

ABSTRACT
Primary Epstein-Barr virus (EBV) infection is the most common cause of infectious mononucleosis, and persistent infection is associated with multiple cancers. EBV vaccine development has focused on the major membrane glycoprotein, gp350, since it is the major target for antibodies that neutralize infection of B cells. However, EBV has tropism for both B cells and epithelial cells, and it is unknown whether serum neutralizing antibodies against B cell infection will provide sufficient protection against virus infection initiated at the oral mucosa. This could be stringently tested by passive antibody transfer and oral virus challenge in the rhesus macaque model for EBV infection. However, only neutralizing monoclonal antibodies (MAbs) against EBV are available, and EBV is unable to infect rhesus macaques because of a host range restriction with an unknown mechanism. We cloned the prototypic EBV-neutralizing antibody, 72A1, and found that recombinant 72A1 did not neutralize rhesus lymphocryptovirus (rhLCV) infection of macaque B cells. Therefore, we constructed a chimeric rhLCV in which the native major membrane glycoprotein was replaced with EBV gp350. This chimeric rhLCV became sensitive to neutralization by the 72A1 MAb, efficiently immortalized macaque B cells in vitro, and successfully established acute and persistent infection after oral inoculation of rhesus macaques. Thus, EBV gp350 can functionally replace rhLCV gp350 and does not restrict rhLCV infection in vitro or in vivo. The chimeric rhLCV enables direct use of an EBV-specific MAb to investigate the effects of serum neutralizing antibodies against B cell infection on oral viral challenge in rhesus macaques.
IMPORTANCE This study asked whether the EBV major membrane glycoprotein could functionally replace the rhLCV major membrane glycoprotein. We found that an rhLCV humanized with EBV gp350 is capable of efficiently immortalizing monkey B cells in vitro and reproduces acute and persistent infection after oral inoculation of macaques. These results advance our understanding of why EBV cannot infect rhesus macaques by proving that viral attachment through gp350 is not the mechanism for EBV host range restriction. Humanization of rhLCV with EBV gp350 also confers susceptibility to a potent EBV-neutralizing MAb and provides a novel and significant enhancement to the rhesus macaque animal model where both the clinical utility and biological role of neutralizing MAbs against B cell or epithelial cell infection can now be directly tested in the most accurate animal model for EBV infection.
INTRODUCTION
Primary Epstein-Barr virus (EBV) infection is the most common cause of infectious mononucleosis (IM), and persistent EBV infection is associated with cancers of B cell origin, e.g., Burkitt lymphoma and Hodgkin lymphoma, as well as epithelial cell origin, e.g., nasopharyngeal carcinoma and gastric carcinoma. A vaccine that prevents EBV-associated diseases would provide significant public health benefits (1).
The major membrane glycoprotein gp350 is considered an attractive vaccine candidate because it binds to the EBV receptor, CD21, and is important for viral attachment to B cells (2, 3). Antibodies that block this interaction can neutralize EBV infection of B cells in tissue culture, and gp350 is believed to be the major target for neutralizing antibodies present in the sera of EBV-infected humans (4–6). However, EBV clearly has tropism for both B cells and epithelial cells, and different viral glycoproteins are used for attachment to epithelial cells, i.e., gH/gL or BMRF2 (7, 8). Determining which cell type is initially infected and where virus is amplified during acute infection has important implications for effective EBV vaccine strategies.
EBV is usually transmitted to humans through the oral cavity, but how EBV penetrates the oral mucosa to initiate acute primary infection remains poorly understood. The traditional model is that incoming virus initially infects epithelial cells in the oral mucosa, where it may undergo lytic viral amplification. In this model, antibodies that neutralize EBV infection of epithelial cells may be more effective at limiting primary EBV infection than neutralizing antibodies that prevent B cell infection. Epithelial cell neutralizing antibodies would have to block only the small amount of incoming virus, whereas B cell neutralizing antibodies would have to block large amounts of virus after it is amplified in epithelial cells and released from the basal surface to infect B cells. Viruses could also be transmitted directly from epithelial cells to B cells through cell-cell interactions, where B cell neutralizing antibodies may not have access to free virions. As an alternative model, incoming virus may infect B cells directly by passing through microfissures in the oral mucosa, where B cell neutralizing antibodies may be very effective at blocking the small amount of incoming virus. These two models are not necessarily mutually exclusive, and a combination of epithelial and B cell neutralizing antibodies may be required to most effectively block incoming virus at the oral mucosa.
