

ABSTRACT
TcpP and ToxR coordinately regulate transcription of toxT, the master regulator of numerous virulence factors in Vibrio cholerae. TcpP and ToxR are membrane-localized transcription factors, each with a periplasmic domain containing two cysteines. In ToxR, these cysteines form an intramolecular disulfide bond and a cysteine-to-serine substitution affects activity. We determined that the two periplasmic cysteines of TcpP also form an intramolecular disulfide bond. Disruption of this intramolecular disulfide bond by mutation of either cysteine resulted in formation of intermolecular disulfide bonds. Furthermore, disruption of the intramolecular disulfide bond in TcpP decreased the stability of TcpP. While the decreased stability of TcpP-C207S resulted in a nearly complete loss of toxT activation and cholera toxin (CT) production, the second cysteine mutant, TcpP-C218S, was partially resistant to proteolytic degradation and maintained ∼50% toxT activation capacity. TcpP-C218S was also TcpH independent, since deletion of tcpH did not affect the stability of TcpP-C218S, whereas wild-type TcpP was degraded in the absence of TcpH. Finally, TcpH was also unstable when intramolecular disulfides could not be formed in TcpP, suggesting that the single periplasmic cysteine in TcpH may assist with disulfide bond formation in TcpP by interacting with the periplasmic cysteines of TcpP. Consistent with this finding, a TcpH-C114S mutant was unable to stabilize TcpP and was itself unstable. Our findings demonstrate a periplasmic disulfide bond in TcpP is critical for TcpP stability and virulence gene expression.
IMPORTANCE The Vibrio cholerae transcription factor TcpP, in conjunction with ToxR, regulates transcription of toxT, the master regulator of numerous virulence factors in Vibrio cholerae. TcpP is a membrane-localized transcription factor with a periplasmic domain containing two cysteines. We determined that the two periplasmic cysteines of TcpP form an intramolecular disulfide bond and disruption of the intramolecular disulfide bond in TcpP decreased the stability of TcpP and reduced virulence gene expression. Normally TcpH, another membrane-localized periplasmic protein, protects TcpP from degradation. However, we found that TcpH was also unstable when intramolecular disulfides could not be formed in TcpP, indicating that the periplasmic cysteines of TcpP are required for functional interaction with TcpH and that this interaction is required for both TcpP and TcpH stability.
INTRODUCTION
Cholera is caused by ingestion of the aquatic Gram-negative bacterium Vibrio cholerae. The profuse watery diarrhea characteristic of cholera is induced by cholera toxin (CT), an ADP-ribosylating toxin that induces cyclic AMP production in intestinal epithelial cells, resulting in massive secretion of water and electrolytes. Microcolony formation and colonization of the intestine require expression of the toxin-coregulated pilus (TCP) (1). Transcription of the genes encoding CT and TCP, as well as several other virulence factors, is regulated by the transcription activator ToxT (2). toxT expression is regulated by two transmembrane winged-helix-turn-helix (w-HTH) transcription factors, ToxR and TcpP, and their coactivators ToxS and TcpH, respectively (3–10). Based on current models, upon entry into the intestine, environmental signals activate expression of tcpPH (11, 12). TcpPH, along with constitutively expressed ToxRS, activates transcription of toxT, resulting in colonization and CT secretion in a temporally coordinated fashion (13). Under noninducing conditions, TcpP is specifically targeted for degradation by the site 1 periplasmic protease, Tsp, followed by the membrane-localized metalloprotease YaeL (10, 14, 24).
TcpP and ToxR have three distinct domains: an N-terminal cytoplasmic DNA-binding domain, a single membrane-spanning domain, and a C-terminal periplasmic domain. The cytoplasmic domain of TcpP and ToxR is homologous to that of the OmpR/PhoB family of w-HTH transcription factors (15). Mutations in several key residues of the cytoplasmic domains of both TcpP and ToxR that inhibit DNA binding and transcriptional activation have been identified (5, 16–18). TcpP binds to a direct repeat in the toxT promoter just upstream of the predicted RNA polymerase (RNAP) binding site (5, 19), while ToxR binds to a direct repeat, three turns upstream of the TcpP-binding site (5, 20). Based on the location of these binding sites, TcpP likely interacts with RNA polymerase and is the direct activator of toxT. This model is further supported by the fact that overexpressed TcpP can activate toxT in the absence of ToxR, but ToxR cannot activate toxT expression in the absence of TcpP (3–5, 18). Membrane localization of ToxR is required for toxT activation in conjunction with TcpP but not activation of the TcpP-independent ompU promoter (21–23). Thus, ToxR is believed to serve as a coactivator, enhancing transcriptional activation of toxT by promoting TcpP recruitment and/or binding to the toxT promoter (5, 17, 18).
The periplasmic coactivators TcpH and ToxS coordinate with the periplasmic domains of TcpP and ToxR, respectively, for full activation of toxT (3, 4, 6, 8–10). ToxS has been shown to increase ToxR dimerization and transcriptional activation of toxT by ToxR (9), while TcpH is required for TcpP stability and enhances transcriptional activation of toxT by protecting TcpP from degradation (10, 14). The periplasmic domain of TcpP is particularly vulnerable to proteolytic degradation resulting in instability of the entire protein (10, 14). Evidence for the role of the periplasmic domain in TcpP instability was provided when the periplasmic domain of TcpP was fused to ToxR, making a ToxR-TcpPperi chimeric protein. This resulted in an unstable ToxR species unless TcpH was present (10). Conversely, replacement of the periplasmic domain of TcpP with the periplasmic domain of ToxR resulted in increased TcpP stability (10). Proteolysis of TcpP is regulated in a multistep process in which initially the site 1 protease Tsp (tail-specific protease) recognizes the C terminus and cleaves TcpP (24). This partially degraded TcpP, designated TcpP*, is then further cleaved by the membrane-localized metalloprotease YaeL, resulting in complete degradation of TcpP (14). In Escherichia coli, YaeL cleaves RseA, a transmembrane protein that tethers σE to the membrane. Cleavage and subsequent degradation of RseA release σE from the membrane, thereby allowing σE to activate its target promoter (25–27). TcpP is active in the membrane, and cleavage of TcpP by YaeL results in inactivation and degradation (14).
