

Abstract
To adapt to environmental high osmolarity, the budding yeast Saccharomyces cerevisiae activates the Hog1 mitogen-activated protein kinase, which regulates diverse osmoadaptive responses. Hog1 is activated through the high-osmolarity glycerol (HOG) pathway, which consists of independent upstream signaling routes termed the SLN1 branch and the SHO1 branch. Here, we report that the extracellular cysteine-rich (CR) domain of the transmembrane-anchor protein Opy2 binds to the Hkr1-Msb2 homology (HMH) domain of the putative osmosensor Msb2 and that formation of the Opy2-Msb2 complex is essential for osmotic activation of Hog1 through the MSB2 subbranch of the SHO1 branch. By analyzing the phenotypes of mutants with Opy2 cysteine-to-alanine mutations, we deduced that the CR domain forms four intramolecular disulfide bonds. To probe for the potential induction of conformational changes in the Opy2-Msb2 complex by osmostress, we constructed mutants with a site-specific Cys-to-Ala mutation of the Opy2 CR domain and mutants with a Cys substitution of the Msb2 HMH domain. Each of these mutants had a reduced cysteine. These mutants were then combinatorially cross-linked using chemical cross-linkers of different lengths. Cross-linking between Opy2 Cys48 and Msb2 Cys1023 was sensitive to osmotic changes, suggesting that osmostress induced a conformational change. We therefore propose that the Opy2-Msb2 complex might serve as an osmosensor.
INTRODUCTION
Survival of living organisms depends on their ability to adapt to adverse environmental conditions. A well-known example is the response of the budding yeast Saccharomyces cerevisiae to high external osmolarity. When exposed to high osmolarity, yeast cells initiate a coordinated adaptive response that includes the synthesis and accumulation of the compatible osmolyte glycerol, changes in the global pattern of gene expression and protein synthesis, and a temporary arrest of cell cycle progression (1–3). These responses are controlled by the Hog1 mitogen-activated protein kinase (MAPK), which is activated via the high-osmolarity glycerol (HOG) signal pathway. The HOG pathway employs multiple and redundant routes between the input (external high osmolarity) and the output (Hog1-dependent responses). Specifically, the HOG pathway consists of two independent upstream signaling routes termed the SLN1 branch and the SHO1 branch (Fig. 1A). The osmosensor for the SLN1 branch is the sensor histidine kinase Sln1, which transmits the signal through a two-component phosphorelay mechanism to the redundant MAPK kinase kinases (MAPKKKs) Ssk2 and Ssk22 (4, 5). Ssk2/Ssk22 activates the Pbs2 MAPK kinase (MAPKK), which eventually activates the Hog1 MAPK (5–8).
FIG 1.
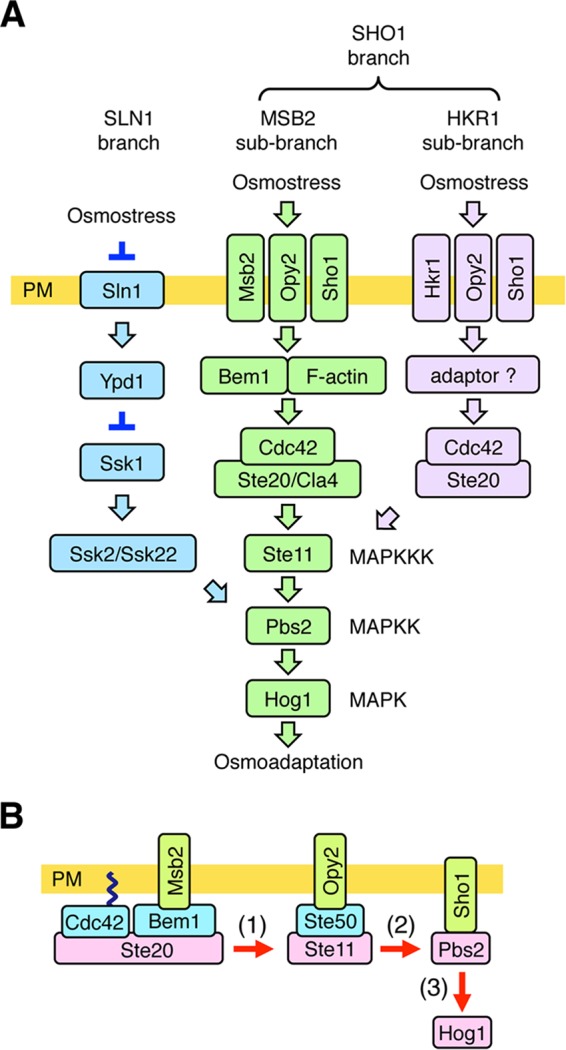
Schematic model of the HOG pathway. (A) Proteins that are involved in the MSB2 subbranch are shown in green. Proteins that are specific to the SLN1 branch are colored blue, and those that are involved in the HKR1 subbranch are colored lavender. The proteins separated by a slash are functionally redundant. Not all the known components are shown. The yellow bar represents the plasma membrane (PM). Arrows indicate activation, whereas the inverted T-shaped bars represent inhibition. (B) A simplified schematic model of the MSB2 subbranch of the HOG pathway. Arrows indicate the flow of the signal by sequential phosphorylation.
When the SLN1 branch is inactivated by the ssk2Δ ssk22Δ double mutation (here abbreviated ssk2/22Δ), yeast can still adapt to high osmolarity using the SHO1 branch. The major osmosensor for the SHO1 branch is the four-transmembrane (TM) protein Sho1 (5, 9). In the SHO1 branch, osmostress activates the Ste11 MAPKKK, which then sequentially activates Pbs2 and Hog1 (10). Thus, Pbs2 and Hog1 are common to both branches. The SHO1 branch can be further divided into the MSB2 and HKR1 subbranches (11, 12). The namesake component of the MSB2 subbranch, Msb2, was originally identified to be a multicopy suppressor of the cdc24 budding defect, suggesting that its function is related to signaling by the Cdc42 small G protein (13). Although Msb2 was implicated in osmoregulation, on the basis of an analysis of global gene expression patterns (14), the role of Msb2 in the HOG pathway has been unclear. We later found that disruption of HKR1 together with MSB2 in the ssk2/22Δ background completely inhibited Hog1 activation, indicating that Hkr1 and Msb2 are functionally redundant (11). Because the signaling mechanisms employed by Hkr1 and Msb2 are significantly different, they were termed the HKR1 and MSB2 subbranches, respectively (12). Msb2, but not Hkr1, also acts at the head of the filamentous growth Kss1 MAPK signaling pathway (15, 16).
An outline of how Hog1 is activated through the MSB2 subbranch is schematically shown in Fig. 1B. The consecutively activating kinases in the Hog1 MAPK cascade, Ste20, Ste11, and Pbs2, are recruited from the cytoplasm to the plasma membrane by the TM proteins Msb2, Opy2, and Sho1, respectively. These TM proteins thereby concentrate the cytoplasmic kinases on the plasma membrane to facilitate their mutual interactions.
Sho1 is composed of four TM segments at the N terminus and a cytoplasmic SH3 domain at the C terminus. The SH3 domain binds to a Pro-rich motif in the Pbs2 MAPKK, thereby localizing Pbs2 to the plasma membrane (5, 17). The 4-TM domain of Sho1 has multiple functions. First, Sho1 homodimerizes at the TM1/TM4 interface and homotrimerizes at the TM2/TM3 interface (9). By reiterative dimerization and trimerization, Sho1 forms large planar oligomers. Second, the TM1/TM4 interface of the Sho1 oligomer binds to Opy2 (9). Third, the TM2/TM3 interface of the Sho1 oligomer binds to Hkr1 (9) and also to Msb2 (11). Finally, the 4-TM domain of Sho1 binds to Ste50 when osmostress is applied. Ste50 is a cytoplasmic adaptor protein that binds simultaneously to both Ste11 and Opy2 (18–24). Thus, the induced Sho1-Ste50 interaction appears to underlie the osmosensor function of Sho1.
The second TM protein in the MSB2 subbranch, Opy2, is a single-path TM protein of 360 amino acids. Its cytoplasmic region contains three (two major and one minor) Ste50 binding sites that are collectively essential for signaling in the SHO1 branch (21, 23, 24). As Ste50 is constitutively bound to Ste11 (19, 20, 22), Opy2 recruits the Ste50/Ste11 complex to the plasma membrane (Fig. 1B). The membrane localization of Ste50 is an important function of Opy2, as an artificial membrane localization of Ste50, by using, for example, the C-terminal prenylation site of Ras2 (Cpr), suppresses the Hog1 activation defect of the opy2Δ mutation (11). However, it is likely that Opy2 has additional roles, as suppression of opy2Δ occurs only when Ste50-Cpr is overexpressed.
The extracellular region of Opy2 is composed of two serine-rich (SR) domains, SR1 (residues 2 to 27) and SR2 (residues 67 to 86), which flank a cysteine-rich (CR) domain (residues 30 to 63). The SR1 domain, which is highly O glycosylated, is functionally dispensable (25). In contrast, the CR domain seems to be important for activating Hog1 through the SHO1 branch (9). Opy2 is also involved in the filamentous growth Kss1 MAPK pathway, and a point mutation in the Opy2 CR domain (C30Y) reduces the activation of Kss1 by glucose starvation (26). The molecular function of the Opy2 CR domain, however, remains unclear.
The third TM protein in the MSB2 subbranch, Msb2, is a single-path TM protein of 1,306 amino acids. Its extracellular region contains a serine- and threonine-rich (STR) domain (residues 49 to 950) that is extensively O glycosylated (11, 15). Removal of the STR domain either by deletion mutations or by proteolytic cleavage converts Msb2 to an activated form (11, 15, 27, 28). The extracellular region of Msb2 also contains a domain (residues 961 to 1117) termed the Hkr1-Msb2 homology (HMH) domain (11). The HMH domain appears to have a positive regulatory function, because deletion of the HMH domain renders Msb2 inactive (11, 15). The cytoplasmic regions of Msb2 functionally interact with the scaffold protein Bem1 (12). Bem1 binds, among other proteins, Ste20 and its activator, Cdc42. Thus, Msb2 recruits activated Ste20 to the plasma membrane.
