (Taz1) or mating type-specific (Atf1) silencing pathway restores silencing in a reciprocal manner at pericentromeres. This observation is in agreement with the previous notion that different heterochromatin domains compete for a limited pool of soluble silencing factors (HP1 in S. pombe and SIR proteins in S. cerevisiae) (Taddei et al. 2009; Tadeo et al. 2013). These results unveil a network of partially redundant pathways that cooperate in heterochromatin silencing. Consequently, integrating multiple pathways and constraining them at the nuclear periphery could be a cellular strategy to gain specificity and lower the risk of initiating heterochromatin at ectopic sites. In addition, this network of distinct silencing pathways could provide more complexity and help to regulate silencing in response to environmental cues.
Mechanism of Lem2-mediated silencing
While Lem2 promotes the association of SHREC with heterochromatin, its loss causes increased binding of the JmjC protein Epe1. Notably, Lem2 and SHREC share the same silencing pathway at telomeres, and their necessity can be bypassed by deleting Epe1 (Fig. 7; Zofall and Grewal 2006). We therefore conclude that one main function of Lem2 is to coordinate the balance between SHREC and the anti-silencing factor Epe1 (Fig. 7E). In this regard, Lem2 resembles CK2 in that it promotes the recruitment of SHREC through the phosphorylation of HP1 proteins (Shimada et al. 2009). However, we found that both pathways work independently of each other, implying that Lem2 affects a different step in the recruitment of SHREC to heterochromatin. In fact, we did not detect a physical interaction between Lem2 and Clr3 by coimmunoprecipitation experiments (data not shown), suggesting that Lem2 contributes indirectly to SHREC binding. However, we cannot exclude the possibility that Lem2 and SHREC interact transiently.
At centromeres, the relationship between Lem2 and SHREC is more complex, as mutants of the individual subunits of SHREC (i.e., clr1Δ and clr3Δ) display disparate genetic interactions with lem2Δ (Supplemental Fig. S10). Moreover, Epe1 appears to have a dual role, as it antagonizes heterochromatin (Braun et al. 2011) but also promotes the generation of siRNAs (Trewick et al. 2007), explaining why the necessity of Lem2 for the repression of pericentromeric transcripts cannot be simply bypassed by deleting Epe1. Thus, this complexity of intertwined pathways at pericentromeres makes it more difficult to assign Lem2 a discrete function. Nonetheless, loss of Lem2 causes a decrease of pericentromeric H3K9me2, particularly when combined with dsh1Δ (Fig. 3B), suggesting that Lem2 influences the establishment or maintenance of this repressive mark. Epe1 shows homology with histone demethylases and has been reported to affect the stability of H3K9me at ectopic heterochromatic domains (Audergon et al. 2015; Ragunathan et al. 2015). Thus, it may be conceivable that Lem2 controls H3K9me and silencing at pericentromeres primarily through regulating Epe1. Finally, it is noteworthy that Lem2 also contributes to the repression of chromatin regions that lack H3K9me (and hence HP1/Epe1). Consequently, its role in silencing is likely not limited to H3K9me and SHREC recruitment but may also affect other silencing pathways.
Concluding remarks
Proteins of the nuclear envelope have moved into the focus of understanding the functional relationship between silencing and the peripheral position of repressed chromatin, particularly with respect to development and cellular differentiation (Meister et al. 2010; Zuleger et al. 2013). The large number of metazoan INM proteins and the resulting complexity of these functional networks make it a challenging task to approach; thus, studying their molecular mechanisms in model organisms with less redundancy might be advantageous. Remarkably, while there are several LAP homologs in S. pombe, only Lem2 appears to be involved in heterochromatin silencing, which allowed us to dissect the functions of LEM domain-mediated tethering and MSC domain-mediated silencing. Given the conserved presence of the LEM and MSC domains in this group of INM proteins across evolution, this mechanism may represent a universal mode of heterochromatin repression.
Materials and methods
Yeast strains, plasmids, and techniques
Standard media and genome engineering methods were used. Strains used in this study are listed in Supplemental Table S1. For the cloning of expression constructs, full-length lem2+ or fragments of lem2+ were amplified by PCR and cloned into the BamHI–PacI site of pREP41x/81x or the SalI–BamHI site of pREP81x-EGFP-Ctag (Supplemental Table S2). Thiamine was used at a concentration of 5 µg/mL to repress expression from nmt1 promoters (pREP41x/81x). 5-FOA medium contained 1 g/L 5′-fluoroorotic acid. If not otherwise indicated, EMM medium was used for silencing reporter growth assays. For RT-qPCR experiments with Lem2 expressed from pREP41x/81x plasmids, cells were initially grown in EMM-Leu and then transferred to rich medium (YES) for four to five generations prior harvest.
