

ABSTRACT
Latency-associated nuclear antigen (LANA) is a conserved, multifunctional protein encoded by members of the rhadinovirus subfamily of gammaherpesviruses, including Kaposi sarcoma-associated herpesvirus (KSHV) and murine gammaherpesvirus 68 (MHV68). We previously demonstrated that MHV68 LANA (mLANA) is required for efficient lytic replication. However, mechanisms by which mLANA facilitates viral replication, including interactions with cellular and viral proteins, are not known. Thus, we performed a mass spectrometry-based interaction screen that defined an mLANA protein-protein interaction network for lytic viral replication consisting of 15 viral proteins and 191 cellular proteins, including 19 interactions previously reported in KSHV LANA interaction studies. We also employed a stable-isotope labeling technique to illuminate high-priority mLANA-interacting host proteins. Among the top prioritized mLANA-binding proteins was a cellular chaperone, heat shock cognate protein 70 (Hsc70). We independently validated the mLANA-Hsc70 interaction through coimmunoprecipitation and in vitro glutathione S-transferase (GST) pulldown assays. Immunofluorescence and cellular fractionation analyses comparing wild-type (WT) to mLANA-null MHV68 infections demonstrated mLANA-dependent recruitment of Hsc70 to nuclei of productively infected cells. Pharmacologic inhibition and small hairpin RNA (shRNA)-mediated knockdown of Hsc70 impaired MHV68 lytic replication, which functionally correlated with impaired viral protein expression, reduced viral DNA replication, and failure to form viral replication complexes. Replication of mLANA-null MHV68 was less affected than that of WT virus by Hsc70 inhibition, which strongly suggests that Hsc70 function in MHV68 lytic replication is at least partially mediated by its interaction with mLANA. Together these experiments identify proteins engaged by mLANA during the MHV68 lytic replication cycle and define a previously unknown role for Hsc70 in facilitating MHV68 lytic replication.
IMPORTANCE Latency-associated nuclear antigen (LANA) is a conserved gamma-2-herpesvirus protein important for latency maintenance and pathogenesis. For MHV68, this includes regulating lytic replication and reactivation. While previous studies of KSHV LANA defined interactions with host cell proteins that impact latency, interactions that facilitate productive viral replication are not known. Thus, we performed a differential proteomics analysis to identify and prioritize cellular and viral proteins that interact with the MHV68 LANA homolog during lytic infection. Among the proteins identified was heat shock cognate protein 70 (Hsc70), which we determined is recruited to host cell nuclei in an mLANA-dependent process. Moreover, Hsc70 facilitates MHV68 protein expression and DNA replication, thus contributing to efficient MHV68 lytic replication. These experiments expand the known LANA-binding proteins to include MHV68 lytic replication and demonstrate a previously unappreciated role for Hsc70 in regulating viral replication.
INTRODUCTION
Gammaherpesviruses (GHVs) are double-stranded DNA viruses that include Epstein-Barr virus (EBV) and Kaposi sarcoma-associated herpesvirus (KSHV), human tumor viruses that cause cancers and lymphoproliferative diseases such as Burkitt's lymphoma, Hodgkin's lymphoma, nasopharyngeal carcinoma, Kaposi sarcoma, and multicentric Castleman's disease, among others (1–3). Similar to the case for other herpesviruses, there are two distinct phases of the GHV infectious cycle: latency and lytic replication. Latency is characterized by the expression of a limited assortment of genes, some of which are oncogenic and bear the capacity to dysregulate the cell cycle and induce cellular transformation in the absence of a competent immune response (4). In contrast, lytic replication is characterized by a temporally regulated expression cascade of immediate early (IE), early (E), and late (L) genes involved in modifying the host cell to enable viral replication and in the manufacture and assembly of new viral particles.
One important viral factor that is expressed and active during both latency and acute viral replication is the latency-associated nuclear antigen (LANA). LANA is a multifunctional protein encoded by ORF73 of gamma-2-herpesviruses (G2HVs) (also known as rhadinoviruses), including KSHV, herpesvirus saimiri, rhesus rhadinovirus, and murine gammaherpesvirus 68 (MHV68) (5). LANA was originally identified as a correlate of KSHV infection in AIDS-related Kaposi sarcoma (KS) lesions, as sera from patients with KS contained antibodies to LANA (6). During latency, LANA facilitates maintenance of the viral genome as an episome, enabling viral genome segregation to daughter cells by anchoring viral DNA to metaphase chromosomes during mitosis (7–12). LANA homologs also function as DNA-binding transcriptional regulators of both cellular and viral genes (13–20). For example, both KSHV and MHV68 LANA proteins (kLANA and mLANA, respectively) engage cognate sequences or LANA-binding sites (LBS) within the terminal repeats (TRs) of the viral genome to repress the activity of a promoter encoded within the TR (15, 17, 21). In addition, kLANA inhibits the functions of host tumor suppressor proteins such as p53, pRb, and glycogen synthase kinase 3-β (GSK3-β), thereby overcoming cell cycle arrest and protecting infected cells against apoptosis (22–25).
However, LANA functions are not limited to latent infection. LANA is transcribed with immediate early kinetics upon G2HV infection of host cells, which suggests a role in productive viral replication (26, 27). Indeed, LANA expression is robust throughout both the KSHV and MHV68 lytic replication cycles (26, 28–32). During MHV68 lytic infection, mLANA regulates viral gene expression, prevents premature cell death, and ultimately is required for efficient viral replication both in culture and in vivo (15, 28, 33, 34). Further, recombinant viruses with point mutations in mLANA that ablate DNA binding also exhibit deregulated gene expression and inefficient viral replication, which demonstrates that the capacity of mLANA to bind DNA is important for lytic replication (15). While less studied, kLANA also regulates gene expression during the KSHV lytic cycle (35).
Given its importance in both acute and latent G2HV infection and its association with disease, understanding LANA function is an area of intense experimental focus, making LANA a prime target for novel treatments of KSHV-related malignancies (36, 37). Since interactions with viral and/or cellular factors are hypothesized to modulate LANA-regulated processes, recent studies have employed proteomics approaches to identify host and/or viral proteins that interact directly with kLANA (38–43). These studies have focused on defining roles for such interactions in latent KSHV infection. Whether similar interactions are shared with other G2HVs such as MHV68 and whether they regulate lytic viral replication are not known.
Here we describe experiments to identify cellular and viral proteins that interact with mLANA to regulate MHV68 lytic infection. We employed a stable-isotope labeling of amino acids in cell culture (SILAC)-based differential proteomics technique to simultaneously elucidate and prioritize mLANA-binding proteins. In addition to expanding the network of intraviral protein-protein interactions for MHV68, we found that mLANA preferentially engaged host proteins associated with splicing and translation, including heat shock cognate protein 70 (Hsc70). Hsc70 was recruited to nuclei of infected cells in an mLANA-dependent manner, and pharmacologic inhibition and small hairpin RNA (shRNA)-mediated depletion of Hsc70 demonstrated that Hsc70 contributes to MHV68 replication by facilitating translation, replication complex formation, and viral DNA replication. The effect of Hsc70 inhibition on viral replication was less pronounced for mLANA-null MHV68, which suggests that Hsc70 function is at least partially mediated through its interaction with mLANA. Together, these findings provide the first analysis of LANA homolog interactions with both viral and cellular proteins during lytic replication and define a role for Hsc70 in promoting MHV68 replication.
MATERIALS AND METHODS
Cells and viruses.
Swiss albino mouse 3T3 fibroblasts, NIH 3T12 fibroblasts, and HEK 293T cells originally were purchased from ATCC. All cells, except those grown in isotopic labeling media, were cultured in Dulbecco's modified Eagle medium (DMEM) (Gibco Life Technologies) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (cMEM). Cells were cultured at 37°C with 5% CO2 and ∼99% humidity. Viruses used in this study include MHV68 expressing mLANA-green fluorescent protein (73.GFP) (28), mLANA-null MHV68 (73.STOP) (15, 33), and wild-type (WT) bacterial artificial chromosome (BAC)-derived MHV68 (44).
Isotopic labeling of cells.
3T3 cells were cultured in DMEM containing either “heavy” l-[13C6]arginine and l-[13C6]lysine or “light” l-[12C6]arginine and l-[12C6]lysine provided in the Pierce SILAC protein quantitation kit (89983) according to the manufacturer's instructions. In addition, “light” and “heavy” media were supplemented with 10% dialyzed FBS (Thermo Scientific Pierce), 100 U penicillin/ml, 100 μg/ml of streptomycin, and 2 mM l-glutamine. Cells were expanded in isotopically labeled media for 10 passages to obtain cells with >95% labeled proteins (45).
Virus infections.
3T3 cells were infected at a multiplicity of infection (MOI) of 5 PFU/cell with either WT MHV68 for “heavy-labeled” cells or 73.GFP MHV68 for “light-labeled” cells. Briefly, viral stocks were diluted in low-volume (2.5-ml) inocula of either “light-labeled” or “heavy-labeled” cMEM and added directly onto labeled cell monolayers seeded the previous day at a density of 1.0 × 106 cells on 15-cm dishes. Plates were incubated at 37°C and rocked gently every 15 min for 1 h. After 1 h, inocula were removed by aspiration, and “light” or “heavy” cMEM was added back to the cultures.
For all other viral infections, 3T3 or 3T12 cells plated the previous day were inoculated with a low volume of virus diluted in cMEM. Plates were rocked every 15 min for 1 h at 37°C, followed by aspiration of the viral inocula. A suitable volume of fresh cMEM was added to the cell monolayer for viral replication. For multistep growth curves, infected cells were frozen at −80°C at the indicated time points. Cells were subjected to freeze-thaw lysis to release progeny virions, and lysates were serially diluted for plaque assays as previously described (46).
Affinity purification of mLANA-GFP from infected fibroblasts.
Both “light-labeled” and “heavy-labeled” 3T3 cells infected with either 73.GFP or wild-type MHV68 were harvested at 18 h postinfection (hpi). Supernatants were aspirated, and cells were washed with 5 ml phosphate-buffered saline (PBS). Cells were trypsinized, resuspended in cMEM to inactivate trypsin, and collected into separate 15-ml conical tubes. Cells were then pelleted at 4°C, washed twice with ice-cold PBS, and pelleted again into separate, preweighed 1.5-ml microcentrifuge tubes. Light and heavy cell pellet masses were determined, mixed 1:1 by cell weight, and snap-frozen in liquid nitrogen. Frozen pellets were cryogenically lysed using five 3-min cycles in a Retsch MM301 Mixer Mill, resuspended in 1 ml of immunoprecipitation (IP) lysis buffer (1% NP-40, 5% glycerol, 25 mM Tris [pH 7.4], 150 mM NaCl, 1 mM EDTA) supplemented with protease and phosphatase inhibitors (Thermo Scientific), and sonicated for three 10-s cycles with a 550 Sonic Dismembrator (Fisher Scientific) to shear cellular DNA. Lysates were centrifuged at 16,000× g for 15 min at 4°C to remove insoluble material. Clarified supernatants were incubated with 2.0 mg of magnetic M-270 Dynabeads (Life Technologies) conjugated with polyclonal goat anti-GFP IgG antibody (Rockland Immunochemicals, Inc., 600-101-215) overnight at 4°C. Beads were isolated using a magnet and washed three times with lysis buffer. Immunoprecipitated proteins were eluted with 0.5 N ammonium hydroxide–5 mM EDTA. Eluted proteins were concentrated by vacuum centrifugation in a Savant SpeedVac (Thermo Scientific), and the concentrate was resuspended in 2× SDS sample buffer. Samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in a 4 to 20% gradient polyacrylamide gel (Bio-Rad), and protein bands were visualized by GelCode Blue staining (Thermo Scientific Pierce).
