

In this study, Akkari et al. show the stage-dependent effects of simultaneously deleting cathepsin B (CtsB) and CtsS in a murine pancreatic neuroendocrine tumor model. They also identified CtsZ as the compensatory protease that regulates the acquired tumor-promoting functions of lesions deficient in both CtsB and CtsS, thus providing insight into a novel mechanism regulating tumorigenesis.
Keywords: invasion, macrophage, tumor microenvironment
Abstract
Proteases are important for regulating multiple tumorigenic processes, including angiogenesis, tumor growth, and invasion. Elevated protease expression is associated with poor patient prognosis across numerous tumor types. Several multigene protease families have been implicated in cancer, including cysteine cathepsins. However, whether individual family members have unique roles or are functionally redundant remains poorly understood. Here we demonstrate stage-dependent effects of simultaneously deleting cathepsin B (CtsB) and CtsS in a murine pancreatic neuroendocrine tumor model. Early in tumorigenesis, the double knockout results in an additive reduction in angiogenic switching, whereas at late stages, several tumorigenic phenotypes are unexpectedly restored to wild-type levels. We identified CtsZ, which is predominantly supplied by tumor-associated macrophages, as the compensatory protease that regulates the acquired tumor-promoting functions of lesions deficient in both CtsB and CtsS. Thus, deletion of multiple cathepsins can lead to stage-dependent, compensatory mechanisms in the tumor microenvironment, which has potential implications for the clinical consideration of selective versus pan-family cathepsin inhibitors in cancer.
Proteases play critical roles in numerous processes essential for normal function in multicellular organisms, and thus proteolysis is highly regulated to avoid aberrant activation and tissue destruction. However, proteases can frequently be dysregulated in cancer as a consequence of increases in mRNA or protein expression, decreased expression of their endogenous inhibitors, or alterations in subcellular localization, among other mechanisms (Lopez-Otin and Bond 2008; Sevenich and Joyce 2014). It has also become evident that these enzymes can be regulated at a network level (Mason and Joyce 2011) in which proteases and their endogenous inhibitors and activators are integrated within a complex interconnected “protease web,” a concept proposed by Overall and colleagues (auf dem Keller et al. 2007; Fortelny et al. 2014).
In recent years, one of the most intensively studied families of proteases in cancer has been the cysteine cathepsin proteases (Olson and Joyce 2015) or cathepsins as we refer to them here. The cathepsin family is comprised of 11 members in humans: cathepsin B (CtsB), CtsC, CtsH, CtsF, CtsK, CtsL, CtsO, CtsS, CtsL2/V, CtsW, and CtsX/Z (Shi et al. 1992; Cao et al. 1994; Santamaria et al. 1998; Deussing et al. 2000; Turk et al. 2012). CtsX/Z has been referred to by both names following its identification and characterization (Nagler and Menard 1998; Santamaria et al. 1998; Nagler et al. 1999; Deussing et al. 2000); however, as the official gene name for this enzyme is CtsZ, we refer to it using this nomenclature throughout. Increased expression and activity of individual cathepsin family members has been reported in multiple cancers and generally correlates with poor patient prognosis (Jedeszko and Sloane 2004; Mohamed and Sloane 2006). Important insights into the regulatory functions of cathepsins in cancer have come from a number of genetically engineered mouse models, including the RIP1-Tag2 (RT2) pancreatic neuroendocrine tumor (PanNET) model, the MMTV-PyMT model of mammary tumorigenesis and lung metastasis, the K14-HPV16 squamous cell carcinoma model, and the KPC model of pancreatic ductal adenocarcinoma (Gocheva et al. 2006, 2010a; Vasiljeva et al. 2006, 2008; Wang et al. 2006; Dennemarker et al. 2010; Sevenich et al. 2010; Gopinathan et al. 2012; Ruffell et al. 2013; Akkari et al. 2014; for review, see Olson and Joyce 2015).
Expression of six of the 11 cathepsins (CtsB, CtsC, CtsH, CtsL, CtsS, and CtsZ) is up-regulated in the multistage RT2 model as PanNET lesions progress from hyperplastic and angiogenic islet precursors to end-stage invasive tumors (Joyce et al. 2004). While cancer cells produce a subset of these cathepsins, tumor-associated macrophages (TAMs) are the major source of these enzymes in the PanNET microenvironment (Gocheva et al. 2006, 2010a,b; Wang and Joyce 2010; Akkari et al. 2014). The number of TAMs increases with tumor progression in both human and murine PanNETs (Gocheva et al. 2010b; Pyonteck et al. 2012), and TAMs have critical roles in promoting essential hallmark capabilities of cancer, including tumor angiogenesis, invasion, and metastasis (Qian and Pollard 2010; Quail and Joyce 2013).
In the RT2 PanNET model, bone marrow transplantation experiments demonstrated that TAM-derived CtsB and CtsS were either predominantly or exclusively responsible for the growth-promoting and invasive functions of these cathepsins (Gocheva et al. 2010b). The tumorigenic functions of CtsZ were derived from both the cancer cell and TAM compartments (Akkari et al. 2014). We showed that the proteolytic activities of CtsB and CtsS drive the dissolution of cell–cell junctions and extracellular matrix degradation in the RT2 model, while CtsZ acts in a proteolytically independent manner by regulating interactions with components of the extracellular matrix (Gocheva et al. 2006; 2010b; Akkari et al. 2014).
We performed a comprehensive series of genetic experiments to individually delete each of the six up-regulated cathepsins in the RT2 model. We identified both unique and shared roles in promoting PanNET malignancy without evident compensatory mechanisms by other cathepsin family members (Gocheva et al. 2006, 2010a,b; Gocheva and Joyce 2007; Akkari et al. 2014). Given that multiple cathepsins have been shown to have tumor-promoting functions in RT2 mice, here we investigated whether combined deletion of individual family members would enhance these effects or rather cause alterations to the protease web that may result in unexpected phenotypes. These are important questions to address for understanding not only the unique versus overlapping functions of multigene family members but also the translational implications of using selective versus pan-family cathepsin inhibitors in cancer.
Results
Combined deletion of CtsB and CtsS reduces angiogenic switching
We reported previously the pronounced effects of individual CtsB or CtsS deletion in blocking multiple aspects of PanNET development and progression (summarized in Table 1; Gocheva et al. 2006). To investigate whether there are additive effects of simultaneously deleting these tumor-promoting cathepsins, we generated CtsB−/−S−/− double-knockout RT2 mice. We first analyzed the number of angiogenic islets at 10.5 wk as an indication of the ability of premalignant lesions to induce neovascularization, which is required for subsequent tumor development and growth (Parangi et al. 1995). This revealed a more pronounced 60% reduction in angiogenic switching in the CtsB−/−S−/− RT2 mice (Fig. 1A) compared with the 24% reduction in either CtsB- or CtsS-null RT2 animals (Table 1), suggesting an additive effect of combined CtsB and CtsS deletion on this early rate-limiting step in multistage tumorigenesis.
Table 1.