Proper disulfide bond formation is important for the function of many periplasmic proteins (28). In V. cholerae, the disulfide isomerase DsbA is required for proper formation and activity of TCP, CT, ToxR, and other virulence factors (29–32). The periplasmic domain of ToxR contains two cysteines that form an intramolecular disulfide bond, and disruption of this periplasmic intramolecular disulfide in ToxR by mutation of one of the cysteines to serine in the classical V. cholerae strain O395 resulted in formation of an intermolecular disulfide, likely between two ToxR molecules (33). The resulting mutant, when expressed from a plasmid, was found to be 30-fold defective in induction of CT expression (33). A recent study in which ToxR was expressed from the chromosome found that mutating both periplasmic cysteines of ToxR in the classical strain O395 led to a dramatic decrease in OmpU production but less than a 2-fold defect in CT production in LB medium at pH 6.5 and 30°C (toxin-inducing conditions [32]). Differences in CT production in these two studies may be due to differences in the effects of replacing one (33) or both (32) cysteines or due to differences between plasmid-expressed and chromosomally expressed ToxR. TcpP also has cysteines at similar positions in its periplasmic domain. In this study, we determined that the TcpP periplasmic cysteines also form an intramolecular disulfide bond. In addition, we examined the role of TcpP periplasmic cysteines in toxT transcriptional activation and found that the intramolecular periplasmic disulfide bond of TcpP is important for TcpP stability and maximal transcriptional activation of toxT and V. cholerae virulence gene expression. Finally, we also found that the stability of the TcpP affector protein TcpH depends upon the presence of the periplasmic cysteines of TcpP and the single periplasmic cysteine of TcpH (C114). This suggests that TcpP and TcpH interact via a transient disulfide bond, while TcpH facilitates proper TcpP folding and intramolecular disulfide bond formation.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All strains and plasmids used in this study are listed in Table S1 in the supplemental material. Specific mutants were generated by site-directed mutagenesis using primers listed in Table S2 in the supplemental material and Pfu Turbo (Stratagene) followed by DpnI digestion as described previously (19). Plasmids containing the mutants were cloned into DH5α. The sequences of all constructs were verified at the University of Michigan DNA Sequencing Core or by Genewiz. Plasmids were then transferred to reporter strains. For chromosomal mutations, the sequence containing the mutation was cloned into the suicide plasmid pKAS32 (34). The plasmids were mated into V. cholerae and chromosomal recombination was selected as described previously (34). The same system was used to delete tcpP from the chromosomes of various V. cholerae protease deletion strains. The locus encoding non-epitope-tagged TcpPH was digested from pMMB207-tcpPH (pEK32 [see Table S1]) using EcoRI and BamHI and cloned into the EcoRI and BamHI sites in a pBAD18 (Kan) plasmid in which the second BamHI site and the NheI site at the upstream end of the multiple-cloning site were removed by site-directed mutagenesis [the new plasmid was designated pGOOD (Kan)].
Culture conditions.
V. cholerae strains were routinely grown overnight in Vc LB (LB containing 5 g/liter of NaCl rather than 10 g/liter) at 37°C. To induce virulence gene expression and promote tcpP expression and stability, all samples were assayed after induction in Vc LB (pH 6.5) at 30°C. Cultures were grown in the presence of 100 μg/ml of ampicillin, 25 μg/ml of chloramphenicol, 100 μg/ml of streptomycin, and 30 μg/ml of kanamycin as needed.
β-Galactosidase assays.
Cultures were diluted from overnight growth in Vc LB at 37°C and induced at 30°C for 4 h in Vc LB (pH 6.5) with 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG) or 0.1% arabinose (as required). To monitor induction from plasmid-based TcpP, the previously described reporter strains EK813 (ΔtcpP toxT-lacZ) and EK1490 (ΔtcpP ΔtoxR toxT-lacZ) (18) were used. For quantification of toxT expression directed by chromosomally encoded tcpP, a plasmid containing a toxT-lacZ reporter was utilized (pTLI2 [see Table S1 in the supplemental material]) (35). Twenty to 100 μl of culture was used to measure β-galactosidase activity as described previously (36). For quantification of ToxR activation of ompU-lacZ and toxT-lacZ, the chromosomal reporter strains EK383 and EK733 were used (4, 22). The optical density at 600 nm (OD600) was determined by spectrophotometry and used to normalize cultures for subsequent Western blot analysis.
Western blotting.
Samples were resuspended in SDS-PAGE sample buffer containing 1 mM dithiothreitol (DTT) adjusted for OD600 and boiled for 5 min before being loaded onto a 10 to 15% polyacrylamide gel. TcpP-herpes simplex virus (HSV) was detected using mouse monoclonal anti-HSV at a 1:10,000 dilution (Novagen) or rabbit polyclonal anti-TcpP at a 1:500 dilution (a kind gift from Victor DiRita). ToxR was detected using rabbit polyclonal anti-ToxR antibodies at a 1:10,000 dilution. TcpH was detected with a rabbit polyclonal antibody generated against two TcpH peptides, TRYQTLPDPSSQK and LIPDYSQSNASRDYN, used at a 1:500 dilution (a kind gift from Victor DiRita). Alkaline phosphatase (AP)-conjugated anti-mouse (Invitrogen) or anti-rabbit (Invitrogen) secondary antibody was used at a 1:3,000 dilution. The intensities of the bands were determined using the freely available software ImageJ.
Production of rabbit anti-ToxR antibodies.
Two rabbits were immunized by Covance with a purified form of ToxR-His6 containing the first 170 amino acids of the ToxR cytoplasmic domain (ToxRcyt2) followed by a tobacco etch virus (TEV) protease cleavage site and C-terminal His6 tag, according to standard procedures. ToxRcyt2-His6 protein was expressed and purified from the E. coli overexpression strain Rosetta (DE3) pLysS (see Table S1 in the supplemental material) following cloning of toxRcyt2 into the pET30b+ vector (Novagen) via NdeI and KpnI restriction sites (see Tables S1 and S2).
Nonreducing Western blotting.
Dilutions (1:50) of overnight cultures were grown in Vc LB broth with 100 μM IPTG for 3 h at 30°C. One milliliter of culture was treated on ice for 15 min with 10 μM iodoacetamide (Sigma) to block free cysteines before being pelleted. The OD600 of the culture was used to determine the appropriate volume of SDS sample buffer with or without DTT for resuspension. Samples were analyzed by Western blotting as described above.
Immunoprecipitation and mass spectrometry.
Membranes containing TcpP-HSV were prepared as previously described (6) and boiled for 5 min in 1% SDS. Samples were diluted 1:100 in 50 mM Tris (pH 7.4)–300 mM NaCl–1% Triton X-100 and centrifuged to remove any precipitates. Mouse monoclonal anti-HSV (Novagen; 1:500 dilution) was used to bind TcpP-HSV to protein A/G-agarose beads (CalBiochem). After washing, the beads were resuspended in sample buffer with or without DTT. Samples were analyzed by Western blotting and Coomassie staining, and selected bands were removed and analyzed by mass spectrometry by the University of Michigan Proteomics Consortium.
TcpP stability in protease deletion strains.
Cultures were induced for 16 h in Vc LB (pH 6.5) with 0.1% arabinose at 30°C. Samples were adjusted for OD600 and analyzed by Western blotting using the antibody against TcpP as described above.
ELISA for cholera toxin production.
Enzyme-linked immunosorbent assays (ELISAs) for CT were performed as described previously (5, 37). The supernatant from induced cultures was allowed to bind to GM1 ganglioside-coated wells in a 96-well plate. Bound CT was then detected using an anti-CT primary antibody and AP-conjugated anti-rabbit secondary antibody. Color development of the substrate para-nitrophenylphosphate (pNPP) was measured after 20 min, and the average of 2 replicate wells was used to determine CT content for each sample.
RESULTS
TcpP forms an intramolecular disulfide bond.