In this work, we studied the role of Msb2 and Opy2 in the Hog1 MAPK pathway. We show that the Msb2 HMH domain binds to the Opy2 CR domain and that the Msb2-Opy2 interaction is essential for Hog1 activation by osmostress. We determined a plausible disulfide bonding pattern in the Opy2 CR domain, as well as further detail regarding the Msb2-Opy2 interaction, using site-directed cysteine substitution mutants and cysteine-specific cross-linking analyses. Our results suggest that the Msb2-Opy2 complex might serve an osmosensing function, as high osmolarity induces a structural change in this complex.
MATERIALS AND METHODS
Buffers and media.
Standard yeast media and genetic procedures were previously described (29, 30). CAD medium consists of 0.67% yeast nitrogen base (Sigma), 2% glucose, 0.5% Casamino Acids (Sigma), and appropriate supplements (20 μg/ml uracil and 40 μg/ml tryptophan), as needed. CARaf medium is the same as CAD medium, except that it contains 2% raffinose in place of glucose. TE buffer contains 10 mM Tris-HCl (pH 7.5) and 1 mM EDTA. Buffer A for coimmunoprecipitation assays contains 50 mM Tris-HCl (pH 7.5), 15 mM EDTA, 15 mM EGTA, 2 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine, 5 μg ml−1 leupeptin, 50 mM NaF, 25 mM β-glycerophosphate, 150 mM NaCl, and an appropriate detergent (usually 0.2% Triton X-100). Buffer A4 for solubilization of membrane fractions is buffer A containing 1% Triton X-100 and 0.2% sodium dodecyl sulfate (SDS). Buffer A9 for quenching the cross-linking reaction of intact cells contains 50 mM Tris-HCl (pH 7.5), 15 mM EDTA, 15 mM EGTA, 1 mM PMSF, 1 mM benzamidine, 5 μg ml−1 leupeptin, 50 mM NaF, 25 mM β-glycerophosphate, 150 mM NaCl, and 10 mM N-ethylmaleimide. Buffer C2 for detergent-free membrane preparation contains 50 mM Tris-HCl (pH 7.2), 15 mM EDTA, 15 mM EGTA, 150 mM NaCl, 1 mM PMSF, 1 mM benzamidine, and 5 μg ml−1 leupeptin. Buffer X for cross-linking contains 50 mM Tris-HCl (pH 7.2) and 15 mM EDTA. Buffer Z for the β-galactosidase assay contains 60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, and 1 mM MgSO4, adjusted to pH 7.0. SDS loading buffer (1×) contains 50 mM Tris-HCl (pH 6.8), 2% SDS, 0.01% bromophenol blue, 10% glycerol, and 700 mM 2-mercaptoethanol (2-ME).
Reagents.
The following reagents were used: the Cys-specific chemical cross-linkers M-1-M through M-17-M (consisting of methanethiosulfonate [MTS] groups connected by alkane chain of various lengths) and M-8-O2-M through M-14-O4-M (consisting of MTS groups connected by oxaalkane chains of various lengths in which each third position is an oxygen) (Toronto Research Chemicals); maleimide-polyethylene glycol 2 (PEG2)-biotin (Thermo Scientific); streptavidin-agarose (Thermo Scientific); and the detergents Brij L23 and Brij 99 (Sigma), Triton X-100 (MP Biochemical), and digitonin (Sigma and Calbiochem). Other chemicals were purchased from Sigma, Wako Pure Chemical, Nacalai Tesque, and BD.
Yeast strains.
All yeast mutants used in this work are derivatives of the S288C strain (Table 1). Gene disruption was carried out using a PCR-based strategy, and missense and intragenic deletion mutations were created by oligonucleotide-based mutagenesis (29).
TABLE 1.
Yeast strains used in this studya
| Strain | Genotype | Reference or source |
|---|---|---|
| KT075 | MATα ura3 leu2 trp1 his3 ssk2::LEU2 ssk22::LEU2 hkr1::natMX4 msb2::kanMX6 pbs2::HIS3 sho1::hphMX4 | 11 |
| KY477 | MATα ura3 leu2 trp1 his3 ssk2::LEU2 ssk22::LEU2 opy2::kanMX6 | 23 |
| KY562-1 | MATα ura3 leu2 trp1 his3 ssk2::LEU2 ssk22::LEU2 msb2::kanMX6 opy2::natMX4 | This study |
| KY595-2 | MATa ura3 leu2 trp1 his3 ssk2::hisG ssk22::hisG hog1::LEU2 fus3::kanMX6 kss1::hphMX4 pmt4::natMX4 | This study |
| TM257 | MATα ura3 leu2 trp1 his3 ssk2::LEU2 ssk22::LEU2 | 5 |
All strains were constructed in our laboratory and are derived from strain S288C.
Plasmid constructs.
Deletion and missense mutants were constructed using PCR-based oligonucleotide mutagenesis, and their sequences were confirmed by nucleotide sequence determination.
(i) Vector plasmids.
The vector plasmids pRS416, p414GAL1, and p416GAL1 have been described previously (31–33).
(ii) Opy2 plasmids.
pRS414-Opy2 (POPY2-OPY2 TRP1 CEN6) and pRS416-Opy2 (POPY2-OPY2 URA3 CEN6) are full-length OPY2 genomic DNA clones that express Opy2 under the control of the OPY2 promoter. p414GAL1-Opy2-GFP (PGAL1-OPY2-GFP TRP1 CEN6) and p416GAL1-Opy2-GFP (PGAL1-OPY2-GFP URA3 CEN6) express C-terminally green fluorescent protein (GFP)-tagged Opy2 under the control of the GAL1 promoter. p414GAL1-Opy2ΔSR1-GFP (PGAL1-OPY2ΔSR1-GFP TRP1 CEN6) and p416GAL1-Opy2ΔSR1-GFP (PGAL1-OPY2ΔSR1-GFP URA3 CEN6) express C-terminally GFP-tagged Opy2ΔSR1 under the control of the GAL1 promoter. p414GAL1-Opy2ΔSR1-myc (PGAL1-OPY2ΔSR1-3×myc TRP1 CEN6) expresses C-terminally 3× Myc-tagged Opy2ΔSR1 under the control of the GAL1 promoter.
(iii) Msb2 plasmids.
p416GAL1-Msb2-2×HA (PGAL1-Msb2-HA/HA URA3 CEN6) and p416GAL1-Msb2ΔHMH-2×HA [PGAL1-Msb2Δ(951-1120)-HA/HA URA3 CEN6] encode two hemagglutinin (HA) tags, one following amino acid position 48 and another at the C terminus. pRS416GAL1-Msb2Δ1-2×HA [PGAL1-Msb2Δ(49-950)-HA/HA URA3 CEN6] and pRS416GAL1-Msb2Δ2-2×HA [PGAL1-Msb2Δ(49-364)-HA/HA URA3 CEN6] encode two HA tags, one at the site of the internal deletion and another at the C terminus. pRS416GAL1-Msb2Δ2-HA [PGAL1-Msb2Δ(49-364)-HA URA3 CEN6] encodes an HA tag at the site of the internal deletion.
Reporter assays.
Reporter assays using the HOG reporter plasmid pRS416-8×CRE-lacZ (8×CRE-lacZ URA3 CEN6) have been described previously (34). In the appropriate figures, the level of 8×CRE-lacZ expression is expressed in Miller units and is presented as an average and standard deviation (SD) for three or more independent samples (35).
In vivo binding assay.
Cell extracts were prepared in buffer A using glass beads, essentially as described previously (29). To immunoprecipitate HA-tagged proteins, a 1,000-μg aliquot of protein extract was first incubated with an appropriate dilution of the anti-HA high-affinity antibody 3F10 (Roche) for 2 h at 4°C, followed by further incubation with 50 μl of protein G beads for 1 h at 4°C. Beads were washed 3 times in buffer A, resuspended in SDS loading buffer, boiled for 5 min, and separated by SDS-PAGE.
Immunoblotting analyses.
Immunoblotting analyses were carried out essentially as described previously (36). The following antibodies were used to detect proteins by immunoblotting: anti-HA antibodies F-7 (Santa Cruz) and 12CA5 (Roche), anti-GFP antibody B-2 (Santa Cruz), anti-Myc antibodies A-14 and 9E10 (Santa Cruz), and anti-Hog1 antibody yC-20 (Santa Cruz). An anti-phospho-p38 antibody (Cell Signaling) was used to detect phosphorylated Hog1. Enhanced chemiluminescence images were digitally captured using a ChemDoc system (Bio-Rad) equipped with a charge-coupled-device camera. Band intensities were quantified using ImageJ software (37).
Preparation of membrane fractions.
The appropriate strain, e.g., KY477 carrying an Opy2-Myc expression plasmid, was grown to log phase in a selective medium. Expression of the Opy2-Myc protein was induced for 2 h with 2% galactose. The membrane fraction was then prepared as follows. Briefly, 20 ml of culture was centrifuged at 2,000 × g for 3 min, and the cell pellet was resuspended in 500 μl of detergent-free buffer C2. Cells were broken by vigorous vortexing with glass beads. Unbroken cells were removed by two rounds of low-speed centrifugation (300 × g for 3 min), and the supernatant fraction (S300) was subjected to a high-speed centrifugation (13,000 × g for 10 min). The precipitated fraction (P13,000) was resuspended in 150 μl of an appropriate buffer (e.g., the TE buffer or buffer X).
Chemical cross-linking of membrane fractions.
Chemical cross-linkers were dissolved in dimethyl sulfoxide (DMSO). In a typical chemical cross-linking reaction, 30 μl of the membrane fraction was incubated with 0.2 mM maleimide-PEG2-biotin (or DMSO alone as control) for 5 min at 30°C. Reactions were terminated by adding DTT to a final concentration of 50 mM. The membrane was precipitated by centrifugation at 13,000 × g for 10 min at 4°C. Pellets were resuspended in 300 μl buffer A4 to solubilize the membrane.
