

ABSTRACT
Global regulator of virulence A (GrvA) is a ToxR-family transcriptional regulator that activates locus of enterocyte effacement (LEE)-dependent adherence in enterohemorrhagic Escherichia coli (EHEC). LEE activation by GrvA requires the Rcs phosphorelay response regulator RcsB and is sensitive to physiologically relevant concentrations of bicarbonate, a known stimulant of virulence systems in intestinal pathogens. This study determines the genomic scale of GrvA-dependent regulation and uncovers details of the molecular mechanism underlying GrvA-dependent regulation of pathogenic mechanisms in EHEC. In a grvA-null background of EHEC strain TW14359, RNA sequencing analysis revealed the altered expression of over 700 genes, including the downregulation of LEE- and non-LEE-encoded effectors and the upregulation of genes for glutamate-dependent acid resistance (GDAR). Upregulation of GDAR genes corresponded with a marked increase in acid resistance. GrvA-dependent regulation of GDAR and the LEE required gadE, the central activator of GDAR genes and a direct repressor of the LEE. Control of gadE by GrvA was further determined to occur through downregulation of the gadE activator GadW. This interaction of GrvA with GadW-GadE represses the acid resistance phenotype, while it concomitantly activates the LEE-dependent adherence and secretion of immune subversion effectors. The results of this study significantly broaden the scope of GrvA-dependent regulation and its role in EHEC pathogenesis.
IMPORTANCE Enterohemorrhagic Escherichia coli (EHEC) is an intestinal human pathogen causing acute hemorrhagic colitis and life-threatening hemolytic-uremic syndrome. For successful transmission and gut colonization, EHEC relies on the glutamate-dependent acid resistance (GDAR) system and a type III secretion apparatus, encoded on the LEE pathogenicity island. This study investigates the mechanism whereby the DNA-binding regulator GrvA coordinates activation of the LEE with repression of GDAR. Investigating how these systems are regulated leads to an understanding of pathogenic behavior and novel strategies aimed at disease prevention and control.
INTRODUCTION
Enterohemorrhagic Escherichia coli (EHEC) O157:H7 is a virulent intestinal pathogen causing foodborne outbreaks of bloody diarrhea (hemorrhagic colitis) and the life-threatening kidney disease hemolytic-uremic syndrome (1–3). Competitive colonization of the intestine by EHEC requires a type III secretion (T3S) mechanism and is characterized by the formation of attaching-and-effacing (A/E) lesions (1, 2). The T3S apparatus is encoded on a pathogenicity island, referred to as the locus of enterocyte effacement (LEE), containing 41 genes organized into five operons (LEE1 to LEE5) (4). The master LEE regulator, Ler, encoded as the first gene of LEE1, positively stimulates LEE transcription by relieving H-NS-mediated repression. Ler further activates the LEE-encoded regulators GrlA and GrlR, which in turn activate and repress ler transcription, respectively (5–10). Numerous non-LEE-encoded regulators also converge on ler and grlA promoters to coordinate LEE-dependent colonization with environmental/physiological cues (11–19).
One such regulator, RcsB, has been shown to both activate and repress LEE transcription and more recently has been shown to be required for induction of the LEE in response to the bicarbonate ion (20, 21). RcsB is the response regulator of the Rcs phosphorelay system, a multicomponent signaling pathway including RcsC (sensor kinase), RcsD (histidine phosphotransferase), and RcsF (outer membrane lipoprotein) (22–26). Upon phosphorylation, RcsB can bind to target promoters as a homodimer and as a heterodimer in conjunction with other regulatory auxiliary proteins, such as RcsA, BglJ, and GadE (27–29). Binding occurs at specific RcsB consensus sites (30, 31), and the location of binding relative to the −35 consensus sequence can dictate whether RcsB positively or negatively affects transcription (32). Tobe et al. (21) demonstrated that upregulation of the LEE effector EspB in response to rcsB overexpression also required the transcriptional regulator GrvA (for global regulator of virulence A). Furthermore, the overexpression of grvA was shown to activate transcription from the ler promoter (LEE1 operon) and to increase adherence to Caco-2 cells in vitro. In a subsequent study by Morgan et al. (20), both rcsB and grvA were shown to be required for bicarbonate induction of the LEE. Collectively, these two studies reveal GrvA to be an activator of Ler that can interact with RcsB to communicate the bicarbonate signal to the LEE colonizing mechanism.
How RcsB controls grvA and how GrvA, in turn, controls LEE expression are not known. GrvA shares homology with ToxR-family protein regulators, such as MarT in Salmonella enterica (30% amino acid identity) and CadC in E. coli (13% amino acid identity). Both MarT and CadC are transcriptional activators, with the latter activating the cadBA operon by displacing H-NS-mediated repression (33). The similarity of GrvA to MarT and CadC largely resides in the N terminus, containing a conserved DNA-binding helix-turn-helix domain (COG3710) and a CheY-like response regulator receiver domain (COG0745). In this study, details of the molecular basis for grvA regulation and for GrvA-dependent control of the LEE and LEE-dependent adherence are determined. Furthermore, global RNA sequencing analysis significantly broadens our knowledge of the role for GrvA in regulation to include genes central to acid resistance and metabolic fitness.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains and plasmids used in this study are listed in Table 1. Strains were stocked at −80°C in glycerol diluted (final concentration, 15%, vol/vol) in Luria broth (LB) and were maintained in LB or on LB with 1.5% agar (LBA). Unless otherwise noted, overnight (18- to 20-h) cultures grown in LB were used to inoculate fresh LB or LB buffered with sodium bicarbonate (44 mM NaHCO3) or fresh Dulbecco's modified Eagle's medium (DMEM; 4 g/liter glucose, 4 mM glutamine, 44 mM NaHCO3, pH 7.4) to a final optical density at 600 nm (OD600) of 0.05. Cultures were grown at 37°C in a rotary shaker (200 rpm) using a 1:10 medium-to-flask volume. Antibiotics (Sigma-Aldrich) were added to the cultures when required.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Reference or source |
|---|---|---|
| Strains | ||
| DH5α | Vector propagation, recA1 endA1 | |
| BL21(DE3)pLysS | BL21 with IPTG-inducible T7 polymerase | 90 |
| TW14359 | Wild-type strain from the 2006 outbreak, western USA | 91 |
| EcRJM-1 | TW14359 ΔescN | 20 |
| EcRJM-6 | TW14359 ΔrcsB | 20 |
| EcRJM-10 | TW14359 rcsB-FLAG | 20 |
| EcRJM-11 | TW14359 ΔgrvA | 20 |
| EcRJM-12 | TW14359 ΔrcsB ΔgrvA | 20 |
| EcRJM-13 | TW14359 tir-FLAG | 20 |
| EcRJM-35 | TW14359 ΔgrlR::kan | 20 |
| EcRJM-72 | TW14359 ΔrcsB/pRJM-20 | 20 |
| EcRJM-73 | TW14359 ΔlacZ | This study |
| EcRJM-74 | TW14359/pRJM-23 | This study |
| EcRJM-75 | TW14359/pRJM-24 | This study |
| EcRJM-76 | TW14359/pRJM-25 | This study |
| EcRJM-77 | TW14359/pRJM-26 | This study |
| EcRJM-78 | TW14359 ΔgrvA/pRJM-23 | This study |
| EcRJM-79 | TW14359 ΔgrvA/pRJM-24 | This study |
| EcRJM-80 | TW14359 ΔgrvA/pRJM-25 | This study |
| EcRJM-81 | TW14359 ΔgrvA/pRJM-26 | This study |
| EcRJM-82 | TW14359 ΔgrvA ΔgadE::kan/pRJM-24 | This study |
| EcRJM-110 | TW14359 ΔgrvA ΔgadE::kan/pRJM-26 | This study |
| EcRJM-83 | TW14359 ΔgrvA/pRJM-22 | This study |
| EcRJM-84 | TW14359 ΔrcsB ΔgrvA/pRJM-22 | This study |
| EcRJM-85 | TW14359 ΔrcsB ΔgrvA/pRJM-20 | This study |
| EcRJM-86 | TW14359 rcsDB-luxE | This study |
| EcRJM-87 | TW14359 grvAB-luxE | This study |
| EcRJM-88 | TW14359 rcsDB-luxE/pluxCDAB3 | This study |
| EcRJM-89 | TW14359 grvAB-luxE/pluxCDAB3 | This study |
| EcRJM-90 | TW14359 ΔgadE::kan | This study |
| EcRJM-91 | TW14359 ΔgrvA ΔgadE::kan | This study |
| EcRJM-92 | TW14359 ΔgadE::kan/pRJM-30 | This study |
| EcRJM-93 | TW14359 ΔgrvA ΔgadE::kan/pRJM-30 | This study |
| EcRJM-94 | TW14359 zinT-FLAG::kan | This study |
| EcRJM-95 | TW14359 ΔgrvA zinT-FLAG::kan | This study |
| EcRJM-96 | TW14359/pRJM-31 | This study |
| EcRJM-97 | EPECa E2348/69/pRJM-31 | This study |
| EcRJM-98 | MG1655/pRJM-31 | This study |
| EcRJM-99 | DH5α/pRJM-31 | This study |
| EcRJM-100 | TW14359/pRJM-32 | This study |
| EcRJM-101 | EPEC E2348/69/pRJM-32 | This study |
| EcRJM-102 | MG1655/pRJM-32 | This study |
| EcRJM-103 | DH5α/pRJM-32 | This study |
| EcRJM-104 | TW14359 rcsDB-luxE/pluxCDAB3/pRJM-21 | This study |
| EcRJM-105 | TW14359 grvAB-luxE/pluxCDAB3/pRJM-21 | This study |
| EcRJM-106 | TW14359 ΔgrvA/pRJM-23 | This study |
| EcRJM-107 | TW14359 ΔgrvA/pRJM-24 | This study |
| EcRJM-108 | TW14359 ΔgrvA/pRJM-25 | This study |
| EcRJM-109 | TW14359 ΔgrvA/pRJM-26 | This study |
| Plasmids | ||
| pACYC177 | Low-copy-no. cloning vector, Ampr Kanr P15A | |
| pBAD22 | Ara-inducible expression vector, Ampr M13 | 92 |
| pMPM-A2 | Low-copy-no. cloning vector, Ampr pMB1/f1 | 36 |
| pMPM-K3 | Low-copy-no. cloning vector, Kanr p15A/f1 | 36 |
| pMPM-T3 | Low-copy-no. cloning vector, Tetr p15A/f1 | 36 |
| pUC19 | High-copy-no. cloning vector, Ampr pMB1 | 93 |
| pRS551 | lac fusion vector, Ampr Kanr lacZ+ ColE1 | |
| pCP20 | FLP recombinase expression vector | 34 |
| pKD4 | Template plasmid for Kan cassette | 34 |
| pKM208 | Bacteriophage λ Red recombinase expression vector | 34 |
| pET-24d | T7 promoter, His tag vector, Kanr f1, pBR322 | Novagen |
| pSU312 | FLAG epitope template, Ampr Kanr, R6K | 94 |
| pRJM-20 | pACYC177-rcsB | 20 |
| pRJM-33 | pACYC177-grvAB | This study |
| pRJM-21 | pMPM-K3-rcsDB | This study |
| pRJM-22 | pMPM-A2-grvA | This study |
| pRJM-23 | pRS551-gadEP(−773, −1)b | This study |
| pRJM-24 | pRS551-gadEP(−320, −1) | This study |
| pRJM-25 | pRS551-gadEP(−516, −276) | This study |
| pRJM-26 | pRS551-gadEP(−773, −497) | This study |
| pRJM-27 | pET-24d-6×His-rcsB | This study |
| pRJM-28 | pMPM-T3-kan | This study |
| pRJM-29 | pMPM-T3-luxE-kan | This study |
| pRJM-30 | pMPM-A2-gadE | This study |
| pRJM-31 | pRS551-grvAP(−268, +38) | This study |
| pRJM-32 | pUC-grvA-6×His | This study |
| placlux8 | placlux8 | 40 |
| pLuxCDAB3 | pLuxCDAB3 | 40 |
EPEC, enteropathogenic E. coli.