To determine whether TcpP, like ToxR, forms primarily intramolecular disulfide bonds, V. cholerae lysates containing pMMB207-expressed TcpP-HSV (TcpP which contains a C-terminal HSV tag) were run on nonreducing SDS-PAGE and analyzed by Western blotting. To preserve disulfide bond formation and prevent additional disulfide bonds from forming during sample preparation, V. cholerae lysates were treated with iodoacetamide as described previously (33). Nonreduced TcpP-HSV ran slightly faster than reduced TcpP-HSV due to the presence of an intramolecular disulfide bond (Fig. 1, lanes 1, 4, and 5). Two fainter bands at approximately 70 kDa were also visible on the nonreduced gel (Fig. 1, lanes 4, and 5); these bands are consistent with formation of intermolecular disulfide bonds between two TcpP molecules. These dimers could be the result of formation of a single intermolecular disulfide bond between two TcpP molecules or two disulfide bonds between two TcpP molecules. To determine whether the disulfide bonds observed were formed between the two periplasmic cysteines present in TcpP, each periplasmic cysteine was mutated to serine (C207S and C218S). Because these mutants were no longer able to form intramolecular disulfide bonds, TcpP-C207S and TcpP-C218S ran either at the same position as reduced TcpP or at a higher molecular mass consistent with a TcpP dimer formed by intermolecular disulfide bonds (Fig. 1, lanes 6 to 9). The TcpP dimer bands migrated slightly differently depending on whether C207 or C218 was present, likely due to differences in the location of the disulfide bond relative to the C terminus of the protein. Since proteins in this family tend to dimerize (21, 33, 38–42), and this band was the expected size of a TcpP dimer, we predicted that this band was likely the result of an intermolecular disulfide bond formed between two TcpP molecules. To confirm this, the complex was isolated by immunoprecipitation and analyzed by mass spectrometry (data not shown). The only peptides detected by mass spectrometry were from TcpP, indicating that this band is the result of a TcpP homodimer (data not shown). When both periplasmic cysteines were mutated to serine, only a single band at the expected size for reduced TcpP was observed (Fig. 1, lanes 10 and 11), confirming that the disulfide bonds observed were formed by the periplasmic cysteines and not any of the four cytoplasmic cysteines in TcpP. The presence of ToxR had no effect on disulfide bond formation (Fig. 1). These data are in agreement with a previous study demonstrating a role for DsbA in the formation of TcpP intramolecular disulfide bonds (43).
FIG 1.

TcpP forms intramolecular and intermolecular disulfide bonds. Lysates of V. cholerae (EK459 ΔtoxR ΔtcpP) expressing pMMB-TcpP-HSV and either pSK or pSK-ToxR-HA (a ToxR-expressing plasmid with a C-terminal HA tag) were incubated with iodoacetamide prior to resuspension in sample buffer with or without DTT. Blots were probed with anti-HSV primary antibody and alkaline phosphatase-conjugated anti-mouse secondary antibody. Mutations in either or both periplasmic cysteines were used to perturb disulfide bond formation. The arrows and diagrams along the side of the blot indicate the state of the periplasmic cysteines in each form of TcpP detected.
In conclusion, the periplasmic cysteines in TcpP form primarily intramolecular disulfide bonds. However, when only one periplasmic cysteine is available, TcpP forms cross-linked homodimers.
TcpP-HSV containing intermolecular disulfide bonds maintains toxT activation.
The TcpP periplasmic cysteine mutants were tested for transcriptional activation of a chromosomal toxT-lacZ reporter to determine whether an intramolecular periplasmic disulfide bond is required for activity. TcpP-C218S was able to activate transcription at >80% of wild-type levels (Fig. 2A). Since TcpP-C218S is not able to form intramolecular disulfide bonds, this demonstrates that the intramolecular disulfide bond is not required for transcriptional activation of toxT. Additionally, the presence of an intermolecular disulfide bond only partially interfered with toxT activation by TcpP-C207S, which maintained 48% activity. TcpP-C207S was also present at about 59% of the levels of wild-type TcpP (Fig. 2B). TcpP-C207S/C218S, on the other hand, was the most defective for toxT activation (13% of the wild type), even though it had a level of protein similar to TcpP-C207S (Fig. 2B). Thus, either intramolecular or intermolecular disulfide bonds in the periplasmic domain of TcpP are required to maintain ∼50% or greater toxT activation capacity.
FIG 2.

TcpP-HSV is able to activate transcription of toxT in the presence of intramolecular or intermolecular periplasmic disulfide bonds. (A) toxT activation by the TcpP-HSV periplasmic cysteine mutants expressed from the pMMB207 plasmid was measured by β-galactosidase assay using chromosomal toxT-lacZ reporters (EK813 and EK1490 [see Table S1 in the supplemental material]) (18). Activation was measured in both the presence (black bars) and the absence (white bars) of chromosomally expressed ToxR. (B) TcpP-HSV expression was monitored by Western blotting using anti-HSV antibody. *, P < 0.005 relative to the value for wild-type TcpP.
TcpP-mediated toxT activation in the absence of ToxR decreased 10-fold, as ToxR facilitates TcpP-mediated toxT activation (5). ToxR-independent activation of toxT by overexpressed TcpP-C207S and TcpP-C218S was at or above wild-type levels (Fig. 2A, white bars), again demonstrating that maintenance of either intramolecular or intermolecular periplasmic disulfide bonds in TcpP is sufficient for toxT activation activity. Mutation of both cysteines, yielding TcpP-C207S/C218S, resulted in the loss of toxT activation by TcpP overexpressed in the absence of ToxR (Fig. 2A), although in this strain background, TcpP-C207S/C218S was particularly unstable, maintaining just 36% of the level of TcpP protein as wild-type TcpP.
Stability of TcpP periplasmic cysteine mutants is greatly reduced when expressed from the chromosome without a C-terminal epitope tag.
Because our initial experiments were performed with C-terminally HSV-tagged TcpP and the C terminus of TcpP is known to be susceptible to periplasmic proteases (10, 14), we determined the consequences of disrupting TcpP periplasmic disulfide bond formation with untagged chromosomally expressed TcpP. tcpP-C207S, tcpP-C218S, or tcpP-C207S/C218S alleles were introduced on the chromosome and toxT activation was measured using a plasmid-based reporter containing a toxT-lacZ fusion. Expression of TcpP-C207S and the TcpP-C207S/C218S mutant resulted in a dramatic decrease in toxT expression, approaching the levels seen in the ΔtcpP strain (Fig. 3). This decrease in activity corresponded to profound TcpP instability, since little to no TcpP-C207S or TcpP-C207S/C218S was detectable by Western blotting (Fig. 3). TcpP-C218S was the most stable of all the chromosomally expressed mutants (although only present at 15% of wild-type levels) and maintained ∼50% of toxT activation relative to wild-type TcpP. While TcpP-C207S was not detectable by Western blotting, it was able to activate toxT expression 1.5-fold above background. These findings indicate that TcpP can be artificially protected from proteolysis by addition of a C-terminal HSV tag, and this likely accounts for the differences observed in stability between the mutants expressed from the chromosome and the pMMB207-tcpP-HSV plasmid (Fig. 2B and 3A). Analysis of chromosomally expressed wild-type TcpP demonstrated that TcpP still forms intramolecular disulfides when expressed from the chromosome (Fig. 3B).
FIG 3.

The instability of chromosomally expressed TcpP cysteine mutants lacking a C-terminal epitope tag results in a toxT activation defect. (A) Transcription activation of toxT was monitored by β-galactosidase assay in V. cholerae strains in which the wild-type allele of tcpP was replaced with alleles encoding TcpP cysteine mutants. toxT activation was monitored using the plasmid-based toxT-lacZ reporter, pTLI2 (35). A Western blot of lysates from the β-galactosidase assay was probed with anti-TcpP antibodies and demonstrated the instability of these constructs. While TcpP-C207S and TcpP-C207S/C218S were undetectable, TcpP-C218S was present at 15% of the level of wild-type TcpP as determined by ImageJ software. *, P < 0.005 relative to the value for wild-type TcpP; #, P < 0.005 relative to the value for the ΔtcpP mutant. (B) The presence of an intramolecular disulfide bond in chromosomally encoded TcpP was confirmed following overnight growth of the wild-type strain O395 and processing of samples with and without DTT.
