

Kim et al. discovered that histone H3 N-terminal tail (H3NT) proteolysis is selectively targeted near transcription start sites of a small group of genes and that most H3NT-cleaved genes displayed significant expression changes during osteoclastogenesis. The principal H3NT protease of osteoclastogenesis is matrix metalloproteinase 9 (MMP-9).
Keywords: histone, chromatin, epigenetic, proteolysis, MMP-9, osteoclast
Abstract
Although limited proteolysis of the histone H3 N-terminal tail (H3NT) is frequently observed during mammalian differentiation, the specific genomic sites targeted for H3NT proteolysis and the functional significance of H3NT cleavage remain largely unknown. Here we report the first method to identify and examine H3NT-cleaved regions in mammals, called chromatin immunoprecipitation (ChIP) of acetylated chromatin (ChIPac). By applying ChIPac combined with deep sequencing (ChIPac-seq) to an established cell model of osteoclast differentiation, we discovered that H3NT proteolysis is selectively targeted near transcription start sites of a small group of genes and that most H3NT-cleaved genes displayed significant expression changes during osteoclastogenesis. We also discovered that the principal H3NT protease of osteoclastogenesis is matrix metalloproteinase 9 (MMP-9). In contrast to other known H3NT proteases, MMP-9 primarily cleaved H3K18-Q19 in vitro and in cells. Furthermore, our results support CBP/p300-mediated acetylation of H3K18 as a central regulator of MMP-9 H3NT protease activity both in vitro and at H3NT cleavage sites during osteoclastogenesis. Importantly, we found that abrogation of H3NT proteolysis impaired osteoclastogenic gene activation concomitant with defective osteoclast differentiation. Our collective results support the necessity of MMP-9-dependent H3NT proteolysis in regulating gene pathways required for proficient osteoclastogenesis.
The basic components that comprise the canonical nucleosome core particle are evolutionarily conserved in all eukaryotes. Within a living cell, however, there exists a remarkable degree of heterogeneity in nucleosome composition that influences chromatin structure and function (Luger et al. 2012). This is exemplified by the various covalent post-translational modifications (PTMs) of the N-terminal tails (NTs) of the core histone proteins (H3, H4, H2A, and H2B) that alter nucleosome composition to regulate fundamental DNA-templated programs such as transcription (Zentner and Henikoff 2013). For example, acetylation of the H3 N-terminal tail (H3NT) can directly facilitate transcription by destabilizing nucleosome structure, whereas methylation of the H3NT can indirectly regulate transcription by binding effector proteins that stimulate or repress transcription (Bannister et al. 2001; Shogren-Knaak et al. 2006). Recent landmark reports determined that differences in the “epigenomic signatures” of several H3NT PTMs are strongly correlated to the cell type-specific gene expression programs observed in >250 normal and diseased human tissues (Polak et al. 2015; Roadmap Epigenomics Consortium et al. 2015). These studies demonstrate that precise alterations of the epigenome are essential in the regulation of gene pathways necessary for the derivation of normal and aberrant cell types.
Altering epigenomic signatures by “erasing” H3NT PTMs can be achieved by several different enzymatic mechanisms. One mechanism is the selective removal of specific H3 PTMs by histone-modifying enzymes such as deacetylases and demethylases (Black et al. 2012; Seto and Yoshida 2014). A more extreme mechanism involves ATP-dependent deposition of a new H3, resulting in the removal of all pre-existing PTMs and associated interacting proteins (Narlikar et al. 2013). An alternative intermediate mechanism between the specific and complete erasure of H3 PTMs is proteolysis of the H3NT, which selectively removes pre-existing H3NT PTMs and associated interacting proteins without affecting the H3 core region (Azad and Tomar 2014; Dhaenens et al. 2015). While retention of the H3 core region preserves nucleosome structure, lack of the H3NT destabilizes intranucleosomal and internucleosomal interactions that may increase DNA accessibility and facilitate factor binding (Allan et al. 1982; Andresen et al. 2013; Nurse et al. 2013). Therefore, H3NT cleavage provides an efficient means to rapidly and drastically alter chromatin structure and function both directly and indirectly.
Although proteolysis of histones within chromatin was first reported >55 years ago, the mechanisms and biological functions of histone cleavage remain largely unknown (Phillips and Johns 1959). Proteolysis of the H3NT has been detected in various single and multicellular eukaryotes, indicating that H3NT cleavage is an evolutionarily conserved process that likely functions in epigenetic regulation (Allis et al. 1980; Bortvin and Winston 1996; Duncan et al. 2008; Pauli et al. 2010; Duarte et al. 2014; Vossaert et al. 2014). Consistent with this, H3NT cleavage is frequently observed during mammalian developmental programs, including embryonic stem cell (ESC) differentiation, mammary gland development, and myogenesis (Duncan et al. 2008; Asp et al. 2011; Khalkhali-Ellis et al. 2014; Vossaert et al. 2014). The recent identification of cathepsins L and D as the principal H3NT proteases during mouse ESC (mESC) differentiation and mammary gland development, respectively, suggests that precursor cells use different H3NT proteases in a differentiation-dependent context (Duncan et al. 2008; Khalkhali-Ellis et al. 2014). While these reports imply that targeted H3NT proteolysis at specific genomic regions facilitates differentiation, the lack of a method to identify H3NT-cleaved regions has precluded significant insights into the mechanistic functions of H3NT proteolysis.
In our recent study examining the regulatory mechanisms of osteoclast differentiation, we observed limited proteolysis of the H3NT in an established ex vivo mouse model of osteoclastogenesis (An et al. 2014). In this study, we demonstrate that matrix metalloproteinase 9 (MMP-9) is the principal H3NT protease of osteoclastogenesis. By developing the first method to map H3NT-cleaved regions in mammals, called chromatin immunoprecipitation (ChIP) of acetylated chromatin (ChIPac), we discovered the selective targeting of MMP-9-dependent H3NT proteolysis near transcription start sites (TSSs) of osteoclastogenic genes during differentiation. Consistent with H3NT cleavage-dependent gene activation reported in Saccharomyces cerevisiae, we found that H3NT proteolysis was strongly correlated with transcriptional activation (Santos-Rosa et al. 2009). Abrogation of MMP-9-dependent H3NT proteolysis resulted in impaired osteoclastogenic gene activation and defective osteoclast differentiation. Therefore, our collective results support a model in which MMP-9-dependent H3NT proteolysis at osteoclastogenic genes facilitates their activation necessary for proficient osteoclast differentiation.
Results
Proteolysis of the H3NT during osteoclastogenesis
Since the epigenetic mechanisms that regulate mammalian osteoclastogenesis are largely unknown, an established ex vivo cell model was used to examine possible histone PTM changes during differentiation. Primary osteoclast precursor (OCP) cells derived from adult mouse long bone were cultured with the osteoclastogenic factor RANKL to induce synchronous osteoclast differentiation (An et al. 2014). As shown in Figure 1A, nearly all 3-d OCP-induced cells were mononuclear preosteoclasts that fuse to form large multinuclear osteoclasts by day 5. Nuclei were isolated from OCP-induced cells at these time points, and chromatin was extracted. Western blot analysis revealed an unexpected differentiation-dependent fast-migrating H3 band in chromatin of preosteoclasts, with elevated levels detected in osteoclasts (Fig. 1B). Because an H3 C-terminal (H3CT) antibody was used in the Western blot analysis, the observed fast-migrating H3 band indicates specific proteolysis of the H3NT. Fast-migrating bands of other nucleosome core histones were not observed, demonstrating the selective, but limited, proteolysis of the H3NT during osteoclastogenesis (Fig. 1B).
Figure 1.
