

ABSTRACT
Recent breakthroughs in next-generation sequencing technologies have led to the identification of small noncoding RNAs (sRNAs) as a new important class of regulatory molecules. In prokaryotes, sRNAs are often bound to the chaperone protein Hfq, which allows them to interact with their partner mRNA(s). We screened the genome of the zoonotic and human pathogen Brucella suis 1330 for the presence of this class of RNAs. We designed a coimmunoprecipitation strategy that relies on the use of Hfq as a bait to enrich the sample with sRNAs and eventually their target mRNAs. By deep sequencing analysis of the Hfq-bound transcripts, we identified a number of mRNAs and 33 sRNA candidates associated with Hfq. The expression of 10 sRNAs in the early stationary growth phase was experimentally confirmed by Northern blotting and/or reverse transcriptase PCR.
IMPORTANCE Brucella organisms are facultative intracellular pathogens that use stealth strategies to avoid host defenses. Adaptation to the host environment requires tight control of gene expression. Recently, small noncoding RNAs (sRNAs) and the sRNA chaperone Hfq have been shown to play a role in the fine-tuning of gene expression. Here we have used RNA sequencing to identify RNAs associated with the B. suis Hfq protein. We have identified a novel list of 33 sRNAs and 62 Hfq-associated mRNAs for future studies aiming to understand the intracellular lifestyle of this pathogen.
INTRODUCTION
Brucella organisms are Gram-negative, facultative, intracellular pathogens responsible for a major zoonosis. The genus classically comprised 6 species: B. suis, B. abortus, B. melitensis, B. ovis, B. canis, and B. neotomae. However, over the last 20 years, several new species have been reported, including B. ceti, B. pinnipedialis, B. microti, B. inopinata, and, most recently, B. papionis (1–10). B. melitensis, B. abortus, and certain biovars of B. suis are the major causes of human brucellosis.
Animal brucellosis causes abortion and infertility, and the disease can be transmitted to humans in contact with infected animals or their contaminated products (11, 12). Although several countries have succeeded in eradicating the disease in cattle and small ruminants, there are still small pockets of infection throughout Europe and the United States, and the disease is still endemic in many countries in South America, Africa, Asia, and the Middle East, where it is a serious public health and economic problem (13, 14).
The brucellae are facultative intracellular pathogens that can survive and multiply in both professional and nonprofessional phagocytes (15, 16). This is dependent on several virulence factors, including the VirB type IV secretion system (T4SS). One of the first virulence factors to be identified was the RNA chaperone protein Hfq (17). An hfq deletion mutant of B. abortus is more sensitive to H2O2 and less resistant to acid stress during stationary-phase growth. The hfq mutant fails to replicate in macrophages and is rapidly cleared from the spleens and livers of infected BALB/c mice. Since the discovery of its role in Brucella virulence, Hfq has been shown to be essential for the virulence of many extra- and intracellular Gram-negative pathogens, including Escherichia coli, Salmonella enterica, Vibrio cholerae, Bordetella pertussis, and Legionella pneumophila (18–22; R. Roop, G. Robertson, V. Grippe, M. Kovach, K. LeVier, S. Hagius, J. Walker, N. Booth, T. Fulton, and P. Elzer, presented at the 53rd Annual Brucellosis Research Conference, 2000).
Hfq is an Sm-like RNA binding protein that forms a hexameric ring structure containing multiple RNA binding sites (23). Hfq binds to both small noncoding RNAs (sRNAs) and their target mRNAs and facilitates their interaction by the formation of short imperfect base pairing (23–25). This interaction often results in the repression of target mRNAs by blocking the ribosome binding site (RBS) or by recruiting ribonucleases to initiate mRNA decay (26), but in several cases, it also leads to target activation by freeing self-inhibitory mRNA structures or stabilizing target mRNAs (27). Bacterial sRNAs range between 50 and 250 nucleotides in length and typically include a small stretch of conserved bases located toward the 5′ end, referred to as the “seed” sequence (28). The seed is required to base pair with one or multiple target mRNAs, generally on their 5′ ends, including the RBS region and early coding sequences (23, 29–32). Small RNAs have their own transcription start sites (TSSs) and ρ-independent terminators that are intrinsic to their sequence. Most sRNAs are encoded in the intergenic regions (IGRs) as independent transcripts, but they can be encoded elsewhere in the bacterial genome, including the 5′ and 3′ untranslated regions (UTRs) of coding genes or their antisense regions (33, 34).
While >200 sRNAs have been experimentally validated in model bacteria such as E. coli and S. enterica (34–38), only 4 sRNAs have been studied in Brucella so far. These sRNAs include AbcR1 and AbcR2 (39), orthologues of SmrC15 and SmrC16 of Rhizobium etli, and AbcR1 and AbcR2 of Agrobacterium tumefaciens. These two sRNAs are required for full virulence of B. abortus 2308. An abcR1 abcR2 double mutant shows a decreased level of survival in cultured macrophages and a defect in the colonization of the spleens of infected mice (39). The other two sRNAs were reported recently. BSR0602 has been described to regulate gntR, encoding a transcriptional regulator that plays a role in the virulence of B. melitensis (40, 41). Another cis-encoded sRNA, BsrH, has been shown to repress hemH and the expression of its encoded ferrochelatase, but it had no impact on virulence (42).
The identification of sRNAs has long been challenging for several reasons. Their small size and low expression levels made them difficult to analyze by using classical biochemical methods. Their localization in the genome, sometimes antisense to existing genes, made them undetectable in bioinformatics-based searches (43). In the last decade, several new approaches have been used to identify bacterial sRNAs. One strategy consists of using Hfq as a bait to enrich sRNAs by coimmunoprecipitation of the chaperone and all RNA species bound to it (25, 44). This technique is now coupled to strand-specific cDNA library generation and deep sequencing to identify sRNAs with an unprecedented resolution. It has been successfully used to identify Salmonella sRNAs and at least doubled the number of known sRNAs in a single study (36). Here we have chosen B. suis 1330 as a representative member of the Brucella genus. Our aim was to identify as-yet-undescribed noncoding RNAs that might be related to virulence or stress adaptation through their association with Hfq in this pathogen.
MATERIALS AND METHODS
Bacterial strains.
Brucella suis 1330 (ATCC 23444T) and derivatives were grown in Trypticase soy (TS) broth, and E. coli DH5α was grown in L broth supplemented with antibiotics as required. All manipulation of live Brucella cells was performed in a biosafety level 3 (BSL-3) containment laboratory in a class II microbiological safety cabinet with centrifugation in sealed aerosol-free rotors.
Introduction of 3×FLAG-tagged Hfq into Brucella by allelic replacement.