Comparative analyses of multiple tumorigenic processes in the RT2 genotypes listed at the top of each column
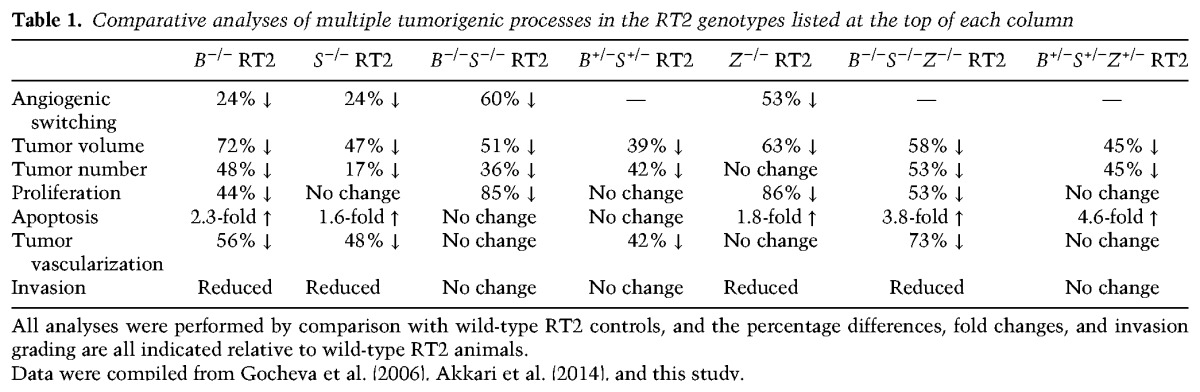
Figure 1.

Simultaneous deletion of CtsB and CtsS reduces angiogenic switching and tumor growth but does not affect tumor invasion. (A) Angiogenic switching was assessed in 10.5-wk-old wild-type RT2 or CtsB−/−S−/− RT2 mice (n = 10 mice for both genotypes) by manually counting the number of angiogenic islets in the pancreas. The graph shows the average number of angiogenic islets per mouse. (B) Cumulative tumor volume, represented as the sum of the volumes of all tumors per mouse, was calculated for 13.5-wk-old wild-type RT2 (n = 57) and CtsB−/−S−/− RT2 (n = 46) mice. (C) Graph depicting the average number of tumors per mouse in wild-type and CtsB−/−S−/− RT2 animals at the 13.5-wk endpoint. The following numbers of animals were analyzed per group: wild-type RT2, n = 52; CtsB−/−S−/− RT2, n = 37. (D) Quantitation of Ki67+ cells in wild-type and CtsB−/−S−/− RT2 tumors relative to the total number of DAPI+ cells showed an 85% decrease in cell proliferation in tumors deficient for both CtsB and CtsS. All tumors from five wild-type RT2 and 11 CtsB−/−S−/− RT2 mice were analyzed. (E) Graph showing the proportions of encapsulated, microinvasive (IC1), and invasive (IC2) carcinomas in wild-type RT2 and CtsB−/−S−/− RT2 mice at 13.5 wk. The following numbers of samples were analyzed: wild-type RT2, 18 mice, 97 tumors; CtsB−/−S−/− RT2, 14 mice, 68 tumors. The graphs show mean + SEM. Statistical significance was calculated by unpaired two-tailed Student's t-test (A–D) or using a cumulative logit model with generalized estimating equations to correct for correlations within individual mice (E). (n.s.) Nonsignificant; (***) P < 0.001.
A separate cohort of CtsB−/−S−/− RT2 mice was analyzed at the end-stage time point of 13.5 wk, in which we found a significant reduction in both tumor number and tumor volume compared with wild-type RT2 animals (Fig. 1B,C). However, deletion of both cathepsins did not result in additional impairment of overall tumor burden compared with the individual CtsB−/− or CtsS−/− RT2 mice (Table 1), in contrast to the additive effect of combined deletion on angiogenic switching at 10.5 wk. Similarly, no further differences in tumor volume or tumor number were observed in CtsB+/−S+/− RT2 animals (Supplemental Fig. S1A,B) compared with the homozygous deletion of either cathepsin.
CtsB−/−S−/− RT2 mice show no difference in tumor invasion, vascularization, or apoptosis compared with wild-type RT2 animals
Characterization of several key tumorigenic properties revealed that while cancer cell proliferation was impaired in CtsB−/−S−/− 13.5-wk tumors (Fig. 1D; Supplemental Fig. S1C), apoptosis levels were not significantly altered (Supplemental Fig. S1D). This is in contrast to both the single CtsB−/− and CtsS−/− tumors, where a significant increase in apoptosis was observed (Table 1). Similarly, analysis of the spectrum of invasive tumors showed no significant difference between CtsB−/−S−/− and wild-type RT2 mice (Fig. 1E; Supplemental Fig. S1E), while the single deletion of these genes resulted in a marked impairment in invasion (Table 1; Gocheva et al. 2006). Finally, analysis of a panel of vascular parameters, including vessel branching, thickness, length, and pericyte coverage, revealed no differences between CtsB−/−S−/− and wild-type tumors (Supplemental Fig. S1F,G). This result contrasts with the impaired tumor vascularization in the individual knockouts (Table 1; Gocheva et al. 2006) and the significant decrease in angiogenic switching observed in CtsB−/−S−/− RT2 animals at 10.5 wk of age (Fig. 1A).
Collectively, these data indicate defined stage-dependent effects of combined CtsB and CtsS deletion on PanNET progression by comparison with either cathepsin knockout alone. The additive effects observed in the double knockouts were restricted to the premalignant stage, where the number of angiogenic lesions was substantially decreased by comparison with either cathepsin knockout alone. In contrast, the subsequent processes of tumor growth, vascularization, and invasion were either not further impaired in CtsB−/−S−/− RT2 mice compared with single knockouts or, unexpectedly, not altered compared with wild-type RT2 animals (Table 1).
Altered protease expression in CtsB−/−S−/− tumors
A concern that is frequently raised in the generation and analysis of knockouts for members of large gene families (as for the cathepsins presented here) is that functional compensation by other family members may occur to take over the role of the deleted genes, potentially masking the resultant phenotype. While we have not observed this phenomenon in previous analyses of individual Cts mutants in RT2 tumorigenesis (Gocheva et al. 2006, 2010a,b; Akkari et al. 2014), the adaptive selection resulting from simultaneously deleting more than one cathepsin family member may give rise to conditions that are more favorable to functional compensation.
As the additive, tumor-limiting phenotype we observed at the early angiogenic islet stage in CtsB−/−S−/− RT2 tumorigenesis is reversed in end-stage lesions, we hypothesized that up-regulation and functional compensation by another factor, potentially a protease, might specifically occur later in tumor progression in the absence of both CtsB and CtsS to paradoxically promote malignancy. To investigate the potential mediators of this proposed compensation, we analyzed the expression levels of a panel of proteases and protease inhibitors that have previously been shown to be critical in cancer progression (Affara et al. 2009; Shchors et al. 2013; Sevenich and Joyce 2014). Analyses of mRNA expression for a panel of cathepsins, cathepsin inhibitors, and several matrix metalloproteinases (MMPs) were performed in 13.5-wk end-stage wild-type, CtsB−/−S−/− and CtsB+/−S+/− tumors (Fig. 2A; Supplemental Fig. S2A,B). This revealed a significant up-regulation of CtsZ and Mmp13 in CtsB−/−S−/− tumors, while Mmp3 and Mmp9 expression was down-regulated (Fig. 2A). The increase in CtsZ expression in CtsB−/−S−/− tumors was also confirmed at the protein level (Fig. 2B).
Figure 2.