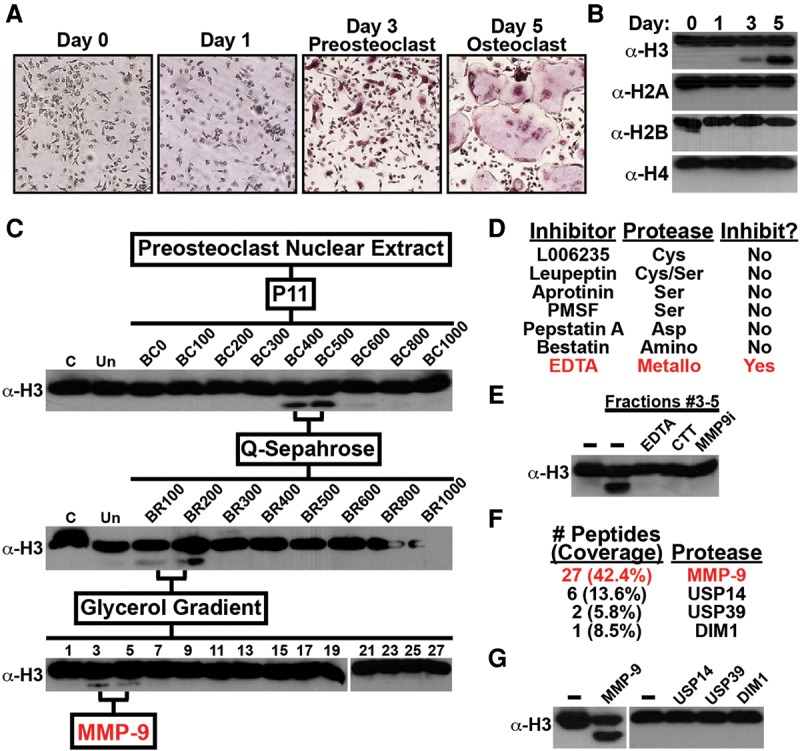
Identification of MMP-9 as a novel H3NT protease. (A) Primary mouse OCP cells were cultured with RANKL to induce osteoclastogenesis. OCP-induced cells were fixed, stained for TRAP (tartrate-resistant acid phosphatase), and photographed under a light microscope (10×) at the indicated days. TRAP-positive cells containing three or more nuclei represent osteoclasts. (B) Western blot analysis of chromatin extracted from OCP-induced cells using antibodies recognizing the C-terminal histone regions, as indicated. (C) Nuclear extracts from 3-d OCP-induced cells were fractionated on a P11 column by increased salt concentration, as indicated, and the in vitro H3NT cleavage assay was performed to identify H3NT active fractions by Western blot analysis using the H3CT antibody. The indicated H3NT active fractions were fractionated on a Q-Sepharose column followed by a 10%–40% glycerol gradient. (D) H3NT cleavage assays of purified fractions #3–5 treated with different protease family inhibitors as indicated. (E) H3NT cleavage assays of purified fractions #3–5 treated with metalloproteinase inhibitor EDTA or CTT or a selective MMP-9 inhibitor. (F) Proteomic analysis of known proteases identified in purified fractions #3–5. (G) H3NT cleavage assays using recombinant MMP-9, USP14, USP39, and DIM1. See also Supplemental Figure 1.
MMP-9 is the principal H3NT protease in preosteoclasts
To identify the protease responsible for H3NT cleavage observed in preosteoclasts, nuclear extracts from 3-d OCP-induced cells were fractionated by a series of chromatography steps (Fig. 1C). An in vitro H3NT cleavage assay was developed using a recombinant H3 (rH3) substrate and Western blot analysis with the H3CT antibody to track and isolate nuclear fractions containing H3NT protease activity. Initial fractionation by P11 chromatography revealed that H3NT protease activity was largely restricted to two sequential fractions, BC400 and BC500, suggesting that a single protease cleaves the H3NT in preosteoclasts. These fractions were combined and fractionated on a Q-Sepharose column where, again, H3NT protease activity was largely restricted to two sequential fractions, BR100 and BR200. The fractions were combined for glycerol gradient sedimentation, resulting in the final purification of fractions (#3–5) containing H3NT protease activity. These purified fractions were initially incubated with different protease inhibitors to identify the family of proteases responsible for H3NT cleavage. Various serine and cysteine protease inhibitors failed to inhibit H3NT protease activity, which was highly unexpected, as these families were reported to proteolyze the H3NT in yeast and mice, respectively (Fig. 1D; Duncan et al. 2008; Khalkhali-Ellis et al. 2014; Xue et al. 2014). Further screening demonstrated that only metalloproteinase inhibition could abrogate H3NT cleavage activity (Fig. 1E). Consistent with these results, proteomic analysis of the purified fractions revealed that MMP-9 was the predominant of the four known proteases identified (Fig. 1F). In vitro H3NT cleavage assays using these recombinant proteases demonstrated that only rMMP-9 possessed H3NT protease activity (Fig. 1G). The purified fractions and rMMP-9 displayed similar H3NT protease activities for octamer and nucleosome array substrates as well as for H3.1, H3.2, and H3.3 substrates (Supplemental Fig. 1A,B). Incubation of the purified fractions with a selective MMP-9 inhibitor abolished H3NT proteolysis, confirming that MMP-9 activity is required for H3NT cleavage in vitro (Fig. 1E). These findings identify MMP-9 as a novel H3NT protease and the principal H3NT protease in preosteoclasts.
MMP-9 activity is required for H3NT proteolysis during osteoclastogenesis
MMPs are a large diverse family of zinc-dependent endopeptidases that function to remodel the pericellular space via proteolysis of extracellular matrix proteins (Nagase et al. 2006). MMP-9 and MMP-2 comprise the gelatinase subfamily of MMPs and are differentially expressed in osteoclasts and osteoblasts, respectively. MMP-9 is synthesized as an inactive/latent 92-kDa proenzyme and subsequently converted to an 82-kDa active form by proteolysis of its inhibitory N-terminal prodomain. Since MMP-9 is a secretory protein, our results demonstrating nuclear MMP-9 activity were unexpected. To address this discrepancy, MMP-9 localization dynamics were examined during osteoclastogenesis. Western blot analysis demonstrated that MMP-9 was absent in control OCP cells, but both the proform and active form of MMP-9 were detected in the nuclear compartment of 1-d OCP-induced cells (Fig. 2A; Supplemental Fig. 2A). MMP-9 protein abundance was maximal by day 3 after induction and was sustained in 5-d OCP-induced cells. Immunofluorescence microscopy confirmed the progressive nuclear accumulation of MMP-9 in OCP-induced cells (Supplemental Fig. 2B). Gelatin zymography was performed using nuclear extracts isolated from OCP-induced cells to assess nuclear-specific MMP-9 gelatinase activity. Nuclear gelatinase activity mirrored the progressive nuclear accumulation of MMP-9 during osteoclastogenesis (Fig. 2B). These results indicate that maximal MMP-9 nuclear abundance and activity observed in 3-d OCP-induced cells directly correlate with H3NT proteolysis. To test the dependence of H3NT cleavage on MMP-9 during osteoclastogenesis, OCP cells were transduced with a control or MMP-9-specific shRNA to deplete MMP-9 prior to induction (Fig. 2C). MMP-9 depletion impeded nuclear gelatinase activity in OCP-induced cells concurrent with the significant and sustained impairment of H3NT proteolysis (Fig. 2B,D). The continuous impairment of H3NT cleavage was similarly observed in OCP-induced cells treated with a selective MMP-9 inhibitor (Fig. 2D). These collective results support the dependence of H3NT proteolysis on nuclear MMP-9 activity during osteoclastogenesis.
Figure 2.