The introduction of a chromosomal 3×FLAG-tagged hfq allele in Brucella was done by unmarked allelic replacement using a suicide vector and SacB-dependent sucrose counterselection as described previously by Patey et al. (45). A 3×FLAG tag was incorporated into the B. suis hfq gene at the carboxy terminus. The hfq gene and ∼1 kb of the upstream flanking region were amplified by using oligonucleotide primers hfq-Up-FLAG-For and hfq-Up-FLAG-Rev (all primers are listed in Table S1 in the supplemental material), B. suis 1330 genomic DNA, and Pfx supermix (Invitrogen). The hfq-Up-FLAG-Rev primer contains 11 codons of the 3×FLAG tag up to the next-to-last codon of the B. suis hfq gene. The stop codon of the hfq gene and 1 kb of the downstream flanking region were amplified by using oligonucleotide primers hfq-Dn-FLAG-For and hfq-Dn-FLAG-Rev. The hfq-Dn-FLAG-For primer contains the remaining 11 codons of the 3×FLAG tag and the last codon of the B. suis hfq gene. The upstream fragment was digested with BamHI, and the downstream fragment was digested with PstI. Following enzyme digestion and agarose gel purification, each fragment was treated with polynucleotide kinase (PNK; Monserate Biotechnology Group, San Diego, CA). The DNA fragments were then ligated into BamHI/PstI-digested pNTPS138 (46) using T4 DNA ligase (Monserate Biotechnology Group, San Diego, CA). The resulting plasmid construct, named phfq3xFLAG, was verified by DNA sequence analysis. B. suis 1330 was electroporated with phfq3xFLAG, and the first recombination events were selected as kanamycin-resistant colonies. A second recombination event was selected by using sucrose sensitivity, as described previously (45). Sucrose-resistant, kanamycin-sensitive colonies were checked for replacement of wild-type (WT) hfq by PCR on genomic DNA (using primers hfq wt 1b, hfq wt 2, and hfq 3 × FLAG 1b).
Total RNA extraction, RT-PCR, and Northern blotting.
Total RNA was extracted from B. suis 1330 cells grown in TS agar to late log phase (optical density at 600 nm [OD600] of 1) with TRIzol. For reverse transcriptase PCR (RT-PCR), reverse transcription was performed by using the Bio-Rad iScript Select cDNA synthesis kit. Northern blotting for small RNAs was performed as described previously (45), using 32P-labeled oligonucleotide probes (see Table S1 in the supplemental material).
Coimmunoprecipitation.
Hfq coimmunoprecipitation (co-IP) experiments were performed according to established protocols, with minor modifications, using lysates of 1330::hfq-3xFLAG cells and wild-type B. suis 1330 cells as a control to determine the level of enrichment of RNAs in the co-IP (36). Bacteria were grown in TS broth with vigorous aeration to an OD of 1 and then chilled on ice. After harvesting by centrifugation (3,000 × g for 15 min) at 4°C, bacterial cells (∼50 ODs) of each strain were washed with ice-cold lysis buffer (20 mM Tris [pH 8.0], 150 mM KCl, 1 mM MgCl2, 1 mM dithiothreitol [DTT]). Bacteria were lysed by three cycles of freezing in liquid nitrogen and thawing on ice, followed by mechanical disruption with glass beads. Pellets were suspended in 800 μl of lysis buffer, and 800 μl of glass beads (0.1-mm diameter; Roth) was added. The mix was vortexed in 30-s bursts with breaks of 30 s for chilling on ice for a total of 15 min. The lysate was then clarified by centrifugation (15,000 × g for 15 min at 4°C). Before immunoprecipitation, the presence of Hfq in the lysate was confirmed by Western blotting using the anti-FLAG antibody (for 1330::hfq-3xFLAG lysates). For immunoprecipitation, the supernatants were transferred into new tubes and incubated with 35 μl of anti-FLAG antibody (Sigma) on a rotating wheel for 30 min at 4°C. For the pulldown, 75 μl of protein A-Sepharose (Sigma) (prewashed in 1 ml lysis buffer) was added, and the tubes were rocked again for 30 min at 4°C. The Sepharose beads were then washed five times with 500 μl of lysis buffer and suspended in 500 μl of the same buffer before phenol-chloroform extraction and ethanol precipitation of RNA.
Western blot analysis.
Samples were boiled in Laemmli buffer, separated on 15% SDS-PAGE gels, and then transferred onto a Biodyne B nylon membrane (Thermo). The membrane was incubated in a blocking buffer (Sigma) for 1 h. FLAG-tagged Hfq was detected by using the M2 anti-FLAG monoclonal antibody (Sigma), followed by a goat anti-mouse antibody coupled to IRDye 800CW (Licor). The membrane was then scanned by using a Licor Odyssey infrared scanner.
Cell culture and infections.
Murine J774 macrophage-like cells (ATCC) were maintained and infected with Brucella by using a standard gentamicin protection assay (47, 48).
cDNA construction.
The RNA samples from both control co-IP and Hfq co-IP were processed for cDNA construction at Vertis AG according to standard procedures (18). Briefly, RNA samples were first treated with poly(A) polymerase to add a poly(A) tail at the 3′ ends and then treated with tobacco acid pyrophosphatase (TAP) to convert the 5′-triphosphate ends to 5′-monophosphate (5′-P) ends prior to 5′-RNA adaptor ligation. First-strand cDNA synthesis was performed by using an oligo(dT) adaptor primer and Moloney murine leukemia virus (M-MLV) reverse transcriptase. The resulting cDNA was barcoded and amplified to ∼30 to 50 ng/μl by using a high-fidelity DNA polymerase. After purification using the Agencourt AMPure XP kit (Beckman Coulter Genomics), cDNA was pooled and sequenced on an Illumina HiSeq platform.
Deep sequencing analysis.
Raw sequencing reads were quality checked by using FastQC (49); the low-quality sequences (Phred score of <28) were trimmed before downstream analyses. The remaining reads were mapped to two circular chromosomes of B. suis 1330 (GenBank accession numbers NC_004310.3 and NC_004311.2) by using READemption pipeline v0.34 with the “-poly_a_clipping” option. The read coverage and read counts per gene were generated by READemption with default parameters (50, 51). Rho-independent terminators were predicted with TransTermHP v2.09 using default parameters with the “-bag” option (50). The predicted terminators with high confidence scores (≥65) were annotated in the B. suis genome for read count analysis. All intergenic regions of >30 bp were flagged as IGRs and were used for read count analysis.
Nucleotide sequence accession numbers.
The transcriptome sequencing (RNA-seq) data have been deposited in the Gene Expression Omnibus (GEO) with accession number GSE73621 (accession number GSM1899587 for the negative control and accession number GSM1899588 for FLAG-tagged Hfq).
RESULTS
Chromosomal expression of the 3×FLAG-tagged Hfq protein in Brucella.