Analysis of protease gene expression in CtsB−/−S−/− RT2 tumors identifies macrophage-derived CtsZ as a potential compensatory factor. (A) The mRNA expression level of CtsZ, Mmp3, Mmp9, and Mmp13 was determined by quantitative PCR (qPCR) in wild-type RT2 and CtsB−/−S−/− RT2 whole tumors (end-stage, 13.5 wk). This analysis demonstrated increased expression of CtsZ and Mmp13 and decreased Mmp3 and Mmp9 expression in the CtsB−/−S−/− RT2 tumors. Three to nine independent tumors per genotype were used for analysis. (B) Representative protein extracts from wild-type RT2 tumors (n = 5) and CtsB−/−S−/− RT2 tumors (n = 4), at 13.5 wk, were analyzed for CtsZ expression. Actin served as a loading control. Quantification of CtsZ normalized to the loading control using ImageJ software showed a significant increase in protein expression in CtsB−/−S−/− RT2 tumors. n = 7 replicate experiments and 31 independent tumors. (C) mRNA expression level of CtsZ, Mmp3, Mmp9, and Mmp13 was determined by qPCR in premalignant angiogenic islets (A.I.) from wild-type RT2 and CtsB−/−S−/− RT2 mice at 10.5 wk of age. No significant changes in expression of CtsZ, Mmp3, or Mmp13 were observed at this early stage of tumorigenesis, while Mmp9 expression was increased. Three to five independent sets of pooled angiogenic islets per genotype. (D) mRNA expression level of CtsZ was determined by qPCR analysis of bone marrow-derived macrophages (BMDMs) prepared from tumor-bearing wild-type RT2 (n = 3) and CtsB−/−S−/− RT2 (n = 4) animals, which revealed a significant increase in CtsZ expression in CtsB−/−S−/− RT2 BMDMs. (E) Tumors from wild-type RT2 or CtsB−/−S−/− RT2 mice were sorted into a mixed population of live cells (DAPI−), cancer cells (CD45− CD31− F4/80−) or TAMs (CD45+ CD31− F4/80+). Expression of CtsZ mRNA was determined by qPCR, and the level is depicted relative to the live-cell fraction. Up-regulation of CtsZ expression was found specifically in the TAM compartment and not in cancer cells. mRNA expression determined by qPCR was normalized to Ubiquitin C for each sample in A and C–E. Graphs show mean ± SEM. Statistical significance was calculated by unpaired two-tailed Student's t-test. (n.s.) Nonsignificant; (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Interestingly, CtsZ deletion significantly decreases RT2 tumor invasion (Akkari et al. 2014), while Mmp9 ablation has been reported to unexpectedly promote tumor invasion in two independent PanNET mouse models (Shchors et al. 2013), including the RT2 model used in this study. Other MMPs, including Mmp3 and Mmp13, have been implicated in the regulation of tumor development and invasion (Kessenbrock et al. 2015); however, there have been no correlative studies linking these two MMPs with PanNET malignancy. In sum, these results suggest that the increase in CtsZ expression and decrease in Mmp9 expression observed in CtsB−/−S−/− tumors may represent compensatory molecular mechanisms used to promote tumor invasion.
Given the stage-specificity of the effects observed in CtsB−/−S−/− RT2 mice, we hypothesized that the compensatory expression changes in the genes identified above might occur specifically during the transition from angiogenic islets to neoplastic lesions. Analysis of CtsZ, Mmp3, and Mmp13 mRNA expression in premalignant CtsB−/−S−/− angiogenic islets showed no significant differences compared with wild-type angiogenic islets (Fig. 2C), while Mmp9 mRNA expression was increased in CtsB−/−S−/− angiogenic islets compared with wild type (Fig. 2C). This is in contrast to the differential gene expression observed between these genotypes in end-stage tumors (Fig. 2A), suggesting that alterations in the expression of multiple factors indeed occur late in the course of CtsB−/−S−/− tumor progression to potentially promote several tumorigenic processes, including invasion.
CtsZ levels are elevated specifically in CtsB−/−S−/− RT2 macrophages
We recently identified CtsZ as an important regulator of PanNET development and malignancy (Akkari et al. 2014). Given that CtsZ is specifically up-regulated during the transition from angiogenic islets to neoplastic lesions in CtsB−/−S−/− RT2 animals among the proteases analyzed above, we subsequently focused on this protease as a candidate mediator of the compensatory phenotype in CtsB−/−S−/− tumors. CtsZ is supplied from both cancer cells and TAMs and promotes tumor invasion through both intrinsic and extrinsic regulation of cancer cell motility (Akkari et al. 2014). We showed previously that infiltrating TAMs in RT2 tumors predominantly originate from the bone marrow (Gocheva et al. 2010b). Thus, to determine the specific cell type responsible for CtsZ up-regulation in end-stage CtsB−/−S−/− tumors, we first analyzed CtsZ expression in bone marrow-derived macrophages (BMDMs) from tumor-bearing mice. Interestingly, we found that CtsZ levels were increased in BMDMs from CtsB−/−S−/− RT2 mice compared with their wild-type RT2 counterparts (Fig. 2D), while mRNA expression of Mmp9 and Mmp3 was down-regulated, and Mmp13 did not change significantly (Supplemental Fig. S2C).
Furthermore, when we sorted cancer cells and TAMs from wild-type and CtsB−/−S−/− tumors and analyzed CtsZ expression in each cell population, we found that only TAMs from CtsB−/−S−/− tumors showed a significant up-regulation of CtsZ expression, while no differences were observed in cancer cells (Fig. 2E). Analysis of Mmp3, Mmp9, and Mmp13 showed no significant differences in mRNA expression in either cell compartment (Supplemental Fig. S2D).
We next sought to determine the molecular mechanisms underlying the specific up-regulation of CtsZ in CtsB−/−S−/− RT2 BMDMs. Comparison of the CtsZ promoter with the promoters of the other cathepsin family members up-regulated during wild-type RT2 tumorigenesis (CtsB, CtsC, CtsH, CtsL, and CtsS) revealed nine transcription factor motifs (TFMs) specific to CtsZ (Fig. 3A,B; Supplemental Table 1). We focused our interest on one of these, NFκB, as it is a key orchestrator of innate immunity and inflammation (Karin 2006). NFκB activity in TAMs has been shown to regulate and maintain macrophage activation and modulate tumor-promoting phenotypes in response to diverse environmental cues, including stress signals, inflammatory cytokines, and infection (Karin and Greten 2005; Hagemann et al. 2008). Interestingly, we detected a significant increase in nuclear expression of the NFκB subunit p65 (RelA) in CtsB−/−S−/− RT2 BMDMs compared with their wild-type RT2 counterparts (Fig. 3C,D).
Figure 3.