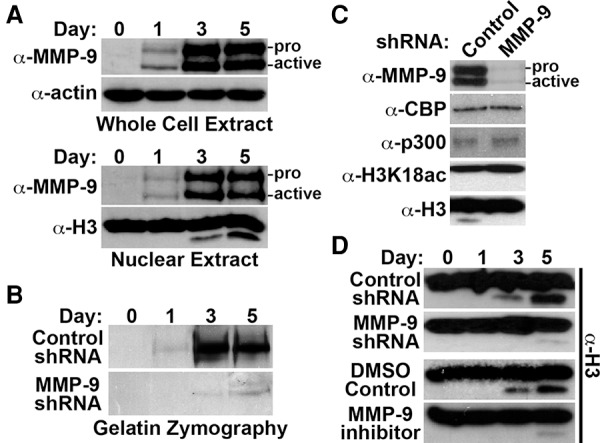
MMP-9-dependent H3NT proteolysis during osteoclastogenesis. (A) Western blot analysis for MMP-9 using whole-cell (top) or nuclear (bottom) lysates isolated from OCP-induced cells at the indicated days. Proform and active MMP-9 are indicated. (B) Gelatin zymography of nuclear lysates isolated from OCP-induced cells expressing a control (top) or MMP-9-specific (bottom) shRNA at the indicated days. (C) Western blot analysis of nuclear lysates from 3-d OCP-induced cells expressing a control (left) or MMP-9-specific (right) shRNA using the indicated antibodies. (D) OCP cells transduced with a control or MMP-9-specific shRNA (top) or treated with DMSO control or a selective MMP-9 inhibitor (bottom). H3NT cleavage was assessed at the indicated days after induction. See also Supplemental Figure 2.
H3K18 is the primary primary cleavage (P1) site of MMP-9
Previous reports demonstrated that H3A21 is the P1 site for cysteine and serine proteases; however, the H3NT P1 site of the MMP-9 metalloproteinase was unknown (Duncan et al. 2008; Santos-Rosa et al. 2009). In silico analysis of histone H3 identified K18 as the only potential P1 site on the H3NT, which was also predicted to be cleaved by MMP-9 (Supplemental Fig. 3A; Song et al. 2012). To test these predictions, liquid chromatography-tandem mass spectrometry (LC-MS/MS) of the gel-excised rH3-cleaved product generated by rMMP-9 was performed (Fig. 3A). Peptide fragments containing H3 residues prior to Q19 were not identified in the proteomic analysis, supporting H3K18 as the major P1 site of MMP-9. This result suggested that the sequence flanking H3K18 is a putative MMP-9 consensus site. To test this in vitro, rH3-containing mutated residues predicted to be necessary for MMP-9 activity were used as substrates in the H3NT cleavage assay (Fig. 3B; Supplemental Fig. 3B). Mutation of rH3 at (P1, K18R; P1′, Q19G) or flanking (P3, P16G; P3′, A21F) the cleavage site ablated H3NT proteolysis by rMMP-9, confirming that the H3 amino acid 16–21 sequence is an MMP-9 consensus site in vitro (Fig. 3C). These results suggested that similar H3 mutations would ablate H3NT proteolysis during osteoclastogenesis. To test this, C-terminal Flag tag fusions of wild-type H3 or the H3 mutants were transduced in OCP cells prior to induction. Western blot analysis of chromatin extracts confirmed proteolysis of wild-type H3-Flag in OCP-induced cells (Fig. 3D). Although mutant H3-Flag proteins were also readily detected in chromatin extracts, each mutation of the MMP-9 consensus sequence abrogated H3NT proteolysis in OCP-induced cells. These findings support MMP-9 as the primary protease that directly cleaves H3K18-Q19 during osteoclastogenesis.
Figure 3.

H3K18 is the principal site of MMP-9 proteolysis. (A) Peptide sequences identified by LC-MS/MS of the gel-excised rH3 product (H3Δ19) generated by rMMP-9. (B) Putative MMP-9 cleavage site identified in the H3NT (amino acids 16–21) in silico. Amino acids that are more (red) or less (blue) conserved in the canonical MMP-9 consensus sequence are indicated. (C) H3NT cleavage assays using rMMP-9 and rH3 wild-type or mutant substrates as indicated. (D) OCP cells were transduced with wild-type or mutant H3-Flag. Chromatin was isolated on the indicated days after induction for Western blot analysis using a Flag antibody to specifically detect proteolysis of H3-Flag. See also Supplemental Figure 3.
Acetylation of H3K18 augments MMP-9 activity
Analysis of the canonical MMP-9 consensus sequence indicated that MMP-9 preferentially cleaves uncharged residues over charged residues at the P1 site (Supplemental Fig. 3B). Based on this, we hypothesized that neutralizing the H3K18 charge by acetylation would augment MMP-9 activity. As predicted, kinetic analysis of MMP-9 activity using H3 peptide substrates confirmed that K18ac selectively amplified rMMP-9 activity, resulting in a threefold increase of cleaved H3 compared with unmodified H3 or H3K27me1 peptides (Fig. 4A). Consistent with these results, H3NT cleavage assays using rH3 acetyl-lysine analogs as substrates demonstrated that the specific acetylation of K18 robustly increased H3NT proteolysis by rMMP-9 (Fig. 4B; Shogren-Knaak and Peterson 2004). MMP-9 activity with rH3 methyl-lysine analogs at K4, K9, K27, or K36 was nearly identical to rH3 control in all cases (Supplemental Fig. 4A). These findings suggested that the acetylation of H3K18 facilitates MMP-9 activity in chromatin. To test this, nucleosome arrays containing unmodified rH3, the rH3 acetyl-lysine analogs, or modified native histones purified from HeLa cells were generated in vitro and used as substrates in the H3NT cleavage assay (Kim et al. 2013a). Remarkably, unmodified nucleosome array substrates were completely resistant to rMMP-9 protease activity, in contrast to unmodified rH3 and octamer substrates (Fig. 4B,C; Supplemental Fig. 1). Conversely, rMMP-9 displayed robust activity for modified native nucleosome arrays, supporting the dependence of MMP-9 H3NT protease activity on specific histone PTMs in chromatin (Fig. 4C). Consistent with this, H3NT cleavage assays confirmed that the specific acetylation of H3K18 was both necessary and sufficient for H3NT cleavage by rMMP-9 in a nucleosome array context (Fig. 4B). These results suggested that acetylation of H3K18 was required for MMP-9-dependent H3NT cleavage during osteoclastogenesis. To test this, OCP cells were transduced with a control shRNA or an shRNA to deplete the CBP/p300 acetyltransferases, which are responsible for the majority of H3K18ac, prior to induction (Henry et al. 2013). Diminishment of H3K18ac in CBP/p300-depleted OCP-induced cells resulted in the sustained impairment of H3NT proteolysis without perturbing the nuclear abundance of active MMP-9 (Fig. 4D,E). Identical results were obtained from OCP-induced cells treated with a selective CBP/p300 inhibitor (Fig. 4E). Depletion of the known H3K18 acetyltransferase GCN5, Tip60, or MOZ did not impair H3NT proteolysis during osteoclastogenesis, in contrast to CBP/p300 depletion (Supplemental Fig. 4B,C). These collective results support the regulation of MMP-9-dependent H3NT cleavage during osteoclastogenesis by the CBP/p300-mediated acetylation of H3K18.
Figure 4.

Acetylation status of H3K18 regulates MMP-9 activity. (A) The indicated H3 peptides (amino acids 10–35) were incubated with increasing concentrations of rMMP-9 (X-axis), and proteolysis was measured fluorometrically by reaction of free amino groups with fluorescamine (Y-axis). The Vmax and Km values were obtained by nonlinear regression fit. (B) H3NT cleavage assays using rMMP-9 and the indicated rH3 acetyl-lysine analog substrates either alone (top) or as reconstituted nucleosome arrays (bottom). (C) H3NT cleavage assays using rMMP-9 and recombinant (top) or native (bottom) nucleosome array substrates. (D) Western blot analysis of nuclear lysates from 3-d OCP-induced cells expressing a control or CBP/p300-specific shRNA using the indicated antibodies. (E) OCP cells transduced with a control or CBP/p300-specific shRNA (top) or treated with DMSO control or a selective CBP/p300 inhibitor (bottom). H3NT cleavage was assessed at the indicated days after induction. See also Supplemental Figure 4.
