

ABSTRACT
Human cytomegalovirus (HCMV) is the major viral cause of birth defects and a serious problem in immunocompromised individuals and has been associated with atherosclerosis. Previous studies have shown that the induction of autophagy can inhibit the replication of several different types of DNA and RNA viruses. The goal of the work presented here was to determine whether constitutive activation of autophagy would also block replication of HCMV. Most prior studies have used agents that induce autophagy via inhibition of the mTOR pathway. However, since HCMV infection alters the sensitivity of mTOR kinase-containing complexes to inhibitors, we sought an alternative method of inducing autophagy. We chose to use trehalose, a nontoxic naturally occurring disaccharide that is found in plants, insects, microorganisms, and invertebrates but not in mammals and that induces autophagy by an mTOR-independent mechanism. Given the many different cell targets of HCMV, we proceeded to determine whether trehalose would inhibit HCMV infection in human fibroblasts, aortic artery endothelial cells, and neural cells derived from human embryonic stem cells. We found that in all of these cell types, trehalose induces autophagy and inhibits HCMV gene expression and production of cell-free virus. Treatment of HCMV-infected neural cells with trehalose also inhibited production of cell-associated virus and partially blocked the reduction in neurite growth and cytomegaly. These results suggest that activation of autophagy by the natural sugar trehalose or other safe mTOR-independent agents might provide a novel therapeutic approach for treating HCMV disease.
IMPORTANCE HCMV infects multiple cell types in vivo, establishes lifelong persistence in the host, and can cause serious health problems for fetuses and immunocompromised individuals. HCMV, like all other persistent pathogens, has to finely tune its interplay with the host cellular machinery to replicate efficiently and evade detection by the immune system. In this study, we investigated whether modulation of autophagy, a host pathway necessary for the recycling of nutrients and removal of protein aggregates, misfolded proteins, and pathogens, could be used to target HCMV. We found that autophagy could be significantly increased by treatment with the nontoxic, natural disaccharide trehalose. Importantly, trehalose had a profound inhibitory effect on viral gene expression and strongly impaired viral spread. These data constitute a proof-of-concept for the use of natural products targeting host pathways rather than the virus itself, thus reducing the risk of the development of resistance to treatment.
INTRODUCTION
Human cytomegalovirus (HCMV) infects multiple cell types, including endothelial, smooth muscle, epithelial, and fibroblast cells, monocytes/macrophages, bone marrow progenitor cells, and cells of the neural lineage, and establishes lifelong persistence in the host (for a review, see reference 1). Seroprevalence is high in the general population, and in most healthy individuals, the infection is asymptomatic. However, it is the major viral cause of birth defects and poses a serious problem for immunocompromised individuals. Between 0.5% to 2.5% of newborns are infected, and most infants who are symptomatic at birth (∼5%) have permanent sequelae (sensorineural hearing loss, microcephaly, chorioretinitis, and motor disabilities). A significant number of asymptomatic infants (∼15%) also later develop disabilities, most frequently hearing loss. The infection of the fetus can result from the primary infection of a seronegative woman as well as from recurrent or reinfection of a seropositive woman. HCMV has also been implicated in many epidemiological, clinical, animal, and in vitro studies as playing a role in vascular diseases and atherosclerosis (2–8). In addition, there is evidence for association of HCMV with cancer, particularly glioblastoma (for a review, see reference 9). This large spectrum of clinical problems associated with HCMV has made it imperative to develop strategies for prevention and treatment. The classical approach is to derive a drug that will target an essential viral protein, but this invariably leads to selection for drug-resistant viral mutants. More recently, consideration has been given to finding a cellular pathway that can be harnessed to inhibit the virus without damaging the host cell.
One cellular process that has received increasing attention in recent years is autophagy. There are three main types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy (which will be referred to as autophagy here) is a highly conserved intracellular degradation pathway whereby double-membrane vesicles engulf regions of the cytoplasm (including organelles, protein aggregates, misfolded proteins, and pathogens) and deliver the cytoplasmic contents to the lysosome for digestion and recycling of resulting metabolites, such as amino acids and fatty acids, that can be used to produce ATP for cell survival (10). This pathway is distinct from the ubiquitin-proteasome degradation pathway and is essential for maintaining cell viability during periods of stress or nutrient deprivation. There are over 30 autophagy-related genes (ATG), with beclin-1 playing a key role. The process begins with the formation of isolated membranes in the cytoplasm called phagophores. These membranes elongate and surround the cytoplasmic cargo to form the double-membrane vesicles referred to as autophagosomes. During this stage, the microtubule-associated protein 1 light chain 3 (LC3-I) is covalently linked to phosphatidylethanolamine (PE) and is incorporated into autophagosome membranes. This lipidation process converts cytosolic LC3-I into the active, autophagosome membrane-bound form, LC3-II. Autophagosomes move bidirectionally along the microtubules in a dynein-dynactin motor complex-dependent manner with preferential movement toward the microtubule organizing center where there is a perinuclear concentration of lysosomes (11). Ultimately, the autophagosomes fuse with the acidic lysosomes to form the autolysosome.
There are several macroautophagy-inducing agents (for a review, see reference 12). Some drugs, such as rapamycin, inhibit the mTOR (mammalian target of rapamycin) pathway, which negatively regulates autophagy. There are also several agents that activate autophagy by mTOR-independent pathways. One potent inducer of autophagy is trehalose. Trehalose is a nontoxic, disaccharide found in plants, insects, microorganisms, and invertebrates but not in mammals. In organisms that produce trehalose, it functions to stabilize proteins and membranes and to protect proteins from unfolding, aggregation, and degradation induced by stressful conditions. In Japan, trehalose is added to many food products, and in the United States it has been used as a stabilizer in several therapeutic monoclonal antibody products. Trehalose can inhibit polyglutamine aggregation in vitro (13), and the protective function of trehalose is also manifest when trehalose is introduced exogenously into mammalian cells. Treatment of cells with trehalose can induce the clearance of mutant huntingtin and alpha-synuclein (14) and is able to decrease the level of toxic aggregates and reduce morbidity and mortality in animal models of Huntington disease, Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS) (13, 15–18). It is known that neural tube defects are greatly increased in the offspring of women with pregestational diabetes. In a mouse model of maternal diabetes that leads to autophagy impairment in neuroepithelial cells and neural tube defects in the embryo, administration of trehalose in the drinking water prevents these neural tube defects (19, 20). Although most studies have examined trehalose in the context of inducing macroautophagy, there is some evidence that it can also induce CMA and counter the effects of the proteasome inhibitor epoxomicin, including elevated reactive oxygen species (ROS), cleaved caspase 3, increased levels of ubiquitinated proteins, and mitochondrial dysfunction and altered morphology (21, 22). Importantly, trehalose is nontoxic. Daily administration of trehalose intragastrically at a level as high as 5 g per kg of body weight had no effect in mice as measured by body weight, food consumption, sperm abnormality test, bone marrow cell micronucleus assay, organ coefficients, or hematological parameters (23).
In the last few years, there have been multiple studies on the role of autophagy in augmenting or inhibiting replication of viruses (for a review, see references 24 and 25). An example of a specific antiviral action is virophagy, whereby activation of autophagy leads to the sequestering of cytoplasmic virions or subviral components in autophagosomes and their subsequent degradation following fusion of the autophagosomes with lysosomes. Additionally, the transfer of viral nucleic acids via autophagy to endosomal compartments where they engage specific Toll-like receptors (TLRs) can activate innate defense responses. Autophagic vesicles can also deliver viral antigens to major histocompatibility complex (MHC) class I and class II complexes for antigen presentation and immune activation. In some cases where autophagy plays an antiviral role, viruses have developed ways to avoid its deleterious effects. For example, viruses can subvert the signaling pathways that activate or inhibit autophagy, the most common of which involves the negative regulator mTOR. Other viruses actually co-opt specific autophagy proteins or steps of the autophagy pathway to promote their own replication. For some viruses, particularly RNA viruses, it is suggested that the membranes derived from the formation of the phagophore are required for assembly of the viral replication complexes, but for the most part the mechanisms by which autophagy facilitates viral replication remain to be elucidated.
Given both the proviral and antiviral roles of autophagy, it is not surprising that during some viral infections there are both activation and inhibition of autophagy, albeit at different times in the viral life cycle. There also appears to be cell type specificity in these effects. Induction of autophagy by a virus does not necessarily require viral gene expression, and the simple interaction of the virus with the host cell membrane may be sufficient. In fact, it appears that activation of autophagy occurs very early during HCMV infection of fibroblasts and does not require de novo protein synthesis since UV-irradiated virus induces autophagy (26). This induction appears to be transient, and at later times in the infection it has been reported that autophagy may be downregulated through the interaction of the viral gene product TRS1 with beclin-1 (27, 28).
In this report, we have examined whether activation of autophagy by trehalose will inhibit HCMV infection. We chose to use trehalose because it acts independently of the mTOR pathway, and HCMV infection is able to alter the sensitivity of mTOR complexes to inhibitors. To date, the interplay of autophagy and HCMV infection has been examined only in fibroblasts. Recognizing that this is only one of the cell types infected in vivo, we have extended these studies to other biologically relevant cells, including human aortic endothelial cells (HAECs) and neural cells derived via directed differentiation of embryonic stem cells. We find that trehalose induces formation of autophagosomes and autophagic flux in all of these cell types. Moreover, in the presence of trehalose, there is significant inhibition of HCMV gene expression and virus production. In HCMV-infected neurons, there is a significant defect in neurite growth, and treatment with trehalose partially blocks this decrease in neurite length and reduces the extent of HCMV-induced cytomegaly. These results suggest that activation of autophagy might provide a novel therapeutic approach to prevent HCMV infection and pathogenesis.
MATERIALS AND METHODS
Cell culture.
Human foreskin fibroblasts (HFFs) were obtained from the American Type Culture Collection and maintained as previously described (29). Human aortic endothelial cells (HAECs) were maintained in culture in T175 flasks as previously described by DuRose et al. (29). H9-derived human neural stem cells (NSCs) were cultured as described by Li et al. (30) and Belzile et al. (31). Approval from the institutional review board (IRB) was obtained for the use of H9 embryonic stem cells. For differentiation to midbrain dopaminergic (mDA) neurons, H9 NSCs were first patterned for 10 days in Geltrex (Invitrogen)-coated flasks in neuronal differentiation medium (Dulbecco's modified Eagle's medium [DMEM]/F12 medium with N2, B27 supplement without vitamin A, minimal essential medium [MEM] nonessential amino acids, 0.2 mM vitamin C, 300 ng/ml cyclic AMP [cAMP], 100 units/ml penicillin, and 100 μg/ml streptomycin) supplemented with 100 ng/ml Sonic hedgehog (R&D Systems) and 100 ng/ml FGF8b (Invitrogen). Patterned cells were then harvested with Accutase (Invitrogen) and reseeded in dishes coated with poly-l-ornithine and Geltrex and then differentiated in neuronal differentiation medium supplemented with 10 ng/ml brain-derived neurotrophic factor (BDNF), 10 ng/ml glial cell-derived neurotrophic factor (GDNF), 1 ng/ml transforming growth factor β3 (TGF-β3), 10 ng/ml IGF1, and 0.5 mM dibutyryl-cAMP (db-cAMP) as previously described (31). Medium was replaced every other day. For most experiments, neurons were differentiated for 8 days, and the resulting neuronal cultures are thereafter referred to as day 8 mDA neurons; they represent a heterogeneous developing population of young neurons as well as neural stem cells. For neurite outgrowth experiments, mDA neurons were differentiated for 21 days.