Analysis of TFMs in the CtsZ promoter reveals a unique NFκB motif and elevated nuclear p65 levels in CtsB−/−S−/− RT2 macrophages. (A) Venn diagram demonstrating the overlap of predicted TFMs present in the CtsZ promoter compared with the CtsB, CtsC, CtsH, CtsL, and CtsS promoters. There are nine TFMs present in the CtsZ promoter that were not identified in any of these other cathepsin family members. Meanwhile, 83 TFMs were present in at least one of the other cathepsins but absent in the CtsZ promoter (see Supplemental Table 1 for a full list of TFMs). (B) The CtsZ promoter is shown with exons and the 5′ untranslated region (UTR) on the top bar and nine TFMs listed below. An NFκB consensus site is highlighted in red upstream of the 5′ UTR. (C) Wild-type RT2 or CtsB−/−S−/− RT2 BMDMs were subjected to subcellular fractionation, and lysates of the nuclear fraction (top two panels) or cytoplasmic fraction (bottom two panels) were isolated for immunoblotting of the NFκB subunit p65 and lamin A/C or p65 and actin. Results are representative of n = 3 independent biological replicates. (D) Quantification of p65—normalized to lamin A/C or actin for the nuclear and cytoplasmic fractions, respectively, using ImageJ software—showed a significant increase in p65 expression in the nucleus of CtsB−/−S−/− RT2 BMDMs. n = 3 replicate experiments. Statistical significance was calculated by unpaired two-tailed Student's t-test. (*) P < 0.05; (**) P < 0.01.
Together, these results indicate that CtsZ is up-regulated in BMDMs in CtsB−/−S−/− RT2 mice, potentially via increased NFκB nuclear translocation, prior to infiltration of these cells to the tumor site and their maturation into TAMs. Therefore, we uncovered a novel compensatory mechanism that is manifest at the level of normal, noncancerous cells in the tumor microenvironment rather than intrinsic to the cancer cells.
Macrophages accumulate in CtsB−/−S−/− tumors in association with increased CtsZ expression and enhanced tumor invasion
To confirm that TAMs are responsible for the overall increase in CtsZ expression, which could potentially promote tumor invasion, we characterized TAM numbers and their infiltrative pattern within CtsB−/−S−/− tumors. Interestingly, immunofluorescence analysis of the macrophage marker Iba1 co-stained with CtsZ showed a pronounced TAM accumulation particularly at the invasive margins of CtsB−/−S−/− tumors compared with wild-type lesions (Fig. 4A). Quantification of Iba1+ and CtsZ+ cells throughout whole tumors revealed a 2.0-fold increase in the number of TAMs and a 2.8-fold increase in CtsZ+ cells in CtsB−/−S−/− tumors compared with wild-type RT2 tumors (Fig. 4B,C). Analysis of mRNA expression of Cd68, another macrophage marker, supported the immunofluorescence data, showing a significant increase in CtsB−/−S−/− tumors compared with wild-type lesions (Fig. 4D). No alterations were detected in CtsB+/−S+/− tumors (Supplemental Fig. S3A–C), indicating that complete ablation of both CtsB and CtsS is necessary to trigger the increase in TAM numbers and the up-regulation of CtsZ. Consistent with this finding, tumor invasion in CtsB+/−S+/− mice was significantly reduced compared with CtsB−/−S−/− animals (Supplemental Fig. 1E). We found that the accumulation of macrophages at the margins of CtsB−/−S−/− tumors correlates with their increased invasive properties (Fig. 4E). While wild-type tumors accumulate TAMs in the intermediate invasive stage (IC1), CtsB−/−S−/− IC1 tumors show higher macrophage numbers, a feature that is maintained in the most invasive IC2 lesions (Fig. 4E, F).
Figure 4.

Macrophages expressing high levels of CtsZ infiltrate CtsB−/−S−/− tumors. (A) CtsZ is highly expressed in CtsB−/−S−/− RT2 tumors, and these tumors also show elevated macrophage numbers. Antibodies against CtsZ (red) and Iba1, a macrophage-specific marker (green), were used to identify cells expressing CtsZ and the presence of TAMs in wild-type RT2 and CtsB−/−S−/− RT2 tumors. CtsZ+ macrophages are particularly concentrated in the invasive edge of CtsB−/−S−/− RT2 tumors (evident in the lower images in the bottom panels). (B) Quantification of CtsZ+ cells in wild-type RT2 (n = 62) and CtsB−/−S−/− RT2 (n = 33) tumors relative to the total number of DAPI+ cells showed a significant increase in CtsB−/−S−/− RT2 tumors. (C) Quantification of Iba+ TAMs in wild-type RT2 (n = 108) and CtsB−/−S−/− RT2 (n = 28) tumors relative to the total number of DAPI+ cells showed a significant increase in TAMs in CtsB−/−S−/− RT2 tumors. (D) mRNA expression of the macrophage marker Cd68 was determined by qPCR analysis of wild-type RT2 (n = 4) and CtsB−/−S−/− RT2 (n = 6) whole tumors and revealed a significant increase in expression in CtsB−/−S−/− RT2 mice, consistent with the results in C. (E) Graph showing the macrophage content within and at the margin of individual tumors from wild-type RT2 and CtsB−/−S−/− RT2 mice, as determined by Iba1+ staining and according to each tumor invasive grade: encapsulated, IC1, or IC2 (as specified in Fig. 1E). (F) Low-magnification images of wild-type RT2 and CtsB−/−S−/− RT2 tumors stained with the macrophage-specific marker Iba1 (green) or hematoxylin and eosin (H&E), identifying the accumulation of TAMs at the margin of invasive CtsB−/−S−/− tumors compared with wild-type invasive tumors. (G,H) mRNA expression of the chemoattractants Csf-1 (G) and Ccl5 (H) was determined by qPCR analysis of wild-type RT2 (n = 4–5) and CtsB−/−S−/− RT2 (n = 5) whole tumors, which revealed a significant increase in expression of both genes in CtsB−/−S−/− RT2 mice. mRNA expression determined by qPCR was normalized to Ubiquitin C for each sample in D, G, and H. The graphs show mean ± SEM. Statistical significance was calculated by unpaired two-tailed Student's t-test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. Bars: A, 50 µm; F, 200 µm.
To investigate how CtsB−/−S−/− tumors evoke compensatory mechanisms to recruit macrophages, we examined the mRNA expression levels of a panel of cytokines and chemokines involved in myeloid cell recruitment (Ben-Baruch 2006; Mantovani et al. 2010). We analyzed the expression of Csf-1, Ccl2, Ccl5, Cxcl2, Cxcl14, Cxcl15, and Vegf-a in wild-type, CtsB−/−S−/,− and CtsB+/−S+/− tumors (Fig. 4G,H; Supplemental Fig. S3D–F). Interestingly, we found that expression of Csf-1 and Ccl5, two well-described monocyte/macrophage chemoattractants (Robinson et al. 2003; Hamilton 2008), was significantly increased specifically in CtsB−/−S−/− tumors, while the other chemokines analyzed did not change significantly.
Together, these results establish a critical role for TAMs in providing high levels of CtsZ in the microenvironment of CtsB−/−S−/− tumors. TAM accumulation correlates with the increased invasiveness of CtsB−/−S−/− tumors and up-regulation of specific factors involved in macrophage recruitment, suggesting critical alterations to the TME in CtsB−/−S−/− animals to mobilize these cells. Given the frequent association between TAMs and tumor invasion (Gocheva et al. 2010b; Qian and Pollard 2010), this may represent a compensatory mechanism to overcome the combined loss of CtsB and CtsS, resulting in the promotion of invasion at later stages of PanNET malignancy.