The numbers indicate the positions in gadEP.
Genetic manipulations and complementation.
Primers used for genetic manipulation and complementation are available upon request. For construction of deletion mutants, the bacteriophage λ Red-assisted one-step deletion method adapted for EHEC was used as described previously (20, 34, 35). Complementation of rcsB was performed using vector pRJM-20, as previously described (20). Additionally, a fragment containing the entire rcsDB operon, including the rcsD promoter region (nucleotide positions 3041665 to 3046191; GenBank accession number NC_013008.1), was cloned into BamHI/XbaI-digested low-copy-number expression vector pMPM-K3 (Kanr) (36) using primers RcsB−3732/BamHI and RcsB+794/XbaI to produce pRJM-21. To complement grvA, a PCR product corresponding to the grvA open reading frame (ORF) and including a 267-bp upstream region of grvA containing the predicted promoter (nucleotide positions 1281984 to 1283134; GenBank accession number NC_013008.1) was produced using primers GrvA−268/XhoI and GrvA+884/BamHI and cloned into XhoI/BamHI-digested low-copy-number expression vector pMPM-A2 (Ampr), creating pRJM-22. To complement gadE, a PCR product corresponding to the gadE ORF and 762 bp of the sequence upstream of gadE (containing the native promoters, nucleotide positions 4458857 to 4460195) was produced using primers GadE−763/XbaI and GadE+577/BamHI and cloned into the XbaI/BamHI-digested vector pMPM-A2 to create pRJM-30. Confirmation of genetic constructs was done using a combination of restriction mapping and DNA sequencing (MWG Operon).
RNA purification and qRT-PCR.
The primers used for quantitative real-time PCR (qRT-PCR) are available upon request. RNA purification, cDNA synthesis, qRT-PCR cycling conditions, and data analysis followed previously described protocols (37) using a Realplex2 Mastercycler instrument (Eppendorf). Cycle threshold (CT) data were normalized to the level of rrsH (16S rRNA gene) expression, and normalized cycle threshold (ΔCT) values were transformed to arbitrary transcript expression levels using 2−ΔCT/10−6, as described previously (38, 39). Expression levels were compared statistically by the appropriate t test or by Tukey's honestly significant difference (HSD) test following a significant F test (n ≥ 3, α = 0.05; R software, version 3.1.0).
Construction of a single-copy grvAB-luxE operon fusion.
A strain containing a single-copy chromosome-plasmid luxE reporter fusion was constructed using a protocol adapted from that of Shimizu et al. (40). To make a grvAB-luxE fusion, the kan cassette and flanking FLP recombination target sites were amplified from pKD4 (34) using primers pKD4forward/SacI and pKD4reverse/BamHI. This product was SacI/BamHI digested and cloned into the BamHI/SacI site of pMPM-T3 to produce pMPM-T3-kan. A XhoI/BamHI-digested PCR product containing the luxE gene and native ribosome binding site was amplified from placlux8 (40) using primers LuxE-18/XhoI and LuxE+1,450/BamHI and cloned into the XhoI/BamHI site of similarly digested pMPM-T3-kan to create pMPM-T3-luxE-kan. The luxE-kan PCR product amplified from pMPM-T3-luxE-kan using primers GrvA+1,283/LuxE and GrvA+1,431/P2 and Phusion high-fidelity DNA polymerase (NEB) was fused to a region 13 bp downstream of the grvAB operon using bacteriophage λ Red recombination (34). Kanamycin resistance cassettes were removed using FLP recombinase as described previously (34), and the grvAB-luxE construct was validated using a combination of restriction mapping and DNA sequencing (MWG Operon). The lux operon genes luxCDAB were constitutively expressed in trans in TW14359 grvAB-luxE from pluxCDAB3 (40, 41).
Luciferase activity.
Overnight cultures grown in LB and DMEM were used to inoculate fresh LB or DMEM, respectively, to an initial OD600 of 0.05. To measure the effect of sodium bicarbonate (NaHCO3) on grvA expression, LB test cultures were grown with and without addition of 44 mM NaHCO3 (all DMEM cultures contained 44 mM NaHCO3). Cultures (0.2 ml) were inoculated into 96-well, clear-bottom white-walled plates (catalog number 655098; Greiner Bio-One) and incubated at 37°C in a rotary shaker (200 rpm). Luciferase and OD600 measurements were taken every hour for 10 h using a BioTek Synergy 2 plate reader (1 s integration) prewarmed to 37°C. Mean luciferase activity was compared between treatments (medium and NaHCO3 effects) using the appropriate t test (n = 4, α = 0.05; R software, version 3.1.0).
EMSAs.
Electrophoretic mobility shift assays (EMSAs) were performed as previously described (42). Briefly, the forward primers RcsB+4/NcoI and RcsB+682/XhoI, used to PCR amplify rcsB from TW14359 DNA, contained a new start codon and an N-terminal 6×His epitope tag, and the PCR product was cloned into the NcoI/XhoI sites of similarly digested vector pET-24d to create pRJM-27. N-terminal 6×His-RcsB fusion proteins have wild-type RcsB activity (43). 6×His-RcsB was purified from strain BL21(DE3)pLysS using Ni-nitrilotriacetic acid spin columns (Qiagen) according to the manufacturer's protocol. Briefly, overnight LB cultures containing 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and kanamycin (50 μg/ml) grown at 18°C (200 rpm) were pelleted and resuspended in 1.5 ml of lysis buffer (10 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, pH 8.0) and lysed by sonication for 3 min (50% amplitude, 10-s intervals, 30-s pause) while they were chilled on ice. The lysate was centrifuged at 21,000 × g to remove insoluble cellular debris, and the supernatant was used for column purification and subsequently eluted with elution buffer (500 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, pH 8.0). Purified 6×His-RcsB protein was quantified using a Bradford protein assay. Five prime biotin-labeled primers (Integrated DNA Technologies oligonucleotides) were used to generate a probe containing the grvA promoter (grvAP) region, including putative RcsB consensus binding sites (Frag-2F and Frag-2R) (Fig. 1B). An additional upstream grvA promoter probe with no predicted RcsB consensus sequence was amplified for use as a negative control (Frag-1F and Frag-1R) (Fig. 1B). Biotin-labeled probes were gel purified using QIAquick gel extraction kits (Qiagen) and diluted to a concentration of 0.5 ng/μl. All EMSAs were performed using a LightShift chemiluminescent EMSA kit (Thermo Pierce) according to the manufacturer's specifications. Prior to electrophoresis, purified 6×His-RcsB and biotin-labeled fragments were coincubated for 40 min in binding buffer (1× binding buffer, 50 ng/μl sheared salmon sperm DNA, 2.5% glycerol, 0.05% NP-40, 5 mM MgCl2, 50 mM KCl, 1 mM EDTA) at room temperature. Samples were loaded into a prerun and cooled 8% native polyacrylamide gel and run at 20 V/cm for 1 h. Following electrophoresis, biotin-labeled DNA was transferred (25 V for 20 min) to a Biodyne B precut modified nylon membrane (Thermo Pierce) and UV cross-linked for 60 s. Biotin-labeled DNA fragments were detected using streptavidin-horseradish peroxidase-conjugated antibodies (1:300) in blocking buffer, and the membranes were washed, equilibrated, and subsequently detected using luminol/enhancer solutions in a ChemiDoc XRS+ imaging system and Image Lab (version 3.0) software (Bio-Rad).
FIG 1.

Rcs response regulator RcsB directly activates transcription from the grvAB promoter. (A) (Left) Average luciferase activity (in relative light units [RLU]) plotted as a function of time for a single-copy grvAB-luxE fusion grown in DMEM, LB, or LB supplemented with 44 mM NaHCO3. Asterisks denote significance by Student's t test (*, P < 0.05; **, P < 0.01; n ≥ 3). Plots at each time point did not vary from the mean by more than 5%. (Right) Growth and grvAB promoter expression (luciferase activity) (bottom) on LB agar plates for strain TW14359 grvAB-luxE/pluxCDAB3 transformed with empty vector pMPM-K3 (left) or pMPM::rcsDB (right). (B) (Left) Map of the grvAB promoter and flanking regions in strain TW14359. Fragments for EMSA (Frag-1 and Frag-2) and the respective primers for probe generation are indicated. Frag-1 is the negative control; Frag-2 contains two putative RcsB binding sites (Box 1 and Box 2). (Right) Alignment of box 1 from the grvAB promoter with experimentally proven RcsB homodimer boxes for rprA and osmC promoters. Conserved bases are in bold and underlined. (C) EMSA for RcsB binding to Frag-2 of the grvAB promoter. The wedge denotes the increasing amount of RcsB added (1 to 12 μg). For Frag-1 (control), RcsB was added at 0 μg (lane −) and 12 μg (lane +).
Construction of lacZ transcriptional fusions and β-galactosidase assays.
Construction of a grvAP-lacZ reporter transcriptional fusion followed a previously described protocol using pRS551 (Table 1) (20). Briefly, a 306-bp PCR product containing the predicted grvA promoter was amplified from TW14359 genomic DNA using primers GrvA−268/EcoRI and GrvA+38/BamHI, BamHI/EcoRI digested, and then cloned into similarly digested pRS551, resulting in pRJM-31. Four different gadEP-lacZ promoter fusions were created by cloning BamHI/EcoRI-digested PCR products into similarly digested pRS551 using primers GadE−320/EcoRI and GadE−1/BamHI, GadE−276/EcoRI and GadE−516/BamHI, GadE−773/EcoRI and GadE−497/BamHI, and GadE−773/EcoRI and GadE−1/BamHI. β-Galactosidase activity (Miller units) was measured as previously described (38, 44), and the numbers of Miller units were compared using the appropriate t test or by Tukey's HSD test following a significant F test (n ≥ 3, α = 0.05; R software, version 3.1.0).
RNA sequencing and data analysis.
RNA extractions were performed as described above for qRT-PCR, except that residual genomic DNA was digested using Turbo DNase (catalog number AM2238; Ambion) following the manufacturer's protocol. Three independent RNA samples for each strain (TW14359 and TW14359 ΔgrvA) were pooled into a single sample (n = 1) in a 1:1:1 ratio to account for intersample variability, and rRNA was removed using a MICROBExpress bacterial mRNA enrichment kit (catalog number AM1905; Ambion) according to the manufacturer's instructions. The integrity and concentration of purified and enriched mRNA were determined using an Agilent 2100 bioanalyzer (Agilent Technologies). RNA sequencing (RNA-seq) was performed as previously described (45). Briefly, mRNA was converted to strand-specific cDNA using an Ion Total RNA-Seq kit (version 2; catalog number 4475936; Ion Torrent), and samples were sequenced using an Ion Personal Genome Machine (PGM) system (Ion Torrent). Sequence mapping and data analysis were performed using CLC Genomics Workbench (version 6) software (CLC Bio). Subsequent reads per kilobase per million mapped reads (RPKM) values were converted to the fold change for TW14359 ΔgrvA relative to the value for wild-type strain TW14359. Circular plots were created using Circos (version 0.65) software (http://www.circos.ca) (46).
Intestinal cell adherence assays.