Culture medium for each cell type was supplemented with trehalose (Sigma-Aldrich) at the concentration indicated in the figure legends, and medium was filtered. Control medium was filtered in the same manner. The lysosomal degradation inhibitor bafilomycin A1 (Enzo Life Science) was prepared in dimethyl sulfoxide (DMSO) and added to the medium at a concentration of 100 nM at 2 h prior to cell harvest to measure autophagic flux. DMSO was added to the medium of nontreated cells as a negative control.
Viral production, titration, and infection.
The HCMV clinical strain TB40E was maintained in HAECs as previously described (29). Infectious supernatant from HAECs was passaged once in HFFs to obtain higher titers. After centrifugation at 1,000 × g to remove cell debris, virions were pelleted by ultracentrifugation through a sorbitol cushion as described previously (31). Titers of concentrated virus stocks were determined by plaque assay in HFFs. Multiplicities of infection (MOIs) were calculated from the PFU counts in HFFs. An equivalent volume of concentrated supernatant from uninfected cells was used in control mock infections.
HFFs, H9 mDA neurons, and HAECs were infected with HCMV in the absence or presence of trehalose at the MOI indicated in the figure legends or mock infected with concentrated tissue culture supernatants. Unless noted otherwise, H9 day 8 mDA neurons were infected directly after differentiation without replating the cells. HAECs were infected 24 h after replating whereas contact-synchronized HFFs were infected concurrently with replating. H9 mDA neurons and HAECs were extensively washed with phosphate-buffered saline (PBS) at 6 h postinfection (hpi) and covered with fresh growth medium. The HFFs were extensively washed with PBS at 16 hpi to allow time for the cells to adhere, and fresh growth medium was added to the cells. In all experiments, culture medium with or without trehalose was replaced every 48 h.
Analysis of cell-associated and released virus.
Cell-associated virus was collected from cells infected in the presence or absence of trehalose at various times postinfection (p.i.). Cells were washed with PBS, trypsinized, quenched with medium, and centrifuged at 500 × g. The pellets were washed twice more before storage at −80°C. Pellets were later resuspended in ice-cold PBS and sonicated for 3 to 4 min on ice using a Misonix Sonicator 3000 with a cup horn attachment as previously described (29). Trypan blue was used to assess cell lysis. For released virus, medium was collected from each sample noted above and centrifuged at 500 × g to pellet cell debris. Cell-free supernatant was stored, and titers were determined as previously described (29).
Viral spread assays.
HFFs and H9-derived day 8 neurons were infected with TB40E at an MOI of 0.025 (in the presence or absence of 100 mM trehalose) on coverslips. In both cases, the viral inoculum was removed at 24 hpi, and medium was changed every other day thereafter. Cells were fixed at various times p.i. with paraformaldehyde, and viral spread was analyzed by immunofluorescence. A monoclonal antibody that detects both immediate early (IE) proteins IE1-72 (UL123) and IE2-86 (UL122) (CH160; Virusys) and an antibody to UL57 (CH167; Virusys) were used. Nuclei were stained with Hoechst 33342. Pictures were acquired using a Nikon epifluorescence microscope equipped with a charge-coupled-device (CCD) camera as described previously (31).
Western blotting.
Cells were collected, washed twice with PBS, and resuspended in radioimmunoprecipitation assay (RIPA) lysis and extraction buffer (Pierce, Rockford, IL) supplemented with 1% SDS and 1× Halt protease and phosphatase inhibitor cocktail (Pierce, Rockford, IL). Lysates were sonicated to shear genomic DNA, and the protein was quantified by bicinchoninic acid (BCA) protein assay reagent (Thermo Scientific, Rockford, IL). Lysates were run on 8, 10, or 13% polyacrylamide SDS gels, transferred to nitrocellulose membranes, and probed with the following mouse monoclonal antibodies in 5% dry milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T): anti-UL57 (CH167), anti-IE1/IE2 (CH160), anti-UL44, and anti-UL99 (all from Virusys), as well as anti-IE2 (Chemicon). A mouse anti-α-tubulin antibody was used to control for loading (Sigma-Aldrich). For LC3B, lysates were transferred to polyvinylidene difluoride (PVDF) membranes as described previously (32) and probed with an antibody specific for LC3B (Cell Signaling Technology) in 5% bovine serum albumin (BSA) in TBS-T. The LC3B antibody recognizes both native (LC3B-I) and lipid-associated (LC3B-II) forms of LC3B.
Detection of LC3B puncta by immunofluorescence.
HFFs were infected with TB40E at an MOI of 0.5 (in the presence or absence of 100 mM trehalose) upon seeding on coverslips as described above. H9-derived day 8 neuronal cultures differentiated on coverslips were infected with TB40E at an MOI of 0.5 in the presence or absence of 100 mM trehalose. At the time points p.i. indicated on the figures, cells were first fixed with formaldehyde and then permeabilized with saponin for 15 min at room temperature (Perm/Wash; BD Biosciences). Cells were stained using mouse monoclonal antibodies against LC3B and IE1 (obtained from Bill Britt at the University of Alabama) in Perm/Wash. Nuclei were stained with Hoechst 33342. Pictures of at least six independent fields were acquired using a Leica SP5 confocal microscopy system as described previously (31).
LC3B-GFP-RFP flux analysis in fixed samples.
HFFs were seeded to 70% confluence on coverslips and transduced 24 h later with a baculovirus expressing LC3B fused to the acid-sensitive green fluorescent protein (emerald GFP) and acid-insensitive red fluorescent protein (TagRFP) (Premo Autophagy Tandem Sensor RFP-GFP LC3B kit; Life Technologies). H9 day 8 neurons differentiated on coverslips were transduced similarly. At 1 day postransduction, cells were infected with TB40E at an MOI of 3 (or mock infected) in the presence or absence of 100 mM trehalose. At the time points indicated in the figures, cells were fixed with paraformaldehyde and mounted with Antifade Prolong Gold containing 4′,6′-diamidino-2-phenylindole (DAPI; Life Technologies). Images were acquired using an UltraView Vox spinning-disk microscopy system (Perkin-Elmer) equipped with a Hamamatsu camera. Stacks of 0.4-μm-thick slices were acquired for each condition. At least eight independent fields (containing 1 to 5 cells) were acquired for each condition with a 40× objective. Three-dimensional (3D) reconstruction and rendering were performed using Volocity software.
Immunofluorescence of day 8 and day 21 neural cultures with specific antibodies.
Neuronal cultures were fixed at day 8 and day 21 after differentiation was initiated, permeabilized in blocking buffer (0.1% Triton in PBS supplemented with 10% normal goat serum), and stained overnight with primary antibodies (diluted in blocking buffer) as described previously (31). After extensive washes with PBS, cells were incubated with secondary antibodies conjugated with Alexa Fluor dyes (Life Technologies), and nuclei were stained with Hoechst 33342. Antibodies used were against class III β-tubulin (βIII-tubulin) (clone Tuj1; Covance), Sox2 (Millipore), nestin (Abcam), and glial fibrillary acidic protein (GFAP; Dako). Images were acquired by confocal microscopy with a 20× objective using a Leica SP5 resonance scanning system. At least six representative fields were acquired for each condition. Images were minimally enhanced using ImageJ to facilitate visualization.
Neurite outgrowth assays.
H9 neurons were harvested at day 21 postdifferentiation with 0.25% trypsin supplemented with 0.5 mM EDTA. Trypsin was inactivated with defined trypsin inhibitors (Life Technologies), and cells were passed through a 100-μm-pore-size cell strainer to remove residual large aggregates. A total of 5,000 cells were seeded in wells of a Costar 96-well black clear-bottom plate coated with poly-l-ornithine and Geltrex in mDA differentiation medium. One day after seeding, cells were mock infected or infected with TB40E in the presence or absence of trehalose, as indicated in figure legends. Medium with and without trehalose was changed at 24 hpi and every other day thereafter. At day 7 p.i., cells were fixed, permeabilized, and stained with antibodies against βIII-tubulin (Tuj1) and IE1/IE2 (CH160). Nuclei were stained with Hoechst 33342. Images were acquired by a CellInsight automated high-content imaging platform. Approximately 2,500 βIII-tubulin-positive (βIII-tubulin+) cells (five wells with 500 cells in each) were acquired for each condition. Cell bodies and neurites were modeled, and analysis of neurite length was performed on βIII-tubulin+ cells using a neuronal profiling algorithm (Cellomics BioApplication). For the infected samples, analysis was performed on IE-positive (IE+) cells. The average and median total neurite lengths were computed along with other parameters such as cell body area. Statistical significance was determined by parametric (one-way analysis of variance [ANOVA] combined with Bonferroni's test) and nonparametric (Kruskal-Wallis test combined with Dunn's multiple-comparison test) methods.
RESULTS
Trehalose induces autophagosome formation in infected HFFs.
Trehalose is a nontoxic disaccharide that is able to induce autophagy via an mTOR-independent pathway. In this first set of experiments, we determined the effect of HCMV infection on autophagosome formation and whether treatment with trehalose could modulate these effects. One method used to assess autophagy involves measuring the relative levels of both native (LC3B-I, 18 kDa) and lipid-associated (LC3B-II, 16 kDa) forms of LC3B by Western blotting. During autophagy, the cytosolic LC3B-I form is conjugated to phosphatidylethanolamine to form LC3B-II. LC3B-II binds to the membranes of the phagophore, autophagosome, and autolysosome, where it is degraded. Confluence-synchronized HFFs were incubated in medium in the presence or absence of 100 mM trehalose and mock infected or infected with the clinical strain TB40E at a high MOI (MOI of 3) so that all cells were infected. Lysates were harvested at 24 and 72 hpi and analyzed by Western blotting with antibody to LC3B (Fig. 1). Short and long exposures of the blot are shown to facilitate visualization of both LC3B forms. HCMV infection increased the levels of LC3B-II at 24 hpi, but these levels dropped at 72 hpi such that they were only slightly higher in the infected cells. This is consistent with prior work showing that HCMV transiently induces autophagy early in the infection (26–28). Incubation of cells with 100 mM trehalose from the beginning of the infection further increased LC3B-II levels in both the mock-infected and HCMV-infected cells at 24 and 72 hpi, with a greater increase observed in the infected cells.