Deletion of CtsZ in CtsB−/−S−/− RT2 mice decreases tumor invasion and reduces TAM recruitment
To determine whether the observed CtsZ up-regulation in CtsB−/−S−/− tumors contributes functionally to enhanced malignancy, we generated CtsB+/−S+/−Z+/− RT2 and CtsB−/−S−/−Z−/− RT2 animals and assessed the consequences of deleting these three proteases on multiple tumorigenic processes (Table 1). Analysis of cumulative tumor volume changes in the triple-knockout RT2 mice showed a 58% reduction in tumor burden and a 53% decrease in tumor number compared with wild-type RT2 animals (Fig. 5A,B). These observations are in accordance with our results above that deletion of multiple cathepsins does not lead to additive effects on tumor growth. As observed for the combined deletion of CtsB and CtsS, heterozygous deletion revealed effects on tumor burden and number comparable with that of the complete removal of all three cathepsins (Table 1; Supplemental Fig. S4A,B).
Figure 5.

Deletion of CtsZ in CtsB−/−S−/− RT2 animals reduces macrophage infiltration and tumor invasion. (A) Graph depicting the cumulative tumor burden, represented as the sum of the volumes of all tumors per mouse, in wild-type RT2 and CtsB−/−S−/−Z−/− RT2 animals at the 13.5-wk endpoint. Numbers of mice per group were as follows: wild-type RT2, n = 57; CtsB−/−S−/−Z−/− RT2, n = 14. (B) Graph depicting the average number of tumors per mouse in wild-type RT2 and CtsB−/−S−/−Z−/− RT2 animals at 13.5 wk. The following numbers of animals were analyzed per group: wild-type RT2, n = 58; CtsB−/−S−/−Z−/− RT2, n = 18. (C) Graph showing the proportions of encapsulated, microinvasive, and invasive carcinomas in CtsB−/−S−/− RT2 and CtsB−/−S−/−Z−/− RT2 mice at 13.5 wk. The following number of mice were analyzed: CtsB−/−S−/− RT2, 14 mice, 68 tumors; CtsB−/−S−/−Z−/− RT2, 13 mice, 32 tumors. (D) Quantitation of cleaved caspase-3 (CC3) staining in wild-type RT2 and CtsB−/−S−/−Z−/− RT2 tumors relative to the total number of DAPI+ cells revealed a significant 3.6-fold increase in apoptosis in tumors deficient for CtsB, CtsS, and CtsZ. Tumors from 11 wild-type RT2 and seven CtsB−/−S−/−Z−/− RT2 mice were analyzed. (E) Quantitation of Ki67 staining in wild-type RT2 (n = 5) and CtsB−/−S−/−Z−/− RT2 (n = 7) tumors relative to the total number of DAPI+ cells. This revealed a 53% decrease in cell proliferation in tumors simultaneously deficient for CtsB, CtsS, and CtsZ. (F) Quantification of CD31+ endothelial cells in wild-type RT2 (n = 4) and CtsB−/−S−/−Z−/− RT2 (n = 10) tumors relative to the total number of DAPI+ cells, as determined by immunostaining of tissue sections. This analysis revealed a significant decrease in tumor vascularization in CtsB−/−S−/−Z−/− RT2 mice. (G) Quantification of Iba+ macrophages in wild-type RT2 (n = 108), CtsB−/−S−/− RT2 (n = 28), and CtsB−/−S−/−Z−/− RT2 (n = 14) tumors relative to the total number of DAPI+ cells. This analysis revealed that the increase in TAM numbers observed in CtsB−/−S−/− RT2 tumors is reversed when CtsZ is deleted in these tumors.
Analysis of tumor grade in CtsB−/−S−/−Z−/− RT2 animals revealed a significant reduction in the incidence of invasive carcinomas compared with CtsB−/−S−/− RT2 mice (Fig. 5C; Supplemental Fig. S4C), thus supporting our hypothesis that CtsZ mediates the invasive phenotype observed in CtsB−/−S−/− RT2 animals. Quantitation of cleaved caspase-3-positive (CC3+) cells demonstrated a 3.8-fold and 5.7-fold increase in apoptosis in CtsB−/−S−/−Z−/− tumors compared with wild-type and CtsB−/−S−/− tumors, respectively (Fig. 5D; Supplemental Figs. S1D, S4D). Cell proliferation in CtsB−/−S−/−Z−/− and CtsB+/−S+/−Z+/− tumors was significantly reduced compared with wild-type RT2 (Fig. 5E; Supplemental Fig. S4E), although no further decrease was observed compared with CtsB−/−S−/− lesions (Table 1). Vascularization was also impaired in CtsB−/−S−/−Z−/− tumors compared with either wild-type lesions (Fig. 5F; Supplemental Fig. S4F) or CtsB−/−S−/− lesions (Table 1).
We next asked whether the diminished invasive properties of CtsB−/−S−/−Z−/− tumors correlated with a change in TAM numbers and performed staining of Iba1+ macrophages in the different genotypes analyzed in this study. Indeed, the increase in TAMs observed in CtsB−/−S−/− end-stage lesions was reversed in CtsB−/−S−/−Z−/− tumors (Fig. 5G; Supplemental Fig. S4G). Expression of the chemoattractant factors Ccl5 and Csf-1 was similar in CtsB−/−S−/−Z−/− tumors and CtsB−/−S−/− tumors, while CtsZ−/− RT2 tumors, which are poorly invasive and show reduced TAM numbers (Akkari et al. 2014), did not show significant changes in Ccl5 and Csf-1 levels (Supplemental Fig. S4H,I). These results suggest that the reduced TAM numbers in the triple-knockout mice is a consequence specifically of CtsZ deletion, resulting in impaired BMDM recruitment and reduced tumor invasion.
Discussion
Our results show that functional compensation occurs when multiple cathepsin proteases are targeted in PanNETs. Interestingly, this occurs in a stage-specific manner. Significant synergistic effects were observed early in the tumorigenic process, where the simultaneous deletion of both CtsB and CtsS led to a 60% reduction in angiogenic switching. This was the most pronounced effect observed at this early stage in multistage RT2 tumorigenesis for any of the cathepsin knockouts previously reported (Table 1). However, at end stage, no additive anti-tumorigenic effects were evident. There was no difference in proliferation, apoptosis or angiogenesis in CtsB−/−S−/− tumors when compared with single-knockout animals. Furthermore, the invasive capacity of the CtsB−/−S−/− lesions was actually comparable with wild-type tumors, revealing adaptive compensatory mechanisms counteracting loss of both cathepsins via macrophage-derived CtsZ.
Interestingly, up-regulation of CtsZ also occurs in CtsB−/− PyMT mammary tumors, specifically at the cancer cell surface (Vasiljeva et al. 2006; Sevenich et al. 2010). However, in our study, compensation by CtsZ was observed only when multiple cathepsins were deleted and was largely mediated by accumulation of macrophages in the tumor microenvironment. The increased number of TAMs at the tumor margin is potentially a result of elevated expression of the chemoattractive factors Csf-1 and Ccl5, associated with the enhanced capacity of macrophages to respond to these signals via increased expression of CtsZ. Thus, in the absence of CtsB and CtsS, the ability to degrade ECM components that favors tumor invasiveness may be compromised, and the recruitment of TAMs expressing and secreting high levels of CtsZ provides an alternative mode of tumor invasion.