Mass spectrometry.
The entire gel lane of mLANA-GFP-interacting proteins was cut into 24 2-mm sections. Enzymatic digestion of proteins was performed by (i) reduction of disulfide bonds using Tris (2-carboxyethyl)phosphine hydrochloride, (ii) alkylation of reduced Cys residues with iodoacetamide, and (iii) digestion of proteins with trypsin. The reaction was stopped by adding formic acid. Tryptic peptides from the 24 gel bands were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) with a Thermo LTQ-XL mass spectrometer coupled to an Eksigent nanoLC-2D. For data analysis, collected MS/MS spectra were searched against the International Protein Index (IPI) database with the taxonomy selected for Homo sapiens and for Mus musculus with the MuridHerpesVirus refseq sequences using Mascot Distiller (Matrix Sciences). Mass spectra were also searched using the Mascot UniProt database. Mascot results were uploaded into Scaffold 3 (version 3.00.01) for viewing protein and peptide information, and protein IDs were searched using the Con_ipi.MOUSE.v3.82_ConMuridHerpesVirusRefSeq database. Mascot Distiller was used to determine the fraction of peptides containing light isotopes of arginine and lysine. The light/heavy isotope fractions for each protein were manually verified. Reported results are from a single experiment.
Plasmids and transfections.
pMSCV-mLANA-GFP and pCMV14-mLANA-FLAG were previously described (28). Viral open reading frames (ORFs) 21, 25, 52, 57, 59, 62, and M3 were amplified from the MHV68 BAC by PCR and cloned into pCR-Blunt vectors (Life Technologies) following the manufacturer's instructions. All pCR-Blunt vector sequences were validated, and the inserted viral ORFs were then shuttled to the mammalian expression vector pCMV14-3XFLAG (Sigma-Aldrich) using viral gene-specific primers modified to encode restriction site overhangs. The expression vector pTAG2B-ORF75B encoding the viral ORF75B was a gift from Laurie T. Krug (Stony Brook University). pGEX-4T-3 (Addgene) was utilized as a control in glutathione S-transferase (GST) pulldown assays. Hsc70 was amplified from cDNA derived from 3T3 cells using primers hsc71mrna_F (5′-GAACGCGGAGGCAGCTGCCTGGC-3′) and hsc71mrna_R (5′-GGGTCCCTGTGGAACAAAGCT-3′) followed by a nested reaction using primers hsc71nest_F (5′-GCAACCATGTCTAAGGGACCTGCAGTT-3′) and hsc71nest_R (5′-GGACTGACTTAATCCACCTCTTCAATGGT-3′). The PCR product was reamplified to append att sites using primers hsc70pENTR_F (5′-CACCATGTCTAAGGGACCTGCAGTTGGCA-3′) and hsc70pENTR_R (5′-ATCCACCTCTTCAATGGTGGGGCCTGAA-3′) for cloning into pENTR (Life Technologies). Hsc70 cDNA was amplified from pENTR-Hsc70 and appended with EcoRI and BamHI restriction sites vector using primers hsc71EcoRI_F (5′-GCGAATTCAATGTCTAAGGGACCTGCAGT-3′) and hsc71BamHI_R (5′-GCGGATCCATCCACCTCTTCAATGGTGGGGCCT-3′) and cloned into pCMV14-3XFLAG to generate pCMV14-Hsc70-FLAG. To generate pDEST24-mLANA-GST, mLANA was amplified from pMSCV-mLANA-GFP using the primers pENTR_F (5′-CACCATGCCCACATCCCCACCGACTACA-3′) and pENTR_R (5′-GGCTGTCTGAGACCCTTGTCCCTGTT-3′) and cloned into pENTR-SD-TOPO (Life Technologies). mLANA was then transferred into pDEST24 (Life Technologies) using Gateway LR Clonase II enzyme mix (Life Technologies). The fidelity of all cloned constructs was verified by DNA sequencing. Transfections were performed using Lipofectamine (Life Technologies) according to the manufacturer's instructions. Transfected cells were incubated at 37°C for 24 or 48 h to allow protein expression prior to immunoprecipitation or immunofluorescence assays (IFAs).
Antibodies, antibody bead coupling, and drug treatments.
Primary antibodies used in this study include goat polyclonal anti-GFP (1:2,000 dilution; Rockland Immunochemicals, Inc., 600-101-215), mouse monoclonal anti-FLAG (1:1,000 dilution; Sigma-Aldrich, F3165), horseradish peroxidase (HRP)-conjugated rabbit anti-FLAG (1:2,000 dilution; Sigma-Aldrich, A8592), rabbit monoclonal anti-Hsc70 (1:2,000 dilution; Cell Signaling Technology, D12F2), rabbit monoclonal anti-histone H3 (1:2,000 dilution; Cell Signaling Technology, 9715), rabbit polyclonal mLANA antiserum (1:2,000 dilution) (15), mouse polyclonal MHV68 antiserum (1:1,000 dilution) (46), rabbit monoclonal anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase) (1:1,000 dilution; Cell Signaling Technology, 2118), rabbit polyclonal ORF57 antiserum (1:2,000 dilution) (47), chicken anti-ORF59 IgY (1:250 dilution) (46), mouse monoclonal anti-β-actin (1:5,000 dilution; Sigma-Aldrich, A2228), and mouse monoclonal anti-GST–HRP (1:1,000 dilution; Santa Cruz Biotechnology, sc-138). Fluorophore-conjugated secondary antibodies used in this study include Alexa Fluor donkey anti-goat 488, Alexa Fluor goat anti-mouse 568, and Alexa Fluor goat anti-chicken 568 (Life Technologies). Polyclonal GFP antibody (50 μg) was coupled to 10 mg of epoxy magnetic M-270 Dynabeads (Life Technologies) according to the manufacturer's instructions. For drug treatments, the Hsp70/Hsc70 inhibitor VER-155008 (Sigma-Aldrich) (48) was dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 10 mM. Cells were pretreated with VER-155008 for 1 h prior to infection and retreated after viral adsorption. DMSO treatments served as vehicle controls.
IPs.
HEK 293T cells transfected with the indicated plasmids were lysed in immunoprecipitation (IP) lysis buffer supplemented with phosphatase and protease inhibitors (Thermo Scientific). Cell lysates were sonicated on ice for three 10-s cycles at 30% output on a 550 Sonic Dismembrator (Thermo Fisher). Lysates were cleared by centrifugation (16,000× g) at 4°C for 15 min to remove insoluble material and transferred to new microcentrifuge tubes. Ten percent of the supernatant volume for each sample was removed and saved for input controls. The remainder of the lysate was incubated with polyclonal anti-GFP antibody (1 μg) and rotated overnight at 4°C. Immune complexes were captured with protein A/G Plus-agarose beads (Santa Cruz Biotechnology, Inc., sc-2003) for 1 h at room temperature. Beads were precipitated by centrifugation and washed three times with lysis buffer. Immunoprecipitated proteins were eluted from beads by addition of 2× Laemmli buffer (49).
Immunoblot analyses.
Immunoblot analyses were performed essentially as previously described (46, 50). Briefly, protein samples were harvested by disrupting cells with either radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 20 mM Tris, 2 mM EDTA, 1% NP-40, and 0.25% deoxycholate supplemented with phosphatase and protease inhibitors) or IP lysis buffer. Processed samples were resolved by SDS-PAGE, transferred to nitrocellulose membranes (Thermo Scientific), and probed with the indicated primary antibodies. Horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch) and SuperSignal Pico West ECL reagent (Thermo Scientific) or Clarity ECL reagent (Bio-Rad) were used for detection. Data were captured using a Bio-Rad ChemiDoc MP imaging system.
GST pulldown assays.
Arabinose-inducible BL21-AI competent Escherichia coli (Life Technologies) transformed with pDEST24-mLANA-GST (mLANA-GST) or isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible BL21(DE3)/pLysS E. coli (Monserate Biotechnology Group) transformed with pGEX-4T-3 (GST) was grown overnight at 37°C. The next day, a 200-μl aliquot of each culture was transferred to a flask containing 20 ml Luria broth (LB) supplemented with ampicillin and was incubated for 3 h at 37°C to allow for logarithmic growth (optical density [OD] = 0.6). Fifty percent of each culture was transferred to a separate flask for induction. mLANA-GST cultures were induced with a final concentration of 0.2% l-arabinose (Sigma-Aldrich) for 3 h. GST cultures were induced with 1 mM IPTG. Induced cultures were pelleted by centrifugation, resuspended in ice-cold PBS, and sonicated for four 30-s cycles (40% output), followed by addition of 1% Triton X-100. Cell debris was removed by centrifugation at maximum speed. Clarified lysates were incubated with glutathione-agarose beads (Sigma-Aldrich, G4510) for 2 h to pull down mLANA-GST or GST. Captured complexes were washed 3 times with PBS and split equally for separate overnight incubations at 4°C in lysates of 293T cells expressing mLANA-FLAG, ORF57-FLAG, ORF59-FLAG, or Hsc70-FLAG. Complexes were pelleted by centrifugation and washed three times with PBS. Samples were resuspended and boiled in 2× Laemmli sample buffer. Samples were resolved by SDS-PAGE and probed for the presence of FLAG-tagged and GFP-tagged proteins in immunoblot analyses.
Cell viability assay.
Cell viability assays were performed essentially as previously described (28). Briefly, cells were treated for 48 h with the indicated drugs and/or chemicals, washed three times with PBS, and fixed in 10% formalin for 15 min at room temperature. Cells were washed with PBS and stained with 0.1% crystal violet for 2 h at room temperature. Excess stain was removed by washing three times in deionized water, and samples were allowed to dry. Retained crystal violet was extracted by addition of 10% acetic acid, diluted 1:2, and transferred to microtiter plates. Absorbances were measured at 570 nm in a FLUOstar Omega plate reader (BMG Labtech).
IFAs.
Immunofluorescence assays (IFAs) were performed as previously described (28, 46). Briefly, transfected and/or infected cells plated on glass coverslips the previous day were washed with PBS and fixed in 10% phosphate-buffered formalin. Cells were permeabilized for 15 min with 0.5% Triton-X 100 in Tris-buffered saline (TBS) at room temperature. Supernatants were aspirated, and cells were incubated in blocking buffer (5% bovine serum albumin [BSA], 1% normal donkey serum, and 0.1% Triton-X 100 in TBS) for 30 min at room temperature. Blocking buffer was removed, and cells were incubated for 1 h with the indicated primary antibodies diluted in blocking buffer. Samples were washed three times with wash buffer (0.1% Triton X-100 in TBS). Fluorophore-conjugated secondary antibodies specific to primary antibodies diluted in blocking buffer were added to the samples for 1 h in the dark at room temperature. Samples were washed three times with wash buffer, and glass coverslips were mounted on slides using ProLong Antifade Gold reagent with DAPI (4′,6′-diamidino-2-phenylindole) (Sigma-Aldrich) to stain DNA. Samples were visualized under a magnification of ×20 or ×60 by fluorescence microscopy using an Eclipse Ti-U fluorescence microscope (Nikon). Images were acquired with a D5-QilMc digital camera and analyzed using NIS-Elements software (Nikon).
Nuclear and cytoplasmic fractionations.