The maintenance and culture of HT-29 colonic intestinal cells were performed as previously described (20). Adherence competition indexes were determined using the method of Gabbianelli et al. (47). Briefly, overnight DMEM cultures were used to inoculate fresh DMEM to an OD600 of 0.05, and these were then incubated for 3 h at 37°C with shaking (200 rpm). Cultures were then diluted to an OD600 of 0.5 and mixed in a 1:1 ratio (test strain/control strain), and 0.2 ml of each was used to inoculate HT-29 cells in 6-well culture plates. A control strain incapable of utilizing lactose (a Lac− strain) was constructed by deleting the entire lacZ ORF in TW14359 (strain EcRJM-73) using the bacteriophage λ Red approach and deletion primers LacZ−1/P1 and LacZ+3115/P2 (primer sequences are available upon request). Each plate was gently mixed before centrifugation at 500 × g for 5 min and then incubated as described above. Following 3 h of incubation, each well was washed four times using sterile phosphate-buffered saline (PBS) to remove nonadherent cells, and adherent cells were removed using 500 μl of 0.1% Triton X-100. Cells were enumerated (number of CFU per milliliter) by serial dilution in PBS and plating onto MacConkey agar, which is differential for lactose utilization. After overnight growth on MacConkey agar, pink (Lac-positive [Lac+]) colonies were scored as the numbers of CFU per milliliter for the test strain, whereas white (Lac-negative [Lac−]) colonies were scored as the numbers of CFU per milliliter for strain TW14359 ΔlacZ. The competitive index was derived by dividing the numbers of CFU per milliliter of the test strain by the numbers of CFU per milliliter of TW14359 ΔlacZ, and the indexes were compared statistically using the appropriate t test (n ≥ 3, α = 0.05; R software, version 3.1.0).
Test for acid resistance.
Acid resistance by the glutamate-dependent system was measured as described previously (37) with slight adaptations. Mid-exponential-phase (OD600 = 0.5) DMEM cultures were inoculated to a 106-CFU/ml final cell density in E minimal glucose (EG) medium with or without 5.7 mM l-glutamate at pH 7 (control) or acidified with HCl (pH 2). For cell count (number of CFU per milliliter) and percent survival determinations, samples were serially diluted in PBS (pH 7), plated onto LBA, and incubated overnight at 37°C.
RESULTS
Transcription of grvA is growth phase dependent and directly activated by RcsB.
Previous work has shown the response regulator of the rcs phosphorelay system RcsB to be an activator of global regulator of virulence A (grvA) and both factors to be required for full bicarbonate induction of the LEE-encoded regulator Ler and subsequent stimulation of LEE expression in EHEC during exponential-phase growth (20, 21). RcsB is predicted to act upstream of grvA in the regulation of ler, yet the mechanism underlying the control of grvA by RcsB is unknown.
The level of transcription of grvA in reporter strain TW14359 grvAB-luxE∕pluxCDAB3 (EcRJM-89) (Table 1) grown in DMEM with sodium bicarbonate (44 mM) was the highest during early exponential-phase growth, rapidly declining as cultures transitioned into postexponential phase (Fig. 1A). When grown in LB, the level of grvA transcription was similar to that observed when grown in DMEM; however, the levels were consistently lower over the first 7 h. Addition of sodium bicarbonate (44 mM) to LB, previously shown to upregulate RcsB levels (20), had no significant impact on grvA transcription, while overexpression of rcsDB in trans in TW14359 grvAB-luxE∕pluxCDAB3 substantially increased the level of grvA transcription when grown on LB agar in the absence of sodium bicarbonate (Fig. 1A). Thus, the addition of bicarbonate alone is not sufficient for upregulation of grvA. That overexpression of rcsB increased the level of transcription from the grvA promoter suggests that RcsB may directly activate grvA transcription. In support of this, two putative tandem RcsB binding sites proximal to a predicted −35 site (nucleotides 1282968 to 1282982 in TW14359) of the grvA promoter (grvAP) have been described previously (21, 31) (Fig. 1B, Box 1 and Box 2). To test for interactions between RcsB and the grvA promoter, purified 6×His-tagged RcsB was coincubated with a fragment of the core grvABP element, including putative RcsB binding sites (Fig. 1B, Frag-2) and analyzed using electrophoretic mobility shift assays. As anticipated, the 235-bp grvABP fragment was visibly shifted with the addition of increasing amounts (1 to 12 μg) of 6×His-RcsB, while no shift was observed for the control fragment (Fig. 1B, Frag-1, and C). These findings suggest that RcsB is a direct transcriptional activator of grvA and that activation occurs at the grvA core promoter region containing at least one putative RcsB binding site.
GrvA is a regulator of pathogenic mechanisms and metabolic fitness in EHEC.
grvA is required for LEE activation by the Rcs phosphorelay system in EHEC (21). How, in turn, GrvA upregulates LEE expression and the scope of genes under the control of GrvA are unknown. To explore this, the transcriptomes of EHEC O157:H7 strain TW14359 and its grvA isogenic derivative (TW14359 ΔgrvA) were measured by RNA-seq analysis during exponential-phase growth (OD600 = 0.5) in LEE-inducing medium (DMEM).
In TW14359 ΔgrvA, 765 genes were altered in expression >2-fold above the RPKM cutoff compared to their expression in TW14359; of these, 264 were upregulated and 501 were downregulated (Fig. 2; unpublished data). Known virulence-associated genes made up 6% of the genes whose expression was altered (8 upregulated, 38 downregulated). The majority of these were LEE-encoded structural proteins and secreted effectors, consistent with the role of GrvA as a positive regulator of LEE expression (Fig. 2A and B). Twenty-one LEE genes were downregulated in TW14359 ΔgrvA, including the LEE-encoded regulator ler, type III secretion translocon genes espA, espB, espD, and eaeA (encoding intimin), and the secreted effector tir (translocated intimin receptor) (Table 2). Five non-LEE-encoded effectors (nleC, nleA, nleH2, nleF, and nleE) were also downregulated in TW14359 ΔgrvA. Genes encoded within the 15-kb genomic acid fitness island (AFI), the products of which are required for survival at low pH (48), were upregulated in TW14359 ΔgrvA compared to their level of regulation in TW14359, in which the expression of AFI genes was barely detectable (Table 2). Most notably, genes of the glutamate-dependent acid resistance (GDAR) system were upregulated, including gadA, gadE, gadW, gadX, and the non-AFI-encoded gadCB operon. Thus, GrvA activates the LEE colonization mechanism while repressing acid resistance systems.
FIG 2.

The GrvA regulon in EHEC strain TW14359. (A) Circular plot of RNA-seq data for EHEC strains TW14359 and TW14359 ΔgrvA. Nucleotide positions are based on the TW14359 annotation (GenBank accession number NC_013008.1), indicated by the black outer ring. The heat map denotes the fold change in RPKM for TW14359 ΔgrvA relative to the level of expression in TW14359 for each gene. The two inner histograms indicate the RPKM values of TW14359 (black) and TW14359 ΔgrvA (red). Plots were generated using Circos (version 0.65; http://www.circos.ca). (B) Number of genes in grvA whose expression is altered relative to their expression in TW14359 plotted for each ontological group. The levels of expression of a total of 765 genes were altered by ≥2-fold; upregulation and downregulation are indicated.
TABLE 2.
RNA-seq GrvA transcriptome
| Gene identifiera | Gene name | Fold change in ΔgrvA mutantb | RPKMc |
Gene product description | |
|---|---|---|---|---|---|
| Wild type | ΔgrvA mutant | ||||
| LEE pathogenicity island genes and genes for non-LEE-encoded effectors | |||||
| ECSP_4665 | espF | −3.1 | 16.5 | 5.4 | LEE-encoded effector |
| ECSP_4666 | orf29 | −3.4 | 11.9 | 3.5 | LEE-encoded protein |
| ECSP_4667 | escF | −5.8 | 15.5 | 2.7 | LEE-encoded protein |
| ECSP_4668 | cesD2 | −8.3 | 46.3 | 5.6 | Predicted chaperone |
| ECSP_4669 | espB | −6.6 | 658.8 | 100.5 | Secreted protein EspB |
| ECSP_4670 | espD | −4.5 | 208.3 | 46.4 | Secreted protein EspD |
| ECSP_4671 | espA | −4.4 | 165.3 | 37.7 | Secreted protein EspA |
| ECSP_4672 | sepL | −4.5 | 46.8 | 10.3 | LEE-encoded T3S component |
| ECSP_4673 | escD | −4.0 | 4.8 | 1.2 | LEE-encoded T3S component |
| ECSP_4674 | eaeA | −4.8 | 75.0 | 15.8 | Intimin adherence protein |
| ECSP_4675 | cesT | −5.9 | 105.0 | 17.7 | Chaperone |
| ECSP_4676 | tir | −4.0 | 186.7 | 46.6 | Translocated intimin receptor |
| ECSP_4677 | map | −4.4 | 27.7 | 6.3 | LEE-encoded effector |
| ECSP_4678 | cesF | ND (−)d | 3.4 | <10−4 | Chaperone |
| ECSP_4679 | espH | −4.8 | 22.9 | 4.8 | LEE-encoded effector |
| ECSP_4680 | escQ | −1.9 | 9.7 | 5.0 | LEE-encoded protein |
| ECSP_4681 | orf16 | −8.1 | 14.7 | 1.8 | LEE-encoded protein |
| ECSP_4682 | orf15 | −1.2 | 5.9 | 4.9 | LEE-encoded protein |
| ECSP_4683 | escN | −2.7 | 16.8 | 6.2 | LEE-encoded ATPase |
| ECSP_4684 | escV | −2.3 | 11.6 | 5.1 | LEE-encoded protein |
| ECSP_4685 | orf12 | −1.3 | 2.6 | 2.0 | LEE-encoded protein |
| ECSP_4686 | espZ | −3.5 | 78.4 | 22.6 | LEE-encoded effector |
| ECSP_4687 | escI | −1.3 | 12.7 | 9.9 | LEE-encoded protein |
| ECSP_4688 | escJ | −2.8 | 18.4 | 6.6 | LEE-encoded protein |
| ECSP_4689 | sepD | −1.3 | 2.2 | 1.7 | LEE-encoded protein |
| ECSP_4690 | escC | −1.7 | 7.8 | 4.5 | T3S needle complex subunit |
| ECSP_4691 | cesD | −1.1 | 6.5 | 6.0 | LEE-encoded protein |
| ECSP_4692 | grlA | −1.1 | 15.3 | 13.8 | Global regulator of LEE, activator |
| ECSP_4693 | grlR | −1.9 | 11.4 | 5.9 | Global regulator of LEE, repressor |