FIG 1.
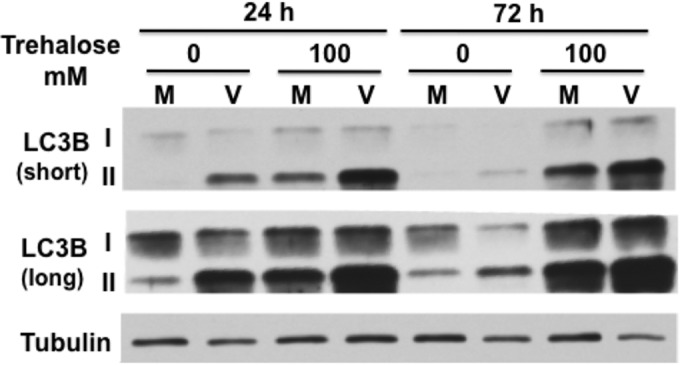
Trehalose increases LC3B-II levels in uninfected and infected HFFs. Synchronized HFFs were infected with TB40E at an MOI of 3 (V) or mock infected (M) upon seeding in the presence or absence of 100 mM trehalose. Cells were harvested at the indicated time points, and extracts were analyzed by Western blotting using a rabbit polyclonal antibody against LC3B. Both native 18-kDa (I) and lipid-associated 16-kDa (II) forms of LC3B are detectable using this antibody. An antibody against α-tubulin was used to control for loading. Short and long exposures are shown. This experiment was repeated at least twice.
To confirm the results obtained by Western blotting, immunofluorescence assays (IFAs) were performed to monitor the effect of the infection on the levels of LC3B puncta (autophagosomes). To avoid potential artifacts with overexpression of transiently transfected GFP-LC3, we used a monoclonal antibody to LC3B to detect the endogenous protein. Confluence-synchronized HFFs were incubated in medium in the presence or absence of 100 mM trehalose and mock infected or infected with TB40E at an MOI of 0.5 so that not all cells would be infected. The cells were fixed at 24 hpi and 72 hpi, permeabilized with saponin to remove free LC3B, and then stained with monoclonal antibodies against LC3B and the HCMV IE1. Figure 2A shows that mock-infected cells that were not incubated with trehalose (Fig. 2A, NT) showed diffuse LC3B staining, with some puncta visible at both the 24-h and 72-h time points. Treatment of the mock-infected cells with trehalose (Fig. 2A, TRE) for 24 h resulted in a significant increase in the level of LC3B fluorescence as well as the number of puncta (as shown in Fig. 2C by the percentage of cells with >15 LC3B puncta) (P < 0.01), and there was an even greater increase after 72 h (P < 0.01). In these experiments, because there was an elevated basal level of autophagy in the HFFs, we used a threshold of 15 LC3B puncta per cell to facilitate quantification of the effects of viral infection and trehalose treatment on autophagosome formation (Fig. 1 and 2A). As expected, there was no HCMV IE1 protein detected in the mock-infected cells.
FIG 2.

Autophagosome formation in HCMV-infected cells is induced by trehalose. Synchronized HFFs were mock infected (A) or infected with TB40E at an MOI of 0.5 (B) upon seeding on coverslips in the absence (not treated, NT) or presence (TRE) of 100 mM trehalose. Cells were fixed at the indicated time points and permeabilized with saponin as described in Materials and Methods. Cells were stained with mouse monoclonal antibodies against LC3B (green) and IE1 (red). Nuclei were counterstained with Hoechst 33342 (blue). Pictures were acquired by confocal microscopy. Representative images are shown for each condition. Scale bar, 100 μm. (C) Quantification of the percentage of cells represented in panels A and B with more than 15 LC3B puncta. At least five fields (with approximately 25 cells in each) were counted for each condition. Histograms represent the means (bars), and the standard deviations (error bars) of the five fields are shown. Statistical significance was determined by one-way ANOVA combined with Tukey's multiple-comparison test (**, P < 0.01 versus results for mock uninfected and infected IE+ untreated cells; *, P < 0.05 versus results for infected IE+ untreated cells only). This experiment was repeated at least twice.
In the infected cells, we observed several different patterns of LC3B staining (Fig. 2B). To illustrate this, two different fields are shown for the 24-hpi and 72-hpi time points in the absence (Fig. 2B, NT) and presence of 100 mM trehalose (Fig. 2B, TRE). Cells that express the viral IE1 protein are shown with red nuclei and at both 24 hpi and 72 hpi show barely detectable levels of green LC3B staining. One group of cells lacking expression of IE1 had a pattern of staining similar to that observed for the mock-infected cells. However, another group of IE1-negative (IE−) cells showed what appeared to be aggregates of LC3B in the cytoplasm. This pattern of staining was particularly striking at 72 hpi when some cells were present in clusters. It should be noted that while saponin allowed the optimal staining of LC3B puncta, it did not provide high levels of sensitivity for IE1. It was therefore not possible to distinguish between true IE1-negative cells and cells with low levels of IE1. However, quantification of puncta revealed a significantly higher percentage of cells in the IE1-negative population with greater than 15 LC3B puncta than in mock-infected untreated cells (Fig. 2C, Mock NT) at 72 hpi (P < 0.01) and in infected IE1-positive untreated cells (Fig. 2C, Infected IE+ NT) at both 24 hpi and 72 hpi (P < 0.05 at 24 hpi and P < 0.01 at 72 hpi). We suspect that this increase in LC3B aggregates in some of the IE1-negative cells at 24 hpi may have been due to the autophagy-inducing properties of noninfectious virions or viral double-stranded DNA (dsDNA) as described previously (26, 28). When the infection progressed and the levels of IE1 became detectable, the aggregates were rarely observed, explaining the very low numbers of puncta in the cells showing strong IE1 staining (Fig. 2C). Nevertheless, treatment with trehalose significantly augmented the numbers of puncta in the IE1-positive and IE1-negative cells in the infected cultures (P < 0.01 compared to numbers in untreated cells), particularly at 72 hpi, suggesting that trehalose was able to override any reduction of autophagy in infected cells.
Trehalose increases the number of autophagosomes and autolysosomes in infected HFFs.
Although the above analysis of the steady-state levels of LC3B-II by Western blotting and of LC3B positive (LC3B+) puncta by immunostaining showed that trehalose enhanced autophagosome formation in both mock-infected and infected cells, it did not reveal whether trehalose increased autophagic flux (i.e., whether it increased the number of autophagosomes fusing to lysosomes and degrading their cargoes). Several means were used to address this question. First, the flux of LC3B-II was monitored by Western blotting following a 2-h pulse with bafilomycin A1, which blocks autolysosome acidification via inhibition of the vacuolar H+ ATPase and prevents the turnover of LC3B-II.
Figure 3 shows that in mock-infected cells that were not treated with trehalose, the bafilomycin pulse increased the levels LC3B-II compared to levels in nonpulsed cells, reflecting the levels of LC3B-II that were degraded in the autolysosome in 2 h. The bafilomycin pulse also resulted in increased LC3B-II in cells that were treated with 50 mM and 100 mM trehalose for 24 h. However, after 72 h of exposure to trehalose (50 mM and 100 mM), although the levels of LC3B-II were very high, the relative increase after the bafilomycin pulse was less than that at 24 h, suggesting that after prolonged treatment with trehalose, there might be less autophagic flux. This could be due to reduced autophagosome turnover or to the inability of the turnover to match the increased rate of autophagosome formation.
FIG 3.

Trehalose increases the basal levels of autophagy and maintains autophagic flux in infected HFFs. Synchronized HFFs were infected with TB40E at an MOI of 3 (or mock infected) upon seeding in the absence or presence of trehalose (50 mM or 100 mM). At the indicated time points, cells were pulsed with bafilomycin A1 (or with DMSO as a control) for 2 h prior to harvest. Cell extracts were analyzed by Western blotting using antibodies against LC3B. An antibody against α-tubulin was used to control for loading. Repeated samples on each gel were used to calibrate blotting and exposures between the different gels. This experiment was repeated at least twice.
The results in cells infected with TB40E at an MOI of 3 were comparable to those in the mock-infected cells, except that cells that were not treated with trehalose showed a relatively greater increase in the ratio of LC3B-II/LC3B-I after a bafilomycin pulse at 72 hpi than at 24 hpi. However, it should be noted that as shown in Fig. 1, the basal level of LC3B-II in the infected cells at 72 hpi was lower than it was at 24 hpi. Nevertheless, these results showed that there was autophagic flux in nontreated infected cells at both 24 and 72 hpi and that trehalose treatment increased the basal levels of autophagy and maintained autophagic flux. Similar results were obtained when HFFs were infected with the HCMV laboratory strain AD169 (data not shown). It should be noted that although the levels of the cellular protein SQSTM1/p62 are often used as an indicator of autophagic flux, we were reluctant to include this in our Western blot assays in view of a number of recent reports that have shown that its level is regulated by a number of factors and that the results of Western blot analysis may be incorrectly interpreted (32–34).
Therefore, as a second method to monitor autophagic flux, we took advantage of the Premo Autophagy Tandem Sensor RFP-GFP LC3B kit (Life Technologies) in which LC3B is fused to the acid-sensitive green fluorescent protein (emerald GFP) and acid-insensitive red fluorescence protein (TagRFP). LC3B with this tandem labeling is expressed by baculovirus, which has high transduction efficiency and minimal toxicity and does not replicate in mammalian cells. With this protein, autophagosomes (which at neutral pH display both the GFP and RFP) appear yellow, and autolysosomes (which at acidic pH display only the RFP) appear red and thus can be distinguished. HFFs were transduced with the baculovirus expressing the RFP-GFP-LC3B construct and 24 h later were mock infected or infected at an MOI of 3 in the presence or absence of 100 mM trehalose. At 24 hpi and 72 hpi, the coverslips containing the cells were fixed as described by the manufacturer, and images were acquired using an UltraView Vox spinning-disk microscopy system (PerkinElmer) equipped with a Hamamatsu camera. Stacks of 0.4-μm-thick slices were acquired for each condition. At least eight independent fields were acquired for each condition with a 40× objective, and 3D reconstruction and rendering were performed using Volocity software.