MMP-9 is required for osteoclastogenic gene activation and proficient osteoclastogenesis
It was previously reported that H3NT cleavage facilitates gene activation in yeast during sporulation, suggesting that MMP-9-dependent H3NT proteolysis similarly functions in gene activation during osteoclastogenesis (Santos-Rosa et al. 2009). To test this, we sought to identify the genes regulated by MMP-9 using RNA sequencing (RNA-seq) of total mRNA isolated from control or MMP-9-depleted 3-d OCP-induced cells (Fig. 2C). Comparative transcriptome analysis revealed 1131 differentially expressed genes between the samples (Supplemental Table 1; Supplemental Fig. 5A,B). More than 67% of these genes displayed significantly reduced expression in MMP-9-depleted cells, supporting a function for MMP-9 in gene activation during osteoclastogenesis (Fig. 5A). Gene ontology analysis revealed that many of these genes are regulatory components of osteoclastogenic signaling pathways, including the RANKL, AMPK, and VEGF pathways (Supplemental Fig. 5C). In addition, gene set enrichment analysis (GSEA) demonstrated that the expression of a large set of RANKL and bone remodeling pathway genes was significantly reduced in MMP-9-depleted cells compared with control cells (Fig. 5B). Examination of the leading edge subset of these genes identified 26 canonical osteoclastogenic genes that required MMP-9 for their activation, including Nfatc1, Lif, and Xpr1 (Fig. 5C; Takayanagi et al. 2002; Bozec et al. 2008; Sharma et al. 2010). Importantly, the diminished H3NT cleavage and defective osteoclastogenic gene activation observed in MMP-9-depleted or MMP-9-inhibited 5-d OCP-induced cells were concurrent with a significant reduction of mature osteoclasts compared with control cells (Fig. 5D; Supplemental Fig. 5D). Similar results obtained from CBP/p300-depleted or CBP/p300-inhibited 5-d OCP-induced cells further support the dependence of osteoclastogenic gene activation and proficient osteoclast differentiation on H3NT proteolysis (Fig. 5D; Supplemental Fig. 5D).
Figure 5.
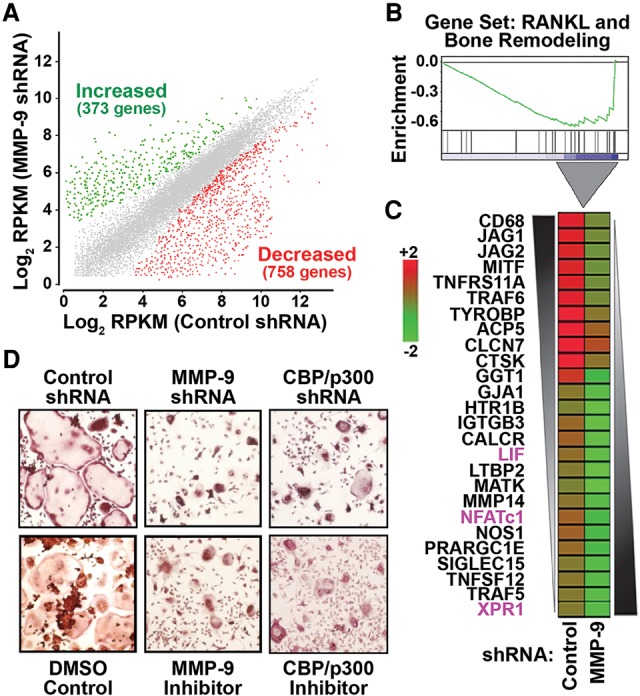
MMP-9 is required for osteoclastogenic gene activation and proficient osteoclast differentiation. (A) Scatter plot of RNA-seq normalized RPKM (reads per kilobase per million mapped reads) levels of annotated genes from 3-d OCP-induced cells expressing a control (X-axis) or MMP-9-specific (Y-axis) shRNA. Genes with significantly increased (green) or decreased (red) expression in MMP-9-depleted cells relative to control cells were defined based on false discovery rate (FDR)-adjusted P < 0.05 and log2 ratio ± 0.7. (B) GSEA of the 758 genes displaying impaired activation in MMP-9-depleted cells (X-axis) were ranked by increasing expression differences (green curve) relative to control cells (Y-axis). (C) Expression differences of the indicated 26 osteoclastogenic genes identified in Figure 6B between control (left) and MMP-9p-depleted (right) 3-d OCP-induced cells displayed as a heat map in the pseudo-color scale indicated. (D) Visualization (10×) of TRAP-stained 5-d OCP-induced cells expressing a control, MMP-9-specific, or CBP/p300-specific shRNA (top) or treated with DMSO control, a selective MMP-9 inhibitor, or a selective CBP/p300 inhibitor (bottom). See also Supplemental Figure 5 and Supplemental Table 1.
ChIPac combined with deep sequencing (ChIPac-seq): a novel approach to identify H3NT-cleaved regions
The results suggested that MMP-9-dependent H3NT proteolysis directly regulates gene expression during osteoclastogenesis, but the possible indirect effects of MMP-9 depletion on gene expression, independent of H3NT cleavage, could not be distinguished by transcriptomic analysis. Therefore, we sought to determine the genomic sites targeted for H3NT proteolysis during osteoclastogenesis to directly investigate the role of H3NT cleavage in transcriptional regulation; however, a method to identify H3NT-cleaved sites in mammalian cells has not been reported. We hypothesized that mapping these sites could be achieved by comparative ChIP-seq analysis of the H3NT between control and MMP-9-depleted 3-d OCP-induced cells that exhibit or lack H3NT cleavage, respectively (Fig. 2D). Computational identification of genomic regions displaying significantly reduced H3NT enrichment in control versus MMP-9-depleted cells would indicate those regions selectively targeted for H3NT proteolysis during osteoclastogenesis. This ChIP approach requires an antibody that has specific affinity for the H3NT of all H3 proteins, is not dependent on or inhibited by existing H3 PTMs, and is validated for ChIP applications; however, such an antibody is currently unavailable to the best of our knowledge. To bypass this technical barrier, established biochemical techniques were used to develop a novel method, called ChIPac, for identification and examination of H3NT-cleaved regions (Fig. 6A). We reasoned that an H3K14 acetyl-specific antibody satisfies the criteria above for ChIP of the H3NT following complete lysine acetylation of cross-linked chromatin in vitro by acetic anhydride. First, cells were fixed with methylene blue to cross-link chromatin after brief exposure to white light (Tuite and Kelly 1993). Because formaldehyde reacts with lysine ε-amino side chains, which likely precludes complete lysine acetylation by acetic anhydride, methylene blue was used as an alternative for cell fixation (Metz et al. 2004). Chromatin was isolated, and the efficiency of cross-linking was confirmed by SDS–chloroform–isoamyl alcohol (SDS-CIA) prior to sonication (Supplemental Fig. 6A; Lalwani et al. 1990). Fragmented chromatin was then treated with acetic anhydride to completely acetylate all unmodified lysine residues in vitro (Supplemental Fig. 6B; Nakayasu et al. 2014). ChIPac using an H3K14 acetyl-specific antibody selectively enriched H3NT-containing chromatin and simultaneously excluded chromatin lacking the H3NT. ChIPac using an H3CT antibody was performed in parallel as the normalization control. H3NT-cleaved regions were identified by the significant reduction in H3K14ac enrichment relative to the control as determined by quantitative PCR (qPCR) or next-generation (NextGen) sequencing analysis.
Figure 6.