In order to identify new sRNAs in Brucella, we employed a previously established coimmunoprecipitation strategy to pull down Hfq and the associated transcripts (25, 36). First, we replaced the wild-type hfq allele in the B. suis 1330 genome with a 3×FLAG-tagged allele (see Materials and Methods), and one isolate was selected and assigned the collection number bIN417 (called 1330::hfq-3xFLAG here). Expression of the FLAG-tagged hfq allele was confirmed by Western blotting using an anti-FLAG antibody (see Fig. S1A in the supplemental material). The introduction of C-terminal 3×FLAG to Hfq did not affect bacterial growth and physiology (data not shown), and the 3×FLAG-tagged mutant strain exhibited no detectable defect in virulence; it was able to invade and replicate in J774 macrophages at wild-type levels (see Fig. S1B in the supplemental material).
Enrichment of Hfq-associated transcripts by coimmunoprecipitation.
Wild-type 1330 and 1330::hfq-3xFLAG cells were grown in TS medium to late logarithmic phase (OD600 of 1), and equal amounts of cells (50 ODs) were lysed and subjected to pulldown as described in detail in Materials and Methods. The successful recovery of sRNAs was confirmed by the presence of AbcR1 and AbcR2 in the samples, detected by RT-PCR using gene-specific oligonucleotides described previously by Caswell et al. (data not shown) (39). Strand-specific cDNA libraries were constructed by using 130 ng of RNA samples from both WT control co-IP and Hfq co-IP and were subjected to deep sequencing using Illumina technology.
Transcriptome-wide analyses of RNA sequencing results.
A total of ∼5 million short reads (100 bp in length) were generated by Illumina sequencing for each cDNA library (Fig. 1). Most sequencing reads had high-quality scores (Phred score of >28) and were long enough (≥20 bp) for mapping analysis after the removal of sequence adaptors. Mapping was performed by using the READemption RNA-seq analysis pipeline (50), and the majority of reads were successfully mapped back to the B. suis 1330 chromosomes (GenBank accession numbers NC_004310 and NC_004311). As expected, most of the short reads were derived from abundant rRNA/tRNA transcripts (Fig. 1), but a large proportion of reads mapped to coding sequences (CDSs) and IGRs. The Hfq co-IP library contained far more reads from CDSs and IGRs than did the WT co-IP library (Fig. 1), suggesting a successful enrichment of Hfq-associated mRNAs and putative noncoding RNA transcripts located in the IGR by the Hfq co-IP procedure.
FIG 1.

Classification of reads from negative-control and Hfq co-IP samples.
To identify these Hfq-bound transcripts, read counts for all known genes annotated in the Brucella genome were calculated, and read coverage profiles were generated and loaded into a genome browser (Integrated Genome Browser [IGB] [52]) for direct visual inspection. We found that 62 genes were at least 3-fold enriched in the Hfq co-IP (ratio of read counts in Hfq co-IP/WT co-IP), suggesting that these are mRNA targets of Hfq in Brucella (Fig. 2; see also Table S2 in the supplemental material).
FIG 2.

Enrichment of sRNA candidates in Hfq co-IPs. EF, enrichment factor.
Identification of Hfq-associated sRNAs.
We manually inspected the Brucella genome for potential sRNA candidates by using IGB. In our analysis, we found that manual screening of the sequencing data was the most effective way to identify candidate sRNAs. Available tools such as DESeq are able to calculate expression levels and differential expression for annotated transcripts only (53). When we are looking for new transcripts, they must be annotated “de novo” by using tools such as Cufflinks that work best with eukaryotic data and fail to accurately annotate transcripts from prokaryotic data (54).
Candidate sRNAs showed high enrichment in the Hfq co-IP relative to the WT control and were identified and documented for their nucleotide sequence, length, orientation, localization, and flanking genes (Table 1). Although there is no clear consensus on the minimum number of cDNA reads needed to consider a transcript biologically relevant, we avoided selecting transcripts that had <20 cDNA reads, a threshold below which the risk of artifacts and background noise increases. This manual analysis yielded a list of 33 candidate Hfq-associated sRNAs (Table 1 and Fig. 3).
TABLE 1.
List of Hfq-associated sRNAs
| sRNAa | Chromosome | Size (nt) | Orientationb | 5′–3′ (nt) | ECc | IGR analysis | Term analysis | Northern/RT-PCR analysis |
|---|---|---|---|---|---|---|---|---|
| BSnc010 | I | 48 | < > < | 868844–868891 | 13 | Yes | Yes | No |
| BSnc082 | I | 49 | < > > | 1522140–1522188 | 8 | Yes | No | No |
| BSnc112 | I | 74 | < < < | 264807–264734 | 12 | Yes | No | No |
| BSnc113 | I | 147 | < < > | 288902–288756 | 67 | Yes | No | No |
| BSnc114 | I | 93 | < < < | 293000–292908 | 35 | Yes | No | No |
| BSnc115 | I | 91 | > < > | 338240–338150 | 13 | No | No | Yes |
| BSnc118 | I | 152 | > < > | 798065–797914 | 43 | Yes | No | Yes |
| BSnc119 | I | 90 | > < > | 953526–953437 | 30 | Yes | No | Yes |
| BSnc120 | I | 89 | > < < | 967105–967018 | 18 | No | Yes | Yes |
| BSnc121 | I | 116 | < < > | 1150053–1149938 | No | No | Yes | |
| BSnc126 | I | 135 | < < > | 1415823–1415689 | No | No | No | |
| BSnc128 | I | 113 | < < < | 1459614–1459502 | 26 | Yes | Yes | No |
| BSnc132 | I | 113 | < < > | 1606503–1606391 | Yes | No | No | |
| BSnc135 | I | 91 | < < < | 1648872–1648782 | 5 | No | No | Yes |
| BSnc140 | I | 94 | < < > | 1781160–1781067 | Yes | No | Yes | |
| BSnc144 | I | 89 | < < < | 1803634–1803546 | No | No | No | |
| BSnc145 | I | 71 | < < < | 1814562–1814492 | Yes | No | No | |
| BSnc148 | II | 66 | < > < | 521046–521111 | 5 | Yes | No | No |
| BSnc149 | II | 155 | < > < | 605989–606143 | 15 | Yes | No | Yes |
| BSnc150 | II | 44 | > > > | 1083352–1083395 | 11 | Yes | No | Yes |
| BSnc159 | II | 52 | < > < | 178758–178809 | 35 | Yes | No | Yes |
| BSnc166 | II | 62 | > > > | 392433–392494 | No | No | No | |
| BSnc167 | II | 126 | > > > | 398361–398486 | 63 | Yes | No | No |
| BSnc175 | II | 71 | < > > | 464600–464670 | 16 | Yes | No | No |
| BSnc235 | II | 81 | < < > | 8941–8861 | 24 | Yes | No | No |
| BSnc239 | II | 58 | < < > | 361504–361477 | 7 | Yes | No | No |
| BSnc246 | II | 68 | < < < | 549470–549403 | 7 | Yes | No | No |
| BSnc247 | II | 88 | < < > | 568177–568090 | No | No | No | |
| BSnc249 | II | 40 | < < > | 729876–729836 | 14 | No | No | No |
| BSnc251 | II | 55 | < < < | 844677–844623 | No | Yes | No | |
| BSnc253 | II | 143 | < < > | 1017459–1017317 | 33 | Yes | No | No |
| BSnc254 | II | 88 | < < > | 1064397–1064310 | 9 | Yes | No | No |
| BSnc258 | I | 51 | < < < | 287507–287457 | 14 | No | No | No |
Underlined sRNAs were confirmed by using Rockhopper.