Figure 4 shows two representative fields for each condition. At both 24 and 72 hpi, most autophagosomes in nontreated mock-infected cells (Fig. 4A) as well as infected cells (Fig. 4B) had a neutral pH (shown in yellow) although there were occasional acidic LC3B+ red puncta. In contrast, treatment of mock-infected cells with trehalose greatly increased the number of puncta. There appeared to be acidification of approximately half of the autophagosomes at 24 h, with the majority appearing red at 72 h. In the case of the infected cells, trehalose treatment also greatly increased the number of puncta and their acidification. Taken together, these data show that treatment of both mock-infected and infected cells with trehalose greatly increases the basal levels of LC3B-II, and there is maintenance of autophagic flux.
FIG 4.

Trehalose promotes acidification of autophagosomes in infected HFFs. HFFs were transduced with a baculovirus expressing LC3B fused to GFP (green) and RFP (red). At 1 day postransduction, cells were mock infected (A) or infected with TB40E at an MOI of 3 (B) upon seeding on coverslips in the absence (not treated, NT) or presence (TRE) of 100 mM trehalose. At the indicated time points, cells were fixed, and nuclei were counterstained with Hoechst 33342. Z-stacks composed of individual 0.4-μm-thick slices were acquired at high magnification using a spinning-disk microscope. Between 8 and 10 fields were acquired for each condition. Representative phenotypes are shown. Colocalization (yellow) between the direct fluorescence of GFP and RFP results from a neutral pH, whereas red puncta correspond to a more acidic environment in which the GFP signal was quenched. Scale bar, 100 μm. This experiment was repeated at least twice.
Trehalose inhibits HCMV replication in HFFs.
To determine whether there were specific effects of increasing amounts of trehalose (50 mM and 100 mM) and increased autophagy on viral gene expression, we assayed by Western blotting the expression of selected IE and early and late viral proteins (Fig. 5A). In the experiment shown, treatment with 50 mM trehalose had only minimal effects on the IE proteins (IE1-72 and IE2-86), the early protein UL44, and the late proteins pp28/UL99 and IE2 proteins IE2-60 and IE2-40 although there was a delay in expression. However, in other experiments we have observed greater inhibition with 50 mM trehalose (data not shown). In all experiments, there was a substantial reduction in levels of all of these proteins when the cells were treated with 100 mM trehalose. Interestingly, trehalose treatment at both 50 mM and 100 mM almost completely abrogated the expression of the early protein UL57, which is required for viral DNA replication. Overall, the effects of trehalose were found to be dose dependent, with 100 mM trehalose treatment resulting in the greater inhibition of viral gene expression.
FIG 5.

Trehalose inhibits HCMV gene expression, virus production, and viral spread in HFFs. (A) Synchronized HFFs were infected with TB40E at an MOI of 0.5 upon seeding in the absence or presence of 50 mM or 100 mM trehalose. Cell extracts prepared at 24, 72, and 120 hpi were analyzed by Western blotting using antibodies against HCMV IE1/IE2 (CH160), IE2-86, late IE2 proteins IE2-60 and IE2-40, UL44, UL57, and UL99. An antibody against α-tubulin was used to control for loading. The experiment was repeated three times, and a representative Western blot is shown. (B) Synchronized HFFs were infected with TB40E at an MOI of 0.5 or 3 upon seeding in the absence or presence of 100 mM trehalose. At the indicated time points, cell-associated and cell-free virus were prepared as described in Materials and Methods. Titers of viral preparations were determined by plaque assays. Graphs represent titer results obtained from a representative experiment. Relevant fold decreases in titers in trehalose-treated cells, relative to those nontreated cells, are indicated. Dotted lines represent assay limits of detection. (C) Synchronized HFFs were infected with TB40E at an MOI of 0.025 upon seeding on coverslips in the absence (not treated, NT) or presence of 100 mM trehalose. Cells were fixed at 24 and 168 hpi and stained with mouse monoclonal antibodies against HCMV IE (green) and UL57 (red) proteins. Nuclei were counterstained with Hoechst 33342 (blue). Images were acquired by epifluorescence microscopy. Representative images of phenotypes obtained at 24 and 168 hpi are shown. Scale bar, 100 μm. Each experiment was repeated three times.
We further assessed whether the trehalose-mediated increase in autophagy in infected HFFs would inhibit the production of infectious virus. In the first set of experiments, we measured by standard plaque assays the amount of both cell-free and cell-associated virus produced in the presence or absence of 100 mM trehalose (Fig. 5B). Cells were infected at either a low MOI (MOI of 0.5) or high MOI (MOI of 3). At both MOIs, treatment of infected cells with 100 mM trehalose greatly reduced titers of cell-free virus at 96 hpi (>200-fold at an MOI of 0.5 and 425-fold at an MOI of 3) and at 120 hpi (60-fold at an MOI of 0.5 and 135-fold at an MOI of 3). Surprisingly, there appeared to be no significant effect of trehalose on the production of cell-associated virus at either a low or high MOI. In the second set of experiments, we looked at the effect of trehalose on viral spread. Cells were infected at a low MOI and maintained in the presence or absence of 100 mM trehalose (Fig. 5C). The cells were harvested at 24 hpi and 168 hpi and assayed by immunofluorescence with antibodies to the IE proteins IE1/IE2 and the early protein UL57. Figure 5C shows that the numbers of cells positive for the IE proteins were comparable in the presence and absence of trehalose at 24 hpi, showing that there was no effect on the initiation of the infection. However, at 168 hpi, treatment of the cells with trehalose significantly inhibited viral spread as measured by the size of IE+ plaques. These results show that trehalose greatly impaired the release and spread of infectious virus.
Treatment of human aortic endothelial cells with trehalose inhibits viral replication.
To investigate whether a similar inhibition of HCMV infection would occur in other cell types, we next extended these studies to human aortic endothelial cells (HAECs), a physiologically relevant cell type involved in the development of atherosclerosis. By Western blotting, we found that infection of HAECs with TB40E at a high MOI increased, albeit modestly, the levels of LC3B-II at 24 hpi and 72 hpi compared to levels in mock-infected cells (Fig. 6A). As was observed for the HFFs, treatment with 100 mM trehalose significantly increased the levels of LC3B-II compared to levels in nontreated cells in both mock-infected and infected cells although the increase at 24 hpi was less for the virus-infected cells.
FIG 6.

Trehalose increases the basal levels of autophagy and maintains autophagic flux in infected aortic endothelial cells. Human aortic endothelial cells (HAECs) were infected with TB40E at an MOI of 3 (or mock infected) 1 day after seeding in the absence or presence of 50 or 100 mM trehalose. (A) At the indicated time points, cell extracts were prepared and analyzed by Western blotting using antibodies against LC3B. Short and long exposures are shown. (B) At the indicated time points, cells were pulsed with bafilomycin A1 (or with DMSO as a control) for 2 h prior to harvest. Cell extracts were analyzed by Western blotting using antibodies against LC3B. An antibody against α-tubulin was used to control for loading. Repeated samples on each gel were used to calibrate blotting and exposures between the different gels. Each experiment was repeated at least twice.
To evaluate whether treatment with trehalose increased autophagic flux, we assessed the levels of LC3B-II by Western blotting following a 2-h pulse with bafilomycin A1 just prior to harvest at 24 hpi and 72 hpi as described above (Fig. 6B). We also measured these levels after treatment with both 50 mM and 100 mM trehalose and showed that the increased LC3B-II levels in mock-infected and infected cells (MOI of 3) at 24 hpi and 72 hpi were comparable at both doses of trehalose. In both the presence and absence of trehalose, the bafilomycin pulse increased the levels LC3B-II in mock-infected and infected cells, indicating turnover of the protein. However, after 72 h of exposure to trehalose (50 mM and 100 mM) and a 2-h pulse of bafilomycin, the increase in the levels of LC3B-II in mock-infected cells was less than at 24 h, as had been observed with the mock-infected HFFs, suggesting that after prolonged treatment with trehalose, there might be less autophagic flux. We also noted that for the infected cells that were not treated with trehalose, there was a greater increase in the ratio of LC3B-II/LC3B-I after a bafilomycin pulse at 72 hpi than at 24 hpi.
We next examined whether treatment with trehalose would affect viral infection in endothelial cells as shown for the HFFs. HAECs treated with 50 mM and 100 mM trehalose were infected at an MOI of 0.5, and the expression of selected IE and early and late viral proteins was assayed by Western blotting as described in the legend of Fig. 5. Treatment of HAECs with 50 mM trehalose led to greatly reduced expression of the viral proteins at all stages of the infection (IE proteins IE1-72 and IE2-86, early proteins UL44 and UL57, and the late proteins pp28/UL99 and IE2 proteins IE2-60 and IE2-40) (Fig. 7A). The effect was even greater in the presence of 100 mM trehalose.
FIG 7.

Trehalose inhibits HCMV gene expression and virus production in endothelial cells. HAECs were infected with TB40E at an MOI of 0.5 in the absence or presence of trehalose (50 mM or 100 mM). (A) Cell extracts prepared at 24, 72, and 120 hpi were analyzed by Western blotting using antibodies against HCMV IE1/IE2 (CH160), IE2-86, late IE2 proteins IE2-60 and IE2-40, UL44, UL57, and UL99. An antibody against α-tubulin was used to control for loading. (B) At the indicated time points, cell-free and cell-associated virus was prepared as described in Materials and Methods, and virus titers were determined by plaque assays. Graphs represent titer results obtained from a representative experiment. Dotted lines represent assay limits of detection. Relevant fold decreases in titers from trehalose-treated cells, relative to those of nontreated cells, are indicated. Each experiment was repeated at least twice.
To determine if the trehalose-mediated increase in autophagy in infected HAECs would inhibit the production of infectious virus, we measured by plaque assay the amount of both cell-free and cell-associated virus produced in the presence or absence of 100 mM trehalose when cells were infected at an MOI of 3 (Fig. 7B). We found that treatment of infected cells with 100 mM trehalose greatly reduced titers of cell-free virus at 120 hpi (195-fold). If the infection was allowed to proceed for another 48 h, the fold decrease was less (∼14-fold) (data not shown), suggesting that there was a delay in virus release. There also appeared to be a significant effect of trehalose on the production of cell-associated virus at 72 hpi (245-fold), but at 120 hpi, the differences were less (4-fold).
Trehalose increases autophagosome formation and acidification in infected developing neurons.