Selective targeting of MMP-9-dependent H3NT proteolysis near TSSs during osteoclastogenesis. (A) Schematic of ChIPac-seq. Cells were fixed with methylene blue, and chromatin was extracted. Cross-linked chromatin was sonicated to generate nucleosomes containing full-length (green) and NT-cleaved (yellow) H3. Acetic anhydride was used to artificially acetylate all unmodified lysines (red lollipops). ChIPac was performed for H3CT control to enrich all nucleosomes or for H3K14ac to enrich only H3NT-containing nucleosomes. (B) Comparative ChIPac-seq analyses of control (left) and MMP-9-depleted (right) 3-d OCP-induced cells for enrichment (dark blue) or depletion (light blue) of H3CT control (left) or H3K14ac (right) near TSSs (±4 kb) of all known protein-coding genes. K means clustering identified specific H3NT-cleaved TSSs at either promoters (P), coding regions (CR), or both (P+CR). (C) Average tag density profiles (Y-axis) of each H3NT-cleaved group near TSSs (X-axis) for H3CT and H3K14ac in control and MMP-9-depleted 3-d OCP-induced cells, as indicated. See also Supplemental Figure 6 and Supplemental Table 2.
Specific gene TSSs are targeted for H3NT proteolysis during osteoclastogenesis
ChIPac-seq was performed using control and MMP-9-depleted 3-d OCP-induced cells to identify the specific sites targeted for H3NT proteolysis during osteoclastogenesis. MMP-9-depleted cells that lack H3NT-cleaved chromatin displayed nearly indistinguishable enrichment patterns between H3K14ac and the H3CT control, as predicted (Fig. 6B; Supplemental Figs. 6C,D). These important results validated the ChIPac approach to purify all H3NT-containing chromatin using the H3K14ac-specific antibody. The capability of ChIPac-seq to identify specific H3NT-cleaved regions was confirmed in control 3-d OCP-induced cells, which resulted in the identification of 1233 regions displaying significantly reduced H3K14ac enrichment relative to MMP-9-depleted cells. The maximal peak of H3K14ac depletion within each H3NT-cleaved region and the gene nearest this peak were determined (Supplemental Table 2; Kim et al. 2013b). Our subsequent computational analyses focused on the ±4-kb region near gene TSSs, since H3NT cleavage was previously reported at gene promoters in yeast (Santos-Rosa et al. 2009). K means clustering of TSSs revealed that <8% of all protein-coding genes exhibited H3K14ac depletion near TSSs, indicating that these sites are selectively targeted for H3NT proteolysis during osteoclastogenesis (Fig. 6B; Supplemental Fig. 6C,D). Strikingly, these genes partitioned into three distinct groups based on the location of H3NT cleavage: promoter-specific (P; 31%), coding region-specific (CR; 42%), or both (P+CR; 27%). Profiling the average tag densities in each group showed that H3NT cleavage typically peaks ∼2 kb from TSSs (Fig. 6C). Gene pathway analysis indicated that H3NT-cleaved genes are significantly linked to the RANKL and bone remodeling pathways (Supplemental Fig. 6E). Several canonical osteoclastogenic genes were identified within each H3NT-cleaved group, including Nfatc1 (P), Lif (CR), and Xpr1 (P+CR). Notably, MMP-9 was required for H3NT cleavage near Nfatc1, Lif, and Xpr1 TSSs and their concurrent activation during osteoclastogenesis, suggesting that H3NT proteolysis facilitates gene activation (Supplemental Fig. 6C).
H3NT cleavage correlates with gene activation
To broadly examine the association between H3NT cleavage and gene expression, the RNA-seq data were analyzed for the 1233 H3NT-cleaved genes identified in control and MMP-9-depleted 3-d OCP-induced cells. Comparative analyses revealed that most H3NT-cleaved genes (>53%) displayed significant expression differences in control versus MMP-9-depleted cells (Fig. 7A). The majority of these genes (>82%), including Nfatc1, Lif, and Xpr1, exhibited significantly reduced expression in MMP-9-depleted cells, indicating a strong functional correlation between MMP-9 and gene activation (Fig. 7A). To further investigate the MMP-9-dependent activation of these genes during osteoclastogenesis, RT-qPCR was performed in OCP cells transduced with a control or MMP-9-specific shRNA or treated with DMSO control or a selective MMP-9 inhibitor and cultured with or without RANKL for 3 d. Ablation of H3NT cleavage in MMP-9-depleted or MMP-9-inhibited 3-d OCP-induced cells significantly impaired Nfatc1, Lif, and Xpr1 activation, suggesting that H3NT proteolysis directly facilitates their activation during osteoclastogenesis (Figs. 2D, 7B; Supplemental Fig. 7). We reasoned that repeating these experiments in CBP/p300-depleted or CBP/p300-inhibited 3-d OCP cells would test this hypothesis, as nonacetylated H3K18 impedes MMP-9 H3NT protease activity (Fig. 4). Consistent with the hypothesis, diminishment of H3K18 acetylation by CBP/p300 depletion or inhibition ablated H3NT cleavage and significantly impaired Nfatc1, Lif, and Xpr1 activation, identical to the effects of MMP-9 depletion, but without altering nuclear abundance of the active MMP-9 enzyme (Figs. 4D,E, 7B; Supplemental Fig. 7). These results further support the direct function of H3NT cleavage in gene activation and suggest that CBP/p300-mediated acetylation of H3K18 is a key regulator of MMP-9 H3NT protease activity during osteoclastogenesis.
Figure 7.

CBP/p300-mediated acetylation of H3K18 regulates the sites and extent of MMP-9 H3NT proteolysis. (A, left) Distribution of all H3NT-cleaved genes displaying no change (blue) or significantly changed expression (red) between MMP-9-depleted and control 3-d OCP-induced cells. (Right) Distribution of H3NT-cleaved genes dependent on MMP-9 for activation (green) or repression (black) in 3-d OCP-induced cells. (B) Fold expression changes (Y-axis) of Nfatc1, Lif, and Xpr1 normalized to β-actin in 3-d OCP-induced cells expressing a control (blue), MMP-9-specific (red), or CBP/p300-specific (green) shRNA relative to noninduced OCP cells. (C) ChIPac was performed with H3K14ac-specific (H3NT) (left) and H3CT control (right) antibodies in 3-d control (−) or RANKL-induced (+) OCP cells expressing a control (blue), MMP-9-specific (red), or CBP/p300-specific (green) shRNA (X-axis). qPCR was performed at the promoters (P) (left) and coding regions (CR) (right) of Nfatc1 (P-cleaved), Lif (CR-cleaved), and Xpr1 (P+C-cleaved). Enrichments were plotted relative to noninduced control shRNA OCP cells (first column) (Y-axis). The stars indicate H3NT cleavage. (D) ChIP-qPCR analysis of H3K18ac as described above. The stars indicate significantly increased H3K18ac enrichment. See also Supplemental Figure 7.
The sites and extent of H3NT proteolysis are directly linked to H3K18 acetylation
Identification of the specific H3NT-cleaved sites at Nfatc1 (P), Lif (CR), and Xpr1 (P+CR) allowed us to directly examine the necessity of H3K18 acetylation on MMP-9 H3NT protease activity and gene activation during osteoclastogenesis. OCP cells were transduced with a control, MMP-9-specific, or CBP/p300-specific shRNA; cultured with or without RANKL for 3 d; and then processed for ChIPac or conventional ChIP using an H3K18ac-specific antibody. Subsequent qPCR confirmed the site-specific H3NT cleavage at Nfatc1 (P), Lif (CR), and Xpr1 (P+CR) in induced versus noninduced control shRNA cells (Fig. 7C). In contrast, H3K18ac enrichment at all sites examined was remarkably similar between induced and noninduced control shRNA cells (Fig. 7D). These results and the findings above suggested that H3NT cleavage sites are selectively targeted for H3K18 acetylation to activate MMP-9 H3K18-Q19 proteolysis, resulting in a tailless H3 that is exempt from enrichment by the H3K18ac ChIP. Consistent with this model, significantly increased H3K18ac enrichment at each H3NT cleavage site, but not the flanking noncleavage sites, was concurrent with the ablation of H3NT cleavage at these sites in MMP-9-depleted 3-d OCP-induced cells (Fig. 7C,D). Notably, the relative increase of H3K18ac at each H3NT cleavage site observed in MMP-9-depleted cells was directly proportional to the relative decrease of H3K14ac observed in control shRNA cells, suggesting that H3K18 acetylation directly regulates the sites and extent of MMP-9 H3NT protease activity during osteoclastogenesis. Consistent with this, preclusion of H3K18 acetylation at each H3NT cleavage site was concurrent with the loss of H3NT cleavage at these sites in CBP/p300-depleted 3-d OCP-induced cells despite the presence of active MMP-9 in the nucleus (Figs. 4D, 7C,D). The collective results indicate that the targeted CBP/p300-dependent acetylation of H3K18 at Nfatc1 (P), Lif (CR), and Xpr1 (P+CR) is prerequisite but insufficient for their expression, thereby further supporting the necessity of H3NT proteolysis for their activation during osteoclastogenesis.