Tumor cell-derived CtsZ modulates migration and invasion in a proteolytically independent manner in the RT2 PanNET model (Akkari et al. 2014). Whether the proinvasive role of TAM-derived CtsZ requires its carboxypeptidase activity remains to be elucidated. This is currently challenging to address due to limited options available to specifically target the catalytic activity of CtsZ. In light of our recent results reporting the interaction of macrophage-secreted CtsZ with the cancer cell surface via integrins (Akkari et al. 2014), we infer that proteolysis may not be strictly necessary for CtsZ-dependent invasion in CtsB−/−S−/− tumors. These results thus underscore the plasticity of adaptive behavior that tumors exhibit when rendered deficient in critical mediators of malignancy using tumor cell-intrinsic and/or cell-extrinsic mechanisms.
Expression of a subset of MMPs is also altered in CtsB−/−S−/− tumors. Previous reports suggest a mechanism through which resistance to MMP inhibitors involves elevated cathepsin activity, dependent on a change in the abundance of inflammatory cells supplying these proteases (Shchors et al. 2013). Thus, our data share parallels with these results and show that deletion of multiple cathepsin proteases can conversely alter MMP expression, possibly via perturbation of the protease web. However, we infer that these MMP expression changes ultimately have limited functional roles to compensate for the loss of CtsB and CtsS, as further deletion of CtsZ fully reverts the invasive phenotype of CtsB−/−S−/− tumors.
While several studies report the pleiotropic roles of MMPs and cathepsins in promoting tumor progression in different types of cancer (Coussens et al. 2002; Mohamed and Sloane 2006), certain members of these protease classes also display tumor-suppressive functions (Dufour and Overall 2013; Olson and Joyce 2015). For instance, CtsL deletion enhances skin carcinogenesis in the K14-HPV16 and DMBA/TPA mouse models (Dennemarker et al. 2010; Benavides et al. 2012), and the protease MMP8 exerts anti-metastatic activity in the MDA-MB-231 breast cancer model (Soria-Valles et al. 2014). The multifaceted roles of MMPs as enhancers or inhibitors of tumorigenesis (Dufour and Overall 2013) can partly explain the failure of broad-spectrum molecules targeting this protease family and highlight the need for tissue-dependent, stage-specific analysis of protease functions prior to using pan-family inhibitors. In the RT2 PanNET model, all up-regulated cathepsins have been characterized as protumorigenic, with the exception of CtsC, which had no effect when deleted (Gocheva et al. 2006, 2010b; Akkari et al. 2014).
Our finding that CtsZ is up-regulated in response to the simultaneous deletion of CtsB and CtsS has potential translational implications in considering cathepsin inhibitors for the treatment of cancer patients. Our data would argue that selective inhibitors directed against specific subsets of cathepsins might lead to compensation by untargeted family members or other protease classes and therefore may not ultimately be as effective as broad-spectrum inhibitors that target the entire family. Indeed, treatment of RT2 mice with pan-cathepsin inhibitors (e.g., JPM-OEt and VBY-825) results in a pronounced reduction in multiple hallmarks of cancer, including tumor growth, vascularity, and invasion (Joyce et al. 2004; Elie et al. 2010), supporting this viewpoint. Moreover, we report that heterozygous deletion of multiple cathepsins results in impaired tumor growth to an extent similar to complete ablation yet does not lead to compensatory alterations in the expression of other proteases. The accumulation of TAMs evident in CtsB−/−S−/− tumors is not observed in heterozygous lesions, and the increase in Ccl5 and Csf-1 chemoattractants or CtsZ up-regulation is also not observed. Consequently, this failure to develop adaptive compensatory mechanisms is associated with less invasive lesions in CtsB+/−S+/− animals compared with CtsB−/−S−/− mice. This could indicate that partial blockade of cathepsins (as achieved by pharmacological inhibitors) may be preferable to their complete ablation.
In sum, perturbations to the protease web have the potential to result in unexpected phenotypes that may not only complicate interpretation of genetic analyses but also interfere with the efficacy of selectively targeted protease inhibitors. The selective pressure to recalibrate the protease web may be particularly evident in cancer given the heterogeneity and plasticity of the tumor microenvironment, which would facilitate rapid adaptive compensation. These points should therefore be carefully considered when therapeutically targeting protease families in cancer.
Materials and methods
Mouse strains
The generation of RT2 (Hanahan 1985), CtsB-null (Halangk et al. 2000), CtsS-null (Shi et al. 1999), and CtsZ-null (Sevenich et al. 2010) mice and each of the single constitutive cathepsin-null RT2 lines (Gocheva et al. 2006; Akkari et al. 2014) has been reported previously. β-Actin GFP transgenic mice in the C57BL/6 background (Okabe et al. 1997) were purchased from Jackson Laboratories. All animal studies were performed using protocols approved by the Animal Care Committee at Memorial Sloan Kettering Cancer Center.
Tissue processing and immunostaining
For angiogenic switching analyses, RT2 mice were sacrificed at 10.5 wk of age by heart perfusion with 10 mL of PBS under anesthesia. The pancreas was then removed and minced gently with scissors. Following a collagenase P (Sigma) digestion, the number of angiogenic islets, distinguishable by their red and hemorrhagic appearance, was counted under a dissecting microscope, as previously described (Parangi et al. 1995). Angiogenic switching ability was determined by comparing the average of the sum of all angiogenic islets per mouse between the different genotypes.
Tumor volume and number were determined at 13.5 wk of age, and RT2 mice were sacrificed by heart perfusion with PBS followed by 10% zinc-buffered formalin. Tissues were removed, placed in 30% sucrose overnight and embedded in OCT (Tissue-Tek). The dimensions of the tumors were measured as previously described (Joyce et al. 2004). Tumor burden was represented as the sum of the volumes of all tumors per mouse.
For the phenotypic characterization of the tumors, frozen sections (10 µm thick) were cut on a cryostat and analyzed by immunostaining. Immunofluorescence staining on frozen sections from RT2 animals was performed after sections were dried for 30 min at room temperature, rehydrated in PBS for 10 min, preincubated with 1× PNB blocking buffer (PerkinElmer Life Sciences) for 1 h, and then incubated with the primary antibody of interest overnight at 4°C. The following anti-mouse antibodies were used: rat CD31 (1:200; BD Pharmingen), goat CtsZ (1:1000; R&D Systems), rat CD68 (1:1000; Serotec), and rabbit Iba-1 (1:1000; Wako). The corresponding secondary antibodies were used at a 1:500 dilution and incubated for 1 h at room temperature. DAPI (1:5000; Invitrogen) was used to label the nuclei, and the slides were mounted in ProLong Gold mounting medium (Invitrogen). The tissues were observed under a Carl Zeiss Axioimager Z1 microscope, and images were acquired with Axiovision using an Apotome (Zeiss) or with TissueFAXS (TissueGnostics).
To analyze proliferation rates in the tumors, slides were stained with a rabbit anti-mouse Ki67 antibody (1:200; Vector Laboratories), and the numbers of Ki67+ cells relative to total cell number were determined using TissueQuest. For apoptosis analysis, the frozen tissues were stained using rabbit anti-mouse CC3 (1:500; Cell Signaling Technology). The number of CC3+ cells was counted using TissueQuest, and the percentage of cell death was calculated as the percentage of the total cells per tumor. For invasion grading, hematoxylin and eosin (H&E) staining was performed, and the lesions were graded as previously described (Lopez and Hanahan 2002) following a double-blind protocol and independently assessed by two investigators (L. Akkari/V. Gocheva and J.A. Joyce). The numbers of tumors and mice analyzed are specified in the figure legends. In order to include the quantitation of peripheral macrophages in Figure 4E, a dilated peritumoral region 500 μm in diameter was used to calculate the total Iba1+ area and the total DAPI+ area within this region. For analysis of angiogenesis, tumors from 13.5-wk-old RT2 mice from the indicated genotypes were stained with a rat anti-CD31 antibody, and the vessel area was calculated as the ratio of CD31+ endothelial cells divided by the DAPI+ tumor area as detected by pixel intensity and analyzed using Volocity imaging software.