3T3 cells were either mock infected or infected with 73.GFP or 73.STOP at an MOI of 5 PFU/cell. Cells were washed, scraped into PBS, and pelleted at 18 hpi. Cells were swelled in hypotonic buffer (10 mM HEPES [pH 7.9], 10 mM KCl, and 1.5 mM MgCl2 supplemented with protease and phosphatase inhibitors) on ice for 15 min, followed by addition of NP-40 (0.5% final concentration) to disrupt the plasma membrane. Nuclei were pelleted at 500× g for 5 min, and supernatants containing the cytoplasmic fraction were collected. The nuclear pellet was resuspended in 100 μl RIPA buffer and centrifuged for 15 min at 4°C to pellet the insoluble nuclear fraction. The insoluble fraction was resuspended in 2× Laemmli sample buffer. Samples were resolved by SDS-PAGE and subjected to immunoblot analyses.
qPCR and RT-PCR.
Quantitative PCR (qPCR) to quantify viral genomes was performed as previously described (46). Briefly, cells pretreated for 1 h with DMSO or VER-155008 (50 μM) were infected with MHV68 at an MOI of 5 PFU/cell. After adsorption, cells were washed 3 times with PBS to remove cell-free virus. Medium containing either DMSO, VER-155008, or phosphonoacetic acid (PAA) was then added to the cells. Total DNA from infected cells was isolated using a Qiagen DNeasy kit at 4 h, considered the “eclipse” phase and serving as a viral DNA input control, and at 18 h after adsorption. One hundred nanograms of total DNA was diluted in RT2 SYBR green Fluor qPCR Mastermix (Qiagen) and analyzed by quantitative real-time PCR in an Applied Biosystems StepOne Plus thermocycler using primers specific to a sequence within the viral ORF25 gene or primers specific to the cellular GAPDH gene (46). The experiment was performed in technical duplicate for biological duplicate samples. Data were analyzed using the ΔΔCT method (51) where ΔΔCT = [CT(ORF25) − CT(GAPDH)4 h] − [CT(ORF25) − CT(GAPDH)18 h]. Data represent the fold change in viral DNA at 18 h relative to 4 h and normalized to the GAPDH values based on 2−ΔΔCT calculations.
Quantitative reverse-transcription PCR (qRT-PCR) was performed as previously described (28). Briefly, 3T3 cells pretreated with vehicle control (DMSO) or VER-155008 (50 μM) were mock infected or infected with WT MHV68 at an MOI of 5 PFU/cell in the presence or absence of VER-155008. Total RNA from uninfected and infected cells was harvested at the indicated time points postinfection using the Qiagen RNeasy kit (Qiagen). Purified RNA was treated with DNase I (New England BioLabs) for 10 min at 37°C to digest contaminating DNA. One microgram of RNA from each sample was reverse transcribed to cDNA using random hexamers and the SuperScript first-strand synthesis system for RT-PCR (Invitrogen) according to the manufacturer's instructions. Quantitative PCR was performed on the resultant cDNA to quantify transcript levels of the viral genes ORF50, ORF73, ORF59, and ORF25 and the cellular GAPDH gene for each sample utilizing the RT2 SYBR green Fluor qPCR Mastermix (Qiagen). Target genes were detected using primer pairs for ORF50 (28), ORF25 and the GAPDH gene (46), 73PCR1 (5′-ACACAACCTCAGGCAAAAC-3′) and 73PCR2 (5′-CCTTCAACATCAACATCTGG-3′) for ORF73, and 59PCR1 (5′-ATGCAGACCTTCCAGCTTGAC-3′) and 59PCR2 (5′-CTCTTCCAAGGGAGCTTGCG-3′) for ORF59.
shRNA knockdown of Hsc70.
3T12 cells (2 × 105) were cotransfected with MHV68 BAC and either 3 μg of control pLKO1.1 puro vector (Sigma-Aldrich) or pLKO encoding Hsc70-specific small hairpin RNA (shRNA): Hsc70 shRNA1 (TRCN0000098667), Hsc70 shRNA2 (TRCN0000304876) (Sigma-Aldrich), or a combination of both. Viral supernatants and cellular lysates from transfected cells were collected at 72 h and 96 h posttransfection for plaque assays and immunoblot analyses, respectively. Knockdown of Hsc70 was confirmed by immunoblot analysis.
Bioinformatics analyses.
Cellular protein-protein interaction networks for MS-identified proteins were mapped utilizing version 9.05 of the STRING resource (52). The viral protein interaction subnetwork was assembled using Cytoscape 2.8 (53). Previously reported MHV68 intraviral protein-protein interactions were extracted from reference 54 and integrated into the Cytoscape map. Disconnected nodes were not included in the Cytoscape output.
Statistics.
All statistical analyses of data were performed using GraphPad Prism software (GraphPad Software, San Diego, CA). Statistical significance was determined by a two-tailed unpaired Student t test with a confidence level of 95%.
RESULTS
mLANA binding to DNA is necessary for LANA function in MHV68 lytic replication (15). However, whether cellular and/or viral factors are engaged by mLANA to facilitate lytic replication is not known. In order to identify potential cofactors for mLANA in controlling the MHV68 lytic cycle, we performed a mass spectrometry-based interaction screen coupled with a differential proteomics technique known as I-DIRT (isotopic differentiation of interactions as random or targeted [45, 55, 56]) to identify viral and cellular proteins that interact with mLANA during MHV68 lytic replication. The experimental approach is schematized in Fig. 1A.
FIG 1.

Application of I-DIRT to identify mLANA-binding proteins in lytic MHV68 infection. (A) Overview of the I-DIRT method. Light or heavy isotopically labeled 3T3 fibroblasts were infected with either recombinant MHV68 expressing mLANA-GFP (73.GFP) or wild-type MHV68 (WT) at an MOI of 5 PFU/cell. At 18 h postinfection, intact cells were mixed at an equivalent ratio by weight, cryogenically disrupted, and resuspended in lysis buffer. mLANA-GFP and its interactors were immunoprecipitated using GFP antiserum covalently coupled to magnetic beads and purified for mass spectrometry. Isotopically light and heavy peptide fractions for a given protein interaction were calculated and scored as specific or nonspecific interactions based on a ratio calculated for common nonspecific protein contaminants. (B) mLANA-interacting proteins with both light and heavy peptides were classified as specific or nonspecific by I-DIRT. The fraction of light isotopic peptides containing [6-12C]arginine and [6-12C]lysine was calculated by dividing the peak area of the light peptide spectrum of a given protein by the sum of the peak areas for light and heavy peptide spectra for the same protein (ratio light/[light plus heavy]). The nonspecific threshold (0.6918 L/L+H, black dashed line) was determined by calculating the mean light fraction of ribosomal proteins that are common contaminants in interaction screens. Proteins with a light isotopic fraction greater than one standard deviation from the mean ribosomal threshold (>0.7528 L/L+H, dashed red line) are scored as specific interactions (red bars), while those with a fraction less than one standard deviation from the mean are classified as nonspecific interactions (black bars).
For these experiments, 3T3 fibroblasts were labeled with “light” or “heavy” isotopic forms of l-arginine and l-lysine according to SILAC protocols (57). De novo infection of 3T3 cells in culture with MHV68 is ideal for such experiments, because mLANA regulates the MHV68 lytic replication cycle in 3T3 fibroblasts (28). “Light-labeled” cells were infected with an MHV68 recombinant virus that expresses an mLANA-GFP fusion protein (73.GFP [28]), thus providing in GFP a means for monitoring infection efficiency and an affinity tag for purification of mLANA and associating proteins. As the comparative control for I-DIRT, “heavy-labeled” cells were infected in parallel with WT MHV68 (which expresses untagged mLANA). Infected cells were used as the comparative control to account for virus-induced changes in host protein quantities and/or phosphorylation status (50). Cells were harvested at 18 h postinfection, a time point at which mLANA expression is high (28), and the I-DIRT procedure was performed, whereby mLANA-GFP protein complexes were isolated by immunoaffinity and protein identities and SILAC content were determined by in-gel digestion followed by LC-MS/MS (GeLC-MS/MS).
A total of 206 cellular and viral proteins that copurified with mLANA-GFP were identified with the following criteria: 5% protein threshold, minimum of 2 peptides, and 50% peptide threshold (see Table S1 in the supplemental material). I-DIRT analysis, which relies on biophysical principles of protein-protein interaction specificity, was applied to all proteins identified by mass spectrometry to select for those that contained peptides comprised of both light and heavy isotopic arginine and lysine. Proteins that are common exogenous contaminants, such as keratin, were excluded from further analysis. I-DIRT “hits” were validated, and their light peptide fraction [light peptides/(light plus heavy peptides) (L/L+H)] was calculated (45, 56). The specificity threshold was calculated using the mean light fraction of 21 ribosomal proteins (0.6918 L/L+H), which are common abundant intracellular contaminants in protein-protein interaction studies (45, 56), plus one standard deviation (±0.0610 L/L+H). Thus, proteins with a mean light fraction below 0.7528 L/L+H were considered “nonspecific” interactions (Fig. 1B; see Table S2 in the supplemental material). It is important to note that not all peptides identified contained both heavy and light labels and thus were not amenable to I-DIRT-based prioritization. Abundant proteins that lacked SILAC labels could be considered for future studies but were not prioritized for functional validation or analysis in the current work.
Defining an mLANA viral protein interaction network.
Of the 15 viral proteins identified, three scored as specific by I-DIRT analysis: immediate early protein ORF57 (31), chemokine-binding protein M3 (58), and tegument protein ORF52 (Fig. 1B) (59). To evaluate whether these and other viral proteins identified by mass spectrometry were capable of binding mLANA, we generated FLAG-tagged versions of candidate interactors and tested their capacity to bind mLANA in coimmunoprecipitation experiments. We were unable to detect expression of the ORF21 and ORF25 constructs generated for these analyses, so they were not included in the experiment whose results are shown in Fig. 2. Following cotransfection of 293T cells with mLANA-GFP and each of the FLAG-tagged candidate proteins, we immunoprecipitated mLANA-GFP and performed immunoblot analyses to detect FLAG. Previous studies have demonstrated that mLANA homo-oligomerizes (15, 60, 61), so we used FLAG-tagged mLANA as a positive control. As expected, proteins that scored as nonspecific in I-DIRT analyses did not coprecipitate with mLANA. Though we consider it unlikely, it remains possible that proteins such as ORF62 and ORF75B with lower expression levels were simply below detection limits for this assay. Corroborating the I-DIRT analysis, ORF52 efficiently coprecipitated with mLANA, while none of the “nonspecific” viral proteins were detected (Fig. 2A). This indicates that mLANA and ORF52 are capable of interacting in the absence of other viral proteins. M3, however, did not coprecipitate in cotransfection experiments, which suggests that it is tethered to mLANA through other molecules. Interestingly and despite multiple attempts, we were unable to detect mLANA-GFP in cells cotransfected with ORF57 expression constructs (Fig. 2A). An explanation for this result is not immediately clear. We speculate that the RNA-processing function of ORF57 (62; reviewed in reference 63) may specifically target mLANA-encoding ORF73 transcripts to regulate mLANA expression. Thus, we retested the putative mLANA-ORF57 interaction via GST pulldown assays. GST-tagged mLANA or GST alone was incubated with lysates from 293T cells expressing FLAG-tagged versions of either ORF57, mLANA as a positive control, or ORF59 as a negative control. While mLANA-FLAG was efficiently captured and ORF59 was not, we were unable to detect ORF57 in either GST or mLANA-GST purified complexes, suggesting that an interaction between mLANA and ORF57 either is tethered by viral nucleic acids or requires other viral or cellular factors present during lytic infection (Fig. 2B). We have integrated the mLANA interactions described here with previously described intraviral protein-protein interaction data (54) to provide an updated MHV68 viral protein interaction network (Fig. 2C).