Chemical cross-linking between Opy2 and Msb2 in intact cells.
An exponentially growing yeast culture was centrifuged and resuspended in culture medium at a ratio of 1:10. A chemical cross-linker was added to the concentrated culture. After incubation at 30°C for 5 min, the cells were precipitated by centrifugation. To quench the cross-linking reaction, cell pellets were resuspended in buffer A9, which contains N-ethylmaleimide but no detergent, and incubated on ice for 20 min. The cells were then precipitated by a brief spin, resuspended in buffer A without DTT, and frozen in liquid N2. A total lysate was prepared and subjected to immunoblot analyses.
Other methods.
Other methods, including standard genetic procedures, were as described previously (29–32).
RESULTS
The putative osmosensor Msb2 binds to the membrane-anchor protein Opy2.
Based on the current model of the MSB2 subbranch (Fig. 1B), we hypothesized that Msb2 might interact with the membrane-anchor protein Opy2. To examine whether these proteins interact in vivo, we coexpressed GFP-tagged Opy2ΔSR1 (Opy2ΔSR1-GFP) and HA-tagged Msb2Δ1 (Msb2Δ1-2×HA) in the yeast strain TM257 (ssk2Δ ssk22Δ), and coimmunoprecipitation experiments were conducted. The ΔSR1 and Δ1 mutations were introduced to eliminate the bulk of the glycosylation sites from the respective proteins so that they migrate more uniformly in SDS-PAGE (see Fig. 2B for the Msb2Δ1 mutation and Fig. 3A for the Opy2ΔSR1 mutation). When cell extracts were prepared using a standard lysis buffer containing 0.2% Triton X-100, no binding between Msb2 and Opy2 was detected (Fig. 2A, lane 2). In contrast, when milder detergents, such as digitonin, Brij L23, and Brij 99, were used, robust coprecipitation of Opy2ΔSR1-GFP and Msb2Δ1-2×HA was seen (Fig. 2A, lanes 4, 6, and 8).
FIG 2.

Msb2 binds to Opy2. (A) Effect of detergent on Msb2-Opy2 binding. The yeast strain TM257 (a wild-type strain, except for the presence of nutritional markers) was cotransformed with expression plasmids for Opy2ΔSR1-GFP and Msb2Δ1-2×HA (+) or the vector control (−), both of which were under the control of the GAL1 promoter. Cells were grown in CARaf medium, and expression of the tagged Opy2 and Msb2 was induced by 2% galactose for 2 h. Cell lysates were prepared using buffer A containing the following detergents: 0.2% Triton X-100, 1.0% digitonin, 0.3% Brij L23, or 0.3% Brij 99. Msb2Δ1-2×HA was precipitated from cell lysates using an anti-HA antibody, and coprecipitated Opy2ΔSR1-GFP was probed with an anti-GFP antibody. IP, immunoprecipitation; IB, immunoblotting. (B) Schematic model of Msb2. (Top) The full-length Msb2 molecule. The horizontal black bars represent the structures of Msb2 deletion mutants. (Bottom) An expansion of the HMH domain and its deletion mutants. STR, Ser/Thr-rich domain; HMH, Hkr1-Msb2 homology domain; TM, transmembrane domain. (C) The Msb2 HMH domain is necessary for Msb2-Opy2 interaction. The yeast strain KY562-1 (ssk2Δ ssk22Δ msb2Δ opy2Δ) was cotransformed with expression plasmids for Opy2ΔSR1-GFP and Msb2Δ1-2×HA (no mut., no mutation), the indicated deletion derivatives, or the vector control (vec), all of which were under the control of the GAL1 promoter. Cell growth, preparation of cell lysates, immunoprecipitation, and immunoblotting were as described in the legend to panel B. Buffer A containing 1.0% digitonin was used.
FIG 3.
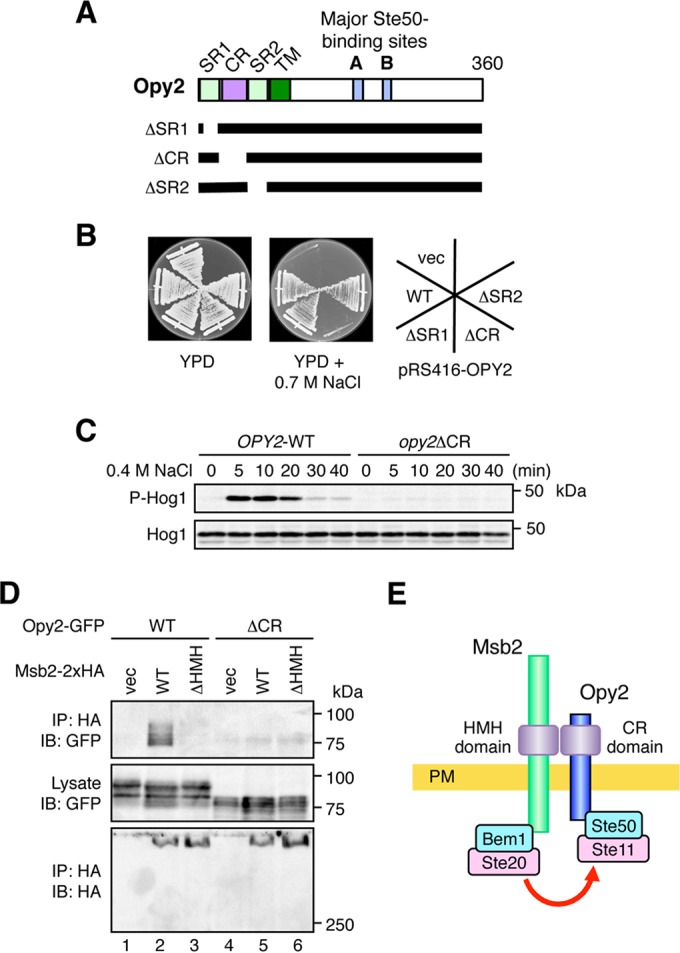
The Opy2 CR domain binds to the Msb2 HMH domain. (A) Schematic model of Opy2. The horizontal black bars represent the structures of the Opy2 deletion mutants. SR1 and SR2, Ser-rich domains 1 and 2, respectively; CR, Cys-rich domain; TM, transmembrane domain. (B) Osmosensitivity of opy2 mutants. The yeast strain KY477 (ssk2Δ ssk22Δ opy2Δ) was transformed with a single-copy plasmid carrying either the wild-type (WT) OPY2 or one of the indicated opy2 deletion constructs. Transformed cells were streaked on yeast extract-peptone-dextrose (YPD) plates in the absence or presence of an osmostressor (0.7 M NaCl). The plates were photographed after 3 days of incubation at 30°C. vec, vector control. (C) Hog1 phosphorylation in response to osmotic stress. KY477 was transformed with either the OPY2 wild-type or opy2ΔCR strain. Cells were collected at the indicated times after addition of 0.4 M NaCl, and the amounts of phosphorylated Hog1 (P-Hog1) and total Hog1 (Hog1) were examined by immunoblotting of the total cell lysate. (D) The Opy2 CR domain is necessary for Msb2-Opy2 interaction. TM257 was cotransformed with expression plasmids for the Opy2-GFP wild type or Opy2ΔCR-GFP (ΔCR), both of which were under the control of the GAL1 promoter, together with Msb2-2×HA (wild type), Msb2-ΔHMH-2×HA, or the vector control. Cell growth, preparation of cell lysates, immunoprecipitation, and immunoblotting were as described in the legend to Fig. 2A. Buffer A containing 1.0% digitonin was used. (E) Schematic model of the Msb2-Opy2 interaction. PM, plasma membrane.
A preliminary analysis indicated that the HMH domain of Msb2 was necessary for its binding to Opy2 (data not shown). To examine which region of the HMH domain is required, we generated a series of short deletion mutants spanning the entire HMH domain (Δ3 through Δ11 in Fig. 2B) and tested their ability to coprecipitate Opy2. Unlike the parental construct (Msb2Δ1-2×HA), none of these HMH domain deletion mutants coprecipitated Opy2 (Fig. 2C), indicating that the entire HMH domain of Msb2 is required for interaction with Opy2.
The Opy2 CR domain binds to the Msb2 HMH domain.
Because the HMH domain is located extracellularly, it likely interacts with an extracellular domain in Opy2. Of the Opy2 extracellular domains (Fig. 3A), we have previously shown that the CR domain is important for osmotic induction of the Hog1-dependent reporter gene (9). We first confirmed that the Opy2 CR domain, but not the SR1 or SR2 domain, is important for adaptation to osmotic stress. For this purpose, we constructed expression plasmids for Opy2 mutants in which one of these domains was deleted (Opy2ΔSR1, Opy2ΔCR, and Opy2ΔSR2). These Opy2 mutants were individually expressed from the OPY2 promoter (POPY2) on a single-copy plasmid in a host strain in which the endogenous OPY2 gene was disrupted (opy2Δ). The host strain also carried the ssk2/22Δ double mutation that inactivated the SLN1 branch. In the absence of plasmid-borne OPY2 expression, this strain cannot survive under high-osmolarity conditions, such as conditions with 0.7 M NaCl (Fig. 3B, vec [vector control]). Expression of wild-type Opy2, Opy2ΔSR1, or Opy2ΔSR2 allowed the strain to survive on high-osmolarity plates. In contrast, cells that express Opy2ΔCR are osmosensitive (Fig. 3B) and cannot activate Hog1 upon osmostress (Fig. 3C). Thus, only the CR domain within the extracellular region of Opy2 is essential for activation of the Hog1 MAPK cascade via the SHO1 branch. We therefore tested if the Opy2 CR domain is required to coprecipitate Msb2. At the same time, we also tested if the full-length Msb2 (containing the glycosylated STR domain) could bind to the full-length Opy2 (containing the glycosylated SR1 domain). Indeed, the full-length Msb2 and Opy2 bound to each other (Fig. 3D, lane 2), and this interaction was dependent on the Msb2 HMH domain (lane 3) and the Opy2 CR domain (lane 5). Thus, we concluded that Msb2 and Opy2 interact through their respective extracellular HMH and CR domains.