Discussion
In this study, we discovered that MMP-9 is a novel H3NT protease required for H3NT proteolysis observed during osteoclastogenesis. Our data demonstrate that RANKL-induced differentiation of primary mouse OCP cells facilitates the progressive nuclear accumulation and activity of MMP-9 concomitant with increased H3NT proteolysis. By applying our novel ChIPac-seq method, we determined that many canonical osteoclastogenic genes are selectively targeted for H3NT proteolysis during osteoclastogenesis. Depletion of MMP-9 or inhibition of MMP-9 activity abrogated H3NT proteolysis concurrent with impaired osteoclastogenic gene activation and defective osteoclast differentiation. Our collective results support a model in which RANKL signaling induces MMP-9-dependent H3NT proteolysis at osteoclastogenic genes to regulate their expression necessary for proficient osteoclast differentiation. Consistent with this, previous reports demonstrated that chemical inhibition of MMP-9 activity in vivo attenuates osteoclast formation in juvenile mouse calvaria, and MMP-9−/− mice display delayed long bone development and defective bone fracture repair (Vu et al. 1998; Colnot et al. 2003; Cackowski et al. 2010; Franco et al. 2011). Although these reports and our results indicate the necessity of MMP-9 activity for osteoclast differentiation, further studies are required to determine whether MMP-9-dependent H3NT proteolysis and/or other functions of MMP-9, independent of H3NT proteolysis, are requisite for proficient osteoclastogenesis.
Despite increasing evidence supporting H3NT proteolysis in epigenetic regulation, advances in the field have been significantly impeded by the lack of a method to define and investigate genomic regions targeted for H3NT proteolysis in mammalian cells. In this study, we describe the development of the ChIPac method as a general approach to map and examine H3NT-cleaved regions. Since H3K14 resides within the H3NT-cleaved fragment and primarily exists in either an unmodified or acetylated state in mammals, an H3K14ac-specific antibody was used to selectively immunoprecipitate H3NT-containing nucleosomes from artificially acetylated cross-linked chromatin. ChIPac-seq in MMP-9-depleted OCP cells that lack H3NT proteolysis validated the proficiency of the H3K14ac antibody to capture all nucleosomes, as the H3K14ac enrichment patterns were nearly identical to H3CT control. Furthermore, ChIPac-seq in control 3-d OCP-induced cells that exhibit limited H3NT proteolysis identified a small number of genomic regions displaying significant reductions in H3K14ac enrichment, indicative of nucleosomes lacking the H3NT. Inhibition of H3NT proteolysis in 3-d OCP-induced cells rescued H3K14ac enrichment, confirming that ChIPac with an H3K14ac-specific antibody identified sites selectively targeted for H3NT proteolysis during osteoclastogenesis. These results validate the ChIPac-seq method to identify H3NT-cleaved sites that most likely can be used in other eukaryotic model systems. Application of ChIPac-seq in this study resulted in the first demonstration of selective H3NT proteolysis at specific genomic sites during mammalian differentiation.
It was previously reported that proteolysis of the H3NT at gene promoters is directly correlated to transcriptional activation during yeast sporulation (Santos-Rosa et al. 2009). In this study, ChIPac-seq similarly identified H3NT-cleaved regions surrounding the TSSs of specific protein-coding genes during osteoclastogenesis. Whereas H3NT proteolysis occurred at yeast promoters, mouse preosteoclasts displayed H3NT-cleaved regions that partitioned as those that were promoter-specific (P), coding region-specific (CR), or both (P+CR). The functional significance for these differences, if any, is unclear. Regardless of H3NT cleavage location (P, CR, or P+CR), the majority of genes targeted for H3NT proteolysis during osteoclast differentiation displayed concomitant significant changes in expression. H3NT proteolysis correlated to transcriptional activation for >82% of these genes during osteoclastogenesis, similar to results in yeast, supporting the direct role of H3NT cleavage in gene activation. Several H3NT-cleaved genes were repressed and many others exhibited little difference in expression during differentiation, suggesting that H3NT proteolysis regulates activation of specific groups of genes in a context-dependent manner. In the context of RANKL signaling in OCP cells, the activation of a group of canonical osteoclastogenic genes necessary for osteoclast differentiation was directly correlated to the selective H3NT proteolysis near their TSSs. Inhibition of H3NT proteolysis at these sites significantly impaired their activation despite continuous RANKL signaling, resulting in defective osteoclast differentiation. Therefore, we postulate that distinct signaling cues induce selective H3NT proteolysis to facilitate the rapid activation of specific developmental genes necessary for proficient differentiation.
Biochemical purification of preosteoclast nuclear extracts identified MMP-9 as a novel H3NT protease. Detailed investigation confirmed that MMP-9 is the principal histone protease in preosteoclasts that selectively cleaves the H3NT in vitro and in cells. Our collective results strongly support a model in which MMP-9 directly cleaves the H3NT to activate genes. However, we cannot exclude the possibility that MMP-9 activity may also regulate unknown factors necessary to facilitate H3NT proteolysis and/or gene activation during osteoclastogenesis. Identification and examination of these factors are required to test the model. Regardless, our results are the first to demonstrate proteolysis of histones by a metalloproteinase that therefore expands the spectrum of histone proteases beyond the canonical serine and cysteine protease families (Dhaenens et al. 2015). While these findings suggest possible histone proteolysis by the remaining 22 MMPs, this has yet to be determined. The P1 site of the yeast PRB1 serine protease and mouse cathepsin cysteine proteases is H3A21 (Duncan et al. 2008; Duarte et al. 2014; Khalkhali-Ellis et al. 2014; Xue et al. 2014). In contrast, in silico analysis predicted H3K18 as the P1 site for MMP-9, which was confirmed experimentally. The MMP-9 and PRB1 endopeptidases generate a single major H3NT-cleaved product, whereas cathepsins generate progressively shorter H3 forms after T22 due to their additional aminopeptidase activity. These enzymatic and substrate differences support MMP-9 as the founding member of a novel class of H3K18 proteases. Future discovery of additional histone proteases will determine whether MMP-9 is the sole H3K18 protease or whether other members also belong to this class.
Based on our findings and previous reports, we postulate that specific classes of H3NT proteases are used in a context-dependent manner to facilitate distinct epigenetic mechanisms due to their particular H3NT proteolytic activities. In the context of mESC differentiation, the cathepsin L H3A21 protease accumulates in the nucleus and is required for H3NT proteolysis (Duncan et al. 2008). The resulting H3Δ22-cleaved product impeded CBX7 chromodomain binding to H3K27me3 in vitro, suggesting that proteolysis of H3 amino acids 1–21 reduces the affinity of repressive H3K27me-binding proteins/complexes, thereby facilitating gene activation in differentiating mESCs. This putative mechanism of epigenetic regulation may be conserved for H3A21 proteases but is less likely to apply to H3K18 proteases, since an H3Δ18 peptide did not impair H3K27me3–CBX7 binding in vitro (Duncan et al. 2008). Although the primary mechanism of epigenetic regulation by H3K18 proteases remains unknown, the contrasting H3NT activities and epigenetic regulatory mechanisms observed between MMP-9 and cathepsins may explain why each protease is selectively used during differentiation of distinct cell types.