FIG 2.

Updating the MHV68 intraviral protein interaction network. (A) Viral proteins identified by MS were validated by coimmunoprecipitation assays with mLANA-GFP. 293T cells were transfected with plasmids encoding the indicated FLAG-tagged viral proteins and mLANA-GFP. At 48 h posttransfection, cells were lysed and mLANA-GFP was immunoprecipitated from lysates using GFP antiserum. Immunoblot analyses were performed with FLAG-specific antibodies to detect coprecipitating viral proteins. (B) GST and mLANA-GST were purified from bacterial lysates and incubated with lysates from 293T cells transfected with plasmids encoding mLANA-FLAG, ORF59-FLAG, or ORF57-FLAG. Complexes were captured using glutathione agarose. Precipitates were washed and resolved by SDS-PAGE. Immunoblot analyses were performed to detect FLAG- and GST-tagged proteins. (C) The mLANA intraviral protein interaction network. Blue nodes represent viral interactions identified in this study, yellow nodes represent viral interactions identified previously (54), green nodes indicate interacting viral proteins identified both in this study and previously (54), square nodes designate interactions validated by coimmunoprecipitation in this study, solid red edges designate specific interactions scored by I-DIRT, dashed red edges designate nonspecific scored interactions, dashed purple edges represent unscored interactions, and solid black edges represent interactions validated in previous studies. WCL, whole-cell lysate; IP, immunoprecipitation; IB, immunoblot.
Defining an mLANA-cellular protein interaction network.
By GeLC-MS/MS, we identified 191 cellular proteins in complex with mLANA. Excluding common protein contaminants, 78 proteins contained both heavy and light peptides and were amenable to I-DIRT analysis. Host proteins that scored as specific interactors included tripartite motif-containing protein 21 (TRIM21), heat shock cognate protein 70 (Hsc70), histone H1.2, serine-arginine rich splicing factors 6 and 7 (SRSF6 and SRSF7), transformer 2-Beta (Tra2B), and eukaryotic translation elongation factor 1 A1 (EF1A1) (Fig. 1B). We next utilized the STRING algorithm (52) to evaluate the interconnectedness of the mLANA cellular interactome (see Fig. S1 in the supplemental material). STRING analyses revealed that host factors identified as mLANA-interacting proteins formed a highly connected network in which ca. 70% of identified proteins were connected. The two most highly connected subnetworks within the global mLANA interaction network included proteins involved in translation and splicing regulatory factors and helicases (see Fig. S1 in the supplemental material). Interestingly, Hsc70, SRSF6, SRSF7, EF1A1, and Tra2B were all closely clustered in the network, suggesting that mLANA engages a multiprotein complex consisting of heat shock proteins, splicing regulatory factors, and translation factors during the lytic replication cycle. It also is worth noting that none of the host proteins identified as specific mLANA-interacting proteins were previously identified as being capable of engaging other MHV68 proteins, including ORF52 (54), suggesting that these interactions are direct and not mediated through other viral proteins.
Of note, 19 mLANA-binding cellular proteins were previously reported as kLANA-interacting factors in other proteomics-based interaction screens (Table 1) (8, 39–43, 64). I-DIRT analyses defined 4 of these proteins as having specific mLANA interactions, while 15 proteins were scored as having nonspecific interactions (Table 1). Interactors that were both conserved and specific included Hsc70, histone H1.2, EF1A1, and β-actin (8, 39, 40). Together, these data reveal a set of cellular factors that interact with mLANA during lytic MHV68 infection, prioritize protein-protein interactions through I-DIRT, evaluate the conserved nature of their interaction with kLANA, and organize them into functional networks.
TABLE 1.
Conserved mLANA interactions scored by I-DIRT
| Gene symbol | Gene name | Molecular mass (kDa) | Assigned spectraa | KSHV LANA interaction reference(s) | L/L+Hb | Specificityc |
|---|---|---|---|---|---|---|
| HSP7C | Heat shock cognate 71-kDa protein | 71 | 30 | 71 | 0.7828 | Specific |
| H12 | Histone H1.2 | 21 | 23 | 8 | 0.7743 | Specific |
| EF1A1 | Elongation factor 1 alpha | 50 | 24 | 40 | 0.7693 | Specific |
| ACTB | Actin, cytoplasmic 1 | 42 | 174 | 39, 40 | 0.7582 | Specific |
| RL12 | Rpl12 60S ribosomal protein L12 | 18 | 16 | 40 | 0.7319 | Nonspecific |
| NPM | Nucleophosmin | 33 | 25 | 40, 41 | 0.7286 | Nonspecific |
| RS4X | 40S ribosomal protein S4 | 30 | 27 | 39 | 0.7282 | Nonspecific |
| ANXA2 | Annexin A2 | 39 | 19 | 40 | 0.7110 | Nonspecific |
| RS5 | 40S ribosomal protein S5 | 20 | 17 | 40 | 0.7027 | Nonspecific |
| TBB5 | Tubulin beta-5 chain | 50 | 46 | 39 | 0.7008 | Nonspecific |
| RS18 | 40S ribosomal protein S18 | 18 | 10 | 40, 41 | 0.6995 | Nonspecific |
| RL11 | 60S ribosomal protein L11 | 20 | 13 | 40 | 0.6964 | Nonspecific |
| HNRPK | Heterogeneous nuclear ribonucleoprotein K | 51 | 6 | 40, 42 | 0.6722 | Nonspecific |
| HS90B | Heat shock protein 90 | 83 | 20 | 40, 71 | 0.6685 | Nonspecific |
| ANXA1 | Annexin A1 | 39 | 9 | 40 | 0.6564 | Nonspecific |
| KPYM | Pyruvate kinase isozyme M2 | 58 | 8 | 40 | 0.6550 | Nonspecific |
| H2AJ | Histone H2A.J | 14 | 14 | 41, 64 | 0.6189 | Nonspecific |
| HNRPM | Heterogeneous nuclear ribonucleoprotein M | 78 | 25 | 41 | 0.6049 | Nonspecific |
| MYH9 | Myosin 9 | 226 | 8 | 39 | 0.5868 | Nonspecific |
Number of tryptic peptides detected for that particular protein.
L/L+H is the fraction of light isotopic peptides, which is defined as the light isotopic peptide abundance divided by a light and heavy isotopic peptide mixture for any given protein.
“Specific” interactions are defined as those interactions which possess an L/L+H value of >0.7528 or one standard deviation above the mean light fraction threshold of 21 ribosomal proteins, which equals 0.6918. Interactions with an L/L+H value of <0.7528 were considered “nonspecific.”
mLANA interacts with Hsc70 and enhances its nuclear accumulation in infected cells.
Based on spectral abundance (30 assigned spectra), I-DIRT specificity (0.7828 L/L+H ratio), and potential functional conservation with KSHV (Table 1), we selected Hsc70 as a high-priority candidate for further study. Hsc70 is a constitutively expressed chaperone belonging to the highly conserved heat shock protein 70 (Hsp70) family (reviewed in reference 65). To validate Hsc70 as a bona fide mLANA-binding protein, we first performed GST pulldown assays to determine if mLANA and Hsc70 interact in the absence of other viral proteins. Purified mLANA-GST fusion protein or GST alone was incubated with lysates from 293T cells that were transfected with expression constructs for FLAG-tagged Hsc70 or mLANA-FLAG as a positive control. Capacities to bind Hsc70 or mLANA were then evaluated in immunoblot analyses. While mLANA-GST pulled down Hsc70 and mLANA, GST alone did not capture either protein (Fig. 3A). To corroborate these findings, we also performed coimmunoprecipitation experiments following cotransfection of 293T cells with Hsc70-FLAG and either mLANA-GFP or GFP alone as a negative control. While GFP alone failed to coprecipitate Hsc70, mLANA-GFP efficiently coprecipitated Hsc70 (Fig. 3B). Together these data indicate that mLANA is capable of interacting with Hsc70 in the absence of other viral proteins.
FIG 3.
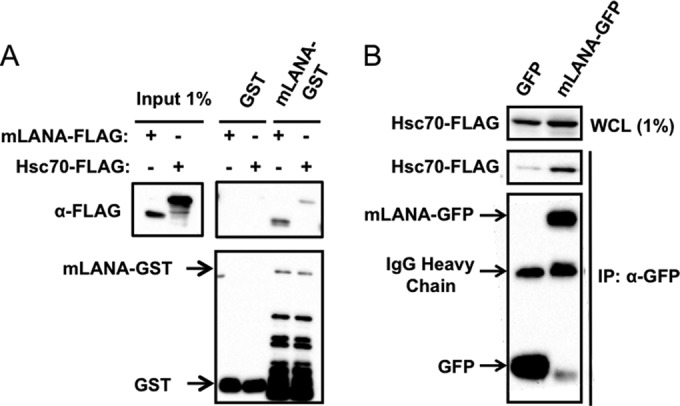
mLANA interacts with Hsc70. (A) GST and mLANA-GST were purified from bacterial lysates and incubated with lysates from 293T cells transfected with plasmids encoding Hsc70-FLAG or mLANA-FLAG. Complexes were captured and resolved by SDS-PAGE. Immunoblot analyses were performed to detect FLAG- and GST-tagged proteins. (B) 293T cells were cotransfected with plasmids encoding Hsc70-FLAG and either GFP or mLANA-GFP. Cells were lysed at 48 h posttransfection, and GFP or mLANA-GFP complexes were immunoprecipitated from cell lysates. Immunoblot analyses were performed to detect the indicated proteins. WCL, whole-cell lysate; IP, immunoprecipitation; IB, immunoblot.
Hsc70 localizes primarily to the cytoplasm but translocates into the nucleus, specifically the nucleolus, under stress conditions such as heat shock (66). Since mLANA is predominantly a nuclear protein (12, 28), we were curious if mLANA would facilitate the recruitment of Hsc70 to nuclei of infected cells. To test this hypothesis, we performed immunofluorescence analyses of 3T12 fibroblasts that were transfected with plasmids encoding Hsc70-FLAG (to facilitate detection) prior to infection with either 73.GFP MHV68 or a yellow fluorescent protein (YFP)-expressing mLANA-null recombinant virus (73.STOP [15]). While Hsc70-FLAG was detected predominantly in the cytoplasm of mock-infected and 73.STOP-infected cells, Hsc70 accumulated in the nuclei of cells infected with LANA-competent MHV68 (Fig. 4A). A cross-sectional analysis of fluorescence intensities confirmed that the strongest Hsc70 signal overlapped with DAPI in the nuclei of cells expressing mLANA-GFP, but the overlap was less prominent in mock-infected cells and cells infected with 73.STOP.
FIG 4.

mLANA enhances nuclear accumulation of Hsc70. (A) 3T12 fibroblasts transfected with plasmids encoding Hsc70-FLAG were mock infected or infected with 73.GFP or 73.STOP MHV68 at an MOI of 5 PFU/cell. Cells were fixed and permeabilized at 18 h postinfection and stained with antibodies specific for GFP and FLAG to visualize LANA and Hsc70 intracellular localization by fluorescence microscopy. DNA was detected with DAPI. White triangles designate infected cells exhibiting nuclear accumulation of Hsc70-FLAG. Fluorescence intensities for GFP, FLAG, and DAPI stains of the region marked within representative cells shown in the insets were plotted for each treatment. (B) 3T3 fibroblasts were mock infected or infected with WT MHV68 or 73.STOP MHV68 at an MOI of 5 PFU/cell. Cells were harvested 18 h postinfection, and cytoplasmic, soluble nuclear, and insoluble nuclear fractions were prepared. Equivalent amounts of protein for each fraction were resolved by SDS-PAGE, and immunoblot analyses were performed to detect the indicated proteins.