To determine whether the defect in TcpP stability, and therefore toxT transcription, resulted in a defect in virulence gene production, CT secretion directed by each chromosomally expressed TcpP mutant was measured. As expected from the toxT activation data (Fig. 3), TcpP-C207S and TcpP-C207S/C218S directed background levels of CT production, a nearly 4,000-fold decrease relative to that of the parental strain O395 (Table 1). TcpP-C207S was able to produce CT levels just above the limit of detection for this assay, indicating that the low levels of toxT induced in this strain (Fig. 3) may be sufficient to induce a small amount of CT production (Table 1). The dramatically reduced levels of CT produced by TcpP-C207S and TcpP-C207S/C218S corresponded to undetectable TcpP protein levels in their respective strains (see Fig. S1 in the supplemental material), as was observed in the toxT activation assay (Fig. 3). TcpP-C218S was the most stable chromosomally expressed cysteine mutant in both the toxT activation and CT assays, although expression levels were still well below the wild-type levels (Fig. 3A; see also Fig. S1). This resulted in a slight (2-fold) defect in toxT-lacZ induction (Fig. 3). This level of toxT expression is sufficient for levels of CT production approaching the wild type, as CT levels were reduced only 6-fold in this strain (1,000-fold above background) (Table 1). Thus, the decrease in toxT expression and CT production by the TcpP periplasmic cysteine mutants corresponds to their relative instability, indicating that defects in toxT transcription and virulence factor production by these mutants are directly related to their decreased stability.
TABLE 1.
Some chromosomally encoded TcpP periplasmic cysteine mutants are severely defective for cholera toxin productiona
| Strain | Amt of CT produced (ng/ml/OD600) |
|---|---|
| O395 | 1,467 ± 583 |
| ΔtcpP ΔtoxR mutant | <0.2 ± 0.2 |
| C207S mutant | 0.39 ± 0.04 |
| C218S mutant | 238 ± 103 |
| C207S C218S mutant | <0.2 ± 0.2 |
Levels of cholera toxin secretion by each chromosomally encoded TcpP periplasmic cysteine mutant were detected by a CT-ELISA on overnight cultures of V. cholerae. CT levels in TcpP-C207S and TcpP-C207S/C218S were not significantly above those in the ΔtcpP ΔtoxR negative-control strain (P = 0.06 for TcpP-C207S). CT was measured in duplicate in two separate experiments (n = 4).Values are means ± standard deviations.
TcpH is unstable in the absence of TcpP.
Since the open reading frames of tcpP and tcpH overlap and the mutation resulting in TcpP-C218S (TGC to TCC) also affects the start codon for tcpH (ATG to ATC), we determined whether expression of TcpH in trans would confer stabilization on any of the chromosomally encoded TcpP periplasmic cysteine mutants. Each chromosomal TcpP periplasmic cysteine mutant was transformed with pACYC or pACYC-TcpH (10), and levels of TcpP and TcpH were assessed by Western blotting. Expression of TcpH in trans was not able to stabilize expression to any of the TcpP periplasmic cysteine mutants (Fig. 4A, lanes 9, 11, and 13). TcpP was stable with or without the TcpH-expressing plasmid since TcpH was also expressed from its normal chromosomal locus (Fig. 4A, lanes 2 and 3).
FIG 4.

TcpP and TcpH require each other for stability. Using chromosomal constructs with and without expression of additional TcpH from pACYC-tcpH (10), only wild-type TcpP was protected from degradation by TcpH (A). Furthermore, TcpH was detected only when expressed in conjunction with wild-type TcpP (B). Using a strain with no endogenous TcpP or TcpH expression (ΔtcpPH), plasmid-based expression of TcpP (pGOOD-tcpP) and TcpH (pACYC-tcpH) from separate plasmids also demonstrated that wild-type TcpP is the only form of TcpP stabilized by TcpH (C) and TcpH was detected only when coexpressed with wild-type TcpP (D). TcpP-C218S was partially stable regardless of the presence of TcpH (C).
To our surprise, in the presence of any of the TcpP periplasmic cysteine mutants, TcpH was unstable, suggesting that TcpH may require the cysteines of TcpP to efficiently interact and maintain both TcpP and TcpH stability (Fig. 4B, lanes 9, 11, and 13). To confirm this result in a strain with no endogenous TcpH expression, a ΔtcpPH mutant was assessed for TcpP and TcpH stability when TcpP and TcpH were expressed from separate plasmids (pGOOD-tcpP and pACYC-tcpH). As seen with the chromosomal mutants, TcpP-C218S was more stable than TcpP-C207S and the presence of TcpH did not affect expression levels of TcpP-C218S (Fig. 4C). In contrast, wild-type TcpP was stabilized only in the presence of TcpH, as reported previously (10, 14) (Fig. 4C). Again, in the absence of TcpP (Fig. 4D, lane 4) or in the presence of the TcpP periplasmic cysteine mutants (Fig. 4D, lanes 7, 9, and 11), TcpH was unstable.
The single periplasmic cysteine of TcpH is required for TcpP and TcpH stability.
TcpH has a single periplasmic cysteine, an unusual arrangement for a periplasmic domain (44). We hypothesized that this cysteine may interact with TcpP, serving a chaperone-like function for TcpP folding and proper disulfide bond formation. A TcpH-C114S derivative was constructed and assessed for its ability to stabilize wild-type TcpP. TcpH-C114S was unable to protect wild-type TcpP from degradation (Fig. 5A). Furthermore, the TcpH-C114S mutant itself was unstable, indicating that this cysteine is required for maintaining a conformation of TcpH that is resistant to periplasmic degradation (Fig. 5B).
FIG 5.
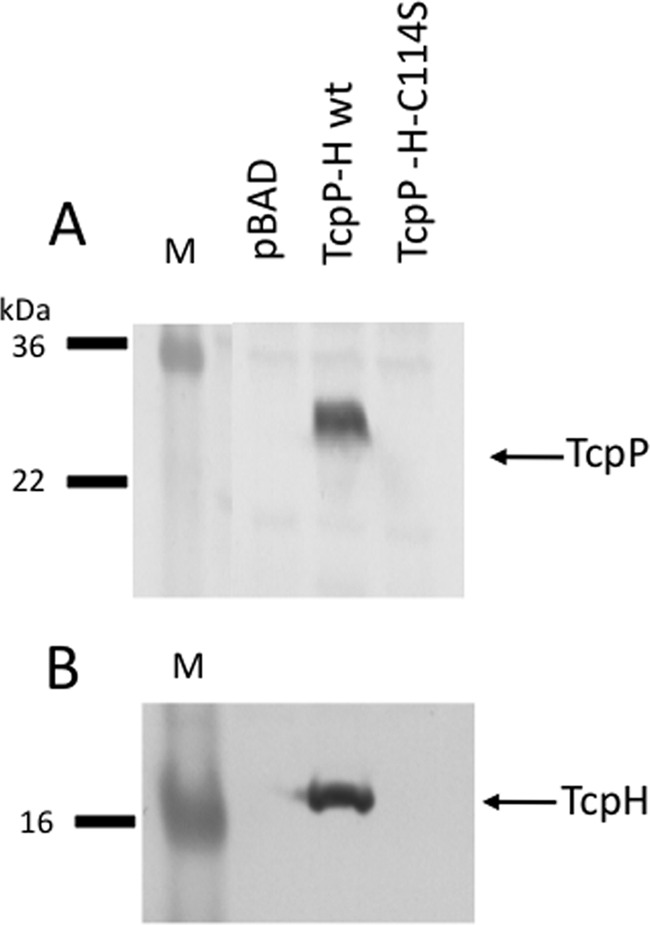
A TcpH-C114S mutant cannot stabilize wild-type TcpP. TcpP and TcpH were cotranscribed from the pBAD18-based plasmid pGOOD-tcpPH encoding either wild-type TcpH or TcpH-C114S. Strains were induced for tcpPH expression with 0.1% arabinose, and levels of TcpP (A) and TcpH (B) were determined by Western blotting.