| ECSP_4694 | etgA | −1.5 | 1.3 | 0.9 | Lytic transglycosylase |
| ECSP_4695 | escU | ND (−) | 1.6 | <10−4 | LEE-encoded protein |
| ECSP_4696 | escT | ND (−) | 2.0 | <10−4 | LEE-encoded protein |
| ECSP_4697 | escS | 4.0 | 0.6 | 2.4 | LEE-encoded protein |
| ECSP_4698 | escR | −1.6 | 4.3 | 2.6 | LEE-encoded protein |
| ECSP_4699 | escL | −2.9 | 12.2 | 4.2 | LEE-encoded protein |
| ECSP_4700 | orf4 | −1.8 | 10.3 | 5.6 | LEE-encoded protein |
| ECSP_4701 | cesA | −1.0 | 2.2 | 2.2 | Chaperone |
| ECSP_4702 | escE | −1.1 | 18.3 | 16.2 | LEE-encoded protein |
| ECSP_4703 | ler | −2.5 | 55.7 | 22.7 | LEE encoded regulator |
| ECSP_4704 | espG | −3.7 | 4.5 | 1.2 | LEE encoded effector |
| ECSP_4705 | rorf1 | −1.0 | 0.6 | 0.6 | LEE-encoded protein |
| ECSP_0061 | espY1 | 2.0 | 0.3 | 0.6 | Non-LEE-encoded effector |
| ECSP_0866 | nleC | −2.3 | 4.3 | 1.9 | Non-LEE-encoded effector |
| ECSP_0868 | nleD | −1.7 | 7.3 | 4.4 | Non-LEE-encoded effector |
| ECSP_1702 | nleA | −4.0 | 1.5 | 0.4 | Non-LEE-encoded effector |
| ECSP_1704 | nleH2 | 2.2 | 3.8 | 8.6 | Non-LEE-encoded effector |
| ECSP_1705 | nleF | −3.7 | 16.2 | 4.4 | Non-LEE-encoded effector |
| ECSP_3954 | nleE | −4.0 | 2.6 | 0.6 | Non-LEE-encoded effector |
| Metabolism and acid resistance genes | |||||
| ECSP_0704 | gltL | −3.9 | 149.2 | 38.4 | Glutamate and aspartate transporter |
| ECSP_0705 | gltK | −3.0 | 31.2 | 10.3 | Glutamate and aspartate transporter |
| ECSP_0706 | gltJ | −2.5 | 29.9 | 12.0 | Glutamate and aspartate transporter |
| ECSP_0707 | gltI | −3.0 | 156.1 | 52.4 | Glutamate and aspartate transporter |
| ECSP_0906 | glnQ | −2.3 | 277.1 | 118.0 | Glutamine transporter subunit |
| ECSP_0907 | glnP | −2.3 | 32.1 | 13.8 | Glutamine transporter subunit |
| ECSP_0908 | glnH | −1.5 | 141.1 | 93.5 | Glutamine transporter subunit |
| ECSP_1977 | gadC | 15.6 | 1.3 | 20.5 | Glutamate:gamma-aminobutyric acid antiporter |
| ECSP_1978 | gadB | 14.6 | 4.1 | 59.5 | Glutamate decarboxylase B |
| ECSP_2312 | astE | −4.0 | 9.7 | 2.4 | Succinylglutamate desuccinylase |
| ECSP_2313 | astB | −5.9 | 10.9 | 1.8 | Succinylarginine dihydrolase |
| ECSP_2314 | astD | −9.1 | 18.3 | 2.0 | Succinylglutamic semialdehyde dehydrogenase |
| ECSP_2315 | astA | ND (−) | 25.5 | <10−4 | Arginine succinyltransferase |
| ECSP_2316 | astC | −66.0 | 52.4 | 0.8 | Succinylornithine transaminase |
| ECSP_2329 | gdhA | −1.8 | 53.4 | 23.0 | Glutamate dehydrogenase |
| ECSP_3185 | argT | −3.5 | 89.1 | 25.3 | Lysine/arginine/ornithine transporter subunit |
| ECSP_4065 | uxaA | 1.7 | 12.6 | 21.4 | Altronate hydrolase |
| ECSP_4066 | uxaC | 1.8 | 7.8 | 13.6 | Uronate isomerase |
| ECSP_4486 | slp | 2.0 | 37.4 | 74.3 | Outer membrane lipoprotein |
| ECSP_4498 | hdeB | ND (+) | <10−4 | 13.6 | Acid resistance protein |
| ECSP_4499 | hdeA | ND (+) | <10−4 | 134.4 | Acid resistance protein |
| ECSP_4500 | hdeD | ND (+) | <10−4 | 11.7 | Acid resistance protein |
| ECSP_4501 | gadE | ND (+) | <10−4 | 10.8 | GDAR activator |
| ECSP_4502 | mdtE | ND (+) | <10−4 | 11.6 | Multidrug resistance efflux transporter |
| ECSP_4503 | mdtF | ND (+) | <10−4 | 12.9 | Multidrug resistance efflux transporter |
| ECSP_4504 | gadW | ND (+) | <10−4 | 9.1 | DNA-binding transcriptional activator |
| ECSP_4505 | 1.0 | <10−4 | 0.0 | Hypothetical protein | |
| ECSP_4506 | gadX | ND (+) | <10−4 | 5.0 | DNA-binding transcriptional dual regulator |
| ECSP_4507 | gadA | ND (+) | <10−4 | 18.3 | Glutamate decarboxylase A |
| ECSP_4508 | yhjA | 1.0 | <10−4 | <10−4 | Cytochrome c peroxidase |
| ECSP_4509 | treF | ND (+) | <10−4 | 3.4 | Cytoplasmic trehalase |
| ECSP_4510 | yhjB | 1.0 | <10−4 | <10−4 | DNA-binding response regulator |
| ECSP_4511 | yhjC | ND (+) | <10−4 | 2.6 | DNA-binding transcriptional regulator |
| ECSP_4512 | yhjD | ND (+) | <10−4 | 5.1 | Conserved inner membrane protein |
| ECSP_4513 | yhjE | 87.3 | 0.2 | 16.3 | Predicted transporter |
| ECSP_4921 | ND | 2.7 | <10−4 | Hypothetical protein | |
| ECSP_4922 | −1.1 | 78.6 | 74.4 | Hypothetical protein | |
| ECSP_4923 | glnG | −3.7 | 83.9 | 22.4 | DNA-binding response regulator |
| ECSP_4924 | glnL | −3.7 | 117.1 | 31.6 | Sensory histidine kinase |
| ECSP_4925 | glnA | −4.3 | 745.3 | 172.4 | Glutamine synthetase |
| ECSP_0518 | glnK | ND (+) | 143.4 | <10−4 | Nitrogen assimilation regulatory protein |
| ECSP_0519 | amtB | −74.8 | 185.6 | 2.7 | Ammonium transporter |
| ECSP_4793 | asnC | −2.0 | 5.2 | 2.6 | DNA-binding transcriptional regulator |
| ECSP_4794 | asnA | −5.0 | 2,647.6 | 532.5 | Asparagine synthetase A |
| ECSP_0721 | asnB | −7.6 | 688.2 | 90.7 | Asparagine synthetase B |
| ECSP_2651 | cbl | −4.8 | 14.1 | 2.9 | DNA-binding transcriptional activator |
| ECSP_2652 | nac | −63.5 | 32.0 | 0.5 | DNA-binding transcriptional dual regulator |
| ECSP_3790 | galR | −2.1 | 13.2 | 6.4 | DNA-binding transcriptional repressor |
| ECSP_3791 | lysA | −4.3 | 23.1 | 5.4 | Diaminopimelate decarboxylase |
| ECSP_3792 | lysR | −2.8 | 2.8 | 1.0 | DNA-binding transcriptional dual regulator |
| Metal stress response and import/export genes | |||||
| ECSP_0334 | ykgL | ND (+) | <10−4 | 1.4 | Predicted protein |
| ECSP_0335 | rpmJ | 170.7 | 1.7 | 284.6 | 50S ribosomal protein L36 |
| ECSP_0336 | rpmE2 | 565.7 | 0.6 | 346.5 | Zn(II)-responsive ribosomal protein |
| ECSP_0622 | cusS | −1.3 | 7.6 | 5.8 | Sensory histidine kinase |
| ECSP_0623 | cusR | 1.1 | 36.3 | 38.9 | DNA-binding response regulator |
| ECSP_0624 | cusC | −1.0 | 15.6 | 15.0 | Copper/silver efflux system |
| ECSP_0625 | cusF | −1.8 | 47.5 | 26.5 | Periplasmic copper-binding protein |
| ECSP_0626 | cusB | −1.3 | 111.6 | 82.8 | Copper/silver efflux system |
| ECSP_0627 | cusA | −1.4 | 67.5 | 48.0 | Copper/silver efflux system |
| ECSP_0731 | fur | 1.7 | 38.2 | 64.5 | DNA-binding regulator |
| ECSP_2430 | yebA | 4.5 | 17.0 | 75.9 | Predicted peptidase |
| ECSP_2431 | znuA | 17.7 | 13.8 | 244.0 | High-affinity zinc uptake system |
| ECSP_2432 | znuC | 2.5 | 4.5 | 18.9 | High-affinity zinc uptake system |
| ECSP_2433 | znuB | 4.2 | 4.6 | 11.4 | High-affinity zinc uptake system |
| ECSP_2579 | zinT | 163.7 | 2.0 | 328.1 | Conserved metal-binding protein |
| ECSP_4263 | zntR | −1.1 | 11.4 | 9.9 | DNA-binding transcriptional activator |
| ECSP_4425 | zntA | −1.4 | 19.9 | 14.3 | Zinc, cobalt, and lead efflux system |
| Genes for other functions | |||||
| ECSP_1968 | ddpF | 1.1 | 2.3 | 2.5 | d-Ala–d-Ala transporter subunit |
| ECSP_1969 | ddpD | 1.5 | 1.0 | 1.4 | d-Ala–d-Ala transporter subunit |
| ECSP_1970 | ddpC | 1.6 | 1.3 | 2.1 | d-Ala–d-Ala transporter subunit |
| ECSP_1971 | ddpB | −6.5 | 3.1 | 0.5 | d-Ala–d-Ala transporter subunit |
| ECSP_1972 | ddpA | −28.7 | 9.3 | 0.3 | d-Ala–d-Ala transporter subunit |
| ECSP_1973 | ddpX | ND (−) | 7.3 | <10−4 | d-Ala–d-Ala dipeptidase |
| ECSP_2574 | yedV | −1.8 | 2.7 | 1.4 | Sensory kinase |
| ECSP_2575 | yedW | 1.0 | 48.2 | 26.5 | DNA-binding regulator |
| ECSP_5434 | yjiA | 2.8 | 12.3 | 34.1 | Predicted GTPase |
| ECSP_5435 | yjiX | 27.8 | 0.7 | 19.5 | Conserved protein |
| ECSP_5436 | yjiY | 3.8 | 64.6 | 246.2 | Predicted protein |
| ECSP_2492 | −2.4 | 1,462.1 | 620.0 | Hypothetical protein | |
| ECSP_4094 | −1.9 | 1,576.0 | 841.5 | Hypothetical protein | |
Based on EHEC O157:H7 strain TW14359 (GenBank accession number NC_013008.1).
Fold change was calculated as the wild-type RPKM value divided by the mutant strain RPKM value.
RPKM, reads per kilobase per million mapped reads.
ND, not determined; (+), upregulated; (−), downregulated.
Twenty-six percent of the genes whose expression was altered (57 upregulated, 140 downregulated) had defined metabolic functions, suggesting a broader role for GrvA in regulation and fitness (Fig. 2B). This included genes for asparagine synthesis (asnA, asnB) and arginine degradation (ast operon) (Table 2). Nine percent (23 upregulated, 45 downregulated) were known or predicted regulators, and of these, global nitrogen metabolism regulators ntrC (also glnG) and nac, as well as cbl for sulfonate utilization, were downregulated. Eight percent (18 upregulated, 42 downregulated) were transport systems, and of these, glutamate (glt operon), glutamine (gln operon, and ammonium (amtB) transporters were downregulated. Thirty-seven percent (112 upregulated, 171 downregulated) had unknown or hypothetical functions (Table 2). Genes for zinc binding and transport, as well as resistance to zinc and other metals, were upregulated in TW14359 ΔgrvA, including znuA, znuCB, zinT (formerly yodA), rpmE2, rpmJ, ykgL, and yebA (49–51).
Alterations in the expression of astC (arginine metabolism), ler (LEE regulator), gadE (acid resistance), and znuC (zinc transport) were validated by qRT-PCR. Complementation of grvA in trans (strain TW14359 ΔgrvA/pgrvA) largely restored wild-type expression for ler and znuA but only partially restored expression for gadE and astC (Table 3). Deletion of rcsB in TW14359 ΔgrvA had no further significant impact on the expression of ler or any other targets tested (Table 3).
TABLE 3.
qRT-PCR validation of RNA-seq data
| Strain | Expression unitsa |
|||
|---|---|---|---|---|
| ler | gadE | astC | znuA | |
| TW14359 | 1,054.77 (51.81) | 0.18 (0.06) | 111.56 (1.55) | 8.02 (2.34) |
| ΔgrvA mutant | 78.28 (21.68) | 61.11 (8.22) | 1.21 (0.67) | 551.15 (150.90) |
| ΔrcsB ΔgrvA mutant | 127.97 (33.65) | 87.87 (4.10) | 2.5 (1.49) | 622.03 (46.05) |
| ΔgrvA pgrvA mutant | 556.98 (180.36) | 12.94 (0.81) | 6.87 (2.24) | 20.46 (8.28) |
For each gene, expression units relative to the level of rrsH expression were calculated, and standard deviations are in parentheses. Gene name identifiers are based on EHEC strain TW14359 (GenBank accession number NC_013008.1).