As an independent and complementary test, we conducted cellular fractionation experiments to quantify levels of endogenous Hsc70 in the nuclear and cytoplasmic compartments of uninfected cells and cells infected with either WT MHV68 or 73.STOP MHV68. While Hsc70 was readily detected in the cytoplasmic fraction for all conditions tested, Hsc70 was only minimally detected in the soluble nuclear fraction of mock-infected and 73.STOP-infected cells (Fig. 4B). In contrast, infection with LANA-competent WT MHV68 resulted in increased detection of Hsc70 in the soluble nuclear fraction of infected cells. Interestingly, although mLANA was detected in both soluble and insoluble nuclear fractions, Hsc70 was not present in the insoluble chromatin fraction under any conditions tested. This suggests that LANA interactions with Hsc70, and perhaps other viral and host factors, are compartmentalized within nuclei of infected cells. Together, these data demonstrate a role for mLANA in enhancing nuclear accumulation of Hsc70 during MHV68 infection.
Hsc70 is necessary for efficient MHV68 lytic replication.
We next sought to define roles for Hsc70 in MHV68 lytic replication. For these experiments, we infected 3T3 fibroblasts in the absence or presence of increasing concentrations of VER-155008 and evaluated viral titers by plaque assay over time. VER-155008 inhibits the ATPase activities of Hsc70 and Hsp70 (48), thereby blocking the chaperone function of these proteins. For all samples, viral titers were equivalent at the 0-h time point, indicating that VER-155008 treatment did not negatively impact viral attachment to cells or plaque assay results (Fig. 5A). Treatment with VER-155008 resulted in a dose-dependent reduction in viral titers, with the highest dose (50 μM) resulting in complete blockade of MHV68 replication at the 24-h time point compared to control-treated cells (Fig. 5A). However, the effect of VER-155008 treatment was not absolute, as cells treated with the highest inhibitor dose (50 μM), exhibited a 5-fold reduction in viral titers relative to controls at 48 h postinfection (Fig. 5A). Importantly, in cytotoxicity assays performed in parallel, VER-155008 treatment did not cause death in either mock-infected or MHV68-infected cells at doses lower than 100 μM (Fig. 5B), thereby demonstrating that reduced viral replication following VER-155008 treatment was not simply due to cell death. These data therefore indicate that a pharmacologic inhibitor of Hsc70/Hsp70 function inhibits MHV68 lytic replication.
FIG 5.

Pharmacologic inhibition of Hsc70 represses MHV68 replication. (A) 3T3 fibroblasts were treated with vehicle (DMSO) or the indicated concentrations of Hsc70 inhibitor VER-155008 for 1 h prior to and during infection with MHV68 at an MOI of 5 PFU/cell. Cells were harvested at the indicated times postinfection, and viral titers were determined by plaque assay. Results are means for triplicate samples. Error bars represent standard deviations. (B) 3T3 fibroblasts were mock infected or infected with MHV68 in the presence of PAA (200 μg/ml) to inhibit viral replication-related cell death and the indicated concentrations of VER-155008. A separate group of cells were treated with cycloheximide (10 ng/ml) and tumor necrosis factor alpha (TNF-α) (0.2 μg/ml) as a positive control for cell death. Cell viability was determined at 48 h posttreatment by crystal violet retention assays. Data represent percentages of cell death relative to mock-treated controls. Results are means for triplicate samples. Error bars represent standard deviations.
Because VER-155008 is capable of inhibiting other members of the Hsp70 chaperone family (48) possibly involved in MHV68 replication, we also tested the effect of shRNA-mediated knockdown as a more specific evaluation of Hsc70 function in MHV68 infection. We were unable to generate stable cell lines in which Hsc70 expression was knocked down, likely due to Hsc70's role in maintaining cellular homeostasis. To overcome this problem, we cotransfected 3T12 fibroblasts with WT MHV68 BAC and either an empty vector control or vectors encoding shRNAs specific to Hsc70. We previously used this method to evaluate the importance of mLANA in lytic replication (34) and roles for c-Jun amino-terminal kinases in MHV68 lytic replication (46). We performed immunoblot analyses and plaque assays at 72 and 96 h posttransfection to determine if Hsc70 knockdown reduced viral protein expression and viral titers, respectively. Transfection efficiency was similar for all samples as assessed by monitoring GFP expressed from the MHV68 BAC at 24 h posttransfection (data not shown). Densitometry analyses revealed 54%, 35%, and 41% reductions in Hsc70 protein levels relative to the empty vector control at 72 h posttransfection for cells transfected with Hsc70 shRNA1, shRNA2, or a combination of both (Fig. 6A), a trend that was maintained at the 96-h time point. Hsc70 knockdown correlated with a drastic reduction in viral protein signal compared to that observed for control shRNA-treated cells (Fig. 6A). The greatest deficit in viral protein detection was observed in cells cotransfected with the combination of two unique shRNAs, which functionally correlates with combined treatment yielding the greatest reduction in Hsc70 protein levels at 96 h posttransfection (Fig. 6A). Consistent with these findings, viral titers were either undetectable or severely reduced in supernatants from cells in which Hsc70 was knocked down (Fig. 6B). These results demonstrate that specific depletion of Hsc70 in MHV68-infected cells is detrimental to viral replication and strongly suggest that effects of VER-155008 treatment on MHV68 replication are due to Hsc70 inhibition. Furthermore, while these data do not provide a direct link between Hsc70 and mLANA in the MHV68 productive replication cycle, it is interesting to note that the results of this experiment phenocopy those obtained following transfection of LANA-null MHV68 (34).
FIG 6.

Hsc70 knockdown impairs viral protein expression and replication. 3T12 fibroblasts were transfected with MHV68 BAC DNA and either empty vector (control) or plasmids encoding shRNAs specific to Hsc70 (shRNA1 and -2) in the combinations shown. (A) Lysates were collected at 72 h and 96 h posttransfection and resolved by SDS-PAGE. Immunoblot analyses were performed to monitor Hsc70 and viral protein levels using antibodies directed against the indicated antigens. Relative Hsc70 expression percentages were determined by densitometry comparisons of vector control- and shRNA-treated cells and are indicated below the Hsc70 blot. (B) Supernatants were collected at the indicated times posttransfection, and viral titers were determined by plaque assay. Results are means for triplicate samples. Error bars represent standard deviations. N.D., no plaques detected.
Hsc70 function facilitates viral protein synthesis.
We next sought to determine if a particular stage of the viral replication cycle was affected by Hsc70 inhibition. We first performed quantitative PCR for representative viral IE (ORF50 and ORF73), E (ORF59), and L (ORF25) transcripts to evaluate the effects of Hsc70 inhibition on viral transcription over time (Fig. 7A). Overall, viral transcription was not greatly impacted by Hsc70 inhibition, as ORF50 and ORF73 transcription remained nearly equivalent in vehicle- and inhibitor-treated cells throughout the time course. This also suggests that treatment of cells with Hsc70 inhibitor probably does not impact viral entry, since IE gene transcription was similar in both inhibitor- and control-treated samples (Fig. 7A). Both ORF59 and ORF25 transcript levels exhibited slight reductions following inhibitor treatment at certain time points but were also largely unaffected. Thus, these results show that Hsc70 function minimally impacts the transcription of IE, E, and L genes throughout the lytic cycle.
FIG 7.

Hsc70 function is necessary for efficient viral protein production. 3T3 fibroblasts were treated with vehicle or VER-155008 (50 μM) for 1 h prior to and during infection with WT MHV68 (MOI = 5). (A) Total RNA was harvested at the indicated times postinfection. Levels of viral ORF50, ORF73, ORF59, and ORF25 and cellular GAPDH transcripts were determined by qRT-PCR. Relative quantities of viral transcripts are depicted as the ratio of the CT value for the GAPDH gene to the CT value of each viral gene for the indicated time point. Results represent the range of data from two independent experiments. Error bars represent standard deviations from means. Black bars, vehicle-treated samples; white bars, samples treated with VER-155008. (B) Cells were harvested at the indicated times postinfection, and proteins were resolved by SDS-PAGE. Immunoblot analyses were performed using antibodies specific for the indicated proteins. M, mock infected; I, infected.
We next performed comparative immunoblot analyses over time during infection to determine if VER-155008 treatment influenced the expression of viral proteins. We observed a reduction in levels of both mLANA and ORF57 at 4 and 8 h postinfection in cells treated with VER-155008 in comparison to untreated cells. However, expression of mLANA recovered to levels observed for untreated controls as infection progressed. ORF57 levels also increased as infection progressed, yet they did not recover fully to levels observed in untreated cells (Fig. 7B). Similarly, levels of early protein ORF59 were diminished in inhibitor-treated cells at 8, 12, and 18 h postinfection compared to untreated infected cells but recovered to nearly normal levels at 24 h postinfection (Fig. 7B). In contrast, detection of late viral structural antigens with MHV68 immune serum was reduced throughout the time course following VER-155008 treatment and was characterized by an apparent absence of certain unspecified viral proteins. Additionally, Hsc70 steady-state protein levels remained constant in all samples and at all time points, regardless of infection or drug treatment, indicating that Hsc70 protein expression is not induced during lytic viral replication. Together, these results indicate that inhibition of Hsc70 impacts MHV68 protein expression, especially late viral proteins detectable with MHV68 immune serum.
Hsc70 function is important for formation of viral replication complexes and influences viral genome replication.
Given that Hsc70 plays a role in virus-induced chaperone-enriched (VICE) domain formation during herpes simplex virus 1 (HSV-1) infection (67, 68) and that late gene expression occurs coincident to viral DNA replication (46), we were curious if Hsc70 inhibition would impact the formation of subnuclear structures associated with MHV68 productive replication. Thus, we infected 3T3 fibroblasts with 73.GFP MHV68 in the presence or absence of VER-155008 and quantified viral replication complexes (vRCs) marked by accumulation of viral DNA polymerase processivity factor ORF59 into subnuclear foci by immunofluorescence microscopy. For this experiment, detection of mLANA-GFP, which was not affected by Hsc70 inhibition in immunoblot analyses at late time points, served as a marker for identifying infected cells. Compared to vehicle-treated controls, we observed a significantly lower frequency of vRCs in inhibitor-treated cells at 18 h postinfection, with approximately 0.8% of cells containing vRCs following Hsc70 inhibition versus approximately 7% in control cells (Fig. 8A and B). These data are consistent with decreased ORF59 protein levels detected in immunoblot analyses at 18 h postinfection in VER-155008-treated cells (Fig. 7B).
FIG 8.

Hsc70 promotes the formation of viral replication complexes and viral DNA replication. (A) 3T3 fibroblasts were treated with vehicle (DMSO) or VER-155008 (50 μM) for 1 h prior to and during infection with 73.GFP MHV68 at an MOI of 5 PFU/cell. Cells were fixed at 18 h postinfection and stained with antibodies directed against GFP to detect mLANA-GFP or ORF59 as a marker for viral replication complexes. DNA was detected with DAPI. Images were analyzed by fluorescence microscopy at a magnification of ×60. White arrowheads indicate representative viral replication complexes. (B) Viral replication complexes from the experiment for panel A were quantified by counting the number of cells containing ORF59 foci and dividing by the total number of cells exhibiting GFP+ nuclei for a field of view at a magnification of ×20. Results represent mean percentages of ORF59+ GFP+ cells for 15 randomly selected fields of view for each condition. Error bars represent standard errors of the means. *, P = 0.0001. (C) 3T3 fibroblasts were infected with MHV68 at an MOI of 5 PFU/cell in the presence of vehicle, VER-155008 (50 μM), or PAA (200 μg/ml). Total DNA was harvested at 4 h and 18 h postinfection, and viral genomes were measured by quantitative PCR. Data represent the change in viral DNA copy number normalized to the cellular GAPDH gene between 4 h and 18 h postinfection using the ΔΔCT method. Results are means for biological duplicate samples analyzed in technical triplicate. Error bars represent standard deviations.