“>” represents a gene on the positive strand, “<” represents a gene on the negative strand, and the order is flank, sRNA gene, flank.
The enrichment coefficient (EC) is the ratio of the mean number of cDNA reads for a given transcript in the sample with respect to the negative control. The enrichment coefficient could not precisely be calculated for transcripts predicted only manually due to the lack of a mean value for the number of cDNA reads.
FIG 3.

Positions of the identified Hfq-associated sRNAs in the genome of B. suis. Graphics represent typical examples of each category of sRNAs found in this study. Lists to the right of each graphic representation represent the identified sRNAs that fall in this category. *, BSnc249 overlaps the 5′ UTR of BRA0746 and is antisense to BRA0747; †, BSnc258 overlaps the 5′ UTR of dht and is antisense to BR0279. For those sRNAs predicted manually, it was not possible to calculate an enrichment coefficient with precision.
To facilitate an unbiased genome-wide analysis, all the IGRs (≥30 bp) in the B. suis 1330 genome were annotated and fed through the READemption algorithm to calculate read counts and enrichment in the Hfq co-IP (Table 1). This analysis provided a list of candidate regions that are likely to contain Hfq-associated sRNAs. The results of this automatic analysis agreed well with the results of our manual analyses. Most manually identified sRNA candidates are enriched in Hfq co-IPs at least 3-fold, with 26 of them showing >10-fold enrichment (Fig. 4). In addition, we used the “Rockhopper” tool for de novo prediction of prokaryotic sRNA genes from the RNA-seq data. Rockhopper independently predicted 15 out of the 33 sRNA genes that were identified manually by using the IGB, again supporting the accuracy of our manual analyses. For these 15 sRNAs, we annotated their sequence boundaries according to the Rockhopper prediction (Table 1) (53).
FIG 4.
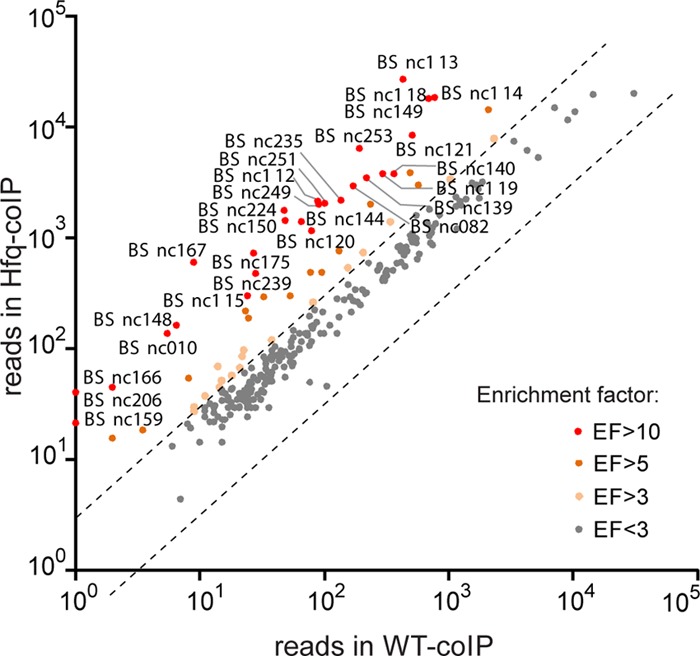
Enrichment of genes in Hfq co-IPs.
Characterization of sRNAs.
The genes encoding the sRNAs identified in this study were found in different locations in the genome. Most of them (22/33) were found within IGRs. sRNAs were also found in the antisense strand of annotated genes, suggesting that they may act as antisense RNA in cis. Several sRNAs largely overlapped 5′ or 3′ UTRs (including start or stop codons), belonging to the emerging class of UTR-derived sRNAs (54). Figure 3 summarizes these results with an example representing each of the cases.
We used both RT-PCR and Northern blotting to detect the expression of the sRNAs, and we have experimentally validated 10 of them. RT-PCR was first applied to sRNAs located within IGRs. With this approach, we were able to confirm the expression of 3 out of 15 tested sRNAs (BSnc115, BSnc118, and BSnc140) (Table 1). This low detection rate is probably due to the following technical obstacles: the short length of the sRNAs and the fact that they can form stable secondary structures (e.g., terminator regions) that hinder reverse transcription. To overcome this, small RNAs were detected by using Northern blotting. This resulted in the successful validation of another eight sRNAs, BSnc118, BSnc119, BSnc120, BSnc121, BSnc135, BSnc149, BSnc150, and BSnc159 (Table 1 and Fig. 5).
FIG 5.

Northern blot analysis of small RNAs in Brucella suis 1330. nt, nucleotides.
Conservation of Hfq-associated sRNAs in the Brucella genus.
Brucella genomes are highly conserved at both the structural and nucleotide sequence levels (4, 55). Using BLASTn searches, we examined the distribution of the sRNAs in the genomes of all Brucella species. As expected, the majority of the queried sRNAs were fully conserved in a panel of 12 strains representing all known species (with the exception of recent frog isolates) (Table 2). A major cause of species- or biovar-specific polymorphisms in the genus is the presence or absence of genomic islands (55). Several sRNAs were found in genomic islands, and this is reflected in their distribution within the genus. For example, the BSnc150, BSnc224, and BSnc245 genes are located in genomic island 5 (GI5) and are therefore missing from early-dividing Brucella as well as B. ovis and B. papionis (56). Similarly, the BSnc239 gene is found in the IncP region, and the BSnc167 gene is found in a block of genes that are missing from the Australian rodent strains (55–57). Adjustment of the BLAST parameters (word size of 10) allowed us to identify the BSnc148 gene in all strains (but with several single nucleotide polymorphisms [SNPs]) and showed that the first 5 nucleotides of the BSnc159 gene are missing in the Australian rodent strains and that there are several SNPs in BO1 and BO2.
TABLE 2.