Flow cytometry and sorting
Wild-type and CtsB−/−S−/− RT2 tumors were isolated and processed for fluorescence-activated cell sorting as previously described (Pyonteck et al. 2012) using the following antibodies: CD31-FITC (1:100; BD Pharmingen), CD45-PE (1:200; BD Pharmingen), anti-F4/80-APC (1:100; Serotec), and DAPI for dead cell exclusion. The cells were sorted on a fluorescence-activated cell sorting Aria flow cytometer (BD Biosciences), and fractions were collected: a mixed population of live cells (DAPI−), purified tumor cells (DAPI− CD31− CD45− F4/80−), and macrophages (DAPI− CD31− CD45+ F4/80+).
Preparation of cell lysates
To obtain whole-cell protein lysates, BMDMs were washed in cold 1× PBS once and then lysed in RIPA buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific). The crude lysates were centrifuged at 13,000 rpm for 12 min, and supernatants containing intracellular proteins were collected for downstream analyses. The NE-PER nuclear and cytoplasmic extraction kit (ThermoFisher Scientific) was used to isolate nuclear and cytoplasmic fractions from RT2 BMDMs (Fig. 3C) according to the manufacturer's instructions.
RNA isolation and quantitative RT–PCR
RNA was isolated with Trizol and DNase-treated, and 1 µg of RNA was used for cDNA synthesis using a high-capacity cDNA reverse transcription kit (Applied Biosystems). TaqMan probes (Applied Biosystems) were used for quantifying expression of CtsB (Mm00514439_m1), CtsZ (Mm00517697_m1), CtsC (Mm00515580_m1), CtsH (Mm00514455_m1), CtsS (Mm00457902_m1), CtsL (Mm00515597_m1), CstC (Mm00438347_m1), Mmp2 (Mm00439502_m1), Mmp3 (Mm00440295_m1), Mmp9 (Mm00600163_m1), Mmp12 (Mm00500554_m1), Mmp13 (Mm00439491_m1), Mmp14 (Mm00485054_m1), Mmp15 (Mm00485062_m1), Csf-1 (Mm00432688_m1), Ccl2 (Mm00441242_m1), Ccl5 (Mm01302428_m1), Cxcl2 (Mm00436450_m1), Cxcl14 (Mm00444699_m1), Cxcl15 (Mm00469712_m1), and Vegf-a (Mm00437304_m1). Ubc (Mm02525934_g1) was used as a housekeeping control.
Promoter analysis
Transcription factor-binding site predictions for the mouse genome (mm9) were downloaded from the SwissRegulon portal (http://swissregulon.unibas.ch/data/mm9/mm9_sites.gff.gz) (Pachkov et al. 2007). TFMs were tabulated 2 kb upstream of and downstream from the transcription start site for CtsB, CtsC, CtsH, CtsL, CtsS, and CtsZ. Promoters in Figure 3B were drawn using the “ggbio” package in R (Yin et al. 2012).
Statistical analysis
Data are presented throughout as mean and SEM. Results were analyzed by unpaired two-tailed Student's t-tests unless otherwise noted and were considered statistically significant if P < 0.05. For the invasion analysis, a cumulative logit model (McCullagh 1980) with generalized estimating equations to correct for correlations within individual mice was used to compare the distribution of tumor types in the control group with the distribution of tumors in the experimental groups.
Supplementary Material
Acknowledgments
We thank Xiaoping Chen for excellent technical support, Oakley Olson for insightful feedback on the manuscript, and members of the Joyce laboratory for comments and discussion. We are grateful to Elyn Reidel of the Memorial Sloan Kettering Cancer Center Biostatistics Department for assistance with statistical analyses of tumor invasion. This research was funded in part through the National Institutes of Health/National Cancer Institute Cancer Center Support Grant P30 CA008748, which supports Memorial Sloan Kettering Cancer Center core facilities. We gratefully acknowledge Dr. Thomas Reinheckel and Dr. Christoph Peters (University of Freiburg) for providing CtsB and CtsZ knockout mice, and Dr. Harold Chapman (University of California at San Francisco) for providing CtsS knockout mice. This research was supported by the American Cancer Society Research Scholar Grant (RSG-12-076-01-LIB), the Gerardo Rosenkranz Neuroendocrine Research Fund (J.A.J.), and fellowships from the American Brain Tumor Association (L.A.), the Geoffrey Beene Cancer Center (V.G.), the Howard Hughes Medical Institute (A.L.G.), and the National Cancer Institute (5F31CA167863-04 to R.L.B.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.270439.115.
References
- Affara NI, Andreu P, Coussens LM. 2009. Delineating protease functions during cancer development. Methods Mol Biol 539: 1–32. [DOI] [PubMed] [Google Scholar]
- Akkari L, Gocheva V, Kester JC, Hunter KE, Quick ML, Sevenich L, Wang HW, Peters C, Tang LH, Klimstra DS, et al. 2014. Distinct functions of macrophage-derived and cancer cell-derived cathepsin Z combine to promote tumor malignancy via interactions with the extracellular matrix. Genes Dev 28: 2134–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- auf dem Keller U, Doucet A, Overall CM. 2007. Protease research in the era of systems biology. Biol Chem 388: 1159–1162. [DOI] [PubMed] [Google Scholar]
- Ben-Baruch A. 2006. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev 25: 357–371. [DOI] [PubMed] [Google Scholar]
- Benavides F, Perez C, Blando J, Contreras O, Shen J, Coussens LM, Fischer SM, Kusewitt DF, DiGiovanni J, Conti CJ. 2012. Protective role of cathepsin L in mouse skin carcinogenesis. Mol Carcinog 51: 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Taggart RT, Berquin IM, Moin K, Fong D, Sloane BF. 1994. Human gastric adenocarcinoma cathepsin B: isolation and sequencing of full-length cDNAs and polymorphisms of the gene. Gene 139: 163–169. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM. 2002. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295: 2387–2392. [DOI] [PubMed] [Google Scholar]
- Dennemarker J, Lohmuller T, Mayerle J, Tacke M, Lerch MM, Coussens LM, Peters C, Reinheckel T. 2010. Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene 29: 1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deussing J, von Olshausen I, Peters C. 2000. Murine and human cathepsin Z: cDNA-cloning, characterization of the genes and chromosomal localization. Biochim Biophys Acta 1491: 93–106. [DOI] [PubMed] [Google Scholar]
- Dufour A, Overall CM. 2013. Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol Sci 34: 233–242. [DOI] [PubMed] [Google Scholar]
- Elie BT, Gocheva V, Shree T, Dalrymple SA, Holsinger LJ, Joyce JA. 2010. Identification and pre-clinical testing of a reversible cathepsin protease inhibitor reveals anti-tumor efficacy in a pancreatic cancer model. Biochimie 92: 1618–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortelny N, Cox JH, Kappelhoff R, Starr AE, Lange PF, Pavlidis P, Overall CM. 2014. Network analyses reveal pervasive functional regulation between proteases in the human protease web. PLoS Biol 12: e1001869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocheva V, Joyce JA. 2007. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 6: 60–64. [DOI] [PubMed] [Google Scholar]
- Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C, Hanahan D, Joyce JA. 2006. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev 20: 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocheva V, Chen X, Peters C, Reinheckel T, Joyce JA. 2010a. Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol Chem 391: 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, Joyce JA. 2010b. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev 24: 241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinathan A, Denicola GM, Frese KK, Cook N, Karreth FA, Mayerle J, Lerch MM, Reinheckel T, Tuveson DA. 2012. Cathepsin B promotes the progression of pancreatic ductal adenocarcinoma in mice. Gut 61: 877–884. [DOI] [PubMed] [Google Scholar]
- Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, Robinson SC, Balkwill FR. 2008. ‘Re-educating’ tumor-associated macrophages by targeting NF-κB. J Exp Med 205: 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, Domschke W, Lippert H, Peters C, Deussing J. 2000. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest 106: 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA. 2008. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 8: 533–544. [DOI] [PubMed] [Google Scholar]
- Hanahan D. 1985. Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315: 115–122. [DOI] [PubMed] [Google Scholar]
- Jedeszko C, Sloane BF. 2004. Cysteine cathepsins in human cancer. Biol Chem 385: 1017–1027. [DOI] [PubMed] [Google Scholar]
- Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M, Hanahan D. 2004. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 5: 443–453. [DOI] [PubMed] [Google Scholar]
- Karin M. 2006. Nuclear factor-κB in cancer development and progression. Nature 441: 431–436. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. 2005. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5: 749–759. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Wang CY, Werb Z. 2015. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol 44-46C: 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T, Hanahan D. 2002. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 1: 339–353. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Bond JS. 2008. Proteases: multifunctional enzymes in life and disease. J Biol Chem 283: 30433–30437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Savino B, Locati M, Zammataro L, Allavena P, Bonecchi R. 2010. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev 21: 27–39. [DOI] [PubMed] [Google Scholar]
- Mason SD, Joyce JA. 2011. Proteolytic networks in cancer. Trends Cell Biol 21: 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh P. 1980. Regression models for ordinal data. J R Stat Soc 42: 109–142. [Google Scholar]
- Mohamed MM, Sloane BF. 2006. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer 6: 764–775. [DOI] [PubMed] [Google Scholar]
- Nagler DK, Menard R. 1998. Human cathepsin X: a novel cysteine protease of the papain family with a very short proregion and unique insertions. FEBS Lett 434: 135–139. [DOI] [PubMed] [Google Scholar]
- Nagler DK, Zhang R, Tam W, Sulea T, Purisima EO, Menard R. 1999. Human cathepsin X: a cysteine protease with unique carboxypeptidase activity. Biochemistry 38: 12648–12654. [DOI] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. 1997. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett 407: 313–319. [DOI] [PubMed] [Google Scholar]
- Olson OC, Joyce JA. 2015. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat Rev Cancer 15: 712–729. [DOI] [PubMed] [Google Scholar]
- Pachkov M, Erb I, Molina N, van Nimwegen E. 2007. SwissRegulon: a database of genome-wide annotations of regulatory sites. Nucleic Acids Res 35: D127–D131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parangi S, Dietrich W, Christofori G, Lander ES, Hanahan D. 1995. Tumor suppressor loci on mouse chromosomes 9 and 16 are lost at distinct stages of tumorigenesis in a transgenic model of islet cell carcinoma. Cancer Res 55: 6071–6076. [PubMed] [Google Scholar]
- Pyonteck SM, Gadea BB, Wang HW, Gocheva V, Hunter KE, Tang LH, Joyce JA. 2012. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene 31: 1459–1467. [DOI] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. 2010. Macrophage diversity enhances tumor progression and metastasis. Cell 141: 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. 2013. Microenvironmental regulation of tumor progression and metastasis. Nat Med 19: 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AE, Balkwill FR. 2003. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res 63: 8360–8365. [PubMed] [Google Scholar]
- Ruffell B, Affara NI, Cottone L, Junankar S, Johansson M, DeNardo DG, Korets L, Reinheckel T, Sloane BF, Bogyo M, et al. 2013. Cathepsin C is a tissue-specific regulator of squamous carcinogenesis. Genes Dev 27: 2086–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria I, Velasco G, Pendas AM, Fueyo A, Lopez-Otin C. 1998. Cathepsin Z, a novel human cysteine proteinase with a short propeptide domain and a unique chromosomal location. J Biol Chem 273: 16816–16823. [DOI] [PubMed] [Google Scholar]
- Sevenich L, Joyce JA. 2014. Pericellular proteolysis in cancer. Genes Dev 28: 2331–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenich L, Schurigt U, Sachse K, Gajda M, Werner F, Muller S, Vasiljeva O, Schwinde A, Klemm N, Deussing J, et al. 2010. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc Natl Acad Sci 107: 2497–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchors K, Nozawa H, Xu J, Rostker F, Swigart-Brown L, Evan G, Hanahan D. 2013. Increased invasiveness of MMP-9-deficient tumors in two mouse models of neuroendocrine tumorigenesis. Oncogene 32: 502–513. [DOI] [PubMed] [Google Scholar]
- Shi GP, Munger JS, Meara JP, Rich DH, Chapman HA. 1992. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J Biol Chem 267: 7258–7262. [PubMed] [Google Scholar]
- Shi GP, Villadangos JA, Dranoff G, Small C, Gu L, Haley KJ, Riese R, Ploegh HL, Chapman HA. 1999. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity 10: 197–206. [DOI] [PubMed] [Google Scholar]
- Soria-Valles C, Gutierrez-Fernandez A, Guiu M, Mari B, Fueyo A, Gomis RR, Lopez-Otin C. 2014. The anti-metastatic activity of collagenase-2 in breast cancer cells is mediated by a signaling pathway involving decorin and miR-21. Oncogene 33: 3054–3063. [DOI] [PubMed] [Google Scholar]
- Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D. 2012. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824: 68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen BS, Almholt K, Bogyo M, et al. 2006. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res 66: 5242–5250. [DOI] [PubMed] [Google Scholar]
- Vasiljeva O, Korovin M, Gajda M, Brodoefel H, Bojic L, Kruger A, Schurigt U, Sevenich L, Turk B, Peters C, et al. 2008. Reduced tumour cell proliferation and delayed development of high-grade mammary carcinomas in cathepsin B-deficient mice. Oncogene 27: 4191–4199. [DOI] [PubMed] [Google Scholar]
- Wang HW, Joyce JA. 2010. Alternative activation of tumor-associated macrophages by IL-4: priming for protumoral functions. Cell Cycle 9: 4824–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Sun J, Kitamoto S, Yang M, Grubb A, Chapman HA, Kalluri R, Shi GP. 2006. Cathepsin S controls angiogenesis and tumor growth via matrix-derived angiogenic factors. J Biol Chem 281: 6020–6029. [DOI] [PubMed] [Google Scholar]
- Yin T, Cook D, Lawrence M. 2012. ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol 13: R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.