GrvA requires acid resistance regulator gadE for control of ler and adherence to intestinal cells.
RNA-seq analysis revealed that deletion of grvA leads to the upregulation of gadE, a known transcriptional repressor of LEE-encoded regulator ler (52, 53). As such, the contribution of gadE to GrvA-dependent control of the LEE and adherence to cultured intestinal epithelial cells was examined.
During exponential-phase growth, gadE transcription is strongly repressed (54, 55). As such, the level of expression of ler in TW14359 ΔgadE only marginally increased compared to that in TW14359 (Fig. 3A). However, when gadE was constitutively expressed in both TW14359 ΔgrvA and TW14359 ΔgrvA ΔgadE, ler expression was reduced to the level of that observed in TW14359 ΔgrvA (Fig. 3A) (P < 0.05).
FIG 3.
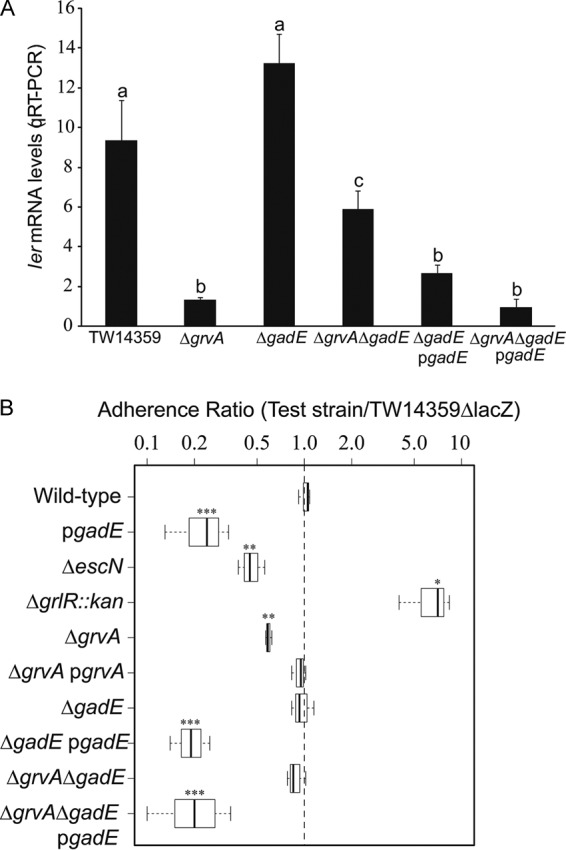
GrvA requires gadE for control of LEE-dependent adherence. (A) ler transcript levels determined by qRT-PCR plotted for TW14359, TW14359 ΔgrvA, TW14359 ΔgadE, TW14359 ΔgrvA ΔgadE, and complemented strains TW14359 ΔgadE/pgadE and TW14359 ΔgrvA ΔgadE/pgadE grown in DMEM (OD600 = 0.5). Plots which differ by lowercase letter differ significantly by Tukey's HSD test following a significant F test (n ≥ 3, P < 0.05). Error bars denote standard deviations. (B) Adherence ratio box plot for test strains (TW14359 and genetic derivatives) relative to control strain TW14359 ΔlacZ when cultured with HT-29 colonic epithelial cells (see the text for details). Box plot boundaries represent the 25th and 75th percentiles, whiskers represent the maximum and minimum values, and the mean adherence ratio is given by the horizontal line. Asterisks denote a significant difference compared with the results for TW14359, as determined by t test (n ≥ 3; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
The effect of the grvA and gadE interaction on in vitro adherence was determined using a competition assay. Strains were coincubated in a 1:1 ratio with HT-29 intestinal cells, followed by plating and enumeration on MacConkey differential medium. For a control strain, lacZ was deleted from wild-type strain TW14359, and thus, the strain was Lac− and colonies on MacConkey agar were white, whereas the test strains (TW14359 and genetic derivatives) were Lac+, producing pink colonies. For each test strain, an adherence index was determined from plate counts as the ratio of the number of CFU per milliliter for the test strain relative to the number of CFU per milliliter for the Lac− wild-type control strain.
In agreement with the ler expression data (Fig. 3A), the adherence index of TW14359 ΔgrvA was significantly decreased compared to the adherence indexes of the wild type, TW14359 ΔgadE, TW14359 ΔgrvA ΔgadE, and grvA-complemented strain TW14359 ΔgrvA/pgrvA (P < 0.05) but not that of type III secretion-defective strain TW14359 ΔescN (Fig. 3B). In strains where gadE was expressed constitutively, the adherence index was uniformly reduced to levels lower than those in all other strains, including TW14359 ΔescN (P < 0.001), suggesting that GadE may impact adherence in both a LEE-dependent and a LEE-independent manner (Fig. 3B). Only in strain TW14359 grlR::kan, in which the LEE is overexpressed (20), was the adherence index increased. The results of these experiments are consistent with the hypothesis that control of the LEE and adherence by GrvA are directed through the repression of gadE.
Negative regulation of gadE by GrvA requires GadW.
GadE is a negative regulator of LEE expression, acting directly on the LEE1 promoter to repress ler transcription (52, 53). In the preceding experiments, the upregulation of ler by GrvA was determined to correspond with gadE repression. Three discrete promoters have been shown to control transcription of the gadE gene in E. coli K-12 MG1655 (56). To better understand the molecular basis underlying the regulation of gadE promoters P1 to P3 by GrvA, the levels of transcription from four different gadE lacZ reporter constructs in TW14359 and TW14359 ΔgrvA were compared during exponential-phase and stationary-phase growth in DMEM (Fig. 4A).
FIG 4.

GrvA represses gadE transcription through GadW, determined from β-galactosidase activity (in Miller units) for gadE-lacZ promoter fusions in TW14359 and genetic derivatives. (A) (Top) Activity (in Miller units) for gadE-lacZ promoter fragments Frag-1 through Frag-4 and the vector control (pRS551) plotted for TW14359 and TW14359 ΔgrvA during exponential-phase (OD600 = 0.5) and stationary-phase (OD600 = 2) growth in DMEM. (Bottom) Cartoon of the gadE promoter and flanking regions. Promoter (P1 to P3) positions and the positions of each cloned promoter fragment (Frag-1 through Frag-4) relative to position +1 of the initiation codon for the gadE ORF are indicated. (B) Activity (in Miller units) plotted for gadE-lacZ promoter fragments Frag-2 (left) and Frag-4 (right) in TW14359 and mutant derivatives. For panels A and B, asterisks denote significant differences by t test (n ≥ 3; *, P < 0.05; **, P < 0.01). Bars denote standard deviations. (C) Activity (in Miller units) plotted for the gadE-lacZ P3 promoter in TW14359 and mutant derivatives. Plots which differ by lowercase letter differ significantly by Tukey's HSD test following a significant F test (n ≥ 3, P < 0.05).
During exponential-phase growth, the level of transcription from gadE-lacZ containing all three gadE promoters (P1 through P3; Frag-1) was significantly increased in TW14359 ΔgrvA compared to that in TW14359 (P < 0.01) (Fig. 4A) (55). Promoter activity from fragments containing the P1 or P3 promoter alone (Frag-2 and Frag-4, respectively) was also significantly higher in TW14359 ΔgrvA (P < 0.05), while activity from the fragment containing only the P2 promoter (Frag-3) did not differ between TW14359 ΔgrvA and TW14359. As anticipated, promoter activity from all fragments increased significantly during stationary-phase growth for TW14359 (P < 0.01) yet increased only slightly from P2 (P < 0.05). For TW14359 ΔgrvA, promoter activity further increased only from P1 during stationary phase (P < 0.01) and was higher than the activity from P1 in TW14359 (Fig. 4A). This was predicted to be due to autoactivation of the P1 promoter by GadE (54), as deletion of gadE in TW14359 ΔgrvA eliminated P1 activation (Frag-2) during exponential-phase growth but had no effect on P3 activity (Frag-4) (Fig. 4B). On the basis of these findings, it is suspected that the GrvA-dependent regulation of gadE transcription is directed solely through the P3 promoter.
Transcription from the gadE P3 promoter is directly controlled by the AFI-encoded regulators GadX and GadW (56), and RNA-seq analysis of TW14359 ΔgrvA revealed the levels of expression of both genes to be elevated compared to their levels of expression in TW14359 (Table 2). It was thus predicted that repression of P3 by GrvA was mediated through one or both of these regulators. To test this, the effect of gadX and gadW deletion in TW14359 ΔgrvA on P3 activity during exponential-phase growth (OD600 = 0.5) in DMEM was measured. Only the deletion of gadW was observed to reduce the level of transcription from P3 (Fig. 4C). Activity from P3 in TW14359 ΔgadW and TW14359 ΔgrvA ΔgadW was reduced significantly compared to that in TW14359 ΔgrvA (P = 0.01 and 0.03, respectively) but not compared to that in TW14359. Conversely, activity from P3 was slightly but significantly increased in TW14359 ΔgrvA ΔgadX compared to that in TW14359 ΔgrvA (P = 0.01) (Fig. 4C). Taken together, these data indicate that GrvA indirectly represses transcription from the gadE P3 promoter during exponential-phase growth in a manner that is dependent on gadW.
GrvA is a novel repressor of glutamate-dependent acid resistance.
The transcription of GDAR genes is growth phase dependent; expression is tightly controlled and low during exponential-phase growth but increases markedly as cells transition into stationary phase (57). Correspondingly, exponential-phase cultures are generally acid susceptible, while stationary-phase cultures are acid resistant. Since deletion of grvA was shown to upregulate GDAR genes (gadA, gadBC, gadE, gadW, gadX) during exponential-phase growth, the contribution of GrvA to GDAR was determined for exponential-phase cultures and was compared to that of the GDAR phenotype of stationary-phase cultures. In addition, since RcsB activates grvA transcription and is a dual regulator of GDAR system genes in E. coli K-12 (28, 58, 59), the interactions of rcsB and grvA in expression of the GDAR phenotype in EHEC O157:H7 were examined.
As expected, no colonies of wild-type TW14359 grown to exponential phase could be recovered on LBA following a 1-h challenge in acidified (pH 2) EG medium (Fig. 5, top). However, for strain TW14359 ΔgrvA, in which GDAR genes are upregulated during exponential-phase growth, 400 CFU/ml, corresponding to 9% of the original inoculum, was recovered. Complementation of TW14359 ΔgrvA with grvA restored acid sensitivity to wild-type levels. GDAR was abrogated following deletion of rcsB in TW14359 ΔgrvA, and complementation with rcsB restored GDAR in TW14359 ΔgrvA ΔrcsB but not in TW14359 ΔrcsB.
FIG 5.
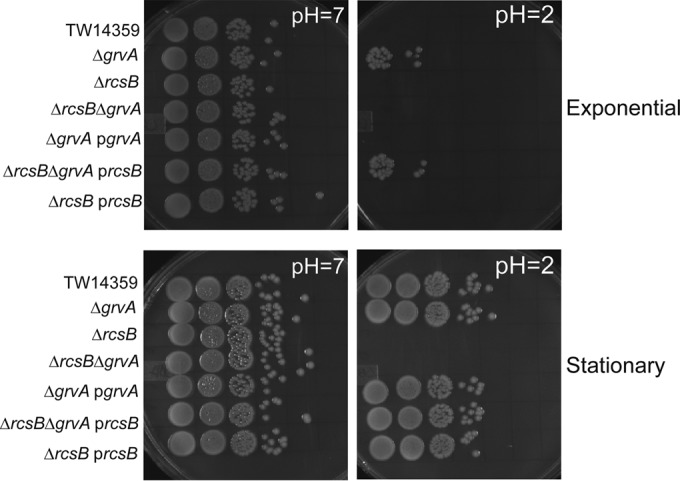
GrvA is a growth phase-dependent repressor of acid resistance. Representative colony-forming units on LB agar for TW14359 and derivative strains following a 1-h challenge in EG medium (pH 7 versus pH 2) are shown. Cultures were tested for acid resistance during exponential-phase (top) and stationary-phase (bottom) growth in DMEM. See the text for details.