Distribution of the identified sRNAs in representative classical and recently described Brucella species
| Noncoding RNA | Presence of noncoding RNA in: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B. suis 1330 | B. papionis NVSL 07-0026 | B. ovis ATCC 25840 | B. neotomae 5K33 | B. microti CCM 4915 | B. abortus 2308 | B. melitensis 16 M | B. ceti Cudo | B. canis ATCC 23365 | Brucella sp. BO2 | Brucella sp. NF 2653 | B. inopinata BO1 | |
| BSnc148 | + | + | + | + | + | + | + | + | + | + | + | + |
| BSnc149 | + | + | + | + | + | + | + | + | + | + | + | + |
| BSnc150 | + | − | − | + | + | + | + | + | + | − | − | − |
| BSnc167 | + | + | + | + | + | + | + | + | + | + | − | + |
| BSnc224 | + | − | − | + | + | + | + | + | + | − | − | − |
| BSnc239 | + | − | − | + | + | − | − | + | + | − | − | − |
| BSnc254 | + | − | − | + | + | + | + | + | + | − | − | − |
Hfq-independent noncoding RNA.
As noted above, not all of the abundant transcripts were enriched in the Hfq co-IP. By querying their sequences with the Rfam database, several transcripts were identified as noncoding RNAs that are highly conserved across the proteobacteria. These transcripts include transfer-messenger RNA (tmRNA), 4.5S RNA, and 6S RNA. 6S RNA acts as a transcriptional regulator controlling adaptation to stationary-phase and stress conditions and was recently implicated in Legionella virulence (58, 59). It is able to bind to RNA polymerase by structurally mimicking a DNA template with an open promoter; analysis of the B. suis 6S RNA sequence indeed revealed a double-stranded RNA structure with open bulges in the center (Fig. 6).
FIG 6.

Brucella suis 6S RNA. (A) Hfq-independent noncoding RNA identified by RNA-seq. (B) Predicted secondary structure of the B. suis 6S RNA.
DISCUSSION
The role of Hfq in bacterial virulence was first observed in Brucella (17). Following the discovery of the pleiotropic role of Hfq and sRNA in gene regulation, many studies have investigated the effect of an hfq mutation on gene expression. In a recent report, Cui et al. used microarray analysis to suggest that 359 genes (∼11% of the genome) in B. melitensis are differentially regulated in an hfq mutant, while 2-dimensional gel electrophoresis coupled with matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry identified 55 proteins with altered expression levels (60). What is not clear, however, is how many of these genes are directly regulated by sRNA/Hfq or through more complex cascades (61). The discovery of sRNA in bacteria has changed the way we look at information flow from DNA to mRNA to protein. It is clear that sRNAs constitute an essential class of regulatory molecules to be explored in organisms like Brucella. Despite the fact that the role of Hfq in bacterial virulence was first observed in Brucella, to date, only four sRNAs have been characterized for this genus (41, 42). Previous studies of Brucella used either low-throughput methods or in silico predictions to identify sRNAs.
Here we employed a high-throughput experimental approach to identify RNA molecules that are associated with an epitope-tagged Hfq protein by RNA-seq (33, 36). This is the first experimental genome-wide analysis of a Brucella transcriptome for the identification of small noncoding RNAs and Hfq targets. This global analysis has revealed 62 Hfq-associated mRNAs as well as 33 candidate sRNA molecules, 10 of which have been experimentally confirmed. Interestingly, many sRNAs identified in this study have not been identified previously and largely escaped previous in silico predictions. For example, no overlap was seen with the 112 sRNAs predicted in B. abortus by Dong et al. (61). Comparison with the B. melitensis sRNAs reported by Wang et al. (41) showed that their BASRCI94 is equivalent to our BSnc010. We also identified a B. suis homologue of BASRCI310 (BSnc081), but it was excluded from our list since the enrichment factor (×2) in the Hfq co-IP was below our cutoff. There was also no overlap between the long lists of sRNA candidates reported by Dong et al. and Wang et al., suggesting that the in silico predictions are not very accurate. However, we have observed overlaps between our B. suis sRNAs and those identified experimentally in B. abortus (C. C. Caswell and R. M. Roop II, unpublished data).
Hfq-associated sRNAs usually bind to trans-encoded target mRNAs by imperfect base pairing, making targets difficult to identify by simple sequence homology. We used TargetRNA, a tool developed by Tjaden (62), to predict their target mRNAs. The results of the predictions are summarized in Table 3. Three sRNAs were predicted to have targets that were previously reported to play a role in the virulence of Brucella. BSnc148 was predicted to regulate the expression of the stationary-phase regulator integration host factor (ihfA). This will have pleiotropic effects and will affect the expression of key virulence factors, including the VirB T4SS, essential for intracellular survival, and erythritol metabolism, thought to be important for the tropism of Brucella for ruminant placenta (63, 64).
TABLE 3.
Predicted targets of the identified Hfq-associated sRNAsa
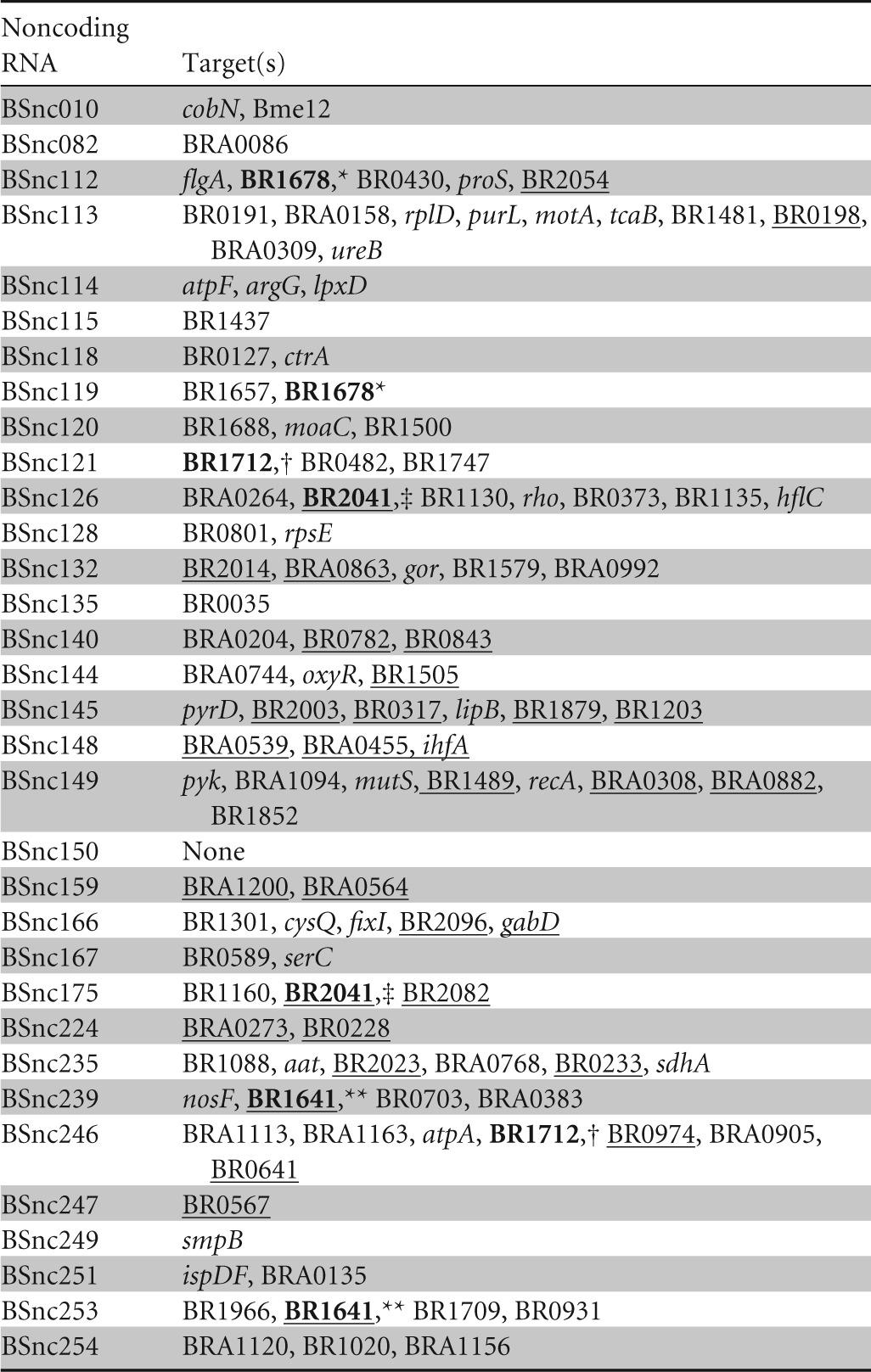
Hypothetical proteins are underlined. The same targets for two different sRNAs are shown in boldface type and have the same symbols (*, **, †, or ‡).