Having established the proautophagic as well as the antiviral effect of trehalose in HFFs and HAECs, we proceeded to investigate whether trehalose would also be effective in inhibiting viral replication in developing neurons, a cell type for which deficiencies in autophagy have been shown to have severe consequences on physiological functions (for a review, see references 12 and 35). Human primitive neural stem cells (pNSCs) were first derived from human H9 embryonic stem cells and then patterned and differentiated under proneurogenic conditions for 8 days (day 8 neurons). At day 8 postdifferentiation, approximately 75% of the cells in the H9 pNSC-derived cultures still expressed some markers of neural stem cells (Sox2 and nestin) (Fig. 8). However, at least 25% of the cells in the culture expressed the neuronal marker βIII-tubulin and displayed nascent to moderately elongated axons. In contrast, there were very few cells expressing the astroglial marker GFAP.
FIG 8.

Differentiation of H9-derived human primitive neural stem cells for 8 days yields a mixed population of stem cells and neurons. H9-derived pNSCs were patterned for 10 days and differentiated on coverslips as described in Materials and Methods. At 8 days postdifferentiation, cells were fixed, permeabilized, and stained with antibodies against βIII-tubulin (red), Sox2 (green), and nestin (cyan) (A) or with antibodies against βIII-tubulin (red) and GFAP (green) (B). Nuclei were counterstained with Hoechst 33342 (blue). Between six and eight fields were acquired for each condition. Representative phenotypes are shown. Scale bar, 100 μm. The experiment was repeated twice.
H9 day 8 neurons in the presence and absence of 100 mM trehalose were infected at a high MOI (MOI of 3) with TB40E, and the levels of both LC3B-I and LC3B-II were monitored by Western blotting as shown in Fig. 1 and 6. In contrast to what was observed in HFFs and HAECs, the steady-state levels of LC3B-II relative to those of LC3B-I were very low in both mock-infected and infected cells (Fig. 9A). In addition, infection of day 8 neurons with TB40E at an MOI of 3 did not increase the levels of lipid-associated LC3B (LC3B-II) at 24 or 72 hpi. Treatment with 100 mM trehalose did increase the amount of LC3B-II in both mock-infected and infected cells, with a greater relative increase seen in the infected cells at 72 hpi. In the trehalose-treated infected cells, the levels of LC3B-II at this later time point were just slightly lower than those of LC3B-I.
FIG 9.

Trehalose increases the basal levels of autophagy and maintains autophagic flux in infected neuronal cultures. H9-derived neurons at day 8 (H9 d8 neurons) postdifferentiation were infected with TB40E at an MOI of 3 (or mock infected) in the absence or presence of trehalose (50 mM or 100 mM). (A) Cells were harvested at the indicated time points, and extracts were analyzed by Western blotting using a rabbit polyclonal antibody against LC3B. An antibody against α-tubulin was used to control for loading. Short and long exposures are shown. (B) At the indicated time points, cells were pulsed with bafilomycin A1 (or with DMSO as a control) for 2 h prior to harvest. Cell extracts were analyzed by Western blotting using antibodies against LC3B. Repeated samples on each gel were used to calibrate blotting and exposures between the different gels. Experiments were repeated at least twice. (C) H9 day 8 neurons were infected with TB40E at an MOI of 0.5 (or mock infected) in the absence (not treated, NT) or presence (TRE) of 100 mM trehalose. Cells were fixed at the indicated time points and permeabilized with saponin as described in Materials and Methods. Cells were stained with mouse monoclonal antibodies against LC3B (green) and IE1 (red). Nuclei were counterstained with Hoechst 33342 (blue). Pictures were acquired by confocal microscopy. Scale bar, 100 μm. Representative images are shown for each condition. (D) Quantification of the percentage of cells in panel C with at least one LC3B punctum. At least five fields (with approximately 150 cells in each) were counted for each condition, and the means (bars) and standard deviations (error bars) of the five fields are shown. Statistical significance was determined with a one-way ANOVA test combined with Tukey's multiple-comparison test (*, P < 0.05 versus results in mock untreated and infected IE+ untreated cells). Each experiment was repeated at least twice.
To determine whether the low levels of LC3B-II were due to decreased autophagy or enhanced autophagic flux, we treated the cells with bafilomycin A1 for 2 h prior to harvest. Figure 9B shows that at both 24 hpi and 72 hpi, the bafilomycin pulse increased the levels of LC3B-II in mock-infected and infected cells, indicating that there was autophagic flux. However, with the exception of the infected cells at 72 hpi, levels of LC3B-I were still significantly greater than those of LC3B-II. This increase in relative levels of LC3B-II was also observed in cells treated with 50 mM and 100 mM trehalose and pulsed with bafilomycin although the increase was less in the mock-infected cells at 72 hpi, indicating that there may have been some inhibition of LC3B-II turnover. The greatest increase in relative LC3B-II levels and autophagic flux was observed in the trehalose-treated infected cells at 72 hpi. Thus, in contrast to what is observed in HFFs, HCMV infection does not appear to transiently induce autophagy at 24 hpi. Taken together, these data suggest that there is significantly less autophagy in the day 8 neurons than in HFFs and HAECs, but autophagic flux is occurring. However, at 72 hpi, autophagic flux is greatly increased in the infected cells, and this level of autophagy is further increased by treatment with trehalose.
To confirm the results obtained by Western blotting, immunofluorescence assays were performed as described in the legend of Fig. 2 to monitor the effect of infection on the levels of endogenous LC3B puncta (autophagosomes) in a culture of differentiating neurons. Day 8 neurons were incubated in medium in the absence or presence of 100 mM trehalose and mock infected or infected with TB40E at an MOI of 0.5. The cells were fixed at 24 hpi and 72 hpi, permeabilized with saponin to remove free LC3B, and then stained with monoclonal antibodies against LC3B and the HCMV IE protein IE1. Importantly for these experiments, as in HFFs, trehalose did not significantly alter the percentage of infected cells at 24 hpi. Figure 9C shows that mock-infected and infected cells showed very few LC3B puncta at both the 24-h and 72-h time points in the absence of trehalose (Fig. 9C, NT). These observations are consistent with several studies describing a very low number of autophagosomes in neurons (36, 37). In accordance with the methods used in these reports, we then monitored the number of neurons positive for LC3B puncta in order to quantify the effects of infection and trehalose treatment on autophagosome formation. As shown in Fig. 9D, only 20 to 25% of nontreated day 8 neurons (mock, IE1−, or IE1+) had one or more LC3B puncta at either time point, and the differences were not statistically significant. However, treatment of the cells with 100 mM trehalose for 24 h resulted in increases in both the level of LC3B fluorescence and the number of puncta (P < 0.05 versus results in mock-untreated, IE1− untreated, and IE1+ untreated cells), and there were even greater increases after 72 h (P < 0.05 relative to results in mock-untreated, IE1− untreated, and IE1+ untreated cells).
To further analyze autophagic flux, day 8 neurons were transduced with the baculovirus expressing the RFP-GFP-LC3B and 24 h later mock infected or infected at an MOI of 3 in the absence (untreated) or presence of 100 mM trehalose. At 24 hpi and 72 hpi, the coverslips containing the cells were fixed, and images were acquired as described in the legend of Fig. 4. At both time points, there were few autophagosomes in mock-infected (Fig. 10A) and infected (Fig. 10B) cells, and most had a neutral pH (indicated in yellow). In contrast, treatment of infected and mock-infected cells with trehalose greatly increased the number of puncta, with both acidic autolysosomes and neutral autophagosomes observed. Consistent with the Western blot results, the greatest increase in yellow and red puncta was observed in the trehalose-treated infected cells at 72 hpi.
FIG 10.

Trehalose promotes acidification of autophagosomes in infected neuronal cultures. H9 day 8 neurons were transduced with a baculovirus expressing LC3B fused to GFP (green) and RFP (red). At 1 day postransduction, cells were mock infected (A) or infected with TB40E at an MOI of 3 (B) in the absence (not treated, NT) or presence (TRE) of 100 mM trehalose. At the indicated time points, cells were fixed, and nuclei were counterstained with Hoechst 33342. Z-stacks composed of individual 0.4-μm-thick slices were acquired at high magnification using a spinning-disk microscope. Between 8 and 10 fields were acquired for each condition. Representative phenotypes are shown. Colocalization (yellow) between the direct fluorescence of GFP and RFP results from a neutral pH, whereas red puncta correspond to a more acidic environment in which the GFP signal was quenched. Scale bar, 25 μm. Each experiment was repeated twice.
Trehalose inhibits HCMV replication in developing neural cultures.
To determine whether there were specific effects of increasing amounts of trehalose (50 mM and 100 mM) and increased autophagy on viral gene expression, we assayed by Western blotting the expression of selected IE and early and late viral proteins (Fig. 11A). Day 8 neurons treated with 50 mM and 100 mM trehalose were infected at an MOI of 0.5, and the expression of viral proteins was assayed by Western blotting. As was the case for HAECs, treatment of day 8 neurons with 50 mM trehalose led to greatly reduced expression of the viral proteins at all stages of the infection, and the effect was greater in the presence of 100 mM trehalose. One notable difference between the effect of trehalose on the infection in HFFs and HAECs was that in the day 8 neurons the expression of UL57 was not completely inhibited at 120 hpi. We also determined the effect of trehalose on production of infectious virus. Treatment with 100 mM trehalose had a significant inhibitory effect on virus production. In two independent experiments, 100 mM trehalose significantly impaired both cell-free virus (more than 200-fold) and cell-associated virus (24- to 56-fold) (Fig. 11B). At this concentration of trehalose, there was also almost complete inhibition of viral spread as assessed by IFA at 168 hpi (Fig. 11C). The boxed areas in the merged and Hoechst-stained panels of Fig. 11C were enlarged to show the relative sizes of the nuclear viral replication centers (labeled IE/UL57).
FIG 11.

Trehalose inhibits HCMV gene expression, virus production, and viral spread in neuronal cultures. H9 day 8 neurons were infected with TB40E at an MOI of 0.5 in the absence or presence of trehalose (50 mM or 100 mM). (A) Cell extracts prepared at 24, 72, and 120 hpi were analyzed by Western blotting using antibodies against HCMV IE1/IE2 (CH160), IE2-86, late IE2 proteins IE2-60 and IE2-40, UL44, UL57, and UL99. An antibody against α-tubulin was used to control for loading. The experiment was repeated three times. (B) At the indicated time points, cell-associated and cell-free virus was obtained from cells infected in the absence (untreated) or presence of 100 mM trehalose as described in Materials and Methods. Titers of viral preparations were determined by plaque assays. Graphs showing titers from two representative experiments are shown. Triplicate assays were performed for each sample. Relevant fold decreases in titers from trehalose-treated cells, relative to levels in nontreated cells, are indicated. Dotted lines represent assay limits of detection. (C) H9 day 8 neurons were infected with TB40E at an MOI of 0.025 in the absence (not treated, NT) or presence of 100 mM trehalose. Cells were fixed at 24, 96, and 168 hpi and stained with mouse monoclonal antibodies against HCMV IE (green) and UL57 (red). Nuclei were counterstained with Hoechst 33342 (blue). Images were acquired by epifluorescence microscopy. Only representative images of phenotypes obtained at 168 hpi are shown. Scale bar, 100 μm. The right panels show enlarged images of the boxed regions.