In stationary phase, during which GDAR is actively expressed (57), deletion and complementation of grvA had no effect on survival in acid, with 100% of the initial inoculum being recovered following a 1-h challenge in acidified EG medium (Fig. 5, bottom). Thus, control of GDAR by GrvA is relegated to exponential-phase growth. In keeping with the requirement for rcsB in stationary-phase GDAR, no growth was observed for TW14359 ΔrcsB and TW14359 ΔgrvA ΔrcsB unless they were complemented with rcsB (Fig. 5, bottom).
DISCUSSION
This study has examined aspects of grvA transcriptional regulation, has defined the GrvA regulon, and has identified genetic determinants underlying GrvA-dependent control of discrete pathogenic mechanisms in EHEC. RcsB, the response regulator of Rcs phosphorelay in E. coli, is predicted to directly activate transcription of grvA, leading to the upregulation of LEE-dependent adherence and repression of glutamate-dependent acid resistance (Fig. 6). While it is not yet clear what cis-acting element(s) is required for RcsB-dependent activation of grvA, this study demonstrated the binding of RcsB to a grvA promoter fragment containing two putative tandem RcsB binding sites located proximal to a predicted σ70 −35 site. These sites share some homology with the RcsAB box consensus sequence described for rcsA, wza, and flhDC promoters (30, 60), yet RcsA, which is highly unstable at elevated temperatures (27), is not required for RcsB-dependent regulation of the LEE (21), and RcsAB generally bind distal to and upstream of the promoter core (31, 61, 62). The binding of RcsB to grvA promoter fragments is more consistent with regulation as a homodimer. RcsB homodimers bind a consensus sequence similar to the RcsAB box and proximal to the −35 site. Of the two putative RcsB binding sites in the grvA promoter, the upstream site (Fig. 1B, Box 1) shares similarities with homodimer sites described for osmC and rprA promoters (63). RcsB has also been shown to negatively regulate grvA expression. In strains of E. coli O157:H7 harboring a 49-bp deletion in rcsB, trans-complementation with wild-type rcsB decreased grvA expression (64). This was observed, however, only during culture at 28°C and correlates with reduced grvA expression during growth at 14°C and 25°C (65). Together, this suggests that regulation of grvA by RcsB is temperature sensitive. There is precedent for this, as repression of flhDC transcription by RcsB occurs at both low and high temperatures. At low temperatures, RcsB partners with RcsA (30), whereas at high temperatures, the LEE-encoded regulator GrlA is required (20).
FIG 6.
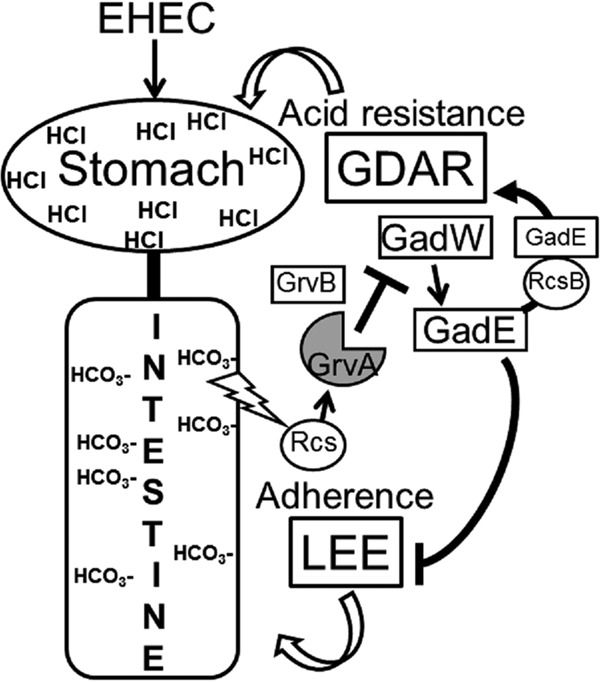
Model predicting GrvA-dependent regulation of acid resistance and LEE-dependent adherence. During exponential-phase growth, GrvA represses acid resistance (GDAR) and activates LEE-dependent adherence. This requires the negative regulation of gadW by GrvA. As GadW is a direct activator of gadE, the central regulator of GDAR, its repression by GrvA downregulates GDAR gene expression (gadE, gadA, gadBC) and acid resistance. GadE also directly represses the LEE-encoded regulator ler, the product of which activates the LEE (5 operons encoding the T3S, other regulators, and toxic effectors). The repression of GadW/GadE by GrvA leads to activation of the LEE and LEE-dependent adherence. Bicarbonate in the intestine is hypothesized to stimulate GrvA-dependent control of GDAR and the LEE in a manner requiring the Rcs phosphorelay system. RcsB activates GDAR as a heterodimer with GadE. The role for GrvB in this regulatory pathway is unknown. GrvA regulates the expression of >700 genes in EHEC O157:H7 strain TW14359. The model is inferred from the results of experiments performed in this and previous studies (20, 21, 52, 53, 55). See the text for further details. GDAR, glutamate-dependent acid resistance; LEE, locus of enterocyte effacement.
Overexpression of grvA increases transcription from the LEE1 promoter and correspondingly leads to increased adherence (21). Consistent with this, RNA-seq analysis of TW14359 ΔgrvA in this study revealed the reduced expression of genes belonging to both the LEE1 and the LEE2-LEE3 operons, including LEE-encoded regulator ler, T3S structural genes (esp genes), and the translocated intimin receptor (tir) gene. This study is the first to show that grvA deletion downregulates non-LEE-encoded T3S effector genes (nle genes). These proteins broadly impact host signaling cascades through inhibition of NF-κB (NleC, NleE, NleD, and NleH2) (66–68) and caspase activation (NleF) (69), as well as interfere with host vesicle trafficking (NleA) (70). How these nle genes are regulated by GrvA is unknown. One hypothesis is through its regulatory effects on LEE expression. Both LEE-encoded activators Ler and GrlA regulate nleA expression (71–73). Moreover, LEE-encoded GrlR has been shown to repress nleB and nleH (71). However, non-LEE-encoded regulators involved in quorum sensing (QseA) and nucleoid structuring (H-NS) have also been implicated in the control of nle gene expression (71, 74). Whatever the mechanism, these findings suggest a more comprehensive role for GrvA in EHEC pathogenesis that includes the coordination of T3S-dependent colonization with immune subversion.
This study identifies GrvA to be a new regulator of glutamate-dependent acid resistance (GDAR) genes (gad genes) (Fig. 6). Deletion of grvA led to the upregulation of gad regulatory (gadE, gadW and gadX) and enzymatic/structural (gadA and gadBC) genes and corresponded with a 100-fold increase in acid survival by the GDAR mechanism. These findings greatly expand the role for GrvA in the pathogenic behavior of EHEC to include the control genes essential for the transmission of this pathogen in acidic food matrices, gastric passage, and a low oral infectious dose. Since grvA is needed for full bicarbonate induction of the LEE (20), it is plausible that it helps to coordinate expression of the LEE and GDAR systems upon entry into the intestine (Fig. 6).
Genes with no direct role in virulence were also shown to be regulated by GrvA. Most notably, glnG (also ntrC) and nac expression was positively regulated by GrvA. Nitrogen regulatory protein C (NtrC) is a transcriptional activator of nitrogen assimilation control (Nac), and together these regulators are responsible for coordinating the expression of genes for nitrogen metabolism under limiting conditions (75). That ntrC and nac are positively controlled by GrvA is consistent with the reduced expression of operons for the utilization of glutamate (glt), glutamine (gln), arginine (ast), and asparagine (asn), as well as for the transport of ammonium (amtB) observed in the TW14359 ΔgrvA background. How nitrogen availability influences GrvA-dependent control of these mechanisms and the importance of grvA to nitrogen assimilation are unknown.
GrvA was shown to activate LEE-encoded master regulator ler through downregulation of gadE, the product of which directly represses ler transcription in EHEC (52, 53, 76). Negative control of gadE by GrvA is predicted to occur indirectly through the P3 promoter, requiring an intact gadW (Fig. 6). This is supported by both gadE-lacZ promoter fusions and RNA-seq analysis in this study. GadW is an AraC-family regulator that acts as a homodimer or, when partnered with GadX, to control the acid fitness genes gadBC, hdeA, hdeB, and gadE (54, 55, 77, 78). The details of how GrvA regulates transcription are not yet known. For the regulation of gadW, there are two predicted promoters encoded in tandem and just upstream of the gadW (gadWP1 and gadWP2) (77). It is suspected that gadWP1 is the target of GrvA repression, as transcripts from this promoter, but not those from gadWP2, were increased in TW14359 ΔgrvA by RNA-seq (data not shown). Multiple trans-acting factors are known to directly influence gadW transcription, including GadE, PhoP, and SdiA (activators) (79–81), as well as GadX, GadW, RutR, Fnr, and H-NS (repressors) (77, 82–84). It is unlikely that gadX contributes to GrvA-dependent repression of gadW, as gadX mutation in TW14359 ΔgrvA was not observed to further alter gadE expression. Save for gadE, no other known regulators of gadW were modified in expression by RNA-seq analysis, and a binding consensus sequence for GrvA has yet to be defined. GrvA shares some homology (29% identity, 79/270 amino acids) with MarT of Salmonella enterica serotype Typhimurium (85–87), most notably, the first 150 residues of the N terminus containing a winged-helix DNA-binding domain. marT is encoded on Salmonella pathogenicity island 3 (SPI3), and its product is a direct transcriptional activator of the misL autotransporter, which binds fibronectin and increases intestinal colonization and invasiveness (88, 89). On the basis of its similarity to CadC of E. coli, MarT is predicted to activate misL by relieving H-NS-mediated repression (33). If the regulation of gadW by GrvA is by a direct mechanism, however, it is unlikely that H-NS is involved, as deletion of grvA increases gadW expression. Future studies will be aimed at identifying cis elements for GrvA-directed transcriptional regulation.