Since Msb2 indirectly interacts with Ste20 via the Bem1 scaffold protein and Opy2 is bound to Ste11 via the Ste50 adaptor protein, Msb2-Opy2 binding might contribute to Hog1 activation by promoting the interaction between Ste20 and Ste11 (Fig. 3E). To examine the possibility that the Msb2-Opy2 interaction is regulated by external osmotic conditions, we studied the Msb2-Opy2 interaction in more detail in the experiments described in the following sections.
Eight conserved cysteine residues in the Opy2 CR domain are engaged in disulfide bonding.
We first focused on analysis of the structure of the Opy2 CR domain. The Opy2 CR domain contains eight cysteine residues that are conserved among Opy2 homologs of diverse yeast and fungal species (Fig. 4A). To examine if the cysteines of the Opy2 CR domain are engaged in disulfide bonding, we used maleimide-PEG2-biotin, which biotinylates only reduced and surface-exposed cysteine residues. Opy2ΔSR1-Myc (which contains the wild-type sequence of the CR domain) was barely biotinylated by this reagent (Fig. 4B, lane 1). However, when Opy2ΔSR1-Myc was pretreated with 2-mercaptoethanol (2-ME), it was robustly biotinylated (Fig. 4B, lanes 2 to 4). Thus, at least one pair of cysteine residues forms a disulfide bond that can be biotinylated after reduction.
FIG 4.
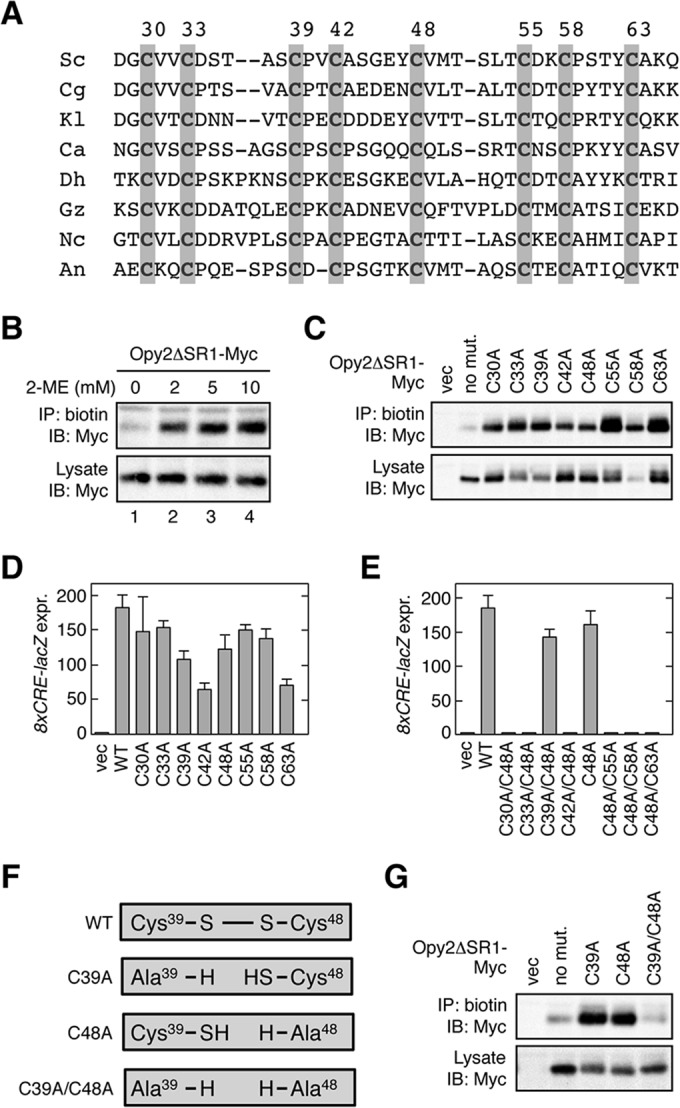
Conserved cysteine residues in the Opy2 CR domain. (A) Comparison of the amino acid sequences of the Opy2 CR domain from various yeast and fungal species. Gray shading highlights conserved cysteine residues. Numbers are amino acid positions in the S. cerevisiae Opy2. Sc, Saccharomyces cerevisiae; Cg, Candida glabrata; Kl, Kluyveromyces lactis; Ca, Candida albicans; Dh, Debaryomyces hansenii; Gz, Gibberella zeae; Nc, Neurospora crassa; An, Aspergillus nidulans. (B) Biotin labeling of reduced cysteine residues in Opy2 using maleimide-PEG2-biotin. KY477 was transformed with a plasmid that expresses Opy2ΔSR1-Myc under the control of the GAL1 promoter. Expression was induced by 2% galactose for 2 h. A membrane fraction was prepared and was incubated with the indicated concentration of 2-ME at 37°C for 20 min. After removal of 2-ME by two rounds of washing, samples were incubated with 0.2 mM maleimide-PEG2-biotin at 30°C for 5 min. The membrane was then solubilized with 1.0% Triton X-100 plus 0.2% SDS. Biotinylated Opy2ΔSR1-Myc was immunoprecipitated using streptavidin-agarose and was detected using an anti-Myc antibody. (C) Biotin labeling of Opy2 Cys-to-Ala mutants using maleimide-PEG2-biotin. KY477 was transformed with the indicated mutants of Opy2ΔSR1-Myc or an empty vector (vec). Expression was induced by 2% galactose for 2 h. A membrane fraction was prepared and incubated with 0.2 mM maleimide-PEG2-biotin at 30°C for 5 min. Detection of biotinylation was as described in the legend to panel B. (D and E) Osmostress-induced expression of the Hog1-specific reporter gene 8×CRE-lacZ. KY477 was cotransformed with the reporter plasmid pRS416-8×CRE-lacZ and a single-copy plasmid carrying the wild-type (WT) OPY2 gene (pRS414-OPY2) or the indicated mutants. Cells were grown in CAD medium and were stimulated with 0.4 M NaCl for 30 min. The activity of β-galactosidase in cell extracts was normalized to the cell density and is expressed as Miller units (35). All reporter assays were carried out in triplicate (or more times) with independent cultures. Error bars represent SDs. expr., expression. (F) Effects of Cys-to-Ala mutations on a disulfide bond. If Cys39 and Cys48 form a disulfide bond in the wild-type Opy2 molecule, either the C39A or the C48A mutation generates a reactive cysteine residue. In contrast, the C39A/C48A double mutant should have no reactive cysteine. (G) Biotin labeling of Opy2 Cys-to-Ala mutants using maleimide-PEG2-biotin. Expression of Opy2ΔSR1-Myc mutants, labeling of cysteines with 0.2 mM maleimide-PEG2-biotin, and detection of biotinylation were conducted as described in the legend to panel B.
We next determined which of the eight cysteines were engaged in disulfide bonding by individually mutating each of the eight cysteines to alanine, followed by maleimide-PEG2-biotin labeling. Mutation of either cysteine of a disulfide-bonded pair to alanine would render the other cysteine accessible to the biotinylation reagent. In fact, mutation of any one of the eight cysteines to alanine allowed efficient biotinylation of the mutants (Fig. 4C), indicating that all eight of the cysteines in the CR domain are engaged in disulfide bonding. All eight mutants with a single Cys-to-Ala mutation were functional, as they could support osmotic induction of the Hog1-dependent reporter gene 8×CRE-lacZ (Fig. 4D). Thus, no single cysteine was absolutely required for Opy2 function. However, when two cysteine residues were simultaneously mutated to alanine, some mutation combinations severely compromised Opy2 function, whereas other combinations had little adverse effect. For example, when the Cys48-to-Ala (C48A) mutation was combined with another Cys-to-Ala mutation, most double mutants were nonfunctional (Fig. 4E). One exception, however, was the C39A/C48A double mutant.
As schematically shown in Fig. 4F, if C39 and C48 formed a disulfide bond, then the C39A/C48A double mutation would not disrupt any additional bond and its phenotype should be similar to that of the mutants with C39A and C48A single mutations. The formation of a disulfide bond between C39 and C48 was more directly examined by biotin labeling using maleimide-PEG2-biotin. Whereas the C39A and C48A single mutants were efficiently biotinylated, the C39A/C48A double mutant was not biotinylated at all (Fig. 4G), indicating that the double mutant has no reduced cysteine. Thus, we concluded that C39 and C48 form a disulfide bond.
Disulfide bonding pattern in the Opy2 CR domain.
We next determined how the remaining six cysteines were paired, based on the above-described finding that at least two disulfide bonds need to be disrupted to inactivate Opy2. When C63A was combined with another Cys-to-Ala mutation, all but the C39A/C63A and C42A/C63A combinations were completely defective (Fig. 5A), suggesting that C63 is disulfide bonded to either C39 or C42. However, as we have shown as described above that C39 is unambiguously paired with C48, we concluded that C63 forms a disulfide bond with C42. That the C39A/C63A double mutant has significant activity is likely due to formation of a compensating bond between C48 and C42, as these cysteine residues are likely to be close to each other within Opy2 (see the summary scheme in Fig. 5E).
FIG 5.