Trehalose significantly protects against neurite growth defects and cytomegaly in infected neurons.
In order to model in vitro whether the antiviral effect of trehalose could potentially translate into therapeutic benefits on fetal central nervous system disease pathologies, we developed a neurite outgrowth assay using H9 embryonic stem cell-derived neurons. H9-derived neurons were harvested at day 21 postdifferentiation and reseeded in a 96-well plate. At day 21 postdifferentiation, there was a modest decrease in the neural stem cell population (compared to day 8 postdifferentiation), as assayed by IFA with antibodies to Sox2 and nestin, but they nevertheless constituted approximately half of the culture (Fig. 12). IFA with an antibody to βIII-tubulin showed that neurons were not significantly increased in numbers compared to the numbers at day 8 (approximately 25%) but showed extensive axon and neurite networks. The remaining cells in day 21 cultures were mainly GFAP+ cells. The process of reseeding the neural cultures resulted in retraction of the axons and permitted regrowth of neurites, similar to results of a neurite outgrowth assay using primary neurons from rodents (38).
FIG 12.

Differentiation of H9-derived human primitive neural stem cells for 21 days yields a mixed population of stem cells, astroglial cells, and mature neurons with long neurites. H9-derived pNSCs were patterned for 10 days and differentiated on coverslips as described in Materials and Methods. At 21 days postdifferentiation, cells were fixed, permeabilized, and stained with antibodies against βIII-tubulin (red), Sox2 (green), and nestin (cyan) (A) or with antibodies against βIII-tubulin (red) and GFAP (green) (B). Nuclei were counterstained with Hoechst 33342 (blue). Between 6 and 8 fields were acquired for each condition. Representative phenotypes are shown. Scale bar, 100 μm. The experiment was repeated twice.
At 24 h postreseeding, neurons were infected with TB40E at an MOI of 0.1, and neurons were imaged 7 days later after being stained with antibodies to HCMV IE1 protein and βIII-tubulin using a nonbiased high-content screening microscopy system. Infection with TB40E induced a significant neurite growth defect in IE+ neurons (Fig. 13A). Automated quantification of total neurite length per neuron revealed an approximately 2-fold decrease in mean total neurite length (112 μm for mock-infected versus 60 μm for infected cells; P < 0.001 by one-way ANOVA combined with Bonferroni's test) (Fig. 13B) and an approximately 3-fold decrease in the median total neurite length (74 μm for mock-infected versus 21 μm for infected cells; P < 0.001 by a Kruskal-Wallis test combined with Dunn's multiple-comparison test) (data not shown). Surprisingly, when mock-infected neurons were treated with trehalose, total neurite length per neuron increased significantly. Mean total neurite length per neuron increased from 112 μm to 161 μm (P < 0.001 by one-way ANOVA combined with Bonferroni's test) (Fig. 13B), and median total neurite length increased from 74 μm to 107 μm (P < 0.001 by a Kruskal-Wallis test combined with Dunn's multiple-comparison test) (data not shown). Importantly, in infected neurons, trehalose also had a neurite length-enhancing effect and significantly restored neurite growth. Mean total neurite length per neuron increased from 60 μm to 92 μm (P < 0.001 by one-way ANOVA combined with Bonferroni's test), and median total neurite length increased from 21 μm to 47 μm (P < 0.001 by a Kruskal-Wallis test combined with Dunn's multiple-comparison test) (data not shown). The mean and median total neurite lengths and distribution of total neurite length for the infected cells were, however, still lower than those of the nontreated mock-infected neurons (P < 0.001).
FIG 13.

Trehalose significantly protects against defects in neurite growth and cytomegaly in infected neurons. (A) H9 neurons differentiated for 21 days were harvested and reseeded in 96-well plates. One day after seeding, neurons were mock infected or infected with TB40E at an MOI of 0.1 in the absence (not treated, NT) or presence of 100 mM trehalose. Cells were fixed at 168 hpi and stained with antibodies against βIII-tubulin (green) and HCMV IE (red). Nuclei were counterstained with Hoechst 33342. For each condition, images (five wells and 25 fields for each well) were acquired using the CellInsight high-content microscopy platform. Representative images are shown. Scale bar, 25 μm. (B and C) Quantification of neurite length was performed on approximately 1,000 neurons for each condition using the Cellomics neuronal profiling algorithm. Histograms represent the mean total neurite length per neuron (bar) with 95% confidence intervals for a representative experiment (B). Statistical significance was determined by one-way ANOVA combined with Bonferroni's multiple-comparison test (*, P < 0.001 versus all other conditions). Cumulative frequency distribution (y axis, cumulative percentage of neurons) of total neurite length per neuron (x axis) highlights the defects in neurite length in infected neurons (shorter neurites are observed at the median of the distribution as well as for the highest quartile) (C). Treatment of infected neurons with 100 mm trehalose partially restored neurite length at 50% of the distribution (median) and completely restored neurons with very long neurites (highest quartile). (D) Quantification of cell body area was performed on approximately 1,000 neurons for each condition using the Cellomics neuronal profiling algorithm. Histograms represent the geometric mean of cell body areas (bars) with 95% confidence intervals (error bars). Statistical significance was determined by one-way ANOVA combined with Bonferroni's multiple-comparison test (*, P < 0.001 versus all other conditions). For all panels, each experiment was repeated three times.
The effect of trehalose and HCMV infection on the distribution of neurite length at the population level can be visualized in a cumulative frequency distribution graph whereby total neurite length is plotted in relation to the cumulative percentage of cells for a given neurite length (Fig. 13C). Infection led to a significant increase in cells with short neurites (first two quartiles) as well as a decrease in the proportion of neurons with very long neurites (highest quartile). Trehalose treatment of infected cells led to a partial increase in neurite length in cells with short neurites and almost completely restored the number of neurons with very long axons.
In addition to its effect on neurite length, trehalose also led to a significant protective effect against HCMV-induced cytomegaly (Fig. 13D) in infected neurons. Infection increased the geometric mean of cytoplasmic area (414 μm2 for mock-infected versus 664 μm2 for infected cells; P < 0.001 by one-way ANOVA combined with Bonferroni's test). Treatment of infected cells with trehalose significantly reduced the geometric mean of cytoplasmic area to 408 μm2 (P < 0.001), well within the range of cytoplasmic area in mock-infected, nontreated cells.
DISCUSSION
In the past few years, there has been increasing interest in the complex interplay between autophagy and viral infection (for a review, see references 25 and 39). Based on multiple studies with many different types of RNA and DNA viruses, it appears that autophagy can serve either a pro- or antiviral role, and viruses can either induce or inhibit the formation of autophagosomes and autophagic flux. However, it is important to appreciate that these opposing effects may be cell type and context specific.
One example of an autophagy-mediated antiviral effect is the finding that induction of autophagy by rapamycin or vitamin D inhibits HIV-1 replication in macrophages (40, 41). A cell-permeable Tat–beclin-1 peptide, which was derived from the region of beclin-1 that was targeted by HIV-1 Nef for inhibition, also activates autophagy and inhibits HIV-1 infection (42). This peptide additionally blocks the replication of Sindbis virus, chikungunya virus, and West Nile virus in vitro and decreases mortality in neonatal mice infected with chikungunya or West Nile virus. Interestingly, induction of autophagy inhibits infection of cells by the alphaherpesvirus herpes simplex virus 1 (HSV-1) but has a proviral effect on replication of the related alphaherpesvirus varicella-zoster virus (VZV) (43–46). It has also been found that induction of autophagy by trehalose in the murine microvascular endothelial MB114 cells enhances survival of cells persistently infected with mouse gammaherpesvirus 68 (47).
Prior studies of the effect of HCMV on autophagy in primary fibroblasts concluded that the virus initially induced autophagy but then inhibited it as the infection progressed (26–28). Based on these studies, our initial hypothesis was that if HCMV impeded autophagy, enhancing autophagy might inhibit the infection. Many inducers of autophagy act through inhibition of the mTOR pathway, which negatively regulates autophagy. However, because HCMV is known to alter the mTOR pathway and maintain mTOR activity under conditions of stress that should be inhibitory, we reasoned that we needed to induce autophagy via an mTOR-independent mechanism (for a review, see reference 48). We also wanted to induce autophagy in a way that would have the lowest probability of toxic effects. For that reason, we decided to use trehalose, a nontoxic disaccharide that has been shown to induce autophagy and to have neuroprotective effects in animal models of Huntington disease, Alzheimer's disease, Parkinson's disease, ALS, and maternal diabetes-associated neural tube defects in the embryo (13, 15–20). In addition to the positive effects of trehalose in neurodegenerative diseases, there is also evidence that trehalose may have a beneficial effect on impaired endothelial cells in ageing arteries, a risk factor for cardiovascular disease (49). We confirmed the finding of other investigators that trehalose did not block the activity of mTOR in infected cells as measured by phosphorylation of 70S6K and eIF4EBP1 (data not shown).
The earlier conclusion that HCMV infection inhibited autophagosome formation in primary fibroblasts was based on redistribution of transiently expressed GFP-LC3, LC3B-II, and p62 accumulation and on electron microscopy (EM) (27). In these experiments, a GFP-LC3 plasmid was transfected into infected MRC-5 cells (derived from human embryonic lung) 24 h prior to harvest. Shown in this previous paper is a figure of an infected cell with high levels of GFP-LC3 distributed in both the nucleus and the cytoplasm. In a subsequent report, similarly high levels of GFP-LC3 or monomeric RFP (mRFP)-GFP-LC3 were observed in the cytoplasm and nucleus of infected cells (as well as uninfected cells) that were transfected with an LC3 expression vector prior to infection (28). In our studies, we specifically avoided using transfected plasmid because overexpression can lead to misleading results, and the stress of transfection can affect autophagy. Transfection of primary cells with plasmids is also very inefficient, and it is difficult to get the majority of the population to express the protein. Therefore, when we expressed exogenous LC3B in the cells, we transduced them with baculovirus, which has minimal toxicity and allows the majority of the cells to express the protein.