REFERENCES
- 1.Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat Rev Microbiol 2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 2.Nataro JP, Kaper JB. 1998. Diarrheagenic Escherichia coli. Clin Microbiol Rev 11:142–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rangel JM, Sparling PH, Crowe C, Griffin PM, Swerdlow DL. 2005. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982-2002. Emerg Infect Dis 11:603–609. doi: 10.3201/eid1104.040739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci U S A 92:1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elliott SJ, Sperandio V, Giron JA, Shin S, Mellies JL, Wainwright L, Hutcheson SW, McDaniel TK, Kaper JB. 2000. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun 68:6115–6126. doi: 10.1128/IAI.68.11.6115-6126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vazquez A, Barba J, Ibarra JA, O'Donnell P, Metalnikov P, Ashman K, Lee S, Goode D, Pawson T, Finlay BB. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci U S A 101:3597–3602. doi: 10.1073/pnas.0400326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berdichevsky T, Friedberg D, Nadler C, Rokney A, Oppenheim A, Rosenshine I. 2005. Ler is a negative autoregulator of the LEE1 operon in enteropathogenic Escherichia coli. J Bacteriol 187:349–357. doi: 10.1128/JB.187.1.349-357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Islam MS, Bingle LE, Pallen MJ, Busby SJ. 2011. Organization of the LEE1 operon regulatory region of enterohaemorrhagic Escherichia coli O157:H7 and activation by GrlA. Mol Microbiol 79:468–483. doi: 10.1111/j.1365-2958.2010.07460.x. [DOI] [PubMed] [Google Scholar]
- 9.Barba J, Bustamante VH, Flores-Valdez MA, Deng W, Finlay BB, Puente JL. 2005. A positive regulatory loop controls expression of the locus of enterocyte effacement-encoded regulators Ler and GrlA. J Bacteriol 187:7918–7930. doi: 10.1128/JB.187.23.7918-7930.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sperandio V, Mellies JL, Delahay RM, Frankel G, Crawford JA, Nguyen W, Kaper JB. 2000. Activation of enteropathogenic Escherichia coli (EPEC) LEE2 and LEE3 operons by Ler. Mol Microbiol 38:781–793. doi: 10.1046/j.1365-2958.2000.02168.x. [DOI] [PubMed] [Google Scholar]
- 11.Russell RM, Sharp FC, Rasko DA, Sperandio V. 2007. QseA and GrlR/GrlA regulation of the locus of enterocyte effacement genes in enterohemorrhagic Escherichia coli. J Bacteriol 189:5387–5392. doi: 10.1128/JB.00553-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharp FC, Sperandio V. 2007. QseA directly activates transcription of LEE1 in enterohemorrhagic Escherichia coli. Infect Immun 75:2432–2440. doi: 10.1128/IAI.02003-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kendall MM, Rasko DA, Sperandio V. 2010. The LysR-type regulator QseA regulates both characterized and putative virulence genes in enterohemorrhagic Escherichia coli O157:H7. Mol Microbiol 76:1306–1321. doi: 10.1111/j.1365-2958.2010.07174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyoda S, Watanabe H. 2004. Positive effects of multiple pch genes on expression of the locus of enterocyte effacement genes and adherence of enterohaemorrhagic Escherichia coli O157:H7 to HEp-2 cells. Microbiology 150:2357–2571. doi: 10.1099/mic.0.27100-0. [DOI] [PubMed] [Google Scholar]
- 15.Flockhart AF, Tree JJ, Xu X, Karpiyevich M, McAteer SP, Rosenblum R, Shaw DJ, Low CJ, Best A, Gannon V, Laing C, Murphy KC, Leong JM, Schneiders T, La Ragione R, Gally DL. 2012. Identification of a novel prophage regulator in Escherichia coli controlling the expression of type III secretion. Mol Microbiol 83:208–223. doi: 10.1111/j.1365-2958.2011.07927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen AM, Kaper JB. 2009. Hfq affects the expression of the LEE pathogenicity island in enterohaemorrhagic Escherichia coli. Mol Microbiol 73:446–465. doi: 10.1111/j.1365-2958.2009.06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luzader DH, Clark DE, Gonyar LA, Kendall MM. 2013. EutR is a direct regulator of genes that contribute to metabolism and virulence in enterohemorrhagic Escherichia coli O157:H7. J Bacteriol 195:4947–4953. doi: 10.1128/JB.00937-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakanishi N, Abe H, Ogura Y, Hayashi T, Tashiro K, Kuhara S, Sugimoto N, Tobe T. 2006. ppGpp with DksA controls gene expression in the locus of enterocyte effacement (LEE) pathogenicity island of enterohaemorrhagic Escherichia coli through activation of two virulence regulatory genes. Mol Microbiol 61:194–205. doi: 10.1111/j.1365-2958.2006.05217.x. [DOI] [PubMed] [Google Scholar]
- 19.Iyoda S, Koizumi N, Satou H, Lu Y, Saitoh T, Ohnishi M, Watanabe H. 2006. The GrlR-GrlA regulatory system coordinately controls the expression of flagellar and LEE-encoded type III protein secretion systems in enterohemorrhagic Escherichia coli. J Bacteriol 188:5682–5692. doi: 10.1128/JB.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan JK, Vendura KW, Stevens SM Jr, Riordan JT. 2013. RcsB determines the locus of enterocyte effacement (LEE) expression and adherence phenotype of Escherichia coli O157:H7 spinach outbreak strain TW14359 and coordinates bicarbonate-dependent LEE activation with repression of motility. Microbiology 159:2342–2353. doi: 10.1099/mic.0.070201-0. [DOI] [PubMed] [Google Scholar]
- 21.Tobe T, Ando H, Ishikawa H, Abe H, Tashiro K, Hayashi T, Kuhara S, Sugimoto N. 2005. Dual regulatory pathways integrating the RcsC-RcsD-RcsB signalling system control enterohaemorrhagic Escherichia coli pathogenicity. Mol Microbiol 58:320–333. doi: 10.1111/j.1365-2958.2005.04828.x. [DOI] [PubMed] [Google Scholar]
- 22.Huang LH, Syu WJ. 2008. GrlA of enterohemorrhagic Escherichia coli O157:H7 activates LEE1 by binding to the promoter region. J Microbiol Immunol Infect 41:9–16. [PubMed] [Google Scholar]
- 23.Stout V, Gottesman S. 1990. RcsB and RcsC: a two-component regulator of capsule synthesis in Escherichia coli. J Bacteriol 172:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottesman S, Stout V. 1991. Regulation of capsular polysaccharide synthesis in Escherichia coli K12. Mol Microbiol 5:1599–1606. doi: 10.1111/j.1365-2958.1991.tb01906.x. [DOI] [PubMed] [Google Scholar]
- 25.Huang YH, Ferrieres L, Clarke DJ. 2006. The role of the Rcs phosphorelay in Enterobacteriaceae. Res Microbiol 157:206–212. doi: 10.1016/j.resmic.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 26.Brill JA, Quinlan-Walshe C, Gottesman S. 1988. Fine-structure mapping and identification of two regulators of capsule synthesis in Escherichia coli K-12. J Bacteriol 170:2599–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gottesman S, Trisler P, Torres-Cabassa A. 1985. Regulation of capsular polysaccharide synthesis in Escherichia coli K-12: characterization of three regulatory genes. J Bacteriol 162:1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson MD, Burton NA, Gutierrez B, Painter K, Lund PA. 2011. RcsB is required for inducible acid resistance in Escherichia coli and acts at gadE-dependent and -independent promoters. J Bacteriol 193:3653–3656. doi: 10.1128/JB.05040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venkatesh GR, Kembou Koungni FC, Paukner A, Stratmann T, Blissenbach B, Schnetz K. 2010. BglJ-RcsB heterodimers relieve repression of the Escherichia coli bgl operon by H-NS. J Bacteriol 192:6456–6464. doi: 10.1128/JB.00807-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francez-Charlot A, Laugel B, Van Gemert A, Dubarry N, Wiorowski F, Castanie-Cornet MP, Gutierrez C, Cam K. 2003. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol Microbiol 49:823–832. [DOI] [PubMed] [Google Scholar]
- 31.Wehland M, Bernhard F. 2000. The RcsAB box. Characterization of a new operator essential for the regulation of exopolysaccharide biosynthesis in enteric bacteria. J Biol Chem 275:7013–7020. [DOI] [PubMed] [Google Scholar]
- 32.Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu Rev Microbiol 59:379–405. doi: 10.1146/annurev.micro.59.050405.101230. [DOI] [PubMed] [Google Scholar]
- 33.Kuper C, Jung K. 2005. CadC-mediated activation of the cadBA promoter in Escherichia coli. J Mol Microbiol Biotechnol 10:26–39. doi: 10.1159/000090346. [DOI] [PubMed] [Google Scholar]
- 34.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lippolis JD, Bayles DO, Reinhardt TA. 2009. Proteomic changes in Escherichia coli when grown in fresh milk versus laboratory media. J Proteome Res 8:149–158. doi: 10.1021/pr800458v. [DOI] [PubMed] [Google Scholar]
- 36.Mayer MP. 1995. A new set of useful cloning and expression vectors derived from pBlueScript. Gene 163:41–46. doi: 10.1016/0378-1119(95)00389-N. [DOI] [PubMed] [Google Scholar]
- 37.Riordan JT, Tietjen JA, Walsh CW, Gustafson JE, Whittam TS. 2010. Inactivation of alternative sigma factor 54 (RpoN) leads to increased acid resistance, and alters locus of enterocyte effacement (LEE) expression in Escherichia coli O157:H7. Microbiology 156:719–730. doi: 10.1099/mic.0.032631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitra A, Fay PA, Morgan JK, Vendura KW, Versaggi SL, Riordan JT. 2012. Sigma factor N, liaison to an ntrC and rpoS dependent regulatory pathway controlling acid resistance and the LEE in enterohemorrhagic Escherichia coli. PLoS One 7:e46288. doi: 10.1371/journal.pone.0046288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.Shimizu T, Ohta Y, Tsutsuki H, Noda M. 2011. Construction of a novel bioluminescent reporter system for investigating Shiga toxin expression of enterohemorrhagic Escherichia coli. Gene 478:1–10. doi: 10.1016/j.gene.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu T, Tsutsuki H, Matsumoto A, Nakaya H, Noda M. 2012. The nitric oxide reductase of enterohaemorrhagic Escherichia coli plays an important role for the survival within macrophages. Mol Microbiol 85:492–512. doi: 10.1111/j.1365-2958.2012.08122.x. [DOI] [PubMed] [Google Scholar]
- 42.Mitra A, Fay PA, Vendura KW, Alla Z, Carroll RK, Shaw LN, Riordan JT. 2014. Sigma(N)-dependent control of acid resistance and the locus of enterocyte effacement in enterohemorrhagic Escherichia coli is activated by acetyl phosphate in a manner requiring flagellar regulator FlhDC and the sigma(S) antagonist FliZ. Microbiologyopen 3:497–512. doi: 10.1002/mbo3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carballes F, Bertrand C, Bouche JP, Cam K. 1999. Regulation of Escherichia coli cell division genes ftsA and ftsZ by the two-component system rcsC-rcsB. Mol Microbiol 34:442–450. doi: 10.1046/j.1365-2958.1999.01605.x. [DOI] [PubMed] [Google Scholar]
- 44.Miller JH. 1972. Assay of β-galactosidase, p 352–355. In Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 45.Weiss A, Ibarra JA, Paoletti J, Carroll RK, Shaw LN. 2014. The delta subunit of RNA polymerase guides promoter selectivity and virulence in Staphylococcus aureus. Infect Immun 82:1424–1435. doi: 10.1128/IAI.01508-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gabbianelli R, Scotti R, Ammendola S, Petrarca P, Nicolini L, Battistoni A. 2011. Role of ZnuABC and ZinT in Escherichia coli O157:H7 zinc acquisition and interaction with epithelial cells. BMC Microbiol 11:36. doi: 10.1186/1471-2180-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foster JW. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol 2:898–907. doi: 10.1038/nrmicro1021. [DOI] [PubMed] [Google Scholar]
- 49.Ferianc P, Farewell A, Nystrom T. 1998. The cadmium-stress stimulon of Escherichia coli K-12. Microbiology 144(Pt 4):1045–1050. doi: 10.1099/00221287-144-4-1045. [DOI] [PubMed] [Google Scholar]
- 50.Kershaw CJ, Brown NL, Hobman JL. 2007. Zinc dependence of zinT (yodA) mutants and binding of zinc, cadmium and mercury by ZinT. Biochem Biophys Res Commun 364:66–71. doi: 10.1016/j.bbrc.2007.09.094. [DOI] [PubMed] [Google Scholar]