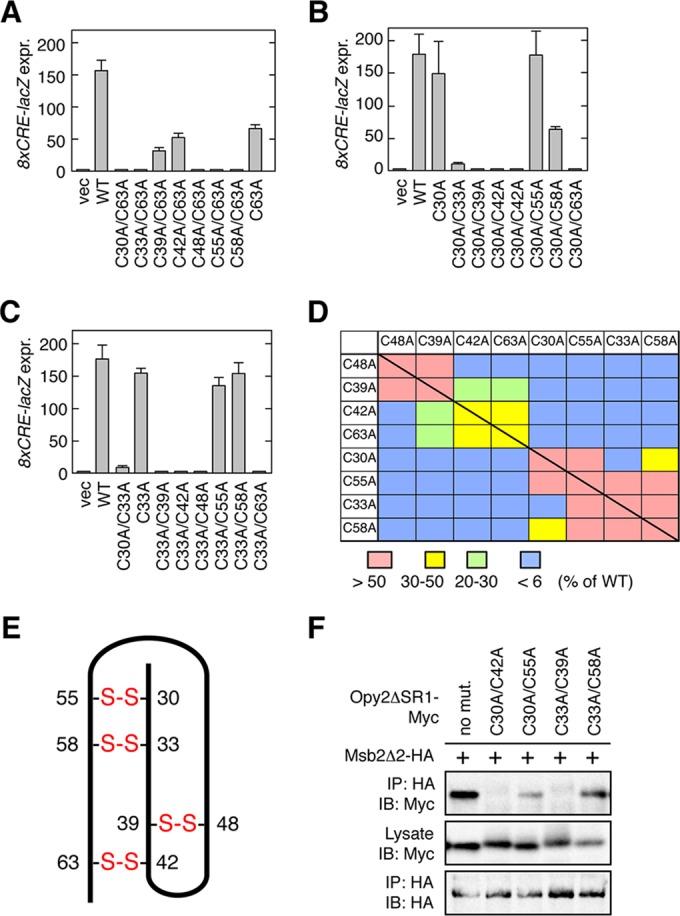
Disulfide bonding pattern of the Opy2 CR domain. (A to C) Osmostress-induced expression of the Hog1-specific reporter gene 8×CRE-lacZ was assayed as described in the legend to Fig. 4D. Error bars represent SDs (n = 3 or 4). (D) Summary of Hog1-specific gene expression assays. Each block (except the blocks on the diagonal line) represents an opy2 double mutant that contains one of the mutations indicated in the top row and one of the mutations indicated in the left column. The blocks on the diagonal line represent single mutants. The strength of reporter gene expression of the mutants was normalized to that of the wild-type control and is indicated in color. (E) Deduced disulfide bonding pattern of the Opy2 CR domain. The bold line represents the peptide chain of the CR domain. The numbers refer to the amino acid positions of the eight cysteine residues. Disulfide bonds are indicated in red. (F) Msb2-Opy2 binding assays. TM257 was cotransformed with expression plasmids for Msb2Δ2-HA (+) and Opy2ΔSR1-Myc (containing the indicated mutations), both of which were under the control of the GAL1 promoter. Cell growth, preparation of cell lysates, immunoprecipitation, and immunoblotting were as described in the legend to Fig. 2A, except for the antibodies used. Buffer A containing 1.0% digitonin was used.
When C30A was combined with another Cys-to-Ala mutation, mutants with all but two combinations of mutations were completely defective (Fig. 5B). Of the two active mutants, the C30A/C55A mutant was as active as the wild type, whereas the activity of the C30A/C58A mutant was substantially compromised. Thus, C30 is most likely disulfide bonded to C55. Finally, when the C33A mutation was combined with another Cys-to-Ala mutation, all mutants except those with the C33A/C55A and C33A/C58A combinations were completely defective (Fig. 5C). As C55 was likely paired to C30, we concluded that C33 is disulfide bonded to C58. That the mutant with C33A/C55A combination had significant activity is likely due to the formation of a compensatory disulfide bond between C30 and C58 (Fig. 5E).
The activities of all possible double C-to-A mutants, including those that are not discussed above, are summarized in Fig. 5D, and the deduced pattern of disulfide bonds are shown in Fig. 5E. These data suggest that the eight cysteines are clustered into two groups within the Opy2 CR domain: the group of C39, C42, C48, and C63 and the group of C30, C33, C55, and C58. A C-to-A mutation of two cysteines belonging to the same group resulted, in general, in less severe functional effects than a double C-to-A mutation involving cysteines from different groups. This effect is likely due to the formation of a compensatory disulfide bond between the two remaining cysteines in the same group. Although the bonding pattern shown in Fig. 5E is highly plausible, it is clear that it must be confirmed by direct chemical analyses in the future.
To test the hypothesis that Msb2-Opy2 binding is essential for osmotic activation of the Hog1 MAPK, we examined Msb2 binding to several Opy2 double C-to-A mutants. Two mutants (C30A/C55A and C33A/C58A) that could activate Hog1 could bind to Msb2, whereas two other mutants (C30A/C42A and C33A/C39A) that could not activate Hog1 could not coprecipitate Msb2 (Fig. 5F). We therefore concluded that Msb2-Opy2 binding via their extracellular domains is essential for Hog1 activation by osmostress.
Direct interaction between the Opy2 CR domain and the Msb2 HMH domain.
If hyperosmolarity affects the way in which Opy2 and Msb2 interact, it could be a potential regulatory mechanism for Hog1 activation. The strength of Msb2-Opy2 binding, however, was not substantially altered by osmostress (Fig. 6A). Thus, we sought to detect more subtle changes in the Msb2-Opy2 interaction that might affect the conformation of the Msb2-Opy2 complex. The strategy that we employed was based on the following logic.
FIG 6.

Direct interaction between the Opy2 CR domain and the Msb2 HMH domain. (A) Effect of osmostress on Msb2-Opy2 binding assays. TM257 was cotransformed with expression plasmids for Opy2ΔSR1-Myc and Msb2Δ2-HA, both of which were under the control of the GAL1 promoter. Cells were grown in CARaf medium, and expression of the tagged Opy2 and Msb2 was induced by 2% galactose for 2 h. The cells were then exposed (+) or not (−) to 1 M NaCl for 5 min. Preparation of cell lysates, immunoprecipitation, and immunoblotting were as described in the legend to Fig. 2A, except for the antibodies used. Buffer A containing 1.0% digitonin was used. (B) Predicted surface accessibility of the amino acid residues in the HMH domain. The HMH domain sequences of Msb2 and Hkr1 are aligned. Residues predicted to be solvent inaccessible are highlighted by gray shading. The blue triangle denotes the site of the 62-amino-acid-residue insertion sequence in Hkr1 relative to the Msb2 sequence. The surface-exposed Ser or Thr residues that were mutated in this work are shown in red. Numbers are amino acid positions in Msb2. (C) Induction of the Hog1-specific reporter gene 8×CRE-lacZ by the hyperactive Msb2Δ1 mutant. TM257 was cotransformed with the reporter plasmid pRS414-8×CRE-lacZ and another plasmid carrying the galactose-inducible Msb2Δ1-2×HA (or its indicated mutants). The cells were grown in CARaf medium, and expression of Msb2Δ1-2×HA was induced by 2% galactose for 2 h. The activity of β-galactosidase in cell extracts was normalized to the cell density and is expressed as Miller units (35). Error bars represent SDs (n = 4). (D) Accessibility of the cysteine residues in Msb2 Ser/Thr-to-Cys mutants to maleimide-PEG2-biotin. TM257 was transformed with a plasmid that expresses Msb2Δ2-2×HA without other mutations (no mut.) or with the indicated mutations under the control of the GAL1 promoter. Expression was induced by 2% galactose for 2 h. Labeling and detection of cysteines accessible to 0.2 mM maleimide-PEG2-biotin were conducted essentially as described in the legend to Fig. 4B. (E and F) Direct disulfide bonding between Opy2 and Msb2. TM257 was cotransformed with a plasmid that expresses the indicated mutants of Opy2ΔSR1-Myc together with another plasmid that expresses the indicated mutants of Msb2Δ2-HA (E) or of Msb2Δ1-2×HA (F). Expression of these proteins is under the control of the GAL1 promoter. The cells were grown in CARaf medium, and expression of the tagged Opy2 and Msb2 was induced by 2% galactose for 2 h. Cell lysates were prepared using buffer A containing 0.2% Triton X-100 with (+) or without (−) 2 mM DTT. HA-tagged Msb2 was precipitated from cell lysates using an anti-HA antibody, and coprecipitated Opy2ΔSR1-Myc was probed with an anti-Myc antibody. Total expression levels of the tagged Opy2 and Msb2 proteins were uniform among the samples but are not shown in panel F to save space. (G) Schematic representation of the direct Opy2-Msb2 interaction. The numbers in the green boxes indicate amino acid positions in Msb2. Blue lines indicate disulfide bonding between Msb2 and Opy2. Other symbols are as described in the legend to Fig. 5E.
If a reactive cysteine residue in the Msb2 HMH domain and another in the Opy2 CR domain are very close to each other in the Msb2-Opy2 complex, they might form a disulfide bond. If so, then conformational changes in the Msb2-Opy2 complex might be detected as changes in the extent of disulfide formation between two cysteine residues. As all eight cysteines in Opy2 are oxidized, they are unreactive. However, when one of the eight cysteines is mutated to alanine, the partner cysteine becomes available for a cross-linking reaction.
As for Msb2, it has only two native cysteine residues: Cys-7 in the signal sequence is absent from the mature molecule, and Cys-1198 in the TM domain is not accessible to reagents in the extracellular medium. Thus, if a surface-exposed residue in the HMH domain is mutated to cysteine, it should be the only cysteine in the entire Msb2 molecule available to a cross-linking reaction. In order to best determine which residues in Msb2 to mutate to cysteine for this cross-linking analysis, we first predicted the surface accessibility of the amino acid residues in the Msb2 HMH domain using the NetSurfP program (http://www.cbs.dtu.dk/services/NetSurfP/)(38). Residues predicted to be solvent nonaccessible (i.e., buried in the hydrophobic core of the domain) are highlighted by shading in Fig. 6B. The predictions for the HMH domain of Msb2 and the homologous domain in Hkr1 are very similar to each other, attaching a high degree of confidence to the prediction. We then individually mutated several Ser and Thr residues that were predicted to be surface exposed to Cys (indicated by red letters in Fig. 6B). These single Cys substitution mutants were functionally indistinguishable from their wild-type counterpart (Fig. 6C). We then examined if those Cys residues could be labeled with maleimide-PEG2-biotin. As expected, the parental Msb2 construct (Msb2Δ2-2×HA) was not biotinylated at all (Fig. 6D, lane 1). In contrast, all of the seven Cys substitution mutants were efficiently biotinylated (Fig. 6D, lanes 2 to 8), indicating that these Cys residues are accessible to the cross-linking reagent.