Rhesus macaques provide an animal model in which important aspects of EBV biology and vaccine development can be stringently investigated. However, macaques cannot be infected with EBV, so the model uses the related herpesvirus, lymphocryptovirus (LCV), naturally infecting rhesus macaques (rhLCV). Sequencing of the rhLCV genome showed a repertoire of viral proteins identical to that of EBV, and all rhLCV proteins studied to date function through the same molecular pathways as their EBV orthologues (9–11). The biology of natural and experimental rhLCV infection in rhesus macaques appears identical to EBV infection of humans (12). Importantly, LCV-naive rhesus macaques can be experimentally infected with rhLCV by oral inoculation, recapitulating the natural route of infection and allowing the potential involvement of epithelial cells within the oral mucosa (12, 13). After inoculation, there is an acute viremia, followed by a lifelong, persistent infection associated with virus-specific humoral and cellular immune responses that closely parallel the human immune responses to EBV infection (14–17). Acute rhLCV infection can induce IM symptoms in some animals, e.g., atypical lymphocytosis, lymphadenopathy, and splenomegaly (12). In addition, immunosuppression of rhesus macaques can lead to rhLCV-induced diseases in both epithelial cells, e.g., oral hairy leukoplakia, and B cells, e.g., lymphoma (18, 19). Thus, rhesus macaques provide a biologically accurate model to investigate the activities of specific immune effectors against oral infection and to test potential vaccine strategies. Specifically, neutralizing antibodies that block B cell or epithelial cell infection could be transfused into naive rhesus macaques, followed by oral challenge with rhLCV, to identify which strategy might be most effective against viral infection via the natural route.
However, no neutralizing monoclonal antibodies (MAbs) targeting rhLCV have been generated. A MAb against EBV gH/gL that neutralizes epithelial cell infection fortuitously cross-reacts with rhLCV gH/gL (rhgH/rhgL) (20). We tested the prototypic MAb against EBV gp350 that neutralizes B cell infection, 72A1 (4), and found that it did not cross-neutralize rhLCV. Therefore, we replaced the major membrane glycoprotein in rhLCV with EBV gp350 and tested whether EBV gp350 could functionally substitute for the native rhLCV glycoprotein and confer sensitivity to neutralization with 72A1.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were approved by the Harvard Medical School (HMS) Institutional Animal Care and Use Committee (IACUC) and were performed in accordance with guidelines from the National Institutes of Health, U.S. Department of Agriculture, and HMS.
Animal infection.
rhLCV-naive rhesus macaques from the extended specific-pathogen-free colony at the New England Primate Research Center were assigned and confirmed to be rhLCV naive shortly before infection. The animals were inoculated with 106 transforming units (TU) of virus applied nontraumatically throughout the oral cavity.
Cell culture.
The 72A1 hybridoma (4) and all lymphoblastoid cell lines (LCLs) were grown in RPMI medium supplemented with 10% fetal calf serum (FCS), 100 units/ml penicillin, and 100 μg/ml streptomycin. HEK-293 cells (ATCC, Manassas, VA) were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin. GH3Δ19 cells (kindly provided by Elliott Kieff [21]), used for gp350 antigen production, were grown in DMEM with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 200 μg/ml G418.
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized whole blood by density centrifugation over a Ficoll gradient, washed 3 times with phosphate-buffered saline (PBS), and counted by flow cytometric analysis using Count Bright counting beads (Life Technologies).
Flow cytometry.
The 72A1 hybridoma was subcloned, and cells growing out from single clones were stained with fluorescein isothiocyanate (FITC)-conjugated mouse anti-kappa (Southern Biotechnology) and phycoerythrin (PE)-conjugated mouse anti-lambda (Southern Biotechnology) antibodies for 30 min at 4°C. The cells were washed twice with PBS containing 1% FCS and 0.1% sodium azide to remove unbound antibody and analyzed on a FACSCalibur cytometer (BD Biosciences).
Sequencing of the 72A1 hybridoma and cloning of variable regions upstream of human constant regions.