Our predicted sRNA targets highlight the importance of Hfq and its associated sRNA in acid resistance. The HdeA protein is required for acid resistance in vitro and has been reported to be regulated by Hfq (65). Interestingly, hdeA is part of an operon with the genes encoding the glutamic acid decarboxylase (GAD) system. Although these genes are not functional in many Brucella species due the accumulation of point mutations, in B. microti, they are functional and are essential for resistance to the very low pH encountered in the stomach (66). We did not identify the hdeA-gad operon in our RNA-seq analysis, probably because we grew our bacteria in rich medium at neutral pH, and we did not find this operon in the predicted sRNA targets. However, BSnc144 was predicted to regulate the expression of OxyR, a transcriptional regulator itself implicated in the regulation of gadB, suggesting an indirect effect of Hfq. Another key factor for surviving stomach acid is urease; one of the predicted targets of BSnc113 was the β-subunit of urease, a multisubunit nickel-containing enzyme that enables B. suis to survive at an extreme pH of 2.0. Future studies are needed to investigate the roles of these sRNAs in acid resistance.
In conclusion, in this study, we have identified 62 mRNAs and 33 sRNA candidates that are associated with Hfq under normal growth conditions in B. suis 1330. We also identified highly conserved noncoding RNAs, including 4.5S RNA, 6S RNA, and tmRNA. This global RNA-seq analysis paved the way for studying posttranscriptional gene regulation in Brucella. Further characterization of Hfq-associated sRNAs will shed light on the Hfq-governed posttranscriptional regulatory network in Brucella and the role of Hfq and sRNA in the virulence and general physiology of Brucella.
Supplementary Material
ACKNOWLEDGMENTS
Work in INSERM U1047 is supported by INSERM, Université Montpellier 1, La Region Languedoc-Roussillon, the French Research National Agency (ANR-09-MIEN-003-01), and the Institute de Veille Sanitaire (CNR-Brucella). Bashir Saadeh was supported by a Ph.D. grant from the Infectiopole Sud. Clayton C. Caswell was supported by funds from the VA-MD College of Veterinary Medicine and the Fralin Life Science Institute at Virginia Tech. Work in the Roop laboratory was funded by a grant (AI48499) from the National Institute of Allergy and Infectious Diseases. Alice Rebecca Wattam was supported by funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract number HHSN272201400027C.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00711-15.
REFERENCES
- 1.Audic S, Lescot M, Claverie JM, Scholz H. 2009. Brucella microti: the genome sequence of an emerging pathogen. BMC Genomics 10:352. doi: 10.1186/1471-2164-10-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De BK, Stauffer L, Koylass MS, Sharp SE, Gee JE, Helsel LO, Steigerwalt AG, Vega R, Clark TA, Daneshvar MI. 2008. Novel Brucella strain (BO1) associated with a prosthetic breast implant infection. J Clin Microbiol 46:43–49. doi: 10.1128/JCM.01494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foster G, Osterman BS, Godfroid J, Jacques I, Cloeckaert A. 2007. Brucella ceti sp. nov. and Brucella pinnipedialis sp. nov. for Brucella strains with cetaceans and seals as their preferred hosts. Int J Syst Evol Microbiol 57:2688–2693. doi: 10.1099/ijs.0.65269-0. [DOI] [PubMed] [Google Scholar]
- 4.O'Callaghan D, Whatmore AM. 2011. Brucella genomics as we enter the multi-genome era. Brief Funct Genomics 10:334–341. doi: 10.1093/bfgp/elr026. [DOI] [PubMed] [Google Scholar]
- 5.Scholz HC, Hofer E, Vergnaud G, Fleche PL, Whatmore AM, Dahouk SA, Pfeffer M, Krüger M, Cloeckaert A, Tomaso H. 2009. Isolation of Brucella microti from mandibular lymph nodes of red foxes, Vulpes vulpes, in lower Austria. Vector Borne Zoonotic Dis 9:153–156. doi: 10.1089/vbz.2008.0036. [DOI] [PubMed] [Google Scholar]
- 6.Scholz HC, Hubalek Z, Sedlacek I, Vergnaud G, Tomaso H, Al Dahouk S, Melzer F, Kampfer P, Neubauer H, Cloeckaert A. 2008. Brucella microti sp. nov., isolated from the common vole Microtus arvalis. Int J Syst Evol Microbiol 58:375–382. doi: 10.1099/ijs.0.65356-0. [DOI] [PubMed] [Google Scholar]
- 7.Scholz HC, Nockler K, Gollner C, Bahn P, Vergnaud G, Tomaso H, Al-Dahouk S, Kampfer P, Cloeckaert A, Maquart M. 2010. Brucella inopinata sp. nov., isolated from a breast implant infection. Int J Syst Evol Microbiol 60:801–808. doi: 10.1099/ijs.0.011148-0. [DOI] [PubMed] [Google Scholar]
- 8.Whatmore AM, Davison N, Cloeckaert A, Al Dahouk S, Zygmunt MS, Brew SD, Perrett LL, Koylass MS, Vergnaud G, Quance C, Scholz HC, Dick EJ, Hubbard G, Schlabritz-Loutsevitch NE. 2014. Brucella papionis sp. nov. isolated from baboons (Papio spp.). Int J Syst Evol Microbiol 64:4120–4128. doi: 10.1099/ijs.0.065482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zygmunt MS, Jacques I, Bernardet N, Cloeckaert A. 2012. Lipopolysaccharide heterogeneity in the atypical group of novel emerging Brucella species. Clin Vaccine Immunol 19:1370–1373. doi: 10.1128/CVI.00300-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zygmunt MS, Maquart M, Bernardet N, Doublet B, Cloeckaert A. 2010. Novel IS711-specific chromosomal locations useful for identification and classification of marine mammal Brucella strains. J Clin Microbiol 48:3765–3769. doi: 10.1128/JCM.01069-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopes L, Nicolino R, Haddad J. 2010. Brucellosis—risk factors and prevalence: a review. Open Vet Sci J 4:72–84. doi: 10.2174/1874318801004010072. [DOI] [Google Scholar]
- 12.Seleem MN, Boyle SM, Sriranganathan N. 2010. Brucellosis: a re-emerging zoonosis. Vet Microbiol 140:392–398. doi: 10.1016/j.vetmic.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 13.Pappas G. 2010. The changing Brucella ecology: novel reservoirs, new threats. Int J Antimicrob Agents 36:S8–S11. doi: 10.1016/j.ijantimicag.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 14.Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. 2006. The new global map of human brucellosis. Lancet Infect Dis 6:91–99. doi: 10.1016/S1473-3099(06)70382-6. [DOI] [PubMed] [Google Scholar]