SGA analyses
Large-scale crosses by SGA were performed as described previously (Verrier et al. 2015) using the Bioneer haploid deletion mutant library (version 3.0) and an imr1L(NcoI)::ura4 reporter strain that harbors an hphMX marker 2 kb upstream for selection. For analysis of the SGA data and epistasis calculation, see Supplemental Fig. S2.
Quantitative ChIP and RT-qPCR analyses
ChIP experiments were performed essentially as described (Braun et al. 2011) except that a Q800R1 sonicator (QSonica) and Dynabeads Protein G (Life Technologies) were used for chromatin shearing (30 min, 30-sec on/off cycles, 90% amplitude) and immunoprecipitation, respectively. To increase the ChIP sensitivity (Fig. 7B), cross-linking was performed by subsequent treatment of 10 mM dimethyl adipimidate and 1.5% formaldehyde as described (Braun et al. 2011). Lem2-GFP, Epe1-Flag, Clr1-Flag, and anti-H3K9me2, were immunoprecipitated with 2–5 µg of antibody (anti-GFP [provided by A. Ladurner, University of Munich], anti-Flag [Sigma, F3165], anti-H3K9me2 [Abcam, ab1220], and anti-H3K9m3 [Millipore, 07-442]) from lysates corresponding to 12–30 OD600 of cells. RT-qPCR experiments were carried out as previously described (Braun et al. 2011).
Immunoprecipitated DNA and cDNAs were quantified by qPCR using Fast SYBR Green Master mix (Life Technologies) and a 7500 Fast real-time PCR system (Applied Biosystems). Primers are listed in Supplemental Tables S3 and S4. For the calculation of mean values and standard deviation from independent experiments, act1+-normalized data sets were standardized against the mean of a sample pool of mutants from each experiment. These results were then shown as relative to the mean value of the wild type (which was set to 1).
Live-cell imaging
Imaging was performed on a Zeiss AxioObserver Z1 confocal spinning-disk microscope equipped with an Evolve 512 (Photometrics) EMM-CCD camera through a Zeiss Alpha Plan/Apo 100×/1.46 oil DIC M27 objective lens (for Fig. 4B and Supplemental Fig. S4A,B, a Zeiss Plan/Apo 63×/1.4 oil immersion objective lens was used). Optical section images were obtained at focus intervals of 0.4 µm. For distance measurements, ImageJ software was used.
Supplementary Material
Acknowledgments
We thank David Arnosti, Guillaume Filion, Stefan Jentsch, Andreas Ladurner, Corey Laverty, and Gyula Timinszky for critical reading of the manuscript; Susan Gasser for valuable comments; and Yasushi Hiraoka for communicating results prior publication. We are grateful to Shelley Sazer for strains, and the Ladurner laboratory for anti-GFP antibody. We also thank Sabine Fischer-Burkart for technical assistance, and Gyula Timinszky for training in live-cell imaging. This work was supported by grants awarded to S.B. from the European Union Network of Excellence EpiGeneSys (HEALTH-2010-257082), the German Research Foundation (BR 3511/3-1), and the Friedrich-Baur Stiftung. S.B. is an Associated Member of the Deutsche Forschungsgemeinschaft Collaborative Research Center 1064 and acknowledges infrastructure support. R.R.B. and S.B. designed the study. R.R.B. constructed plasmids and strains; performed the genetic screens, growth assays, and live-cell imaging; and analyzed with S.B. the SGA data. R.R.B., S.B., P.R.G, M.F., and Z.S. performed RT-qPCR and ChIP experiments. S.B. wrote the manuscript, and all authors contributed to editing.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.271288.115.