During osteoclast differentiation, MMP-9 accumulates in the nucleus concomitant with gelatinase activity and H3NT proteolysis. Consistent with our results, increasing evidence demonstrates MMP-9 nuclear localization and activity in specific cell types and under certain physiological conditions (Mannello and Medda 2012). The mechanisms that facilitate MMP-9 nuclear translocation remain unknown, but the lack of a nuclear localization signal supports the necessity of unidentified MMP-9-associated factors for transport. Our results demonstrate that progressive MMP-9 nuclear accumulation during osteoclastogenesis is directly proportional to the extent of gelatinase activity and H3NT proteolysis. Robust gelatinase activity and H3NT proteolysis were not observed prior to preosteoclast formation despite nuclear localization of the MMP-9 active form shortly after RANKL treatment, suggesting that a threshold of MMP-9 abundance regulates its nuclear protease activity. Furthermore, our results support H3K18 acetylation as a key regulator of MMP-9 activity. The specific acetylation of H3K18 was necessary and sufficient for MMP-9 H3NT proteolysis of nucleosome substrates in vitro, and, in preosteoclasts, the sites and extent of H3NT cleavage were directly proportional to the level of acetylated H3K18. Our results demonstrating that H3NT proteolysis is dependent on CBP/p300 activity but not other canonical H3K18 acetyltransferases support CBP/p300 as a central regulator of H3K18ac-dependent H3NT proteolysis. It was recently reported that CBP recruitment and histone acetylation at the Nfatc1 promoter were directly correlated to Nfatc1 transcriptional activation during osteoclastogenesis, further supporting the necessity of CBP/p300-mediated H3K18 acetylation in regulating H3NT proteolysis and gene activation (Park-Min et al. 2014). Importantly, our results demonstrate that the targeted acetylation of H3K18 at the H3NT cleavage sites of Nfatc1, Lif, and Xpr1 was insufficient to induce their expression during osteoclastogenesis, supporting H3NT cleavage as a prerequisite for their activation. We predict that modulation of H3NT proteolysis by H3K18 acetylation is a conserved regulatory mechanism of many H3NT proteases, as cathepsin L activity was also augmented by H3K18ac in vitro (Duncan et al. 2008).
Previous reports demonstrated that other histone modifications also regulate the activity of H3A21 proteases. Methylation of H3K27 augmented cathepsin L activity in vitro, whereas acetylated H3K23 abrogated H3 proteolysis in a dominant manner (Duncan et al. 2008). Similarly, H3K4me3 suppressed H3NT proteolysis in yeast, consistent with the absence of H3K4me3 in the cleaved H3NT peptides isolated from differentiating mESCs (Santos-Rosa et al. 2009). In contrast to these reports, our results demonstrate that H3K4 methylation, H3K23 acetylation, and H3K27 methylation are insufficient to regulate MMP-9 activity in vitro. These findings suggest that distinct H3NT proteases are differentially regulated by a specific “histone code” that stimulates or inhibits their activity (Strahl and Allis 2000). This may explain in part why only limited H3NT proteolysis is observed during differentiation and why MMP-9-dependent H3NT proteolysis is detected at only a small number of defined regions during osteoclastogenesis.
The H3NT proteases generate a cleaved H3 product that is transiently retained in chromatin during yeast sporulation and mESC differentiation (Duncan et al. 2008; Santos-Rosa et al. 2009). Although the rapid H3NT proteolysis identified at yeast promoters was insufficient for gene activation, the cleaved H3 facilitated subsequent nucleosome eviction at these promoters by unknown mechanisms concomitant with their activation. These findings suggest the similar requirement of cleaved H3-mediated nucleosome eviction for proficient gene activation during mammalian differentiation. However, our results demonstrate that cleaved H3 is retained within chromatin of differentiated osteoclasts and that robust activation of the H3NT-cleaved genes examined during osteoclastogenesis is independent of nucleosome eviction or histone replacement near their TSSs. While retention of cleaved H3 is observed in chromatin of terminally differentiated and senescent cells, the functional significance between retention and eviction/replacement of cleaved H3, as observed in differentiating mESCs and human ESCs, remains unknown (Duncan et al. 2008; Asp et al. 2011; Duarte et al. 2014; Vossaert et al. 2014).
Materials and methods
Cell transduction, inhibition, chromatin extraction, Western blot analysis, antibodies, chromatography, and PCR primers are detailed in the Supplemental Material.
Cell culture
Primary mouse OCP cells were derived, maintained, and differentiated as previously described (An et al. 2014). Cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP) using a TRAP kit (Sigma).
Recombinant proteins and H3 cleavage assays
His-tagged proteins were generated in Rosetta 2 (DE3) pLysS Escherichia coli (Novagen) and purified as previously described (Kim et al. 2008). Recombinant and native histone octamers and nucleosome arrays were prepared as previously described (Kim et al. 2012). Cell extracts or recombinant proteins were incubated with 1 µg of histone octamer or 2 µg of nucleosomes ± protease inhibitors, and H3NT cleavage was determined by Western blot analysis. H3 peptides (amino acids 10–35) were synthesized (EZBiolab) and incubated with rMMP-9 at increasing concentrations. Peptide hydrolysis was measured using a plate chameleon spectrofluorometer (Hidex), and kinetic parameters were determined by Michaelis-Menten analysis. For further details, see the Supplemental Material.
ChIPac-seq
Cells were fixed with methylene blue (Sigma). Cross-linking was confirmed by SDS-CIA of extracted chromatin prior to acetylation with acetic anhydride. Acetylated chromatin was immunoprecipitated with an H3K14ac-specific antibody (Active Motif) or an H3CT antibody (Abcam). Libraries were constructed, sequenced, and aligned to the mm9 reference genome (Li et al. 2008). Each read was extended in the sequencing orientation to a total of 200 bases to infer nucleosome coverage at each genomic position. Signal density was calculated in sliding 1-kb windows, and aligned reads were considered to be within a window of the midpoint of its estimated fragment. Midpoints in each window were counted, and empirical distributions of window counts were created. Genomic bins containing statistically significant regions were identified based on background distribution of randomized reads specific for each sample (Kim et al. 2013b). The ngs.plot was used to calculate the average coverage of TSSs, and composite plots were generated by averaging reads in 200-base-pair (bp) windows (Shen et al. 2014). Peaks were annotated with nonoverlapping genes from Ensembl. K means clustering of tag densities ±4 kb of TSSs was determined in R (http://www.r-project.org). For further details, see the Supplemental Material.
RNA-seq
Libraries were generated from total RNA for sequencing. High-quality reads were aligned to mm9 using TopHat in conjunction with a gene model from Ensembl release 61 (Kim et al. 2013b). Data were quantitated by counting the number of RPKM (reads per kilobase per million mapped reads) (Mortazavi et al. 2008). The values were adjusted globally by matching count distributions at the 75th percentile and then adjusting counts to a uniform distribution between samples. Differential expression was estimated by selecting transcripts that displayed significant changes (P < 0.05) after Benjamini and Hochberg correction using a null model constructed from 1% of transcripts showing the closet average level of observation to estimate experimental noise. The gene list was ranked using fold change criteria. GSEA and leading edge analysis were performed on the ranked list using gene sets from the C2 collection of the GSEA Molecular Signatures Database version 3.0 and several gene sets associated with bone remodeling. For further details, see the Supplemental Material.
Accession numbers
Sequencing data sets were deposited at NCBI Gene Expression Omnibus (GSE72846 and GSE72957).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM84209 to W.A.), the American Cancer Society (RSG1005001 to W.A., and RSG117619 to J.C.R.), a National Cancer Institute Cancer Center Support Grant (P30CA014089), and the National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of Science, ICT and Future Planning [MSIP]) (2015R1A4A1041869).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.268714.115.