- 15.Celli J. 2008. Intracellular localization of Brucella abortus and Francisella tularensis in primary murine macrophages. Methods Mol Biol 431:133–145. [DOI] [PubMed] [Google Scholar]
- 16.Starr T, Child R, Wehrly TD, Hansen B, Hwang S, López-Otin C, Virgin HW, Celli J. 2012. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 11:33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robertson GT, Roop RM. 1999. The Brucella abortus host factor I (HF-I) protein contributes to stress resistance during stationary phase and is a major determinant of virulence in mice. Mol Microbiol 34:690–700. doi: 10.1046/j.1365-2958.1999.01629.x. [DOI] [PubMed] [Google Scholar]
- 18.Chao Y, Vogel J. 2010. The role of Hfq in bacterial pathogens. Curr Opin Microbiol 13:24–33. doi: 10.1016/j.mib.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Guisbert E, Rhodius VA, Ahuja N, Witkin E, Gross CA. 2007. Hfq modulates the σE-mediated envelope stress response and the σ32-mediated cytoplasmic stress response in Escherichia coli. J Bacteriol 189:1963–1973. doi: 10.1128/JB.01243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kajitani M, Kato A, Wada A, Inokuchi Y, Ishihama A. 1994. Regulation of the Escherichia coli hfq gene encoding the host factor for phage Q beta. J Bacteriol 176:531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monteiro C, Papenfort K, Hentrich K, Ahmad I, Le Guyon S, Reimann R, Grantcharova N, Römling U. 2012. Hfq and Hfq-dependent small RNAs are major contributors to multicellular development in Salmonella enterica serovar Typhimurium. RNA Biol 9:489–502. doi: 10.4161/rna.19682. [DOI] [PubMed] [Google Scholar]
- 22.Tsui HCT, Leung HCE, Winkler ME. 1994. Characterization of broadly pleiotropic phenotypes caused by an Hfq insertion mutation in Escherichia coli K-12. Mol Microbiol 13:35–49. doi: 10.1111/j.1365-2958.1994.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 23.Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang A, Altuvia S, Tiwari A, Argaman L, Hengge-Aronis R, Storz G. 1998. The OxyS regulatory RNA represses rpoS translation and binds the Hfq (HF-I) protein. EMBO J 17:6061–6068. doi: 10.1093/emboj/17.20.6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. 2003. Global analysis of small RNA and mRNA targets of Hfq. Mol Microbiol 50:1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x. [DOI] [PubMed] [Google Scholar]
- 26.Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papenfort K, Vanderpool CK. 2015. Target activation by regulatory RNAs in bacteria. FEMS Microbiol Rev 39:362–378. doi: 10.1093/femsre/fuv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papenfort K, Bouvier M, Mika F, Sharma CM, Vogel J. 2010. Evidence for an autonomous 5′ target recognition domain in an Hfq-associated small RNA. Proc Natl Acad Sci U S A 107:20435–20440. doi: 10.1073/pnas.1009784107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fröhlich KS, Vogel J. 2009. Activation of gene expression by small RNA. Curr Opin Microbiol 12:674–682. doi: 10.1016/j.mib.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 30.Marchesini MI, Herrmann CK, Salcedo SP, Gorvel JP, Comerci DJ. 2011. In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell Microbiol 13:1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattick JS, Makunin IV. 2006. Non-coding RNA. Hum Mol Genet 15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 32.Vogel J, Papenfort K. 2006. Small non-coding RNAs and the bacterial outer membrane. Curr Opin Microbiol 9:605–611. doi: 10.1016/j.mib.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. 2012. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 31:4005–4019. doi: 10.1038/emboj.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kröger C, Colgan A, Srikumar S, Händler K, Sivasankaran SK, Hammarlöf DL, Canals R, Grissom JE, Conway T, Hokamp K. 2013. An infection-relevant transcriptomic compendium for Salmonella enterica serovar Typhimurium. Cell Host Microbe 14:683–695. doi: 10.1016/j.chom.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 35.Rivas E, Klein RJ, Jones TA, Eddy SR. 2001. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr Biol 11:1369–1373. doi: 10.1016/S0960-9822(01)00401-8. [DOI] [PubMed] [Google Scholar]
- 36.Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JCD, Vogel J. 2008. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet 4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sittka A, Sharma CM, Rolle K, Vogel J. 2009. Deep sequencing of Salmonella RNA associated with heterologous Hfq proteins in vivo reveals small RNAs as a major target class and identifies RNA processing phenotypes. RNA Biol 6:266–275. doi: 10.4161/rna.6.3.8332. [DOI] [PubMed] [Google Scholar]
- 38.Storz G. 2002. An expanding universe of noncoding RNAs. Science 296:1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- 39.Caswell CC, Gaines JM, Ciborowski P, Smith D, Borchers CH, Roux CM, Sayood K, Dunman PM, Roop RM II. 2012. Identification of two small regulatory RNAs linked to virulence in Brucella abortus 2308. Mol Microbiol 85:345–360. doi: 10.1111/j.1365-2958.2012.08117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haine V, Sinon A, Van Steen F, Rousseau S, Dozot M, Lestrate P, Lambert C, Letesson J-J, De Bolle X. 2005. Systematic targeted mutagenesis of Brucella melitensis 16M reveals a major role for GntR regulators in the control of virulence. Infect Immun 73:5578–5586. doi: 10.1128/IAI.73.9.5578-5586.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Ke Y, Xu J, Wang L, Wang T, Liang H, Zhang W, Gong C, Yuan J, Zhuang Y. 2015. Identification of a novel small non-coding RNA modulating the intracellular survival of Brucella melitensis. Front Microbiol 6:164. doi: 10.3389/fmicb.2015.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng X, Dong H, Wu Q. 2015. A new cis-encoded sRNA, BsrH, regulating the expression of hemH gene in Brucella abortus 2308. FEMS Microbiol Lett 362:1–7. doi: 10.1093/femsle/fnu017. [DOI] [PubMed] [Google Scholar]