To examine the interaction of the Msb2 HMH Cys mutants with the Opy2 CR Cys mutants, we then coexpressed one of the Msb2 single Cys substitution mutants and one of the Opy2 Cys-to-Ala mutants in all possible combinations in an msb2Δ opy2Δ strain. Cell lysates were prepared under nonreducing conditions using a lysis buffer containing 0.2% Triton X-100, and coprecipitation of Opy2 and Msb2 was examined. Under these conditions, Triton X-100 disrupts the noncovalent association between Opy2 and Msb2 (Fig. 2A), and thus, coprecipitation occurs only when Opy2 and Msb2 are covalently bound by disulfide bonding. Under these conditions, Opy2 C48A (which has the reactive Cys39) and Msb2 T986C formed a disulfide bond, as did Opy2 C63A (which has the reactive Cys42) and Msb2 S1023C (Fig. 6E). Their coprecipitation was completely inhibited by inclusion of DTT in the lysis buffer, indicating that they are disulfide bonded. We deduced from these results that Opy2 Cys39 and Msb2 Thr986 are close to each other within the Msb2-Opy2 complex, as are Opy2 Cys42 and Msb2 S1023. By using this strategy, we identified several additional pairs of cysteines in Msb2 and Opy2 that could form disulfide bonds (Fig. 6F), which are schematically summarized in Fig. 6G.
These results suggest that there are specific interfaces on the CR domain and the HMH domain through which they directly interact. However, disulfide bonding of these pairs was neither enhanced nor inhibited by external high osmolarity (Fig. 7A and data not shown). Thus, these limited analyses did not provide any evidence for or against structural changes in the Msb2-Opy2 complex induced by osmostress.
FIG 7.

Probing the Opy2-Msb2 interaction using molecular rulers. (A) Effect of osmostress on direct disulfide bonding between Opy2 and Msb2. TM257 was cotransformed with plasmids that express the indicated mutants of Opy2ΔSR1-Myc and Msb2Δ2-HA under the control of the GAL1 promoter. The cells were exposed to 1 M NaCl for the indicated times. Preparation of cell lysates, immunoprecipitation, and immunoblotting were performed as described in the legend to Fig. 6E. (B) Structures of two molecular rulers. M-8-M, octane-1,8-diyl bismethanethiosulfonate; M-8-O2-M, 3,6-dioxaoctane-1,8-diyl bismethanethiosulfonate. (C and D) Chemical cross-linking between Opy2 and Msb2 using the bifunctional cross-linker M-10-M. KY595-2 was cotransformed with plasmids that express the indicated mutants of Opy2ΔSR1-Myc and Msb2Δ1-2×HA. Expression of these proteins is under the control of the GAL1 promoter. The cells were grown in CARaf medium, and expression of the tagged Opy2 and Msb2 was induced by 2% galactose for 2 h. The intact cells were treated with 0.4 mM M-10-M for 5 min at 30°C. Cell lysates were prepared using lysis buffer containing 0.2% Triton X-100. Immunoprecipitation and immunoblotting were conducted as described in the legend to Fig. 6E. Total expression levels of the epitope-tagged Opy2 and Msb2 proteins were uniform among the samples but are not shown in panel D to save space. (E and F) Cross-linking between Opy2 and Msb2 using bifunctional cross-linkers of different lengths. TM257 was cotransformed with plasmids that express Opy2ΔSR1-Myc-C39A and Msb2Δ2-HA-S1023C. The cells were grown in CARaf medium, and expression of the tagged Opy2 and Msb2 was induced by 2% galactose for 2 h. The intact cells were treated with the indicated bifunctional cross-linkers (0.4 mM) for 5 min at 30°C. Preparation of cell lysates, immunoprecipitation, and immunoblotting were conducted as described in the legend to panel C. (F) Cells were exposed (+) or not (−) to 1 M NaCl during the cross-linking reaction. The intensities of the cross-linked Opy2-Myc (top row) were quantified using ImageJ software (37) and normalized to the intensities of the corresponding total Opy2-Myc (middle row). The intensity of the strongest band was set to 100%, and the intensity of each band relative to that of the strongest band is shown below the top row.
Probing of the Opy2-Msb2 interaction using molecular rulers.
To probe for conformational changes of the Msb2-Opy2 complex, described above, that might involve additional cysteine residues, we used a set of bifunctional thiol-reactive cross-linkers of different spacer lengths (molecular rulers) (39). These cross-linkers are composed of two methanethiosulfonate (MTS) groups connected by an alkane chain of various lengths. Two examples, octane-1,8-diyl bismethanethiosulfonate (M-8-M) and 3,6-dioxaoctane-1,8-diyl bismethanethiosulfonate (M-8-O2-M), are shown in Fig. 7B. These two linkers are similar in length (approximate S-S distance, 13 Å), but M-8-O2-M is more hydrophilic than M-8-M. Using these and other cross-linkers in the set, it is possible to determine the distances between the Cys residues of Opy2 and Msb2 in the presence or absence of osmotic stress.
Initially, each of the eight Opy2 C-to-A mutants (Myc tagged) was combinatorially coexpressed with each of six Msb2 Cys substitution mutants (HA tagged), and their reactive cysteine residues were cross-linked with M-10-M in intact cells. After excess reagents were removed, cell extracts were prepared using a lysis buffer containing Triton X-100 to ensure that only covalently linked Opy2-Msb2 complexes were coimmunoprecipitated. HA-tagged Msb2 was immunoprecipitated, and coimmunoprecipitated Myc-tagged Opy2 was detected by immunoblotting. A typical result (Fig. 7C) shows that Opy2 C48A (which has Cys39 available for reaction) was efficiently cross-linked to Msb2 T986C, T997C, and S1023C but not to three other Msb2 Cys mutants. As seen in the full data set (Fig. 7D), the ability of the various cysteine pairs to be cross-linked was highly variable, suggesting that it is dependent on the distance and other geometric relationships between the two cysteines.
Although Opy2 C39A and Msb2 S1023C could not form a direct disulfide bond (Fig. 6F), they were cross-linked with M-10-M (Fig. 7D). Moreover, as shown in Fig. 7E, Opy2 C39A and Msb2 S1023C were cross-linked by M-5-M (approximate S-S distance, 9.1 Å) or longer cross-linkers but not by M-4-M (approximate S-S distance, 7.8 Å) or a shorter cross-linker. We therefore deduced that Cys48 in Opy2 C39A and Cys1023 in Msb2 S1023C are separated by about 9 Å, when Opy2 and Msb2 form a heterodimer.
We then examined if the distance between these Cys residues could be affected by osmostress. In the presence of 1 M NaCl, cross-linking of these cysteines by M-5-M was slightly decreased (Fig. 7F). This effect was more pronounced when the same pair of cysteine residues was cross-linked with hydrophilic cross-linkers, such as M-8-O2-M and M-11-O3-M. These cross-linkers efficiently cross-linked Opy2 C39A to Msb2 S1023C in the absence of osmostress (Fig. 8A). However, cross-linking by these linkers was strongly inhibited in the presence of 1 M NaCl. Osmostress did not inhibit cross-linking reactions in general, including those that involved either Opy2 C39A or Msb2 S1023C (Fig. 8B). Thus, the observed inhibition was specific to the combination of Opy2 C39A and Msb2 S1023C. Furthermore, their cross-linking was inhibited not only by ionic NaCl (Fig. 8C) but also by nonionic sorbitol (Fig. 8D), indicating that the inhibition was caused by a higher osmolarity rather than by specific ions. Thus, we concluded that osmostress induces specific (though undefined) structural changes at the interface between the CR domain of Opy2 and the HMH domain of Msb2. Speculations over the possible implications of these findings are provided in the Discussion.
FIG 8.

Effect of osmostress on the Opy2-Msb2 interaction. (A and B) Cross-linking of Opy2 and Msb2 using cross-linkers with oxygen substitutions. TM257 was cotransformed with plasmids that express the indicated mutants of Opy2ΔSR1-Myc and Msb2Δ2-HA. The cells were grown in CARaf medium, and expression of the tagged Opy2 and Msb2 was induced by 2% galactose for 2 h. The intact cells were treated with the indicated bifunctional cross-linkers (0.4 mM) in the presence (+) or absence (−) of 1 M NaCl for 5 min at 30°C. Immunoprecipitation and immunoblotting were conducted as described in the legend to Fig. 7C. Quantification of the cross-linked Opy2-Myc was conducted as described in the legend to Fig. 7F. (C and D) Effects of different osmostressors on chemical cross-linking. Opy2ΔSR1-Myc-C39A and Msb2Δ2-HA-S1023C were expressed in TM257 as described in the legend to panel A. The intact cells were treated with 0.4 mM M-8-O2-M in the presence of the indicated concentrations of NaCl (C) or sorbitol (D) for 5 min at 30°C. Immunoprecipitation and immunoblotting were conducted as described in the legend to Fig. 7C.
DISCUSSION
Structure and function of the CR domain of Opy2.
In this study, we showed that the extracellular cysteine-rich (CR) domain of Opy2 directly binds to the HMH domain of Msb2. The compact eight-cysteine motif of Opy2 is well conserved among yeast and fungal species but is not found in any other taxonomic group. A search of the NCBI genome database indicated that homologs of Msb2 with the HMH domain are also found among yeast and fungal species but not in any other taxonomic group. Thus, the binding of Opy2 to Msb2 through their CR and HMH domains is likely to be an evolutionarily conserved signaling mechanism within yeast and fungal species.