In this study, we used three different types of cells that are relevant for HCMV infection in vivo, including fibroblasts, arterial endothelial cells, and neural cells derived via directed differentiation of embryonic stem cells. The results showed that trehalose induced autophagosome formation and autophagic flux in all of these cell types and inhibited HCMV gene expression, virus production, and viral spread. Interestingly, there appeared to be no significant effect of trehalose on the production of cell-associated virus in HFFs and HAECs. In the case of neural cells, there was an inhibitory effect on cell-associated virus, but it was less than that for cell-free virus. Since trehalose is often used for the preservation of proteins and biologic products with lipid membranes, it is possible that it also helped maintain the infectivity of virions bound to cell membranes. The effects of trehalose were dose dependent, with 100 mM trehalose treatment resulting in the greatest inhibition of viral gene expression for all cell types. We also demonstrated that trehalose treatment reduced the cytopathic effects of infection in neural cells, as measured by decreased cytomegaly and virus-induced shortening of neurite length.
With respect to the effects of the infection on autophagy, our data support an early induction of autophagy by HCMV in fibroblasts, but this is most likely due to infection with noninfectious viral particles. However, by several different criteria, we demonstrate that autophagy is not inhibited by the infection in the three different types of cells examined in this study. First, we show that in the presence of bafilomycin A1, endogenous LC3B-II accumulates at 72 hpi in infected primary HFFs, HAECs, and neural cells to at least the same levels as in uninfected cells, suggesting no impairment of autophagic flux. Second, when HFFs are transduced with a baculovirus expressing the RFP-GFP-LC3B construct 24 h prior to infection at an MOI at which all cells are infected, both mock-infected and infected cells show similar patterns of puncta at 24 and 72 hpi (Fig. 4). Most autophagosomes have a neutral pH (Fig. 4, yellow) although acidic red-stained LC3B+ puncta are also observed. When we analyzed endogenous LC3B by IFA in HFFs that were infected at a low MOI, we did observe aggregates of LC3B in some cells that did not appear to be infected based on lack of viral IE staining, but, as noted above, we believe that these cells may have been infected with noninfectious virus and could not proceed to viral gene expression. It is possible that some of the differences in the results that we obtained relative to those obtained by Chaumorcel et al. (27, 28) may be due to their use of MRC5 lung fibroblast cells rather than low-passage-number human foreskin fibroblasts. In this regard, it has been reported that there are differences between MRC5 cells and HFFs in metabolism and susceptibility to drugs that inhibit HCMV infection (50, 51). It should be noted that in accord with our results, McFarlane et al. (26) also found that autophagy did not appear to be inhibited in HCMV-infected primary foreskin fibroblasts. We also did not see similar aggregates by IFA when the neural cultures were infected at a low MOI. Moreover, based on the detection of LC3B-II by Western blotting, the levels of autophagy were comparable in both the mock-infected and infected HAECs and in neural cultures at both 24 hpi and 72 hpi when the cells were infected at a high MOI. It is possible that if we had looked a time points earlier than 24 hpi, we might have observed transient induction of autophagy in these other cell types.
A major question raised by these studies is what is the mechanistic basis by which trehalose inhibits the infection. This will not be an easy problem to address as trehalose appears to have multiple effects on the host cell. Based on the results of trehalose treatment of normal fibroblasts in studies where they served as a control for the effects of trehalose in fibroblasts derived from patients with neurodegenerative diseases (21, 22), trehalose has been found to increase the levels of glutathione (GSH), LC3B-II-positive puncta, beclin-1, and ATG5/ATG12. In addition, trehalose can counter the effects of the proteasome inhibitor epoxomicin, including elevated ROS, cleaved caspase 3, accumulation of ubiquitinated proteins, and altered mitochondrial function and morphology. Interestingly, trehalose also increased CMA, as indicated by increased numbers of LAMP-2A-positive cells and the colocalization of LAMP-2A and HSC70. Thus, the inhibitory effects of trehalose could be the result of increased macroautophagy or CMA or decreased ROS. With respect to the latter possibility, inhibition of ROS appears to inhibit the infection (51). HCMV infection induces the generation of ROS upon entry into the cell, and this event is thought to be necessary for initiation of the virus life cycle since high ROS levels activate NF-κB and lead to transactivation of the viral immediate early promoter. In addition, HCMV infection manipulates the host cell metabolism throughout the infection in ways that would be predicted to result in high levels of ROS (52).
Another mechanism that we have considered is that the virus needs to utilize several of the autophagy proteins, and by inducing autophagy, these proteins are no longer available for alternative virus-associated functions. One possibility is that the virus may need to commandeer selected autophagy proteins to the cytoplasmic assembly center (AC). The viral AC is a membranous structure that forms at a perinuclear location around the centriole and is the site where HCMV acquires its tegument proteins and undergoes its final envelopment. The formation of the viral AC is the result of extensive reorganization of secretory pathway components and involves a large number of cellular and viral proteins (for a review, see references 53 to 55). Cellular proteins associated with the AC suggest multiple membrane sources, including the Golgi compartment, trans-Golgi network, late and early endosomes, and lysosomes; also present are proteins involved in the ESCRT pathway, SNARE family members, Rab GTPases, the endoplasmic reticulum (ER) chaperone glucose-regulated protein (GRP78 or BiP) and mTOR (52, 56–63). At least part of the formation of the AC is mediated by dynein (52, 62). Some of the proteins associated with the viral AC, such as mTOR and BiP, are also implicated in regulation of the macroautophagy pathway.
What is striking about the many EM pictures showing cytoplasmic envelopment of the HCMV capsid is that it appears to be initiated by budding into a vesicle that forms a crescent shape around the capsid and greatly resembles the phagophore that initiates autophagy. The recent use of advanced electron microscopy techniques allows clear visualization of these steps in envelopment (64, 65). Contact of the viral particle with a vesicle membrane initiates the formation of a crescent shape whereby the membrane wraps around the tegumented nucleocapsid. At the final step there is fusion of the vesicle membrane ends, resulting in the enveloped particle now residing within a vesicle. This raises the question of whether specific cellular proteins involved in the initiation of the phagophore are diverted to perform a similar role for envelopment of the viral capsid. If so, by inducing high levels of autophagosome formation and autophagic flux, trehalose may prevent the virus from rerouting these proteins to the viral AC. In support of this hypothesis are the following observations. First, the HCMV protein TRS1 interacts with the autophagy-associated proteins beclin-1 and BiP, thus raising the question of whether TRS1 plays a role in localizing some of the proteins involved in phagophore formation to the viral AC for envelopment of the virus (28, 61). Second, it has been found that Vps34 kinase (class III phosphatidyinositol 3-kinase), which interacts with beclin-1 to form the autophagophores, is required for formation of the large vesicles seen at late times in HCMV-infected fibroblasts and for envelopment of capsids (66). Third, in primary microvascular endothelial cells, viruses lacking UL135 or UL136 have major defects in the secondary envelopment of virus (67, 68). Finally, in HFFs infected with a mutant virus that does not express the tegument protein UL71, secondary envelopment is also affected such that there are few small crescent-shaped vesicles, and, instead, multiple capsids accumulate at the membrane of enlarged multivesicular bodies (65, 69). Interestingly, analogous to our results in trehalose-treated HFFs and HAECs, the UL71 mutation has a large effect on release of virus into the supernatant and on viral spread but only a modest effect on production of cell-associated virus. Also relevant to this hypothesis are the reports that during lytic infection with another herpesvirus, Epstein-Barr virus, autophagy is blocked prior to fusion of the autophagosome with the lysosome, viral particles appear in double-membrane autophagic vesicles in the cytoplasm of the infected cells, and LC3B-II gets incorporated into mature virions (70, 71). Studies are currently in progress to further investigate this question with respect to the HCMV infection.
In summary, we found that trehalose induces autophagy in primary HFFs, HAECs, and neural cells derived from embryonic stem cells. Moreover, in the presence of trehalose, there was significant inhibition of HCMV gene expression, cytopathic effects, and virus production. Trehalose has been approved as a food ingredient in several countries, and in 2000 it was granted generally regarded as safe (GRAS) status by the U.S. FDA. The excellent safety profile of trehalose coupled with the results reported here provide support for the initiation of clinical trials to determine whether trehalose might be used as a safe antiviral in individuals with HCMV disease.
ACKNOWLEDGMENTS
We appreciate the use of the microscopy resources at the UCSD. We thank Bill Britt for providing the IE1 antibody and the members of the Spector lab for their support and comments on the manuscript.
This work was funded by NIH grants AI113443 and NS084037 to D.H.S.