Based on the observation that Hsc70 inhibition curtailed vRC formation and late viral protein detection, we hypothesized that Hsc70 inhibition would also negatively impact viral DNA replication. Indeed, though the effect was not as potent as treatment with the viral DNA polymerase inhibitor PAA, treatment with VER-155008 resulted in more than a 10-fold reduction in viral DNA detected at late times during infection (Fig. 8C).These data therefore strongly suggest that Hsc70 function contributes to the formation of viral replication complexes, thereby enabling viral genome replication, the expression of late viral proteins, and ultimately viral replication.
The impact of Hsc70 inhibition is less pronounced for mLANA-null MHV68.
Having established that inhibition of Hsc70 impairs lytic replication of WT MHV68, we next wanted to define the impact of Hsc70 inhibition on mLANA-null MHV68 infection. We reasoned that if Hsc70 functions in viral replication were mediated through its interaction with mLANA, then mLANA-null virus, which is attenuated to begin with (28, 33), should be less affected by blocking Hsc70 function. To test this hypothesis, we infected 3T3 fibroblasts with either WT or 73.STOP MHV68 in the presence or absence of VER-155008 and quantified viral yields at 24 or 48 h postinfection. As expected, mLANA-null virus exhibited a general deficit in viral yield (∼6-fold) in comparison to WT MHV68 in vehicle-treated cells at both 24 and 48 h postinfection (Fig. 9). Treatment of cells with either 30 or 50 μM VER-155008 significantly decreased yields of WT MHV68 infection by 3.2- or 8.5-fold at 24 h, respectively (Fig. 9A). Hsc70 inhibition reduced yields of WT MHV68 by 4.0-fold (30 μM) and 8.7-fold (50 μM) compared to that in vehicle controls by 48 h postinfection (Fig. 9A). These findings agreed with previous experiments (Fig. 5), and in all cases the effect of Hsc70 inhibition on WT MHV68 replication was significant (Fig. 9A).
FIG 9.

The effect of Hsc70 inhibition on viral replication is less severe for mLANA-null MHV68. 3T3 fibroblasts were treated with vehicle (DMSO) or the indicated concentrations of Hsc70 inhibitor VER-155008 for 1 h prior to and during infection with either WT MHV68 (A) or 73.STOP virus (B) at an MOI of 5 PFU/cell. Cells were harvested at the indicated times postinfection, and viral titers were determined by plaque assay. Results are expressed as the mean viral yield of triplicate samples (viral titers at 24 or 48 h divided by mean viral titers for 0 h) for that particular time point. Error bars represent standard deviations. Statistical significance was determined by performing Student's t test, with a 95% confidence interval comparing the indicated treatments. *, P < 0.05; **, P < 0.01; n.s., not significant (P > 0.05).
In contrast, 73.STOP was less affected by VER-155008 treatment. At both the 24- and 48-h time points, 30 μM VER-155008 had essentially no effect on 73.STOP replication (1.2- and 1.6-fold reductions, respectively; Fig. 9B). Treatment with 50 μM VER-155008 did reduce 73.STOP yields, respectively, by 6.3- and 4.0-fold at 24 and 48 h postinfection; however, the decrease again was less than that observed for WT MHV68 and was not statistically significant for the 24-h time point (P > 0.05) (Fig. 9B). Similar results were obtained following Hsc70 inhibition (50 μM) at 96 h postinfection during low-MOI infections, where WT MHV68 yield was significantly reduced by 7.4-fold, while 73.STOP was reduced by only 4.5-fold, which was not significant (data not shown). It also is interesting to note that Hsc70 inhibition reduces yields of WT MHV68 to levels that are similar to those of mLANA-null virus without inhibitor treatment. Overall, the finding that mLANA-null MHV68 is less sensitive to inhibition of Hsc70 supports the hypothesis that Hsc70 functions in lytic replication are mediated at least in part through its interaction with mLANA. However, given that high concentrations of VER-155008 do impair 73.STOP replication, we speculate that Hsc70 also serves mLANA-independent functions in MHV68 lytic replication.
DISCUSSION
This study represents the first large-scale proteomics analysis to define protein-protein interactions for a LANA homolog during lytic infection. As a total protein set, our studies revealed that mLANA coprecipitates with diverse protein classes, including nucleotide-binding proteins, cytoskeletal components, chaperones, transcription factors, and others (see Table S1 and Fig. S1 in the supplemental material). This diversity may be explained in part by the multifunctional nature of LANA homologs and the promiscuity of their binding to different cellular factors; on the other hand, many of these interactions are also from common contaminants retrieved during affinity purification procedures (69). However, through I-DIRT analyses, we defined a small group of proteins, including Hsc70, splicing factors, and translation regulators, as high-priority candidates for downstream functional analyses.
Among the cellular proteins identified in our screen, we observed a number of factors previously identified and characterized in KSHV LANA interaction screens, suggesting that these interactions are conserved among LANA homologs (Table 1). This is not surprising, as mLANA and kLANA share considerable structural homology, especially in the C termini of the proteins (60, 61, 70). Some of these common interactors have biological roles in latent KSHV infection. For example, heat shock protein 90 (Hsp90) contributes to kLANA stability in latently infected cell lines (71). Also, kLANA interacts with annexin A2 and angiogenin to prevent apoptosis in TIVE-LTC cells (40). Although protein-protein interaction studies for kLANA have focused on elucidating interacting factors in latently infected cells or through peptide arrays utilizing various affinity purification techniques (39, 42), the overlap with mLANA interactors observed during lytic replication suggests that LANA homologs may exploit the functions of such factors in both lytic and latent stages of infection. For example, hnRNPK and other hnRNP proteins, Hsc70, dead-box helicases DHX3 and DHX9, and histone H1 identified in this study (Table 1; see Tables S1 and S2 in the supplemental material) associate with KSHV terminal repeats (TRs) in latently infected cells (22, 72). Thus, it is possible that mLANA and these host factors form complexes at the TRs of the MHV68 genome during lytic infection as well. Moreover, as spontaneous reactivation occurs in KSHV tumor cell lines and KS lesions are mixtures of latent and lytic infection (73), these and other cellular factors could represent ideal drug targets for therapeutic interventions. Finally, though I-DIRT analysis shows that only 4 of the common cellular interactors are classified as “specific” (Table 1), this does not necessarily call into question the relevance of previous studies that addressed potential roles for the remaining proteins in our screen, such as nucleophosmin and annexin A2 in KSHV infection (40, 41). Obviously there are differences in the two LANA proteins in question and in latency versus lytic replication, and, as mentioned above, I-DIRT uses biophysical principles of interaction specificity to help prioritize proteins for downstream analysis but does not necessarily “rule out” those proteins classified as nonspecific. It may be of interest to evaluate other proteins identified as both MHV68 and KSHV LANA-binding proteins in future studies.
In addition to cellular interactions, we also mapped and categorized a network of mLANA interactions with viral proteins, specifically validating mLANA's interaction with ORF52 by coimmunoprecipitation (Fig. 2). Previous studies on ORF52 have characterized it as a late gene product that localizes mostly in the cytoplasm and is necessary for efficient assembly of virions and viral egress (59, 74, 75). These studies have also described an interaction between ORF52 and another tegument protein, ORF42 (75), but did not report an interaction with mLANA. Though we did not attempt to define a biological role for mLANA-ORF52 interactions, it is interesting to note that the EBV homolog of ORF52, BRLF2, accumulates in the nucleus at early time points during EBV reactivation (76). Moreover, BRLF2 protein interaction networks include a cluster of RNA splicing factors, many of which overlap RNA-processing proteins present in mLANA interaction networks described in this study (76). Perhaps ORF52 tethers ORF57 to mLANA through RNA-processing proteins, which are also engaged by ORF57 (62, 76, 77). Our inability to validate other “specific” interactions such as with ORF57 and M3 is consistent with the notion that such proteins engage mLANA by tethering through other proteins or viral DNA present only within infected cells.
As a high-priority mLANA-binding host factor based on I-DIRT score, spectral abundance, and potential conservation with KSHV LANA, we further characterized the interaction between host chaperone Hsc70 and mLANA and provide evidence that Hsc70 function is necessary for MHV68 lytic replication. As a chaperone, Hsc70 maintains protein quality control by mediating the proper folding of nascent or denatured proteins and by preventing the formation of toxic aggregates, especially under cellular stress conditions (reviewed in references 78 and 79). Additionally, Hsc70 and other Hsp70 family members play important roles in infection by diverse viruses (67, 80–82; reviewed in reference 83). For example, Hsc70 is an important component of virus-induced chaperone-enriched (VICE) domains, which form in the nucleus during HSV-1 infection to control nuclear protein quality and prevent buildup of toxic protein aggregates during lytic viral replication (68).
Our data demonstrate that mLANA promotes the nuclear accumulation of Hsc70 during MHV68 infection (Fig. 4). Consistent with this observation, a number of DNA and RNA viruses encode factors that recruit host chaperones to specific cellular compartments to regulate different stages of viral replication and/or to maintain protein quality suitable for viral replication (reviewed in references 83 and 84). For example, Hsc70 is recruited to the nucleoplasm during HSV-1 infection through another multifunctional IE protein, ICP0 (67). While a precise mechanism of Hsc70 recruitment by mLANA is not yet clear, we speculate that mLANA-mediated recruitment of Hsc70 to the nucleus likewise functions to maintain nuclear protein quality control during MHV68 lytic infection in a manner analogous to Hsc70 function in HSV-1 infection (68). It also is possible that Hsc70 is recruited to the nucleus to regulate the proper assembly of nascent MHV68 virions at late time points of infection by mediating the scaffolding and oligomerization of structural proteins. Hsc70's role in mediating virion assembly is well documented for both RNA and DNA viruses. For example, Hsc70 facilitates intracellular virion assembly during hepatitis C virus infection (85) and regulates simian virus 40 (SV40) polyomavirus capsid assembly by binding to the capsid protein VP1 in the nucleus for virion assembly (86).
However, given the multifunctional nature of mLANA (and likely Hsc70), it may prove difficult to directly link mLANA-mediated nuclear accumulation of Hsc70 to driving efficient lytic replication, as other mLANA functions, particularly those involving its capacity to bind DNA, also play an important role in productive infection (15). Indeed, attempts to simply map domains involved in mLANA-Hsc70 interactions proved quite problematic, as mLANA mutagenesis led to drastic variations, both enhancement and reduction, in the efficiency of mLANA protein expression (E. Salinas and J. C. Forrest, unpublished data). Thus, attempts to map mLANA domains involved in nuclear recruitment were inconclusive, and it currently is not clear if mLANA-Hsc70 interactions control viral gene expression and limit infection-related cell death. Nonetheless, the finding that mLANA-null MHV68 is less affected by pharmacologic inhibition of Hsc70 strongly suggests that Hsc70 function during MHV68 lytic replication is at least partially dependent on its interaction with mLANA.