We deduced the connectivity of the eight cysteine residues in Opy2 on the basis of the mutant phenotypes. Clearly, a more direct biochemical determination, for example, using mass spectrometry (40), is desirable. However, the small size and the high cysteine density of the CR domain make it difficult to separate the neighboring cysteines by proteolytic or chemical cleavage, which is necessary for any biochemical method. The availability of the functionally intact Cys-to-Ala mutants of the present study might be helpful in solving this problem in the future.
The deduced disulfide pattern of the CR domain of Opy2 is consistent with the mutant phenotypes observed. For example, both C42A and C63A substantially reduced Opy2 activity, likely because the C42—C63 bond is necessary to prevent the C-terminal end of the CR domain from unfolding. Naturally, the phenotype of the C42A/C63A double mutant is no worse than that of the single mutants. In contrast, disruption of either C30—C55 or C33—C58 had almost no adverse effect. Because these bonds covalently link the same regions of the CR domain, either bond alone is sufficient to maintain the proper conformation. When both bonds are disrupted, as in the C30A/C33A mutant, the mutant has a very low level of activity (Fig. 5C). Curiously, the double mutation C30A/C58A, which is expected to disrupt both disulfide bonds, retains substantial activity. In this double mutant, it is likely that a compensatory disulfide bond is formed between C33 and C55, to maintain the proper conformation.
Extracellular protein-binding domains often contain multiple disulfide bonds. Well-known examples include the Frizzled-like proteins, which contain 5 conserved disulfide bonds (41), and the epidermal growth factor (EGF)-like proteins, which contain 3 conserved disulfide bonds (42). To examine if the Opy2 CR domain is related to any previously recognized cysteine-rich domain, we consulted the classification of small, disulfide-rich protein domains compiled by Cheek et al. (43). According to their classification, the topology of the Opy2 CR domain is identical to that of the protease inhibitor elafin. Elafin contains an eight-cysteine domain termed the whey acidic protein four disulfide core (WFDC). WFDC domains are found in numerous proteins in both vertebrates and invertebrates (44) and consist of approximately 40 to 50 amino acids, including eight conserved cysteine residues (45). A comparison of the elafin WFDC domain with the Opy2 CR domain shows that they are similar in size and have the same topology (Fig. 9). Nonetheless, they are unlikely to be evolutionarily related for a number of reasons. In particular, the fifth and sixth cysteines are always contiguous in the WFDC domains, whereas those in Opy2 homologs are separated by 6 or 7 amino acids (Fig. 4A). Thus, Opy2 defines a new class of disulfide-rich domain.
FIG 9.
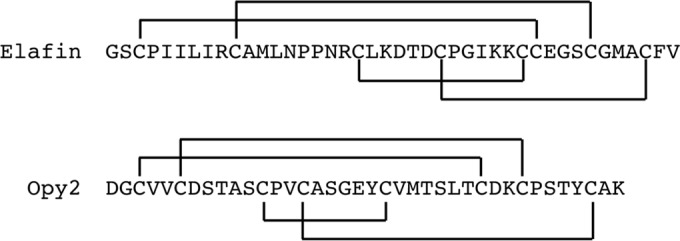
Disulfide connectivity patterns of elafin and Opy2. The disulfide connectivity patterns of the human elafin WFDC domain and the yeast Opy2 CR domain are compared.
Role of the Msb2-Opy2 complex in the Hog1 MAPK cascade.
In this work, we also showed that the Msb2 HMH domain and the Opy2 CR domain directly interact. We considered how this finding might relate to the activation mechanism of the Hog1 MAPK cascade. Accumulating evidence suggests that activation of the Hog1 MAPK through the SHO1 branch involves at least two osmotically regulated steps (activation steps 1 and 2 in Fig. 10). In step 1, Ste11 is activated by the Cdc42-dependent kinase Ste20 (or its paralog, Cla4). Ste20 phosphorylates the N-terminal inhibitory domain of Ste11 to unleash the catalytic activity of the C-terminal kinase domain (46). In step 2, the signal is amplified by enhancing the interaction between activated Ste11 and its substrate, Pbs2 (9). As for the mechanism of step 2, we recently showed that osmostress increases the interaction between Ste50 and Sho1 (9), which should facilitate the interaction between Ste11 and Pbs2. Thus, step 2 is absolutely dependent on the presence of Sho1. In the absence of Sho1, however, there is still some upregulation of the Hog1-dependent genes by osmostress, which requires the presence of Msb2 (14). Here, we propose that this residual response is due to osmotic activation of step 1, which involves Msb2.
FIG 10.
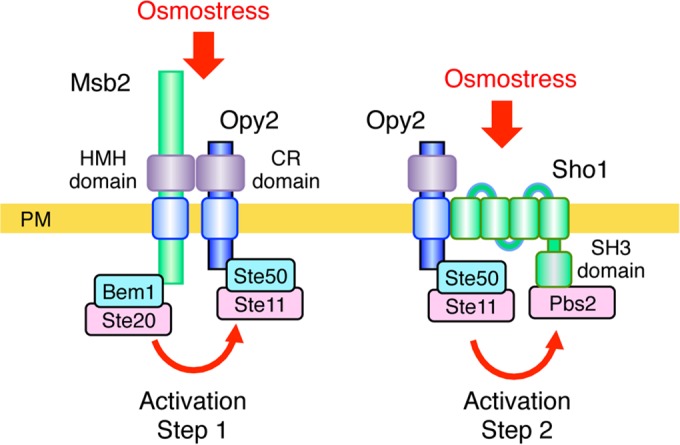
Proposed two-step activation mechanism of the Hog1 MAPK pathway. In activation step 1, increased external osmolarity induces conformational changes in the complex formed between the Msb2 HMH domain and the Opy2 CR domain. This conformational change is transmitted across the plasma membrane (PM) to induce an interaction between the associated Ste20 and Ste11 kinases. In activation step 2, increased osmolarity induces structural changes in the Sho1 TM domains, resulting in an increased interaction between Ste11 and Pbs2 (9).
In step 1, activated Ste20 must interact with Ste11. However, because no direct docking interaction between Ste20 and Ste11 is known, an indirect docking mechanism is likely to play a role in facilitation of efficient activation of Ste11 by Ste20. In theory, the interaction between Msb2 and Opy2 could provide such an indirect docking mechanism, because Msb2 is associated with Ste20 via the cytoplasmic scaffold protein Bem1 (12) and Opy2 is associated with Ste11 via the adaptor protein Ste50 (19–21, 23, 24, 34). Indeed, using an enzymatic in vivo protein proximity assay (the M-track assay), Zuzuarregui et al. (47) demonstrated that Msb2 and Ste11 do interact. The M-track assay does not require that the two proteins directly bind but only that they approach near enough so that their associated histone methyltransferase and histone moiety, respectively, can interact (48). Thus, their findings that the Msb2-Ste11 interaction is abolished in an opy2Δ or a ste50Δ mutant (47) are consistent with the interpretation that Msb2 interacts with Ste11 indirectly through Opy2 and Ste50, as depicted in Fig. 10.
Zuzuarregui et al. also found that the Msb2-Ste11 interaction increased in the presence of osmostress, especially when there was no feedback interference by activated Hog1 (47). On the other hand, we did not find any osmostress-induced increase in Msb2-Opy2 binding. Similarly, there is no evidence to suggest any increase in the strength of Opy2-Ste50, Ste50-Ste11, Msb2-Bem1, or Bem1-Ste20 binding in the presence of osmostress (12, 23, 24). Thus, the increased M-track assay signal between Msb2 and Ste11 likely reflects an osmostress-induced change in the conformation of the Msb2-Opy2 complex, resulting in a change in the proximity of the associated Ste20 and Ste11. Our observation that chemical cross-linking between a specific pair of cysteine residues in Msb2 and Opy2 is highly sensitive to osmostress supports such a conformational change. Thus, we propose that the Msb2-Opy2 complex or, more precisely, the complex between the HMH domain of Msb2 and the CR domain of Opy2 serves an osmosensor function through an induced conformational change. Clearly, further investigation of the dynamic structural properties of the Msb2-Opy2 complex is warranted.
ACKNOWLEDGMENTS
We thank Hiroyuki Fukuda and Akiko Nishimura for helpful discussions, Miho Nagoya for her excellent technical assistance, and Pauline O'Grady for her critical reading and editing of the manuscript.