REFERENCES
- 1.Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2772. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Bentz GL, Yurochko AD. 2008. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and β1 and β3 integrins. Proc Natl Acad Sci U S A 105:5531–5536. doi: 10.1073/pnas.0800037105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berencsi K, Endresz V, Klurfeld D, Kari L, Kritchevsky D, Gönczöl E. 1998. Early atherosclerotic plaques in the aorta following cytomegalovirus infection of mice. Cell Adhes Commun 5:39–47. doi: 10.3109/15419069809005597. [DOI] [PubMed] [Google Scholar]
- 4.Span AH, Grauls G, Bosman F, Boven F, van Boven CP, Bruggeman CA. 1992. Cytomegalovirus infection induces vascular injury in the rat. Atherosclerosis 93:41–52. doi: 10.1016/0021-9150(92)90198-P. [DOI] [PubMed] [Google Scholar]
- 5.Roberts RT, Haan MN, Dowd JB, Aiello AE. 2010. Cytomegalovirus antibody levels, inflammation, and mortality among elderly Latinos over 9 years of follow-up. Am J Epidemiol 172:363–371. doi: 10.1093/aje/kwq177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenfeld ME, Campbell LA. 2011. Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost 106:858–867. doi: 10.1160/TH11-06-0392. [DOI] [PubMed] [Google Scholar]
- 7.Cheng J, Ke Q, Jin Z, Wang H, Kocher O, Morgan JP, Zhang J, Crumpacker CS. 2009. Cytomegalovirus infection causes an increase of arterial blood pressure. PLoS Pathog 5:e1000427. doi: 10.1371/journal.ppat.1000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khoretonenko MV, Leskov IL, Jennings SR, Yurochko AD, Stokes KY. 2010. Cytomegalovirus infection leads to microvascular dysfunction and exacerbates hypercholesterolemia-induced responses. Am J Pathol 177:2134–2144. doi: 10.2353/ajpath.2010.100307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cobbs CS. 2013. Cytomegalovirus and brain tumor: epidemiology, biology and therapeutic aspects. Curr Opin Oncol 25:682–688. doi: 10.1097/CCO.0000000000000005. [DOI] [PubMed] [Google Scholar]
- 10.Feng Y, He D, Yao Z, Klionsky DJ. 2014. The machinery of macroautophagy. Cell Res 24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura S, Noda T, Yoshimori T. 2008. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct 33:109–122. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar S. 2013. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 41:1103–1130. doi: 10.1042/BST20130134. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. 2004. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 14.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. 2007. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 15.Du J, Liang Y, Xu F, Sun B, Wang Z. 2013. Trehalose rescues Alzheimer's disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol 65:1753–1756. doi: 10.1111/jphp.12108. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L, Le W. 2014. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 10:588–602. doi: 10.4161/auto.27710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, Court FA, van Zundert B, Hetz C. 2013. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9:1308–1320. doi: 10.4161/auto.25188. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Guo Y, Wang X, Yu X, Duan W, Hong K, Wang J, Han H, Li C. 2015. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 298:12–25. doi: 10.1016/j.neuroscience.2015.03.061. [DOI] [PubMed] [Google Scholar]
- 19.Correa A, Gilboa SM, Besser LM, Botto LD, Moore CA, Hobbs CA, Cleves MA, Riehle-Colarusso TJ, Waller DK, Reece EA. 2008. Diabetes mellitus and birth defects. Am J Obstet Gynecol 199:237.e1–237.e9. doi: 10.1016/j.ajog.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu C, Li X, Wang F, Weng H, Yang P. 2013. Trehalose prevents neural tube defects by correcting maternal diabetes-suppressed autophagy and neurogenesis. Am J Physiol Endocrinol Metab 305:E667–E678. doi: 10.1152/ajpendo.00185.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casarejos MJ, Perucho J, Lopez-Sendon JL, Garcia de Yebenes J, Bettencourt C, Gomez A, Ruiz C, Heutink P, Rizzu P, Mena MA. 2014. Trehalose improves human fibroblast deficits in a new CHIP-mutation related ataxia. PLoS One 9:e106931. doi: 10.1371/journal.pone.0106931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez-Estevez MA, Casarejos MJ, Lopez Sendon J, Garcia Caldentey J, Ruiz C, Gomez A, Perucho J, de Yebenes JG, Mena MA. 2014. Trehalose reverses cell malfunction in fibroblasts from normal and Huntington's disease patients caused by proteosome inhibition. PLoS One 9:e90202. doi: 10.1371/journal.pone.0090202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Zhang M, Ye H, Lin S, Yang Y, Wang L, Jones G, Trang H. 2013. Multiple toxicity studies of trehalose in mice by intragastric administration. Food Chem 136:485–490. doi: 10.1016/j.foodchem.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 24.Jackson WT. 2015. Viruses and the autophagy pathway. Virology 479–480:450–456. doi: 10.1016/j.virol.2015.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong X, Levine B. 2013. Autophagy and viruses: adversaries or allies? J Innate Immun 5:480–493. doi: 10.1159/000346388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McFarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM. 2011. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J Virol 85:4212–4221. doi: 10.1128/JVI.02435-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaumorcel M, Souquère S, Pierron G, Codogno P, Esclatine A. 2008. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 4:46–53. doi: 10.4161/auto.5184. [DOI] [PubMed] [Google Scholar]
- 28.Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, Esclatine A. 2012. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with beclin 1. J Virol 86:2571–2574. doi: 10.1128/JVI.05746-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DuRose JB, Li J, Chien S, Spector DH. 2012. Infection of vascular endothelial cells with human cytomegalovirus under fluid shear stress reveals preferential entry and spread of virus in flow conditions simulating atheroprone regions of the artery. J Virol 86:13745–13755. doi: 10.1128/JVI.02244-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W, Sun W, Zhang Y, Wei W, Ambasudhan R, Xia P, Talantova M, Lin T, Kim J, Wang X, Kim WR, Lipton SA, Zhang K, Ding S. 2011. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci U S A 108:8299–8304. doi: 10.1073/pnas.1014041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belzile JP, Stark TJ, Yeo GW, Spector DH. 2014. Human cytomegalovirus infection of human embryonic stem cell-derived primitive neural stem cells is restricted at several steps but leads to the persistence of viral DNA. J Virol 88:4021–4039. doi: 10.1128/JVI.03492-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomez-Sanchez R, Pizarro-Estrella E, Yakhine-Diop SM, Rodriguez-Arribas M, Bravo-San Pedro JM, Fuentes JM, Gonzalez-Polo RA. 2015. Routine Western blot to check autophagic flux: cautions and recommendations. Anal Biochem 477:13–20. doi: 10.1016/j.ab.2015.02.020. [DOI] [PubMed] [Google Scholar]
- 34.Sahani MH, Itakura E, Mizushima N. 2014. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 10:431–441. doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nixon RA. 2013. The role of autophagy in neurodegenerative disease. Nat Med 19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 36.Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. 2008. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nikoletopoulou V, Papandreou ME, Tavernarakis N. 2015. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ 22:398–407. doi: 10.1038/cdd.2014.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrill JA, Freudenrich TM, Machacek DW, Stice SL, Mundy WR. 2010. Quantitative assessment of neurite outgrowth in human embryonic stem cell-derived hN2 cells using automated high-content image analysis. Neurotoxicology 31:277–290. doi: 10.1016/j.neuro.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Chiramel AI, Brady NR, Bartenschlager R. 2013. Divergent roles of autophagy in virus infection. Cells 2:83–104. doi: 10.3390/cells2010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell GR, Spector SA. 2011. Hormonally active vitamin D3 (1α,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J Biol Chem 286:18890–18902. doi: 10.1074/jbc.M110.206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell GR, Spector SA. 2012. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog 8:e1002689. doi: 10.1371/journal.ppat.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV, Levine B. 2013. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494:201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yakoub AM, Shukla D. 2015. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci Rep 5:9730. doi: 10.1038/srep09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buckingham EM, Carpenter JE, Jackson W, Grose C. 2014. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J Virol 88:890–902. doi: 10.1128/JVI.02646-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buckingham EM, Carpenter JE, Jackson W, Zerboni L, Arvin AM, Grose C. 2015. Autophagic flux without a block differentiates varicella-zoster virus infection from herpes simplex virus infection. Proc Natl Acad Sci U S A 112:256–261. doi: 10.1073/pnas.1417878112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yakoub AM, Shukla D. 2015. Basal autophagy is required for herpes simplex virus-2 infection. Sci Rep 5:12985. doi: 10.1038/srep12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suarez AL, Kong R, George T, He L, Yue Z, van Dyk LF. 2011. Gammaherpesvirus 68 infection of endothelial cells requires both host autophagy genes and viral oncogenes for optimal survival and persistence. J Virol 85:6293–6308. doi: 10.1128/JVI.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shenk T, Alwine JC. 2014. Human cytomegalovirus: coordinating cellular stress, signaling, and metabolic pathways. Annu Rev Virol 1:355–374. doi: 10.1146/annurev-virology-031413-085425. [DOI] [PubMed] [Google Scholar]
- 49.LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, Seals DR. 2012. Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol 590:3305–3316. doi: 10.1113/jphysiol.2012.229690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. 2011. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog 7:e1002124. doi: 10.1371/journal.ppat.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chou S, Wechel LCV, Marousek GI. 2006. Effect of cell culture conditions on the anticytomegalovirus activity of maribavir. Antimicrob Agents Chemother 50:2557–2559. doi: 10.1128/AAC.00207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clippinger AJ, Maguire TG, Alwine JC. 2011. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J Virol 85:9369–9376. doi: 10.1128/JVI.05102-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alwine JC. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog 8:e1002878. doi: 10.1371/journal.ppat.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Das S, Ortiz DA, Gurczynski SJ, Khan F, Pellett PE. 2014. Identification of human cytomegalovirus genes important for biogenesis of the cytoplasmic virion assembly complex. J Virol 88:9086–9099. doi: 10.1128/JVI.01141-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol 74:975–986. doi: 10.1128/JVI.74.2.975-986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanchez V, Sztul E, Britt WJ. 2000. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-golgi-intermediate compartment. J Virol 74:3842–3851. doi: 10.1128/JVI.74.8.3842-3851.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol 85:5864–5879. doi: 10.1128/JVI.00155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cepeda V, Esteban M, Fraile-Ramos A. 2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell Microbiol 12:386–404. doi: 10.1111/j.1462-5822.2009.01405.x. [DOI] [PubMed] [Google Scholar]
- 60.Cepeda V, Fraile-Ramos A. 2011. A role for the SNARE protein syntaxin 3 in human cytomegalovirus morphogenesis. Cell Microbiol 13:846–858. doi: 10.1111/j.1462-5822.2011.01583.x. [DOI] [PubMed] [Google Scholar]
- 61.Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J Virol 83:11421–11428. doi: 10.1128/JVI.00762-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clippinger AJ, Alwine JC. 2012. Dynein mediates the localization and activation of mTOR in normal and human cytomegalovirus-infected cells. Genes Dev 26:2015–2026. doi: 10.1101/gad.196147.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Indran SV, Britt WJ. 2011. A role for the small GTPase Rab6 in assembly of human cytomegalovirus. J Virol 85:5213–5219. doi: 10.1128/JVI.02605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schauflinger M, Villinger C, Mertens T, Walther P, von Einem J. 2013. Analysis of human cytomegalovirus secondary envelopment by advanced electron microscopy. Cell Microbiol 15:305–314. doi: 10.1111/cmi.12077. [DOI] [PubMed] [Google Scholar]
- 65.Schauflinger M, Fischer D, Schreiber A, Chevillotte M, Walther P, Mertens T, von Einem J. 2011. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J Virol 85:3821–3832. doi: 10.1128/JVI.01540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharon-Friling R, Shenk T. 2014. Human cytomegalovirus pUL37x1-induced calcium flux activates PKCα, inducing altered cell shape and accumulation of cytoplasmic vesicles. Proc Natl Acad Sci U S A 111:E1140–E1148. doi: 10.1073/pnas.1402515111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bughio F, Elliott DA, Goodrum F. 2013. An endothelial cell-specific requirement for the UL133-UL138 locus of human cytomegalovirus for efficient virus maturation. J Virol 87:3062–3075. doi: 10.1128/JVI.02510-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bughio F, Umashankar M, Wilson J, Goodrum F. 2015. Human cytomegalovirus UL135 and UL136 genes are required for postentry tropism in endothelial cells. J Virol 89:6536–6550. doi: 10.1128/JVI.00284-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Womack A, Shenk T. 2010. Human cytomegalovirus tegument protein pUL71 is required for efficient virion egress. mBio 1:e00282-10. doi: 10.1128/mBio.00282-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Granato M, Santarelli R, Farina A, Gonnella R, Lotti LV, Faggioni A, Cirone M. 2014. Epstein-Barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J Virol 88:12715–12726. doi: 10.1128/JVI.02199-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nowag H, Guhl B, Thriene K, Romao S, Ziegler U, Dengjel J, Munz C. 2014. Macroautophagy proteins assist Epstein-Barr virus production and get incorporated into the virus particles. EBioMedicine 1:116–125. doi: 10.1016/j.ebiom.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]