Given that the LANA-null MHV68 replication defect is most evident at low multiplicity, it also is interesting that cotransfection of MHV68 BAC with Hsc70 shRNAs led to reduced viral protein expression and viral replication, which is phenotypically similar to results of BAC transfection experiments with mLANA-null MHV68 (34). Similarly, pharmacologic inhibition of Hsc70 reduced WT MHV68 replication to levels similar to those of mLANA-null virus, which is an interesting correlation. While these findings support a role for mLANA engagement of Hsc70 in MHV68 lytic replication, the finding that high concentrations of VER-155008 reduce replication of mLANA-null virus, albeit less than WT MHV68, leads us to speculate that Hsc70 also performs mLANA-independent functions during the MHV68 lytic cycle. Since a portion of Hsc70 remains in the cytoplasm during WT MHV68 infection and mLANA is predominantly a nuclear protein, it seems likely that Hsc70 promotes efficient viral replication by mediating proper folding of nascent viral peptides or through scaffolding functions that are independent of mLANA. Precisely teasing out mLANA-dependent and -independent functions for Hsc70 may prove difficult.
An analogous case may exist for the HSV-1 protein ICP22, which is necessary both for efficient lytic replication in Vero and human embryonic lung (HEL) cells and for VICE domain formation (87, 88). HSV-1 mutant TF1.5 expresses N-terminally truncated ICP22 that is unable to promote VICE domain formation while not affecting RNA polymerase II (pol II) modification. TF1.5 exhibits a severe replication defect in HEL cells, which is similar to the case for ICP22-null virus, suggesting the importance of VICE domain formation to HSV-1 replication in these cells (87, 88). However, HSV-1 encoding ICP22 mutants such as TF22PS, which remain capable of binding Hsc70 and inducing VICE domain formation but fail to promote RNA pol II modification, also exhibit lytic replication defects in HEL cells (88). These findings demonstrate that the requirement for ICP22 in mediating efficient HSV-1 replication does not depend solely on ICP22's ability to interact with Hsc70 and/or induce VICE domain formation but depends on multiple functions of this protein. Thus, while our data do not provide direct evidence of a functional role for mLANA-Hsc70 interactions in MHV68 lytic replication, they may reveal a functional link for future experimentation.
Nonetheless, the data presented here provide the first demonstration that Hsc70 is important for MHV68 lytic replication (Fig. 4 and 5). The finding that Hsc70 regulates viral protein synthesis during MHV68 infection is consistent with chaperone functions in other viral systems. For example, treatment with the Hsp70 inhibitor quercetin reduces viral protein production during rabies virus infection (72). However, once synthesized, proteins remained stable when translation was inhibited, suggesting that Hsp70 is involved in promoting proper folding of nascent polypeptides (80). Likewise, decreased levels of viral proteins observed following Hsc70 inhibition during MHV68 infection may reflect a disruption in chaperone-mediated folding of newly synthesized viral proteins. Since vRC formation was impaired following VER-155008 treatment, it is reasonable to suggest that Hsc70 is important for the proper expression of viral proteins involved in vRC formation (Fig. 8). It also is likely that the disruption of vRCs caused by Hsc70 inhibition contributes to the decrease in viral DNA replication (Fig. 8C) and consequently a decrease in late viral protein production (Fig. 7). While the subversion of host chaperones by viruses to benefit their replication cycles is a common theme, only a few studies have addressed the role of chaperone proteins in GHV infection, and these focused on latency (71, 89). Thus, this study should provide a rationale for defining roles of chaperones in productive replication of other GHVs.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Forrest laboratory and of the Center for Microbial Pathogenesis and Host Inflammatory Responses for their insightful comments and suggestions. We also thank the UAMS Proteomics Facility for mass spectrometric support. We thank Laurie T. Krug for providing the pTAG2B-ORF75B plasmid. Finally, we thank Paul Ling and Scott Tibbetts for their generous supply of anti-ORF57 and anti-mLANA antibodies, respectively.
J.C.F. was supported by the NIH Center for Microbial Pathogenesis and Host Inflammatory Responses (P20GM103625), the National Cancer Institute (R01CA167065), and startup funds from the Arkansas Biosciences Institute and UAMS College of Medicine. A.J.T. acknowledges funding from National Institutes of Health grants R01GM106024, R33CA173264, P30GM103450, P20GM103429, and UL1TR000039.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02022-15.
REFERENCES
- 1.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2.Wen KW, Damania B. 2010. Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett 289:140–150. doi: 10.1016/j.canlet.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore PS. 2000. The emergence of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). N Engl J Med 343:1411–1413. doi: 10.1056/NEJM200011093431912. [DOI] [PubMed] [Google Scholar]
- 4.Cesarman E. 2011. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett 305:163–174. doi: 10.1016/j.canlet.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Virgin HW IV, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71:5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kedes DH, Lagunoff M, Renne R, Ganem D. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J Clin Invest 100:2606–2610. doi: 10.1172/JCI119804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- 8.Cotter MA II, Robertson ES. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254–264. doi: 10.1006/viro.1999.9999. [DOI] [PubMed] [Google Scholar]
- 9.Ballestas ME, Kaye KM. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J Virol 75:3250–3258. doi: 10.1128/JVI.75.7.3250-3258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verma SC, Robertson ES. 2003. ORF73 of herpesvirus Saimiri strain C488 tethers the viral genome to metaphase chromosomes and binds to cis-acting DNA sequences in the terminal repeats. J Virol 77:12494–12506. doi: 10.1128/JVI.77.23.12494-12506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Si H, Verma SC, Lampson MA, Cai Q, Robertson ES. 2008. Kaposi's sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J Virol 82:6734–6746. doi: 10.1128/JVI.00342-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Habison AC, Beauchemin C, Simas JP, Usherwood EJ, Kaye KM. 2012. Murine gammaherpesvirus 68 LANA acts on terminal repeat DNA to mediate episome persistence. J Virol 86:11863–11876. doi: 10.1128/JVI.01656-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu J, Yang Y, Turner PC, Jain V, McIntyre LM, Renne R. 2014. LANA binds to multiple active viral and cellular promoters and associates with the H3K4 methyltransferase hSET1 complex. PLoS Pathog 10:e1004240. doi: 10.1371/journal.ppat.1004240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mercier A, Arias C, Madrid AS, Holdorf MM, Ganem D. 2014. Site-specific association with host and viral chromatin by Kaposi's sarcoma-associated herpesvirus LANA and its reversal during lytic reactivation. J Virol 88:6762–6777. doi: 10.1128/JVI.00268-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paden CR, Forrest JC, Tibbetts SA, Speck SH. 2012. Unbiased mutagenesis of MHV68 LANA reveals a DNA-binding domain required for LANA function in vitro and in vivo. PLoS Pathog 8:e1002906. doi: 10.1371/journal.ppat.1002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ottinger M, Pliquet D, Christalla T, Frank R, Stewart JP, Schulz TF. 2009. The interaction of the gammaherpesvirus 68 orf73 protein with cellular BET proteins affects the activation of cell cycle promoters. J Virol 83:4423–4434. doi: 10.1128/JVI.02274-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garber AC, Hu J, Renne R. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J Biol Chem 277:27401–27411. doi: 10.1074/jbc.M203489200. [DOI] [PubMed] [Google Scholar]
- 18.Viejo-Borbolla A, Ottinger M, Bruning E, Burger A, Konig R, Kati E, Sheldon JA, Schulz TF. 2005. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J Virol 79:13618–13629. doi: 10.1128/JVI.79.21.13618-13629.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu J, Garber AC, Renne R. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J Virol 76:11677–11687. doi: 10.1128/JVI.76.22.11677-11687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garber AC, Shu MA, Hu J, Renne R. 2001. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Virol 75:7882–7892. doi: 10.1128/JVI.75.17.7882-7892.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renne R, Barry C, Dittmer D, Compitello N, Brown PO, Ganem D. 2001. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Virol 75:458–468. doi: 10.1128/JVI.75.1.458-468.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Si H, Verma SC, Robertson ES. 2006. Proteomic analysis of the Kaposi's sarcoma-associated herpesvirus terminal repeat element binding proteins. J Virol 80:9017–9030. doi: 10.1128/JVI.00297-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borah S, Verma SC, Robertson ES. 2004. ORF73 of herpesvirus saimiri, a viral homolog of Kaposi's sarcoma-associated herpesvirus, modulates the two cellular tumor suppressor proteins p53 and pRb. J Virol 78:10336–10347. doi: 10.1128/JVI.78.19.10336-10347.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujimuro M, Hayward SD. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J Virol 77:8019–8030. doi: 10.1128/JVI.77.14.8019-8030.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friborg J Jr, Kong W, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894. [DOI] [PubMed] [Google Scholar]
- 26.Cheng BY, Zhi J, Santana A, Khan S, Salinas E, Forrest JC, Zheng Y, Jaggi S, Leatherwood J, Krug LT. 2012. Tiled microarray identification of novel viral transcript structures and distinct transcriptional profiles during two modes of productive murine gammaherpesvirus 68 infection. J Virol 86:4340–4357. doi: 10.1128/JVI.05892-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rochford R, Lutzke ML, Alfinito RS, Clavo A, Cardin RD. 2001. Kinetics of murine gammaherpesvirus 68 gene expression following infection of murine cells in culture and in mice. J Virol 75:4955–4963. doi: 10.1128/JVI.75.11.4955-4963.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forrest JC, Paden CR, Allen RD III, Collins J, Speck SH. 2007. ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication. J Virol 81:11957–11971. doi: 10.1128/JVI.00111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krug LT, Pozharskaya VP, Yu Y, Inoue N, Offermann MK. 2004. Inhibition of infection and replication of human herpesvirus 8 in microvascular endothelial cells by alpha interferon and phosphonoformic acid. J Virol 78:8359–8371. doi: 10.1128/JVI.78.15.8359-8371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lan K, Kuppers DA, Verma SC, Sharma N, Murakami M, Robertson ES. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J Virol 79:7453–7465. doi: 10.1128/JVI.79.12.7453-7465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ebrahimi B, Dutia BM, Roberts KL, Garcia-Ramirez JJ, Dickinson P, Stewart JP, Ghazal P, Roy DJ, Nash AA. 2003. Transcriptome profile of murine gammaherpesvirus-68 lytic infection. J Gen Virol 84:99–109. doi: 10.1099/vir.0.18639-0. [DOI] [PubMed] [Google Scholar]
- 32.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol 73:2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moorman NJ, Willer DO, Speck SH. 2003. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J Virol 77:10295–10303. doi: 10.1128/JVI.77.19.10295-10303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paden CR, Forrest JC, Moorman NJ, Speck SH. 2010. Murine gammaherpesvirus 68 LANA is essential for virus reactivation from splenocytes but not long-term carriage of viral genome. J Virol 84:7214–7224. doi: 10.1128/JVI.00133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Q, Zhou F, Ye F, Gao SJ. 2008. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 379:234–244. doi: 10.1016/j.virol.2008.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corte-Real S, Collins C, Aires da Silva F, Simas JP, CFBarbas 3rd, Chang Y, Moore P, Goncalves J. 2005. Intrabodies targeting the Kaposi sarcoma-associated herpesvirus latency antigen inhibit viral persistence in lymphoma cells. Blood 106:3797–3802. doi: 10.1182/blood-2005-04-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C. 2005. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 105:2510–2518. doi: 10.1182/blood-2004-08-3052. [DOI] [PubMed] [Google Scholar]