- 43.McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, Tjaden B. 2013. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 41:e140. doi: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patey G, Qi Z, Bourg G, Baron C, O'Callaghan D. 2006. Swapping of periplasmic domains between Brucella suis VirB8 and a pSB102 VirB8 homologue allows heterologous complementation. Infect Immun 74:4945–4949. doi: 10.1128/IAI.00584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thanbichler M, Iniesta AA, Shapiro L. 2007. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res 35:e137–e137. doi: 10.1093/nar/gkm818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berta P, Bourg G, Hanna N, Saadeh B, Armengaud J, Patey G, O'Callaghan D. 2014. The Brucella suis IbpA heat-shock chaperone is not required for virulence or for expression of the VirB type IV secretion system VirB8 protein. Lett Appl Microbiol 58:564–568. doi: 10.1111/lam.12231. [DOI] [PubMed] [Google Scholar]
- 48.O'Callaghan D, Cazevieille C, Allardet-Servent A, Boschiroli ML, Bourg G, Foulongne V, Frutos P, Kulakov Y, Ramuz M. 1999. A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essential for intracellular survival of Brucella suis. Mol Microbiol 33:1210–1220. [DOI] [PubMed] [Google Scholar]
- 49.Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Babraham Institute, Cambridge, United Kingdom: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- 50.Förstner KU, Vogel J, Sharma CM. 2014. READemption—a tool for the computational analysis of deep-sequencing-based transcriptome data. Bioinformatics 30:3421–3423. doi: 10.1093/bioinformatics/btu533. [DOI] [PubMed] [Google Scholar]
- 51.Kingsford CL, Ayanbule K, Salzberg SL. 2007. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol 8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicol JW, Helt GA, Blanchard SG, Raja A, Loraine AE. 2009. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25:2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tjaden B, Goodwin SS, Opdyke JA, Guillier M, Fu DX, Gottesman S, Storz G. 2006. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res 34:2791–2802. doi: 10.1093/nar/gkl356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caldelari I, Chao Y, Romby P, Vogel J. 2013. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect Med 3:a010298. doi: 10.1101/cshperspect.a010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wattam AR, Inzana TJ, Williams KP, Mane SP, Shukla M, Almeida NF, Dickerman AW, Mason S, Moriyón I, O'Callaghan D, Whatmore AM, Sobral BW, Tiller RV, Hoffmaster AR, Frace MA, De Castro C, Molinaro A, Boyle SM, De BK, Setubal JC. 2012. Comparative genomics of early-diverging Brucella strains reveals a novel lipopolysaccharide biosynthesis pathway. mBio 3:e00246-11. doi: 10.1128/mBio.00246-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wattam AR, Foster JT, Mane SP, Beckstrom-Sternberg SM, Beckstrom-Sternberg JM, Dickerman AW, Keim P, Pearson T, Shukla M, Ward DV. 2014. Comparative phylogenomics and evolution of the Brucellae reveal a path to virulence. J Bacteriol 196:920–930. doi: 10.1128/JB.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lavigne J-P, Vergunst AC, Bourg G, O'Callaghan D. 2005. The IncP island in the genome of Brucella suis 1330 was acquired by site-specific integration. Infect Immun 73:7779–7783. doi: 10.1128/IAI.73.11.7779-7783.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faucher SP, Shuman HA. 2011. Small regulatory RNA and Legionella pneumophila. Front Microbiol 2:98. doi: 10.3389/fmicb.2011.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steuten B, Schneider S, Wagner R. 2014. 6S RNA: recent answers—future questions. Mol Microbiol 91:641–648. doi: 10.1111/mmi.12484. [DOI] [PubMed] [Google Scholar]
- 60.Cui M, Wang T, Xu J, Ke Y, Du X, Yuan X, Wang Z, Gong C, Zhuang Y, Lei S. 2013. Impact of Hfq on global gene expression and intracellular survival in Brucella melitensis. PLoS One 8:e71933. doi: 10.1371/journal.pone.0071933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong H, Peng X, Wang N, Wu Q. 2014. Identification of novel sRNAs in Brucella abortus 2308. FEMS Microbiol Lett 354:119–125. doi: 10.1111/1574-6968.12433. [DOI] [PubMed] [Google Scholar]
- 62.Tjaden B. 2008. TargetRNA: a tool for predicting targets of small RNA action in bacteria. Nucleic Acids Res 36:W109–W113. doi: 10.1093/nar/gkn264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sangari FJ, Agüero J, García-Lobo JM. 2000. The genes for erythritol catabolism are organized as an inducible operon in Brucella abortus. Microbiology 146:487–495. doi: 10.1099/00221287-146-2-487. [DOI] [PubMed] [Google Scholar]
- 64.Sieira R. 2013. Regulation of virulence in Brucella: an eclectic repertoire of transcription factors defines the complex architecture of the virB promoter. Future Microbiol 8:1193–1208. doi: 10.2217/fmb.13.83. [DOI] [PubMed] [Google Scholar]
- 65.Gee JM, Valderas MW, Kovach ME, Grippe VK, Robertson GT, Ng W-L, Richardson JM, Winkler ME, Roop RM. 2005. The Brucella abortus Cu,Zn superoxide dismutase is required for optimal resistance to oxidative killing by murine macrophages and wild-type virulence in experimentally infected mice. Infect Immun 73:2873–2880. doi: 10.1128/IAI.73.5.2873-2880.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Occhialini A, De Bagüés MPJ, Saadeh B, Bastianelli D, Hanna N, De Biase D, Köhler S. 2012. The glutamic acid decarboxylase system of the new species Brucella microti contributes to its acid resistance and to oral infection of mice. J Infect Dis 206:1424–1432. doi: 10.1093/infdis/jis522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.