We have no competing financial interests to declare.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Hohmann S. 2002. Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev 66:300–372. doi: 10.1128/MMBR.66.2.300-372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saito H, Posas F. 2012. Response to hyperosmotic stress. Genetics 192:289–318. doi: 10.1534/genetics.112.140863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Nadal E, Posas F. 2015. Osmostress-induced gene expression—a model to understand how stress-activated protein kinases (SAPKs) regulate transcription. FEBS J 282:3275–3285. doi: 10.1111/febs.13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Posas F, Wurgler-Murphy SM, Maeda T, Witten EA, Thai TC, Saito H. 1996. Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1-YPD1-SSK1 “two-component” osmosensor. Cell 86:865–875. doi: 10.1016/S0092-8674(00)80162-2. [DOI] [PubMed] [Google Scholar]
- 5.Maeda T, Takekawa M, Saito H. 1995. Activation of yeast PBS2 MAPKK by MAPKKKs or by binding of an SH3-containing osmosensor. Science 269:554–558. doi: 10.1126/science.7624781. [DOI] [PubMed] [Google Scholar]
- 6.Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. 1993. An osmosensing signal transduction pathway in yeast. Science 259:1760–1763. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- 7.Maeda T, Wurgler-Murphy SM, Saito H. 1994. A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature 369:242–245. doi: 10.1038/369242a0. [DOI] [PubMed] [Google Scholar]
- 8.Boguslawski G. 1992. PBS2, a yeast gene encoding a putative protein kinase, interacts with the RAS2 pathway and affects osmotic sensitivity of Saccharomyces cerevisiae. J Gen Microbiol 138:2425–2432. doi: 10.1099/00221287-138-11-2425. [DOI] [PubMed] [Google Scholar]
- 9.Tatebayashi K, Yamamoto K, Nagoya M, Takayama T, Nishimura A, Sakurai M, Momma T, Saito H. 2015. Osmosensing and scaffolding functions of the oligomeric four-transmembrane domain osmosensor Sho1. Nat Commun 6:6975. doi: 10.1038/ncomms7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Posas F, Saito H. 1997. Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science 276:1702–1705. doi: 10.1126/science.276.5319.1702. [DOI] [PubMed] [Google Scholar]
- 11.Tatebayashi K, Tanaka K, Yang H-Y, Yamamoto K, Matsushita Y, Tomida T, Imai M, Saito H. 2007. Transmembrane mucins Hkr1 and Msb2 are putative osmosensors in the SHO1 branch of yeast HOG pathway. EMBO J 26:3521–3533. doi: 10.1038/sj.emboj.7601796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka K, Tatebayashi K, Nishimura A, Yamamoto K, Yang H-Y, Saito H. 2014. Yeast osmosensors Hkr1 and Msb2 activate the Hog1 MAPK cascade by different mechanisms. Sci Signal 7:ra21. doi: 10.1126/scisignal.2004780. [DOI] [PubMed] [Google Scholar]
- 13.Bender A, Pringle JR. 1989. Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes including the ras-related gene RSR1. Proc Natl Acad Sci U S A 86:9976–9980. doi: 10.1073/pnas.86.24.9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Rourke SM, Herskowitz I. 2002. A third osmosensing branch in Saccharomyces cerevisiae requires the Msb2 protein and functions in parallel with the Sho1 branch. Mol Cell Biol 22:4739–4749. doi: 10.1128/MCB.22.13.4739-4749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cullen PJ, Sabbagh W Jr, Graham E, Irick MM, van Olden EK, Neal C, Delrow J, Bardwell L, Sprague GF Jr. 2004. A signaling mucin at the head of the Cdc42- and MAPK-dependent filamentous growth pathway in yeast. Genes Dev 18:1695–1708. doi: 10.1101/gad.1178604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitoniak A, Birkaya B, Dionne HM, Vadaie N, Cullen PJ. 2009. The signaling mucins Msb2 and Hkr1 differentially regulate the filamentation mitogen-activated protein kinase pathway and contribute to a multimodal response. Mol Biol Cell 20:3101–3114. doi: 10.1091/mbc.E08-07-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zarrinpar A, Park S-H, Lim WA. 2003. Optimization of specificity in a cellular protein interaction network by negative selection. Nature 426:676–680. doi: 10.1038/nature02178. [DOI] [PubMed] [Google Scholar]
- 18.Ramezani-Rad M. 2003. The role of adaptor protein Ste50-dependent regulation of the MAPKKK Ste11 in multiple signalling pathways of yeast. Curr Genet 43:161–170. [DOI] [PubMed] [Google Scholar]
- 19.Wu C, Leberer E, Thomas DY, Whiteway M. 1999. Functional characterization of the interaction of Ste50p with Ste11p MAPKKK in Saccharomyces cerevisiae. Mol Biol Cell 10:2425–2440. doi: 10.1091/mbc.10.7.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Posas F, Witten EA, Saito H. 1998. Requirement of STE50 for osmostress-induced activation of the STE11 mitogen-activated protein kinase kinase kinase in the high-osmolarity glycerol response pathway. Mol Cell Biol 18:5788–5796. doi: 10.1128/MCB.18.10.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C, Jansen G, Zhang J, Thomas DY, Whiteway M. 2006. Adaptor protein Ste50p links the Ste11p MEKK to the HOG pathway through plasma membrane association. Genes Dev 20:734–746. doi: 10.1101/gad.1375706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Truckses DM, Bloomekatz JE, Thorner J. 2006. The RA domain of Ste50 adaptor protein is required for delivery of Ste11 to the plasma membrane in the filamentous growth signaling pathway of the yeast Saccharomyces cerevisiae. Mol Cell Biol 26:912–928. doi: 10.1128/MCB.26.3.912-928.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto K, Tatebayashi K, Tanaka K, Saito H. 2010. Dynamic control of yeast MAP kinase network by induced association and dissociation between the Ste50 scaffold and the Opy2 membrane anchor. Mol Cell 40:87–98. doi: 10.1016/j.molcel.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Ekiel I, Sulea T, Jansen G, Kowalik M, Minailiuc O, Cheng J, Harcus D, Cygler M, Whiteway M, Wu C. 2009. Binding the atypical RA domain of Ste50p to the unfolded Opy2p cytoplasmic tail is essential for the high-osmolarity glycerol pathway. Mol Biol Cell 20:5117–5126. doi: 10.1091/mbc.E09-07-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H-Y, Tatebayashi K, Yamamoto K, Saito H. 2009. Glycosylation defects activate filamentous growth Kss1 MAPK and inhibit osmoregulatory Hog1 MAPK. EMBO J 28:1380–1391. doi: 10.1038/emboj.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karunanithi S, Cullen PJ. 2012. The filamentous growth MAPK pathway responds to glucose starvation through the Mig1/2 transcriptional repressors in Saccharomyces cerevisiae. Genetics 192:869–887. doi: 10.1534/genetics.112.142661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vadaie N, Dionne H, Akajagbor DS, Nickerson SR, Krysan DJ, Cullen PJ. 2008. Cleavage of the signaling mucin Msb2 by the aspartyl protease Yps1 is required for MAPK activation in yeast. J Cell Biol 181:1073–1081. doi: 10.1083/jcb.200704079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adhikari H, Vadaie N, Chow J, Caccamise LM, Chavel CA, Li B, Bowitch A, Stefan CJ, Cullen PJ. 2015. Role of the unfolded protein response in regulating the mucin-dependent filamentous-growth mitogen-activated protein kinase pathway. Mol Cell Biol 35:1414–1432. doi: 10.1128/MCB.01501-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tatebayashi K, Takekawa M, Saito H. 2003. A docking site determining specificity of Pbs2 MAPKK for Ssk2/Ssk22 MAPKKKs in the yeast HOG pathway. EMBO J 22:3624–3634. doi: 10.1093/emboj/cdg353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose MD, Broach JR. 1991. Cloning genes by complementation in yeast. Methods Enzymol 194:195–230. doi: 10.1016/0076-6879(91)94017-7. [DOI] [PubMed] [Google Scholar]
- 31.Murakami Y, Tatebayashi K, Saito H. 2008. Two adjacent docking sites in the yeast Hog1 mitogen-activated protein (MAP) kinase differentially interact with the Pbs2 MAP kinase kinase and the Ptp2 protein tyrosine phosphatase. Mol Cell Biol 28:2481–2494. doi: 10.1128/MCB.01817-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horie T, Tatebayashi K, Yamada R, Saito H. 2008. Phosphorylated Ssk1 prevents unphosphorylated Ssk1 from activating the Ssk2 MAP kinase kinase kinase in the yeast HOG osmoregulatory pathway. Mol Cell Biol 28:5172–5183. doi: 10.1128/MCB.00589-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mumberg D, Müller R, Funk M. 1994. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res 22:5767–5768. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatebayashi K, Yamamoto K, Tanaka K, Tomida T, Maruoka T, Kasukawa E, Saito H. 2006. Adaptor functions of Cdc42, Ste50, and Sho1 in the yeast osmoregulatory HOG MAPK pathway. EMBO J 25:3033–3044. doi: 10.1038/sj.emboj.7601192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 36.Takekawa M, Saito H. 1998. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 95:521–530. doi: 10.1016/S0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 37.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. 2009. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct Biol 9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loo TW, Clarke DM. 2001. Determining the dimensions of the drug-binding domain of human P-glycoprotein using thiol cross-linking compounds as molecular rulers. J Biol Chem 276:36877–36880. doi: 10.1074/jbc.C100467200. [DOI] [PubMed] [Google Scholar]
- 40.Gorman JJ, Wallis TP, Pitt JJ. 2002. Protein disulfide bond determination by mass spectrometry. Mass Spectrom Rev 21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 41.Dann CE, Hsieh JC, Rattner A, Sharma D, Nathans J, Leahy DJ. 2001. Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature 412:86–90. doi: 10.1038/35083601. [DOI] [PubMed] [Google Scholar]
- 42.Wouters MA, Rigoutsos I, Chu CK, Feng LL, Sparrow DB, Dunwoodie SL. 2005. Evolution of distinct EGF domains with specific functions. Protein Sci 14:1091–1103. doi: 10.1110/ps.041207005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheek S, Krishna SS, Grishin NV. 2006. Structural classification of small, disulfide-rich protein domains. J Mol Biol 359:215–237. doi: 10.1016/j.jmb.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 44.Smith VJ. 2011. Phylogeny of whey acidic protein (WAP) four-disulfide core proteins and their role in lower vertebrates and invertebrates. Biochem Soc Trans 39:1403–1408. doi: 10.1042/BST0391403. [DOI] [PubMed] [Google Scholar]
- 45.Bingle CD. 2011. Towards defining the complement of mammalian WFDC-domain-containing proteins. Biochem Soc Trans 39:1393–1397. doi: 10.1042/BST0391393. [DOI] [PubMed] [Google Scholar]
- 46.Lamson RE, Takahashi S, Winters MJ, Pryciak PM. 2006. Dual role for membrane localization in yeast MAP kinase cascade activation and its contribution to signaling fidelity. Curr Biol 16:618–623. doi: 10.1016/j.cub.2006.02.060. [DOI] [PubMed] [Google Scholar]
- 47.Zuzuarregui A, Li T, Friedmann C, Ammerer G, Alepuz P. 2015. Msb2 is a Ste11 membrane concentrator required for full activation of the HOG pathway. Biochim Biophys Acta 1849:722–730. doi: 10.1016/j.bbagrm.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Zuzuarregui A, Kupka T, Bhatt B, Dohnal I, Mudrak I, Friedmann C, Schüchner S, Frohner IE, Ammerer G, Ogris E. 2012. M-track: detecting short-lived protein-protein interactions in vivo. Nat Methods 9:594–596. doi: 10.1038/nmeth.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]