- 38.Sun R, Liang D, Gao Y, Lan K. 2014. Kaposi's sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J Virol 88:7331–7344. doi: 10.1128/JVI.00596-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen W, Dittmer DP. 2011. Ribosomal protein S6 interacts with the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Virol 85:9495–9505. doi: 10.1128/JVI.02620-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paudel N, Sadagopan S, Balasubramanian S, Chandran B. 2012. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen and angiogenin interact with common host proteins, including annexin A2, which is essential for survival of latently infected cells. J Virol 86:1589–1607. doi: 10.1128/JVI.05754-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaul R, Verma SC, Robertson ES. 2007. Protein complexes associated with the Kaposi's sarcoma-associated herpesvirus-encoded LANA. Virology 364:317–329. doi: 10.1016/j.virol.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shamay M, Liu J, Li R, Liao G, Shen L, Greenway M, Hu S, Zhu J, Xie Z, Ambinder RF, Qian J, Zhu H, Hayward SD. 2012. A protein array screen for Kaposi's sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J Virol 86:5179–5191. doi: 10.1128/JVI.00169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J Virol 76:11596–11604. doi: 10.1128/JVI.76.22.11596-11604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adler H, Messerle M, Wagner M, Koszinowski UH. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol 74:6964–6974. doi: 10.1128/JVI.74.15.6964-6974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smart SK, Mackintosh SG, Edmondson RD, Taverna SD, Tackett AJ. 2009. Mapping the local protein interactome of the NuA3 histone acetyltransferase. Protein Sci 18:1987–1997. doi: 10.1002/pro.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stahl JA, Paden CR, Chavan SS, MacLeod V, Edmondson RD, Speck SH, Forrest JC. 2012. Amplification of JNK signaling is necessary to complete the murine gammaherpesvirus 68 lytic replication cycle. J Virol 86:13253–13262. doi: 10.1128/JVI.01432-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ling PD, Tan J, Sewatanon J, Peng R. 2008. Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J Virol 82:8000–8012. doi: 10.1128/JVI.02752-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, Macias AT, Daniels Z, Geoffroy S, Dopson M, Lavan P, Matassova N, Francis GL, Graham CJ, Parsons R, Wang Y, Padfield A, Comer M, Drysdale MJ, Wood M. 2010. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother Pharmacol 66:535–545. doi: 10.1007/s00280-009-1194-3. [DOI] [PubMed] [Google Scholar]
- 49.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 50.Stahl JA, Chavan SS, Sifford JM, Macleod V, Voth DE, Edmondson RD, Forrest JC. 2013. Phosphoproteomic analyses reveal signaling pathways that facilitate lytic gammaherpesvirus replication. PLoS Pathog 9:e1003583. doi: 10.1371/journal.ppat.1003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. 2011. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee S, Salwinski L, Zhang C, Chu D, Sampankanpanich C, Reyes NA, Vangeloff A, Xing F, Li X, Wu TT, Sahasrabudhe S, Deng H, Lacount DJ, Sun R. 2011. An integrated approach to elucidate the intra-viral and viral-cellular protein interaction networks of a gamma-herpesvirus. PLoS Pathog 7:e1002297. doi: 10.1371/journal.ppat.1002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Byrum S, Smart SK, Larson S, Tackett AJ. 2012. Analysis of stable and transient protein-protein interactions. Methods Mol Biol 833:143–152. doi: 10.1007/978-1-61779-477-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tackett AJ, DeGrasse JA, Sekedat MD, Oeffinger M, Rout MP, Chait BT. 2005. I-DIRT, a general method for distinguishing between specific and nonspecific protein interactions. J Proteome Res 4:1752–1756. doi: 10.1021/pr050225e. [DOI] [PubMed] [Google Scholar]
- 57.Ong SE, Mann M. 2006. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat Protoc 1:2650–2660. [DOI] [PubMed] [Google Scholar]
- 58.van Berkel V, Barrett J, Tiffany HL, Fremont DH, Murphy PM, McFadden G, Speck SH, Virgin HI. 2000. Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J Virol 74:6741–6747. doi: 10.1128/JVI.74.15.6741-6747.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bortz E, Wang L, Jia Q, Wu TT, Whitelegge JP, Deng H, Zhou ZH, Sun R. 2007. Murine gammaherpesvirus 68 ORF52 encodes a tegument protein required for virion morphogenesis in the cytoplasm. J Virol 81:10137–10150. doi: 10.1128/JVI.01233-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Correia B, Cerqueira SA, Beauchemin C, Pires de Miranda M, Li S, Ponnusamy R, Rodrigues L, Schneider TR, Carrondo MA, Kaye KM, Simas JP, McVey CE. 2013. Crystal structure of the gamma-2 herpesvirus LANA DNA binding domain identifies charged surface residues which impact viral latency. PLoS Pathog 9:e1003673. doi: 10.1371/journal.ppat.1003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hellert J, Weidner-Glunde M, Krausze J, Richter U, Adler H, Fedorov R, Pietrek M, Ruckert J, Ritter C, Schulz TF, Luhrs T. 2013. A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog 9:e1003640. doi: 10.1371/journal.ppat.1003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Majerciak V, Yamanegi K, Allemand E, Kruhlak M, Krainer AR, Zheng ZM. 2008. Kaposi's sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J Virol 82:2792–2801. doi: 10.1128/JVI.01856-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jackson BR, Noerenberg M, Whitehouse A. 2012. The Kaposi's sarcoma-associated herpesvirus ORF57 protein and its multiple roles in mRNA biogenesis. Front Microbiol 3:59. doi: 10.3389/fmicb.2012.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 65.Mayer MP, Bukau B. 2005. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Milarski KL, Morimoto RI. 1989. Mutational analysis of the human HSP70 protein: distinct domains for nucleolar localization and adenosine triphosphate binding. J Cell Biol 109:1947–1962. doi: 10.1083/jcb.109.5.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burch AD, Weller SK. 2004. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J Virol 78:7175–7185. doi: 10.1128/JVI.78.13.7175-7185.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Livingston CM, Ifrim MF, Cowan AE, Weller SK. 2009. Virus-induced chaperone-enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog 5:e1000619. doi: 10.1371/journal.ppat.1000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen GI, Gingras AC. 2007. Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods 42:298–305. doi: 10.1016/j.ymeth.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 70.Domsic JF, Chen HS, Lu F, Marmorstein R, Lieberman PM. 2013. Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog 9:e1003672. doi: 10.1371/journal.ppat.1003672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen W, Sin SH, Wen KW, Damania B, Dittmer DP. 2012. Hsp90 inhibitors are efficacious against Kaposi sarcoma by enhancing the degradation of the essential viral gene LANA, of the viral co-receptor EphA2 as well as other client proteins. PLoS Pathog 8:e1003048. doi: 10.1371/journal.ppat.1003048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu J, Liu E, Renne R. 2009. Involvement of SSRP1 in latent replication of Kaposi's sarcoma-associated herpesvirus. J Virol 83:11051–11063. doi: 10.1128/JVI.00907-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, Chang Y. 2000. Differential viral protein expression in Kaposi's sarcoma-associated herpesvirus-infected diseases: Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. Am J Pathol 156:743–749. doi: 10.1016/S0002-9440(10)64940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benach J, Wang L, Chen Y, Ho CK, Lee S, Seetharaman J, Xiao R, Acton TB, Montelione GT, Deng H, Sun R, Tong L. 2007. Structural and functional studies of the abundant tegument protein ORF52 from murine gammaherpesvirus 68. J Biol Chem 282:31534–31541. doi: 10.1074/jbc.M705637200. [DOI] [PubMed] [Google Scholar]
- 75.Wang L, Guo H, Reyes N, Lee S, Bortz E, Guo F, Sun R, Tong L, Deng H. 2012. Distinct domains in ORF52 tegument protein mediate essential functions in murine gammaherpesvirus 68 virion tegumentation and secondary envelopment. J Virol 86:1348–1357. doi: 10.1128/JVI.05497-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Duarte M, Wang L, Calderwood MA, Adelmant G, Ohashi M, Roecklein-Canfield J, Marto JA, Hill DE, Deng H, Johannsen E. 2013. An RS motif within the Epstein-Barr virus BLRF2 tegument protein is phosphorylated by SRPK2 and is important for viral replication. PLoS One 8:e53512. doi: 10.1371/journal.pone.0053512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Majerciak V, Lu M, Li X, Zheng ZM. 2014. Attenuation of the suppressive activity of cellular splicing factor SRSF3 by Kaposi sarcoma-associated herpesvirus ORF57 protein is required for RNA splicing. RNA 20:1747–1758. doi: 10.1261/rna.045500.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Frydman J, Hartl FU. 1996. Principles of chaperone-assisted protein folding: differences between in vitro and in vivo mechanisms. Science 272:1497–1502. doi: 10.1126/science.272.5267.1497. [DOI] [PubMed] [Google Scholar]
- 79.Beckmann RP, Mizzen LE, Welch WJ. 1990. Interaction of Hsp 70 with newly synthesized proteins: implications for protein folding and assembly. Science 248:850–854. doi: 10.1126/science.2188360. [DOI] [PubMed] [Google Scholar]
- 80.Lahaye X, Vidy A, Fouquet B, Blondel D. 2012. Hsp70 protein positively regulates rabies virus infection. J Virol 86:4743–4751. doi: 10.1128/JVI.06501-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bian Z, Xiao A, Cao M, Liu M, Liu S, Jiao Y, Yan W, Qi Z, Zheng Z. 2012. Anti-HBV efficacy of combined siRNAs targeting viral gene and heat shock cognate 70. Virol J 9:275. doi: 10.1186/1743-422X-9-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaufer S, Coffey CM, Parker JS. 2012. The cellular chaperone hsc70 is specifically recruited to reovirus viral factories independently of its chaperone function. J Virol 86:1079–1089. doi: 10.1128/JVI.02662-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sullivan CS, Pipas JM. 2001. The virus-chaperone connection. Virology 287:1–8. doi: 10.1006/viro.2001.1038. [DOI] [PubMed] [Google Scholar]
- 84.Weller SK. 2010. Herpes simplex virus reorganizes the cellular DNA repair and protein quality control machinery. PLoS Pathog 6:e1001105. doi: 10.1371/journal.ppat.1001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khachatoorian R, Ganapathy E, Ahmadieh Y, Wheatley N, Sundberg C, Jung CL, Arumugaswami V, Raychaudhuri S, Dasgupta A, French SW. 2014. The NS5A-binding heat shock proteins HSC70 and HSP70 play distinct roles in the hepatitis C viral life cycle. Virology 454-455:118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chromy LR, Pipas JM, Garcea RL. 2003. Chaperone-mediated in vitro assembly of polyomavirus capsids. Proc Natl Acad Sci U S A 100:10477–10482. doi: 10.1073/pnas.1832245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bastian TW, Livingston CM, Weller SK, Rice SA. 2010. Herpes simplex virus type 1 immediate-early protein ICP22 is required for VICE domain formation during productive viral infection. J Virol 84:2384–2394. doi: 10.1128/JVI.01686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bastian TW, Rice SA. 2009. Identification of sequences in herpes simplex virus type 1 ICP22 that influence RNA polymerase II modification and viral late gene expression. J Virol 83:128–139. doi: 10.1128/JVI.01954-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nayar U, Lu P, Goldstein RL, Vider J, Ballon G, Rodina A, Taldone T, Erdjument-Bromage H, Chomet M, Blasberg R, Melnick A, Cerchietti L, Chiosis G, Wang YL, Cesarman E. 2013. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood 122:2837–2847. doi: 10.1182/blood-2013-01-479972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.