References
- Alfredsson-Timmins J, Kristell C, Henningson F, Lyckman S, Bjerling P. 2009. Reorganization of chromatin is an early response to nitrogen starvation in Schizosaccharomyces pombe. Chromosoma 118: 99–112. [DOI] [PubMed] [Google Scholar]
- Allshire RC, Ekwall K. 2015. Epigenetic regulation of chromatin states in Schizosaccharomyces pombe. Cold Spring Harb Perspect Biol 7: a018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sady B, Madhani HD, Narlikar GJ. 2013. Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Mol Cell 51: 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola M, van Steensel B. 2014. Mechanisms and dynamics of nuclear lamina-genome interactions. Curr Opin Cell Biol 28C: 61–68. [DOI] [PubMed] [Google Scholar]
- Audergon P, Catania S, Kagansky A, Tong P, Shukla M, Pidoux A, Allshire RC. 2015. Restricted epigenetic inheritance of H3K9 methylation. Science 348: 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton LJ, Soshnev AA, Geyer PK. 2015. Networking in the nucleus: a spotlight on LEM-domain proteins. Curr Opin Cell Biol 34: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachner A, Foisner R. 2011. Evolvement of LEM proteins as chromatin tethers at the nuclear periphery. Biochem Soc Trans 39: 1735–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, Garcia JF, Rowley M, Rougemaille M, Shankar S, Madhani HD. 2011. The Cul4–Ddb1(Cdt)² ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell 144: 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo S, Couprie J, Duband-Goulet I, Kondé E, Lin F, Braud S, Gondry M, Gilquin B, Worman HJ, Zinn-Justin S. 2006. The carboxyl-terminal nucleoplasmic region of MAN1 exhibits a DNA binding winged helix domain. J Biol Chem 281: 18208–18215. [DOI] [PubMed] [Google Scholar]
- Chan JNY, Poon BPK, Salvi J, Olsen JB, Emili A, Mekhail K. 2011. Perinuclear cohibin complexes maintain replicative life span via roles at distinct silent chromatin domains. Dev Cell 20: 867–879. [DOI] [PubMed] [Google Scholar]
- Chikashige Y, Yamane M, Okamasa K, Tsutsumi C, Kojidani T, Sato M, Haraguchi T, Hiraoka Y. 2009. Membrane proteins Bqt3 and -4 anchor telomeres to the nuclear envelope to ensure chromosomal bouquet formation. J Cell Biol 187: 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmerle J, Koch AJ, Holaska JM. 2012. The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J Biol Chem 287: 22080–22088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekwall K, Nimmo ER, Javerzat JP, Borgstrøm B, Egel R, Cranston G, Allshire R. 1996. Mutations in the fission yeast silencing factors clr4+ and rik1+ disrupt the localisation of the chromo domain protein Swi6p and impair centromere function. J Cell Sci 109: 2637–2648. [DOI] [PubMed] [Google Scholar]
- Ekwall K, Cranston G, Allshire RC. 1999. Fission yeast mutants that alleviate transcriptional silencing in centromeric flanking repeats and disrupt chromosome segregation. Genetics 153: 1153–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Y, Saito A, Sazer S. 2012. Fission yeast Lem2 and Man1 perform fundamental functions of the animal cell nuclear lamina. Nucleus 3: 60–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Sandoval A, Towbin BD, Kalck V, Cabianca DS, Gaidatzis D, Hauer MH, Geng L, Wang L, Yang T, Wang X, et al. 2015. Perinuclear anchoring of H3K9-methylated chromatin stabilizes induced cell fate in C. elegans embryos. Cell 163: 1333–1347. [DOI] [PubMed] [Google Scholar]
- Grewal SI. 2010. RNAi-dependent formation of heterochromatin and its diverse functions. Curr Opin Genet Dev 20: 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grund SE, Fischer T, Cabal GG, Antúnez O, Pérez-Ortín JE, Hurt E. 2008. The inner nuclear membrane protein Src1 associates with subtelomeric genes and alters their regulated gene expression. J Cell Biol 182: 897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudise S, Figueroa RA, Lindberg R, Larsson V, Hallberg E. 2011. Samp1 is functionally associated with the LINC complex and A-type lamina networks. J Cell Sci 124: 2077–2085. [DOI] [PubMed] [Google Scholar]
- Hall IM, Noma K, Grewal SIS. 2003. RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc Natl Acad Sci 100: 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger F, Taddei A, Neumann FR, Gasser SM. 2004. Methods for visualizing chromatin dynamics in living yeast. Methods Enzymol 375: 345–365. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Hizume K, Kimura H, Takeyasu K, Haraguchi T, Hiraoka Y. 2012. Lamin B receptor recognizes specific modifications of histone H4 in heterochromatin formation. J Biol Chem 287: 42654–42663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka Y, Maekawa H, Asakawa H, Chikashige Y, Kojidani T, Osakada H, Matsuda A, Haraguchi T. 2011. Inner nuclear membrane protein Ima1 is dispensable for intranuclear positioning of centromeres. Genes Cells 16: 1000–1011. [DOI] [PubMed] [Google Scholar]