Degradation of the TcpP periplasmic mutants requires YaeL, with some contributions from DegS and Tsp.
Since YaeL is the known site 2 protease of wild-type TcpP under noninducing conditions (14), we determined whether the degradation of TcpP periplasmic mutants also depends upon YaeL. Thus, we constructed an O395 ΔtcpP strain into which we added back pBAD18-TcpPH plasmids encoding the TcpP periplasmic cysteine mutants (without epitope tags). Upon plasmid-based expression, the untagged TcpP-C207S, TcpP-C218S, and TcpP-C207S/C218S were still unstable (Fig. 6A). A residual degradation band at approximately 15 kDa was observed in the periplasmic cysteine mutant strains, as proteolysis may have been incomplete due to plasmid-based overexpression. This 15-kDa band may represent a partially protease-resistant intermediate containing the portion of TcpP homologous to other winged-HTH proteins of the OmpR/PhoB transcription activator family (15). When these constructs were expressed in a ΔtcpP ΔyaeL background, the TcpP periplasmic cysteine mutants were processed to a protein of ∼20 kDa, previously designated TcpP*, representing a partially proteolyzed product of TcpP in the absence of YaeL (Fig. 6B) (14). This degradation product was previously found upon shifting wild-type O395 cultures to noninducing conditions in a ΔyaeL strain (14). The same pattern was observed when TcpP periplasmic mutants were expressed from the chromosome of a ΔyaeL strain (data not shown). The increased degradation of mutants TcpP-C207S, TcpP-C218S, and TcpP-C207S/C218S to TcpP* in the ΔyaeL background under inducing conditions indicates that they are more vulnerable to cleavage by YaeL than wild-type TcpP. Their susceptibility to degradation under TcpP-inducing conditions is similar to degradation of wild-type TcpP under noninducing conditions.
FIG 6.

TcpP periplasmic cysteine mutants are degraded through the YaeL pathway. (A) Stability of untagged TcpP periplasmic cysteine mutants expressed from pBAD18-based plasmids in a ΔtcpP strain was monitored by Western blotting. (B) Deletion of yaeL from V. cholerae strains expressing TcpP with periplasmic cysteine mutations resulted in accumulation of partially degraded TcpP (designated TcpP*). The stability of the plasmid-expressed TcpP cysteine mutants was also monitored by Western blotting in ΔtcpP ΔdegS (C), ΔtcpP Δptd (D), ΔtcpP ΔdegS Δptd (E), ΔtcpP Δtsp (F), and ΔtcpP ΔdegS Δptd Δtsp (G) strains. TcpP was detected by Western blotting with anti-TcpP as a probe. The percentage of full-length TcpP remaining in each lane relative to wild-type TcpP for each panel is provided below each lane and was determined by ImageJ software (http://rsbweb.nih.gov/ij/).
Cleavage by YaeL is a two-step process, with a site 1 protease initiating cleavage, followed by cleavage by the site 2 protease, YaeL. One site 1 protease candidate is DegS. DegS is the V. cholerae homolog of the site 1 protease responsible for initial cleavage of proteins prior to YaeL cleavage in E. coli (14, 26). Although deletion of DegS in V. cholerae did not result in increased stability of wild-type TcpP under noninducing conditions (14), DegS could be involved in degradation of the more proteolytically sensitive TcpP periplasmic cysteine mutants. Using the pBAD18 expression system, we observed partial stabilization of TcpP-C218S in the ΔtcpP ΔdegS strain (present at 32% of wild-type TcpP levels) but no stabilization of TcpP-C207S or TcpP-C207S/C218S (Fig. 6C). This indicates that although deletion of degS can partially stabilize TcpP-C218S, there is another periplasmic protease that is primarily responsible for the enhanced proteolytic degradation of the TcpP periplasmic cysteine mutants. Another candidate for the site 1 protease mediating degradation of the TcpP periplasmic cysteine mutants is DegP (encoded by ptd and also known as protease-DO). DegP is a periplasmic chaperone responsible for degradation of unstable or misfolded proteins in the periplasm (10, 45, 46). Deletion of ptd resulted in only a modest increase in stability of the TcpP-C218S periplasmic cysteine mutant (Fig. 6D), indicating that Ptd is not the primary site 1 protease for misfolded TcpP. Deletion of both degS and ptd did not give any additional stabilization of the TcpP periplasmic cysteine mutants (Fig. 6E). Finally, it was recently reported that under noninducing conditions, the periplasmic protease Tsp serves as the site 1 protease for TcpP (24). Thus, we measured the stability of the TcpP periplasmic cysteine mutants in a Δtsp background using our pBAD-based TcpPH plasmid expression system. Deletion of tsp led to partial stabilization of TcpP-C218S, similar to the case with the ΔdegS mutant (Fig. 6F) (37% stability relative to that of wild-type TcpP), and slight stabilization of TcpP-C207S (5% stabilization) but no stabilization of the TcpP-C207S/C218S double mutant (Fig. 6F). To determine whether these various periplasmic proteases perform redundant functions, we constructed a ΔdegS Δptd Δtsp triple protease mutant strain. Expression of the TcpP periplasmic cysteine mutants in the ΔdegS Δptd Δtsp strain did not result in any enhanced stabilization over those with the ΔdegS and Δtsp mutants alone (Fig. 6G). Thus, another yet-to-be-identified periplasmic protease or combination of proteases is likely responsible for much of the degradation of the TcpP periplasmic cysteine mutants.
DISCUSSION
The purpose of this study was to determine whether TcpP forms periplasmic disulfide bonds and, if so, to determine whether the disulfide bonds are required for TcpP stability and toxT promoter activation. We found that TcpP, like ToxR, has two periplasmic cysteines that form an intramolecular disulfide bond (Fig. 1 and 3B). Disruption of this disulfide bond by mutation of either of the two TcpP periplasmic cysteines to serine results in an intermolecular disulfide, forming a TcpP homodimer (Fig. 1). An additional faint band can be detected at approximately 50 kDa in both the TcpP-C207S and TcpP-C218S mutants (Fig. 1), which is the predicted size of a TcpP-TcpH heterodimer. TcpH is somewhat unusual in that it is a periplasmic protein with a single cysteine and may serve a chaperone-like function for TcpP (44). TcpP mutants lacking the ability to form an intramolecular disulfide bond are unstable when chromosomally expressed without a C-terminal epitope tag (Fig. 3; see also Fig. S1 in the supplemental material). This instability results in decreased toxT activation and ToxT-dependent CT production (Fig. 3A and Table 1). Disulfide bonds formed with C207 are particularly crucial for stability, since the TcpP-C218S mutant (maintaining C207) is more stable than the TcpP-C207S mutant (Fig. 3A; see also Fig. S1). Since TcpP-C207S is particularly sensitive to proteolytic degradation, formation of a disulfide bond by this residue may partially mask a proteolytic recognition site on TcpP. This would render wild-type TcpP and TcpP-C218S more stable than TcpP-C207S.