- 51.Hensley MP, Gunasekera TS, Easton JA, Sigdel TK, Sugarbaker SA, Klingbeil L, Breece RM, Tierney DL, Crowder MW. 2012. Characterization of Zn(II)-responsive ribosomal proteins YkgM and L31 in E. coli. J Inorg Biochem 111:164–172. doi: 10.1016/j.jinorgbio.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kailasan Vanaja S, Bergholz TM, Whittam TS. 2009. Characterization of the Escherichia coli O157:H7 Sakai GadE regulon. J Bacteriol 191:1868–1877. doi: 10.1128/JB.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tree JJ, Roe AJ, Flockhart A, McAteer SP, Xu X, Shaw D, Mahajan A, Beatson SA, Best A, Lotz S, Woodward MJ, La Ragione R, Murphy KC, Leong JM, Gally DL. 2011. Transcriptional regulators of the GAD acid stress island are carried by effector protein-encoding prophages and indirectly control type III secretion in enterohemorrhagic Escherichia coli O157:H7. Mol Microbiol 80:1349–1365. doi: 10.1111/j.1365-2958.2011.07650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma Z, Masuda N, Foster JW. 2004. Characterization of EvgAS-YdeO-GadE branched regulatory circuit governing glutamate-dependent acid resistance in Escherichia coli. J Bacteriol 186:7378–7389. doi: 10.1128/JB.186.21.7378-7389.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sayed AK, Odom C, Foster JW. 2007. The Escherichia coli AraC-family regulators GadX and GadW activate gadE, the central activator of glutamate-dependent acid resistance. Microbiology 153:2584–2592. doi: 10.1099/mic.0.2007/007005-0. [DOI] [PubMed] [Google Scholar]
- 56.Sayed AK, Foster JW. 2009. A 750 bp sensory integration region directs global control of the Escherichia coli GadE acid resistance regulator. Mol Microbiol 71:1435–1450. doi: 10.1111/j.1365-2958.2009.06614.x. [DOI] [PubMed] [Google Scholar]
- 57.Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. 1999. Control of acid resistance in Escherichia coli. J Bacteriol 181:3525–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Castanie-Cornet MP, Cam K, Bastiat B, Cros A, Bordes P, Gutierrez C. 2010. Acid stress response in Escherichia coli: mechanism of regulation of gadA transcription by RcsB and GadE. Nucleic Acids Res 38:3546–3554. doi: 10.1093/nar/gkq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castanie-Cornet MP, Treffandier H, Francez-Charlot A, Gutierrez C, Cam K. 2007. The glutamate-dependent acid resistance system in Escherichia coli: essential and dual role of the His-Asp phosphorelay RcsCDB/AF. Microbiology 153:238–246. doi: 10.1099/mic.0.29278-0. [DOI] [PubMed] [Google Scholar]
- 60.Pristovsek P, Sengupta K, Lohr F, Schafer B, von Trebra MW, Ruterjans H, Bernhard F. 2003. Structural analysis of the DNA-binding domain of the Erwinia amylovora RcsB protein and its interaction with the RcsAB box. J Biol Chem 278:17752–17759. doi: 10.1074/jbc.M301328200. [DOI] [PubMed] [Google Scholar]
- 61.Stout V. 1996. Identification of the promoter region for the colanic acid polysaccharide biosynthetic genes in Escherichia coli K-12. J Bacteriol 178:4273–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stevenson G, Andrianopoulos K, Hobbs M, Reeves PR. 1996. Organization of the Escherichia coli K-12 gene cluster responsible for production of the extracellular polysaccharide colanic acid. J Bacteriol 178:4885–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sturny R, Cam K, Gutierrez C, Conter A. 2003. NhaR and RcsB independently regulate the osmCp1 promoter of Escherichia coli at overlapping regulatory sites. J Bacteriol 185:4298–4304. doi: 10.1128/JB.185.15.4298-4304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carter MQ, Parker CT, Louie JW, Huynh S, Fagerquist CK, Mandrell RE. 2012. RcsB contributes to the distinct stress fitness among Escherichia coli O157:H7 curli variants of the 1993 hamburger-associated outbreak strains. Appl Environ Microbiol 78:7706–7719. doi: 10.1128/AEM.02157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kocharunchitt C, King T, Gobius K, Bowman JP, Ross T. 2012. Integrated transcriptomic and proteomic analysis of the physiological response of Escherichia coli O157:H7 Sakai to steady-state conditions of cold and water activity stress. Mol Cell Proteomics 11:M111.009019. doi: 10.1074/mcp.M111.009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yen H, Ooka T, Iguchi A, Hayashi T, Sugimoto N, Tobe T. 2010. NleC, a type III secretion protease, compromises NF-kappaB activation by targeting p65/RelA. PLoS Pathog 6:e1001231. doi: 10.1371/journal.ppat.1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao X, Wan F, Mateo K, Callegari E, Wang D, Deng W, Puente J, Li F, Chaussee MS, Finlay BB, Lenardo MJ, Hardwidge PR. 2009. Bacterial effector binding to ribosomal protein s3 subverts NF-kappaB function. PLoS Pathog 5:e1000708. doi: 10.1371/journal.ppat.1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raymond B, Young JC, Pallett M, Endres RG, Clements A, Frankel G. 2013. Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. Trends Microbiol 21:430–441. doi: 10.1016/j.tim.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 69.Blasche S, Mortl M, Steuber H, Siszler G, Nisa S, Schwarz F, Lavrik I, Gronewold TM, Maskos K, Donnenberg MS, Ullmann D, Uetz P, Kogl M. 2013. The E. coli effector protein NleF is a caspase inhibitor. PLoS One 8:e58937. doi: 10.1371/journal.pone.0058937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thanabalasuriar A, Koutsouris A, Weflen A, Mimee M, Hecht G, Gruenheid S. 2010. The bacterial virulence factor NleA is required for the disruption of intestinal tight junctions by enteropathogenic Escherichia coli. Cell Microbiol 12:31–41. doi: 10.1111/j.1462-5822.2009.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Angulo VA, Martinez-Santos VI, Villasenor T, Santana FJ, Huerta-Saquero A, Martinez LC, Jimenez R, Lara-Ochoa C, Tellez-Sosa J, Bustamante VH, Puente JL. 2012. A distinct regulatory sequence is essential for the expression of a subset of nle genes in attaching and effacing Escherichia coli. J Bacteriol 194:5589–5603. doi: 10.1128/JB.00190-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roe AJ, Tysall L, Dransfield T, Wang D, Fraser-Pitt D, Mahajan A, Constandinou C, Inglis N, Downing A, Talbot R, Smith DG, Gally DL. 2007. Analysis of the expression, regulation and export of NleA-E in Escherichia coli O157:H7. Microbiology 153:1350–1360. doi: 10.1099/mic.0.2006/003707-0. [DOI] [PubMed] [Google Scholar]
- 73.Schwidder M, Hensel M, Schmidt H. 2011. Regulation of nleA in Shiga toxin-producing Escherichia coli O84:H4 strain 4795/97. J Bacteriol 193:832–841. doi: 10.1128/JB.00582-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reading NC, Torres AG, Kendall MM, Hughes DT, Yamamoto K, Sperandio V. 2007. A novel two-component signaling system that activates transcription of an enterohemorrhagic Escherichia coli effector involved in remodeling of host actin. J Bacteriol 189:2468–2476. doi: 10.1128/JB.01848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zimmer DP, Soupene E, Lee HL, Wendisch VF, Khodursky AB, Peter BJ, Bender RA, Kustu S. 2000. Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proc Natl Acad Sci U S A 97:14674–14679. doi: 10.1073/pnas.97.26.14674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Branchu P, Matrat S, Vareille M, Garrivier A, Durand A, Crépin S, Harel J, Jubelin G, Gobert AP. 2014. NsrR, GadE, and GadX interplay in repressing expression of the Escherichia coli O157:H7 LEE pathogenicity island in response to nitric oxide. PLoS Pathog 10:e1003874. doi: 10.1371/journal.ppat.1003874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tramonti A, De Canio M, De Biase D. 2008. GadX/GadW-dependent regulation of the Escherichia coli acid fitness island: transcriptional control at the gadY-gadW divergent promoters and identification of four novel 42 bp GadX/GadW-specific binding sites. Mol Microbiol 70:965–982. doi: 10.1111/j.1365-2958.2008.06458.x. [DOI] [PubMed] [Google Scholar]
- 78.Tucker DL, Tucker N, Ma Z, Foster JW, Miranda RL, Cohen PS, Conway T. 2003. Genes of the GadX-GadW regulon in Escherichia coli. J Bacteriol 185:3190–3201. doi: 10.1128/JB.185.10.3190-3201.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dyszel JL, Soares JA, Swearingen MC, Lindsay A, Smith JN, Ahmer BM. 2010. E. coli K-12 and EHEC genes regulated by SdiA. PLoS One 5:e8946. doi: 10.1371/journal.pone.0008946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hommais F, Krin E, Coppee JY, Lacroix C, Yeramian E, Danchin A, Bertin P. 2004. GadE (YhiE): a novel activator involved in the response to acid environment in Escherichia coli. Microbiology 150:61–72. doi: 10.1099/mic.0.26659-0. [DOI] [PubMed] [Google Scholar]
- 81.Zwir I, Shin D, Kato A, Nishino K, Latifi T, Solomon F, Hare JM, Huang H, Groisman EA. 2005. Dissecting the PhoP regulatory network of Escherichia coli and Salmonella enterica. Proc Natl Acad Sci U S A 102:2862–2867. doi: 10.1073/pnas.0408238102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Constantinidou C, Hobman JL, Griffiths L, Patel MD, Penn CW, Cole JA, Overton TW. 2006. A reassessment of the FNR regulon and transcriptomic analysis of the effects of nitrate, nitrite, NarXL, and NarQP as Escherichia coli K12 adapts from aerobic to anaerobic growth. J Biol Chem 281:4802–4815. doi: 10.1074/jbc.M512312200. [DOI] [PubMed] [Google Scholar]
- 83.Krin E, Danchin A, Soutourina O. 2010. Decrypting the H-NS-dependent regulatory cascade of acid stress resistance in Escherichia coli. BMC Microbiol 10:273. doi: 10.1186/1471-2180-10-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shimada T, Hirao K, Kori A, Yamamoto K, Ishihama A. 2007. RutR is the uracil/thymine-sensing master regulator of a set of genes for synthesis and degradation of pyrimidines. Mol Microbiol 66:744–757. doi: 10.1111/j.1365-2958.2007.05954.x. [DOI] [PubMed] [Google Scholar]
- 85.Blanc-Potard AB, Solomon F, Kayser J, Groisman EA. 1999. The SPI-3 pathogenicity island of Salmonella enterica. J Bacteriol 181:998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 87.Tukel C, Akcelik M, de Jong MF, Simsek O, Tsolis RM, Baumler AJ. 2007. MarT activates expression of the MisL autotransporter protein of Salmonella enterica serotype Typhimurium. J Bacteriol 189:3922–3926. doi: 10.1128/JB.01746-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dorsey CW, Laarakker MC, Humphries AD, Weening EH, Baumler AJ. 2005. Salmonella enterica serotype Typhimurium MisL is an intestinal colonization factor that binds fibronectin. Mol Microbiol 57:196–211. doi: 10.1111/j.1365-2958.2005.04666.x. [DOI] [PubMed] [Google Scholar]
- 89.Morgan E, Campbell JD, Rowe SC, Bispham J, Stevens MP, Bowen AJ, Barrow PA, Maskell DJ, Wallis TS. 2004. Identification of host-specific colonization factors of Salmonella enterica serovar Typhimurium. Mol Microbiol 54:994–1010. doi: 10.1111/j.1365-2958.2004.04323.x. [DOI] [PubMed] [Google Scholar]
- 90.Pan SH, Malcolm BA. 2000. Reduced background expression and improved plasmid stability with pET vectors in BL21 (DE3). Biotechniques 29:1234–1238. [DOI] [PubMed] [Google Scholar]
- 91.Manning SD, Motiwala AS, Springman AC, Qi W, Lacher DW, Ouellette LM, Mladonicky JM, Somsel P, Rudrik JT, Dietrich SE, Zhang W, Swaminathan B, Alland D, Whittam TS. 2008. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc Natl Acad Sci U S A 105:4868–4873. doi: 10.1073/pnas.0710834105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 94.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. 2001. Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A 98:15264–15269. doi: 10.1073/pnas.261348198. [DOI] [PMC free article] [PubMed] [Google Scholar]