References
- Allan J, Harborne N, Rau DC, Gould H. 1982. Participation of core histone ‘tails’ in the stabilization of the chromatin solenoid. J Cell Biol 93: 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Bowen JK, Abraham GN, Glover CV, Gorovsky MA. 1980. Proteolytic processing of histone H3 in chromatin: a physiologically regulated event in tetrahymena micronuclei. Cell 20: 55–64. [DOI] [PubMed] [Google Scholar]
- An D, Kim K, Lu W. 2014. Defective entry into mitosis 1 (Dim1) negatively regulates osteoclastogenesis by inhibiting the expression of nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1 (NFATc1). J Biol Chem 289: 24366–24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen K, Jimenez-Useche I, Howell SC, Yuan C, Qiu X. 2013. Solution scattering and FRET studies on nucleosomes reveal DNA unwrapping effects of H3 and H4 tail removal. PLoS One 8: e78587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asp P, Blum R, Vethantham V, Parisi F, Micsinai M, Cheng J, Bowman C, Kluger Y, Dynlacht BD. 2011. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci 108: E149–E158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad GK, Tomar RS. 2014. Proteolytic clipping of histone tails: the emerging role of histone proteases in regulation of various biological processes. Mol Biol Rep 41: 2717–2730. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124. [DOI] [PubMed] [Google Scholar]
- Black JC, Van Rechem C, Whetstine JR. 2012. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 48: 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortvin A, Winston F. 1996. Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science 272: 1473–1476. [DOI] [PubMed] [Google Scholar]
- Bozec A, Bakiri L, Hoebertz A, Eferl R, Schilling AF, Komnenovic V, Scheuch H, Priemel M, Stewart CL, Amling M, et al. 2008. Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature 454: 221–225. [DOI] [PubMed] [Google Scholar]
- Cackowski FC, Anderson JL, Patrene KD, Choksi RJ, Shapiro SD, Windle JJ, Blair HC, Roodman GD. 2010. Osteoclasts are important for bone angiogenesis. Blood 115: 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C, Thompson Z, Miclau T, Werb Z, Helms JA. 2003. Altered fracture repair in the absence of MMP9. Development 130: 4123–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaenens M, Glibert P, Meert P, Vossaert L, Deforce D. 2015. Histone proteolysis: a proposal for categorization into ‘clipping’ and ‘degradation’. Bioessays 37: 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte LF, Young AR, Wang Z, Wu HA, Panda T, Kou Y, Kapoor A, Hasson D, Mills NR, Ma'ayan A, et al. 2014. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat Commun 5: 5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan EM, Muratore-Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF, Allis CD. 2008. Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 135: 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco GC, Kajiya M, Nakanishi T, Ohta K, Rosalen PL, Groppo FC, Ernst CW, Boyesen JL, Bartlett JD, Stashenko P, et al. 2011. Inhibition of matrix metalloproteinase-9 activity by doxycycline ameliorates RANK ligand-induced osteoclast differentiation in vitro and in vivo. Exp Cell Res 317: 1454–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RA, Kuo YM, Andrews AJ. 2013. Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry 52: 5746–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalkhali-Ellis Z, Goossens W, Margaryan NV, Hendrix MJ. 2014. Cleavage of histone 3 by cathepsin D in the involuting mammary gland. PLoS One 9: e103230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Choi J, Heo K, Kim H, Levens D, Kohno K, Johnson EM, Brock HW, An W. 2008. Isolation and characterization of a novel H1.2 complex that acts as a repressor of p53-mediated transcription. J Biol Chem 283: 9113–9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Heo K, Choi J, Jackson S, Kim H, Xiong Y, An W. 2012. Vpr-binding protein antagonizes p53-mediated transcription via direct interaction with H3 tail. Mol Cell Biol 32: 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Kim JM, Kim JS, Choi J, Lee YS, Neamati N, Song JS, Heo K, An W. 2013a. VprBP has intrinsic kinase activity targeting histone H2A and represses gene transcription. Mol Cell 52: 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Punj V, Choi J, Heo K, Kim JM, Laird PW, An W. 2013b. Gene dysregulation by histone variant H2A.Z in bladder cancer. Epigenetics Chromatin 6: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalwani R, Maiti S, Mukherji S. 1990. Visible light induced DNA–protein crosslinking in DNA–histone complex and sarcoma-180 chromatin in the presence of methylene blue. J Photochem Photobiol B 7: 57–73. [DOI] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R. 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Dechassa ML, Tremethick DJ. 2012. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 13: 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannello F, Medda V. 2012. Nuclear localization of matrix metalloproteinases. Prog Histochem Cytochem 47: 27–58. [DOI] [PubMed] [Google Scholar]
- Metz B, Kersten GF, Hoogerhout P, Brugghe HF, Timmermans HA, de Jong A, Meiring H, ten Hove J, Hennink WE, Crommelin DJ, et al. 2004. Identification of formaldehyde-induced modifications in proteins: reactions with model peptides. J Biol Chem 279: 6235–6243. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. 2006. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69: 562–573. [DOI] [PubMed] [Google Scholar]
- Nakayasu ES, Wu S, Sydor MA, Shukla AK, Weitz KK, Moore RJ, Hixson KK, Kim JS, Petyuk VA, Monroe ME, et al. 2014. A method to determine lysine acetylation stoichiometries. Int J Proteomics 2014: 730725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. 2013. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154: 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse NP, Jimenez-Useche I, Smith IT, Yuan C. 2013. Clipping of flexible tails of histones H3 and H4 affects the structure and dynamics of the nucleosome. Biophys J 104: 1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park-Min KH, Lim E, Lee MJ, Park SH, Giannopoulou E, Yarilina A, van der Meulen M, Zhao B, Smithers N, Witherington J, et al. 2014. Inhibition of osteoclastogenesis and inflammatory bone resorption by targeting BET proteins and epigenetic regulation. Nat Commun 5: 5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, van Bemmel JG, Oliveira RA, Itoh T, Shirahige K, van Steensel B, Nasmyth K. 2010. A direct role for cohesin in gene regulation and ecdysone response in Drosophila salivary glands. Curr Biol 20: 1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DM, Johns EW. 1959. A study of the proteinase content and the chromatography of thymus histones. Biochem J 72: 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence MS, Reynolds A, Rynes E, Vlahovicek K, Stamatoyannopoulos JA, et al. 2015. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518: 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Kirmizis A, Nelson C, Bartke T, Saksouk N, Cote J, Kouzarides T. 2009. Histone H3 tail clipping regulates gene expression. Nat Struct Mol Biol 16: 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto E, Yoshida M. 2014. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 6: a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Patntirapong S, Hann S, Hauschka PV. 2010. RANKL–RANK signaling regulates expression of xenotropic and polytropic virus receptor (XPR1) in osteoclasts. Biochem Biophys Res Commun 399: 129–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, Nestler E. 2014. ngs.plot: quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15: 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak MA, Peterson CL. 2004. Creating designer histones by native chemical ligation. Methods Enzymol 375: 62–76. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311: 844–847. [DOI] [PubMed] [Google Scholar]
- Song J, Tan H, Perry AJ, Akutsu T, Webb GI, Whisstock JC, Pike RN. 2012. PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One 7: e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403: 41–45. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al. 2002. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3: 889–901. [DOI] [PubMed] [Google Scholar]
- Tuite EM, Kelly JM. 1993. Photochemical interactions of methylene blue and analogues with DNA and other biological substrates. J Photochem Photobiol B 21: 103–124. [DOI] [PubMed] [Google Scholar]
- Vossaert L, Meert P, Scheerlinck E, Glibert P, Van Roy N, Heindryckx B, De Sutter P, Dhaenens M, Deforce D. 2014. Identification of histone H3 clipping activity in human embryonic stem cells. Stem Cell Res 13: 123–134. [DOI] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. 1998. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93: 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Vashisht AA, Tan Y, Su T, Wohlschlegel JA. 2014. PRB1 is required for clipping of the histone H3 N terminal tail in Saccharomyces cerevisiae. PLoS One 9: e90496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Henikoff S. 2013. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol 20: 259–266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.