It has been previously proposed that in the presence of the bile salt taurocholate in the small intestine, TcpP forms a dimer via a C207-C207 intermolecular disulfide bond (43). Thus, under certain environmental conditions, TcpP may alter its disulfide bonding capacity to favor intermolecular disulfides rather than an intramolecular disulfide bond. One caveat of these studies is that in V. cholerae, the TcpP molecules analyzed by Western blotting to detect monomeric or dimeric TcpP included a C-terminal epitope tag. Thus, the transcription activation activity in V. cholerae by these TcpP periplasmic cysteine mutants may not correlate with the corresponding Western blot analyses that rely on the C-terminal FLAG tag (43). Our findings demonstrate that addition of a C-terminal epitope tag to TcpP artificially stabilizes the protein in the presence of the periplasmic cysteine mutations (Fig. 1 and 2). Thus, it is unclear how the periplasmic mutants in the previous study would have behaved for taurocholate-dependent activation had they included the C-terminal FLAG tag.
One common observation between our studies is that the TcpP-C218S derivative appears to maintain some stability or activity regardless of the presence of a C-terminal epitope tag. Yang et al. found that TcpP-C218S was blind to bile, resulting in constitutive toxT-lux activation with or without taurocholate (43). We consistently found that the TcpP-C218S mutant was more stable than the TcpP-C207S mutant (Fig. 3 and 6; see also Fig. S1 in the supplemental material) and its stability was TcpH independent (Fig. 4). Reinforcing the latter finding, we noted the tcpP-C218S mutation overlaps the start site of tcpH (changing ATG to ATC) and likely decreases translation of tcpH. However, expression of additional TcpH from a plasmid did not further stabilize the TcpP-C218S mutant (or any periplasmic cysteine mutant) (Fig. 4A).
Under inducing conditions, TcpP is protected from degradation by TcpH, allowing it to induce expression of toxT (10, 14). When switched to noninducing conditions, TcpP is cleaved by the recently described site 1 periplasmic protease Tsp (24). This results in further cleavage by YaeL and degradation of TcpP, thus decreasing induction of toxT (14). When the intramolecular disulfide in TcpP is disrupted, TcpP is also readily degraded by the YaeL pathway, even under inducing conditions (Fig. 6). This could be due to decreased interaction with TcpH and/or improper folding of the periplasmic domain.
In the course of assessing the ability of TcpH to stabilize periplasmic cysteine mutants of TcpP, we found that in the absence of TcpP or in the presence of periplasmic cysteine mutants of TcpP, TcpH was also unstable. This suggests that TcpP and TcpH may interact via a disulfide-bonded intermediate between the single periplasmic cysteine of TcpH (C114) and either of the periplasmic cysteines of TcpP as TcpP is secreted through the membrane. We hypothesize that C207 of TcpP would be the first to emerge in the periplasmic space during secretion of TcpP (47); thus, C207 may initially bind to TcpH until C218 of TcpP emerges to allow an intramolecular disulfide bond in TcpP to form (Fig. 7C). According to this model, TcpH would perform a chaperone-like role in assisting with TcpP periplasmic disulfide bond formation and protein folding. Based on the instability of TcpH in the absence of TcpP or in the absence of either periplasmic cysteine of TcpP, this model also suggests that once tcpP transcription is shut down and TcpH has no new TcpP molecules to bind to, it may be degraded. In agreement with the important role of TcpH-C114 in interaction with TcpP and formation of this TcpP/TcpH bond stabilizing TcpPH, when we generated a TcpH-C114S mutant, the protein was unstable. While it is possible that this mutation affected the structural integrity of TcpH, leading to degradation, this is a modest amino acid change and the TcpH degradation phenotype matches that of providing TcpH with forms of TcpP lacking intramolecular disulfide bond formation capability. Unfortunately, attempts to stabilize TcpH-C114S by the addition of an epitope tag did not result in stabilization. The TcpH antibody was designed against two peptides (TRYQTLPDPSSQK and LIPDYSQSNASRDYN) 28 and 11 residues N terminal to the cysteine residue in TcpH. Thus, TcpH-C114 is not part of the epitope used to generate the anti-TcpH antibody.
FIG 7.

Model for disulfide bond- and TcpH-mediated protection of TcpP from degradation. The intramolecular periplasmic disulfide bond, along with TcpH, protects TcpP from degradation under TcpP-inducing conditions. (A) Model showing protection of wild-type TcpP under inducing conditions. TcpH protects TcpP from proteolysis by the site 1 protease Tsp and YaeL. (B) When the intramolecular periplasmic disulfide bond in TcpP is disrupted, TcpH is no longer able to protect TcpP, leaving it vulnerable to degradation by DegS, Tsp, and other, yet-to-be-identified site 1 proteases. This allows for further degradation by the membrane-localized protease YaeL. Based on our data (Fig. 4), TcpH is also degraded when the periplasmic disulfide of TcpP is unable to form. (C) A hypothesized mechanism for TcpP interaction with TcpP via TcpP-C207 and TcpH-C114 as TcpP emerges across the inner membrane is shown. In this model, as newly synthesized TcpP emerges in the periplasm through the SecYEG complex (47). TcpP-C207 emerges first and is engaged by C114 of TcpH until TcpP-C218 emerges and forms an intramolecular disulfide bond with TcpP-C207. Without this interaction, both TcpP and TcpH are degraded. After TcpH facilitates intramolecular disulfide bond formation in one TcpP molecule, TcpH may be degraded or may engage a newly emerging TcpP molecule.
It should be emphasized that at this time, the direct interaction between TcpH-C114 and the periplasmic cysteines of TcpP is speculative. But suggesting such a role for TcpH is instructive for future experiments to determine specific mechanism(s) of TcpP/TcpH interaction.
The periplasmic domain of the TcpP cysteine mutants is degraded in part by the site 1 periplasmic proteases DegS and Tsp (Fig. 6C and F), resulting in production of TcpP* (Fig. 6B). In the presence of YaeL, this intermediate form is cleaved and degraded, preventing detection of TcpP* (Fig. 6A). YaeL is a site 2 protease and therefore is only active on previously cleaved substrates. DegS, a periplasmic protease which is the site 1 protease for YaeL in other systems (26), while not required for proteolytic degradation of wild-type TcpP (14), plays a partial role in degradation of TcpP-C218S but not TcpP-C207S (Fig. 6C). Although proteolysis of the TcpP periplasmic cysteine mutants points to instability in the periplasmic domain, deletion of the gene encoding the general protease typically responsible for degradation of misfolded periplasmic proteins, DegP (45, 46), did not increase stability of the TcpP periplasmic cysteine mutants (Fig. 6D). Finally, the recently described site 1 protease for TcpP degradation under noninducing conditions in V. cholerae, Tsp (24), plays a more modest role in degradation of the TcpP periplasmic cysteine mutants, as deletion of tsp alone or in combination with ΔdegS and Δptd led to only partial stabilization of TcpP-C218S, modest (5%) stabilization of TcpP-C207S, and minimal, if any, stabilization of the TcpP-C207S/C218S double mutant (Fig. 6F and G).