- Hou H, Zhou Z, Wang Y, Wang J, Kallgren SP, Kurchuk T, Miller EA, Chang F, Jia S. 2012. Csi1 links centromeres to the nuclear envelope for centromere clustering. J Cell Biol 199: 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami K, Egelhofer TA, Strome S, Lieb JD. 2010. Caenorhabditis elegans chromosome arms are anchored to the nuclear membrane via discontinuous association with LEM-2. Genome Biol 11: R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Hayashi A, Nakayama J-I, Murakami Y. 2012. A novel RNAi protein, Dsh1, assembles RNAi machinery on chromatin to amplify heterochromatic siRNA. Genes Dev 26: 1811–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mans B, Anantharaman V, Aravind L, Koonin EV. 2004. Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex. Cell Cycle 3: 1625–1650. [DOI] [PubMed] [Google Scholar]
- Margalit A, Brachner A, Gotzmann J, Foisner R, Gruenbaum Y. 2007. Barrier-to-autointegration factor—a BAFfling little protein. Trends Cell Biol 17: 202–208. [DOI] [PubMed] [Google Scholar]
- Mattout A, Pike BL, Towbin BD, Bank EM, Gonzalez-Sandoval A, Stadler MB, Meister P, Gruenbaum Y, Gasser SM. 2011. An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Curr Biol 21: 1603–1614. [DOI] [PubMed] [Google Scholar]
- Meister P, Towbin BD, Pike BL, Ponti A, Gasser SM. 2010. The spatial dynamics of tissue-specific promoters during C. elegans development. Genes Dev 24: 766–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhail K, Seebacher J, Gygi SP, Moazed D. 2008. Role for perinuclear chromosome tethering in maintenance of genome stability. Nature 456: 667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleshko A, Mansfield KM, Burlingame CC, Andrake MD, Shah NR, Katz RA. 2013. The human protein PRR14 tethers heterochromatin to the nuclear lamina during interphase and mitotic exit. Cell Rep 5: 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Jih G, Moazed D. 2015. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348: 1258699–1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A, Dohke K, Sadaie M, Shinmyozu K, Nakayama J-I, Urano T, Murakami Y. 2009. Phosphorylation of Swi6/HP1 regulates transcriptional gene silencing at heterochromatin. Genes Dev 23: 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. 2013. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152: 584–598. [DOI] [PubMed] [Google Scholar]
- Somech R, Shaklai S, Geller O, Amariglio N, Simon AJ, Rechavi G, Gal-Yam EN. 2005. The nuclear-envelope protein and transcriptional repressor LAP2β interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J Cell Sci 118: 4017–4025. [DOI] [PubMed] [Google Scholar]
- Steglich B, Filion GJ, van Steensel B, Ekwall K. 2012. The inner nuclear membrane proteins Man1 and Ima1 link to two different types of chromatin at the nuclear periphery in S. pombe. Nucleus 3: 77–87. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Cam HP, Sugiyama R, Noma K-I, Zofall M, Kobayashi R, Grewal SIS. 2007. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 128: 491–504. [DOI] [PubMed] [Google Scholar]
- Taddei A, Gasser SM. 2012. Structure and function in the budding yeast nucleus. Genetics 192: 107–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Hediger F, Neumann FR, Bauer C, Gasser SM. 2004. Separation of silencing from perinuclear anchoring functions in yeast Ku80, Sir4 and Esc1 proteins. EMBO J 23: 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Van Houwe G, Nagai S, Erb I, van Nimwegen E, Gasser SM. 2009. The functional importance of telomere clustering: global changes in gene expression result from SIR factor dispersion. Genome Res 19: 611–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadeo X, Wang J, Kallgren SP, Liu J, Reddy BD, Qiao F, Jia S. 2013. Elimination of shelterin components bypasses RNAi for pericentric heterochromatin assembly. Genes Dev 27: 2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. 2012. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 150: 934–947. [DOI] [PubMed] [Google Scholar]
- Towbin BD, Gonzalez-Sandoval A, Gasser SM. 2013. Mechanisms of heterochromatin subnuclear localization. Trends Biochem Sci 38: 356–363. [DOI] [PubMed] [Google Scholar]
- Trewick SC, Minc E, Antonelli R, Urano T, Allshire RC. 2007. The JmjC domain protein Epe1 prevents unregulated assembly and disassembly of heterochromatin. EMBO J 26: 4670–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier L, Taglini F, Barrales RR, Webb S, Urano T, Braun S, Bayne EH. 2015. Global regulation of heterochromatin spreading by Leo1. Open Biol 5: 150045–150045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Grewal SIS. 2006. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol Cell 22: 681–692. [DOI] [PubMed] [Google Scholar]
- Zuleger N, Boyle S, Kelly DA, las Heras de JI, Lazou V, Korfali N, Batrakou DG, Randles KN, Morris GE, Harrison DJ, et al. 2013. Specific nuclear envelope transmembrane proteins can promote the location of chromosomes to and from the nuclear periphery. Genome Biol 14: R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.