While this report focused on the periplasmic cysteines of TcpP, it is instructive to compare our findings with TcpP/TcpH to those for the highly homolgous ToxR/ToxS system. Disruption of the periplasmic disulfide bond of ToxR, a protein of similar structure to TcpP in V. cholerae, is reported to not affect stability except potentially under conditions of nutrient limitation and alkaline pH (33, 48). In a ToxR overexpression system, Ottemann and Mekalanos found a 30-fold decrease in CT production when they disrupted the periplasmic disulfide bond by mutating a periplasmic cysteine to serine (33). Using a chromosomally expressed allele, Fengler et al. recently found that mutation of both ToxR periplasmic cysteines prevented ompU expression and allowed for enhanced ompT expression (ompU is activated by ToxR, while ompT is repressed by ToxR) (32) under growth conditions similar to those in the studies by Ottemann and Mekalanos (LB medium). However, chromosomally expressed ToxR lacking both periplasmic cysteines still directed nearly wild-type levels of CT, a process requiring ToxR and TcpP working together (32). Using chromosomally expressed toxR alleles in the V. cholerae classical strain O395, we found that mutation of either periplasmic cysteine in ToxR resulted in a 20% decrease in ompU transcription activation, and mutation of both cysteines in ToxR resulted in a 40% decrease in ompU transcription activation in LB (see Fig. S2 in the supplemental material), similar to the findings of Fengler et al. Disruption of the periplasmic disulfide bond in ToxR did not have much effect on toxT activation when ToxR was expressed from its chromosomal locus, as we observed only a 20% defect in activation of toxT in all of our ToxR periplasmic mutants. This corresponded to no significant defect in CT production in any of the strains we tested, similar to the results obtained by Fengler et al. in both classical and El Tor V. cholerae strains (32). Thus, although ToxR and TcpP contain similar periplasmic intramolecular disulfide bonds, these disulfide bonds appear to play different roles in these proteins. In TcpP, the periplasmic disulfide bond is particularly critical for stability of TcpP and therefore expression of toxT and production of CT. The TcpP periplasmic intramolecular disulfide, in combination with TcpH, enhances stability, allowing TcpP to be present long enough to induce expression of toxT (Fig. 7A). Since TcpH cannot functionally interact with TcpP mutants affecting the periplasmic cysteines (Fig. 4), TcpH is degraded and TcpP is more susceptible to degradation in the presence of such substitutions in TcpP (Fig. 7B). In wild-type V. cholerae, when virulence gene expression is to be turned off, the inherent instability of TcpP and TcpH will then allow for rapid downregulation of cholera toxin, the toxin-coregulated pilus, and other factors (10, 14), while genes directly activated by ToxR (such as ompU) will maintain their expression, as the ToxR periplasmic domain is more stable than that of TcpP (10).
It was recently reported that TcpP-ToxR interactions are also influenced by periplasmic cysteines and that TcpP and ToxR can form intermolecular disulfide bonds under certain environmental conditions, such as low oxygen (49). Given that our data (Fig. 1; see also Fig. S2 in the supplemental material) and those of Fengler et al. (32) indicate that the periplasmic cysteines of ToxR are not required for toxT activation, additional studies are required, under various environmental conditions and in the absence of epitope tags or protein fusions that affect TcpP stability, to determine the importance of TcpP-ToxR disulfide bond formation in virulence gene expression by V. cholerae.
Supplementary Material
ACKNOWLEDGMENTS
We thank Victor DiRita, Jyl Matson, and Wei Ping Teoh for various protease deletion strains and anti-TcpP and anti-TcpH antibodies, as well as for helpful discussions regarding this work.
This work was supported by NIH NIAID R01 AI075087 to E.S.K. and the Frederick G. Novy Fellowship from the University of Michigan Department of Microbiology and Immunology to S.J.M.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00338-15.
REFERENCES
- 1.Taylor DN, Tacket CO, Losonsky G, Castro O, Gutierrez J, Meza R, Nataro JP, Kaper JB, Wassermann SS, Edelman R, Levine MM, Cryz SJ. 1997. Evaluation of a bivalent (CVD 103-HgR/CVD 111) live oral cholera vaccine in adult volunteers from the United States and Peru. Infect Immun 65:3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DiRita VJ, Parsot C, Jander G, Mekalanos JJ. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc Natl Acad Sci U S A 88:5403–5407. doi: 10.1073/pnas.88.12.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murley YM, Carroll PA, Skorupski K, Taylor RK, Calderwood SB. 1999. Differential transcription of the tcpPH operon confers biotype-specific control of the Vibrio cholerae ToxR virulence regulon. Infect Immun 67:5117–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Häse CC, Mekalanos JJ. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 95:730–734. doi: 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krukonis ES, Yu RR, Dirita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol Microbiol 38:67–84. doi: 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- 6.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 7.Peterson KM, Mekalanos JJ. 1988. Characterization of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infect Immun 56:2822–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carroll PA, Tashima KT, Rogers MB, DiRita VJ, Calderwood SB. 1997. Phase variation in tcpH modulates expression of the ToxR regulon in Vibrio cholerae. Mol Microbiol 25:1099–1111. doi: 10.1046/j.1365-2958.1997.5371901.x. [DOI] [PubMed] [Google Scholar]
- 9.DiRita VJ, Mekalanos JJ. 1991. Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell 64:29–37. doi: 10.1016/0092-8674(91)90206-E. [DOI] [PubMed] [Google Scholar]
- 10.Beck NA, Krukonis ES, DiRita VJ. 2004. TcpH influences virulence gene expression in Vibrio cholerae by inhibiting degradation of the transcription activator TcpP. J Bacteriol 186:8309–8316. doi: 10.1128/JB.186.24.8309-8316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skorupski K, Taylor RK. 1999. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol Microbiol 31:763–771. doi: 10.1046/j.1365-2958.1999.01215.x. [DOI] [PubMed] [Google Scholar]
- 12.Kovacikova G, Skorupski K. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J Bacteriol 181:4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee SH, Hava DL, Waldor MK, Camilli A. 1999. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell 99:625–634. doi: 10.1016/S0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- 14.Matson JS, DiRita VJ. 2005. Degradation of the membrane-localized virulence activator TcpP by the YaeL protease in Vibrio cholerae. Proc Natl Acad Sci U S A 102:16403–16408. doi: 10.1073/pnas.0505818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martínez-Hackert E, Stock AM. 1997. Structural relationships in the OmpR family of winged-helix transcription factors. J Mol Biol 269:301–312. doi: 10.1006/jmbi.1997.1065. [DOI] [PubMed] [Google Scholar]
- 16.Ottemann KM, DiRita VJ, Mekalanos JJ. 1992. ToxR proteins with substitutions in residues conserved with OmpR fail to activate transcription from the cholera toxin promoter. J Bacteriol 174:6807–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan SJ, Felek S, Gadwal S, Koropatkin NM, Perry JW, Bryson AB, Krukonis ES. 2011. The two faces of ToxR: activator of ompU, co-regulator of toxT in Vibrio cholerae. Mol Microbiol 81:113–128. doi: 10.1111/j.1365-2958.2011.07681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krukonis ES, DiRita VJ. 2003. DNA binding and ToxR responsiveness by the wing domain of TcpP, an activator of virulence gene expression in Vibrio cholerae. Mol Cell 12:157–165. doi: 10.1016/S1097-2765(03)00222-3. [DOI] [PubMed] [Google Scholar]
- 19.Goss TJ, Seaborn CP, Gray MD, Krukonis ES. 2010. Identification of the TcpP-binding site in the toxT promoter of Vibrio cholerae and the role of ToxR in TcpP-mediated activation. Infect Immun 78:4122–4133. doi: 10.1128/IAI.00566-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goss TJ, Morgan SJ, French EL, Krukonis ES. 2013. ToxR recognizes a direct repeat element in the toxT, ompU, ompT, and ctxA promoters of Vibrio cholerae to regulate transcription. Infect Immun 81:884–895. doi: 10.1128/IAI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dziejman M, Kolmar H, Fritz HJ, Mekalanos JJ. 1999. ToxR co-operative interactions are not modulated by environmental conditions or periplasmic domain conformation. Mol Microbiol 31:305–317. doi: 10.1046/j.1365-2958.1999.01173.x. [DOI] [PubMed] [Google Scholar]
- 22.Crawford JA, Krukonis ES, DiRita VJ. 2003. Membrane localization of the ToxR winged-helix domain is required for TcpP-mediated virulence gene activation in Vibrio cholerae. Mol Microbiol 47:1459–1473. doi: 10.1046/j.1365-2958.2003.03398.x. [DOI] [PubMed] [Google Scholar]
- 23.Kolmar H, Hennecke F, Götze K, Janzer B, Vogt B, Mayer F, Joachim-Fritz H. 1995. Membrane insertion of the bacterial signal transduction protein ToxR and requirements of transcription activation studied by modular replacement of different protein substructures. EMBO J 14:3895–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teoh WP, Matson JS, DiRita VJ. 2015. Regulated intramembrane proteolysis of the virulence activator TcpP in Vibrio cholerae is initiated by the tail-specific protease (Tsp). Mol Microbiol 97:822–831. doi: 10.1111/mmi.13069. [DOI] [PubMed] [Google Scholar]
- 25.Makinoshima H, Glickman MS. 2006. Site-2 proteases in prokaryotes: regulated intramembrane proteolysis expands to microbial pathogenesis. Microbes Infect 8:1882–1888. doi: 10.1016/j.micinf.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 26.Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA. 2002. DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma(E)-dependent extracytoplasmic stress response. Genes Dev 16:2156–2168. doi: 10.1101/gad.1008902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehara K, Ito K, Akiyama Y. 2002. YaeL (EcfE) activates the sigma(E) pathway of stress response through a site-2 cleavage of anti-sigma(E), RseA. Genes Dev 16:2147–2155. doi: 10.1101/gad.1002302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Missiakas D, Raina S. 1997. Protein folding in the bacterial periplasm. J Bacteriol 179:2465–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peek JA, Taylor RK. 1992. Characterization of a periplasmic thiol:disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc Natl Acad Sci U S A 89:6210–6214. doi: 10.1073/pnas.89.13.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu J, McLaughlin S, Freedman RB, Hirst TR. 1993. Cloning and active site mutagenesis of Vibrio cholerae DsbA, a periplasmic enzyme that catalyzes disulfide bond formation. J Biol Chem 268:4326–4330. [PubMed] [Google Scholar]
- 31.Yu J, Webb H, Hirst TR. 1992. A homologue of the Escherichia coli DsbA protein involved in disulfide bond formation is required for enterotoxin biogenesis in Vibrio cholerae. MolMicrobiol 6:1949–1958. [DOI] [PubMed] [Google Scholar]
- 32.Fengler VH, Boritsch EC, Tutz S, Seper A, Ebner H, Roier S, Schild S, Reidl J. 2012. Disulfide bond formation and ToxR activity in Vibrio cholerae. PLoS One 7:e47756. doi: 10.1371/journal.pone.0047756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ottemann KM, Mekalanos JJ. 1996. The ToxR protein of Vibrio cholerae forms homodimers and heterodimers. J Bacteriol 178:156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skorupski K, Taylor RK. 1996. Broad-host-range positive selection vectors for allelic exchange. Gene 169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
- 35.Higgins DE, DiRita VJ. 1994. Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol Microbiol 14:17–29. doi: 10.1111/j.1365-2958.1994.tb01263.x. [DOI] [PubMed] [Google Scholar]
- 36.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 37.Holmes RK, Baine WB, Vasil ML. 1978. Quantitative measurements of cholera enterotoxin in cultures of toxinogenic wild-type and nontoxinogenic mutant strains of Vibrio cholerae by using a sensitive and specific reversed passive hemagglutination assay for cholera enterotoxin [sic]. Infect Immun 19:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dziejman M, Mekalanos JJ. 1994. Analysis of membrane protein interaction: ToxR can dimerize the amino terminus of phage lambda repressor. Mol Microbiol 13:485–494. doi: 10.1111/j.1365-2958.1994.tb00443.x. [DOI] [PubMed] [Google Scholar]
- 39.Hennecke F, Muller A, Meister R, Strelow A, Behrens S. 2005. A ToxR-based two-hybrid system for the detection of periplasmic and cytoplasmic protein-protein interactions in Escherichia coli: minimal requirements for specific DNA binding and transcriptional activation. Protein Eng Des Select 18:477–486. doi: 10.1093/protein/gzi053. [DOI] [PubMed] [Google Scholar]
- 40.Maris AE, Walthers D, Mattison K, Byers N, Kenney LJ. 2005. The response regulator OmpR oligomerizes via beta-sheets to form head-to-head dimers. J Mol Biol 350:843–856. doi: 10.1016/j.jmb.2005.05.057. [DOI] [PubMed] [Google Scholar]
- 41.Blanco AG, Sola M, Gomis-Ruth FX, Coll M. 2002. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10:701–713. doi: 10.1016/S0969-2126(02)00761-X. [DOI] [PubMed] [Google Scholar]
- 42.Okamura H, Hanaoka S, Nagadoi A, Makino K, Nishimura Y. 2000. Structural comparison of the PhoB and OmpR DNA-binding/transactivation domains and the arrangement of PhoB molecules on the phosphate box. J Mol Biol 295:1225–1236. doi: 10.1006/jmbi.1999.3379. [DOI] [PubMed] [Google Scholar]
- 43.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci U S A 110:2348–2353. doi: 10.1073/pnas.1218039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kadokura H, Beckwith J. 2010. Mechanisms of oxidative protein folding in the bacterial cell envelope. Antioxid Redox Signal 13:1231–1246. doi: 10.1089/ars.2010.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clausen T, Southan C, Ehrmann M. 2002. The HtrA family of proteases: implications for protein composition and cell fate. Mol Cell 10:443–455. doi: 10.1016/S1097-2765(02)00658-5. [DOI] [PubMed] [Google Scholar]
- 46.Strauch KL, Johnson K, Beckwith J. 1989. Characterization of degP, a gene required for proteolysis in the cell envelope and essential for growth of Escherichia coli at high temperature. J Bacteriol 171:2689–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalbey RE, Wang P, Kuhn A. 2011. Assembly of bacterial inner membrane proteins. Annu Rev Biochem 80:161–187. doi: 10.1146/annurev-biochem-060409-092524. [DOI] [PubMed] [Google Scholar]
- 48.Almagro-Moreno S, Kim TK, Skorupski K, Taylor RK. 2015. Proteolysis of virulence regulator ToxR is associated with entry of Vibrio cholerae into a dormant state. PLoS Genet 11:e1005145. doi: 10.1371/journal.pgen.1005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan F, Liu Z, Jabeen N, Birdwell LD, Zhu J, Kan B. 2014. Enhanced interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen-limiting conditions. Infect Immun 82:1676–1682. doi: 10.1128/IAI.01377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.