RNA was isolated using RNA-Bee (Tel-Test, Inc.), and immunoglobulin chain variable regions were amplified by rapid amplification of cDNA ends (RACE) PCR using primers specific for the murine IgG1 constant region and Ig(κ)/Ig(λ)1 to -4 constant regions. Sequencing of the cloned immunoglobulin chains revealed two unique heavy-chain variable regions (IgG-H1 and IgG-H2) and two unique light-chain variable regions [one Ig(κ), L1, and one Ig(λ), L2]. Each of the heavy-chain variable regions was cloned upstream of the human IgG1 constant region of pCIRN to create pCIRN-H1 and pCIRN-H2. Each of the light-chain variable regions was similarly cloned upstream of the human Ig(κ) constant region of pEIG to create pEIG-L1 and pEIG-L2. The pCIRN and pEIG plasmids were kindly provided by Lisa Cavacini.
EBV gp350 ELISA.
Recombinant EBV gp350, truncated directly before the transmembrane anchor sequence, was isolated from GH3Δ19 cells by size exclusion chromatography (21). Cells were grown to 70 to 80% confluence and switched to serum-free DMEM for 7 days. The supernatants were precipitated in 60% ammonium sulfate for 1 h at 4°C and pelleted by centrifugation for 10 min at 12,000 × g. The precipitate was resuspended in 100 to 200 μl of PBS and dialyzed overnight against 4 liters of PBS. The dialyzed solution was then fractionated over a Sepharose 4B (Sigma-Aldrich) column, and fractions containing gp350 were identified by immunoblotting with EBV-immune human sera. For each enzyme-linked immunosorbent assay (ELISA), gp350+ fractions were diluted 1:200 in bicarbonate buffer (15 mM Na2CO3, 35 mM NaHCO3, 3 mM NaN3, pH 9.6) and used to coat 96-well polystyrene plates by incubation with 200 μl/well overnight at 4°C. The plates were washed 3 times with PBS–0.1% Tween 20 and blocked for 2 h with blocking solution (PBS, 0.1% Tween 20, 0.3% I-Block [Applied Biosystems]) at room temperature.
Recombinant antibodies were produced by transiently transfecting 293 cells with pCIRN-H1/H2 and pEIG-L1/L2 using Effectene (Life Technologies) according to the manufacturer's protocol. The supernatants were collected 48 h posttransfection, diluted 1:2 in blocking solution, and incubated on ELISA plates for 1 h at room temperature. Sera from an EBV-immune or EBV-naive donor were diluted 1:50 and used as controls. The plates were washed 3 times and incubated with horseradish peroxidase (HRP)-conjugated goat anti-human IgG (Jackson ImmunoResearch) diluted 1:1,000 in blocking solution for 1 h at room temperature. The plates were washed 3 times, and peroxidase activity was detected using SigmaFast OPD (Sigma-Aldrich) according to the manufacturer's protocol. All samples were tested in duplicate.
CFSE neutralization assay.
For neutralization assays, virus and antibody were combined in RPMI in a total volume of 50 μl and incubated for 90 min at 37°C. Two million PBMC were labeled with carboxyfluorescein succinimidyl ester (CFSE) (Life Technologies) according to the manufacturer's instructions, washed, resuspended in 50 μl RPMI, and added to the virus for an additional 90 min at 37°C. The cells were then moved to a 24-well plate and cultured in RPMI for 5 (human PBMC) or 10 (rhesus PBMC) days. Cells were harvested, washed with fluorescence-activated cell sorter (FACS) buffer (PBS, 1% FCS, 0.1% sodium azide), incubated with CD20-allophycocyanin (APC) (BD Biosciences) for 30 min at 4°C, washed, and analyzed by flow cytometry. All assay mixtures included no virus and virus without antibody pretreatment as controls.
Cloning of a chimeric rhLCV-hugp350 in a BAC and generation of an LCL carrying rhLCV-hugp350.
The gp350 open reading frame from the EBV B95-8 strain was inserted in place of rhgp350 in the molecular clone of wild-type (WT) rhLCV (WTr [22]) using a two-step galactokinase (GalK) recombination approach, as previously described (23). In brief, the rhgp350 coding sequence from the translational initiation to termination codons was replaced by the galactokinase gene using lambda recombinase-mediated homologous recombination. Correct insertion and general bacterial artificial chromosome (BAC) integrity were confirmed by PCR, BamHI restriction digestion, and sequencing analysis. Next, the GalK sequence was replaced with the EBV gp350 open reading frame using lambda recombinase-mediated homologous recombination. Correct insertion of an intact EBV gp350 open reading frame was confirmed by nucleotide sequencing, and general BAC integrity was confirmed by PCR and BamHI restriction digestion analysis.
The rhLCV-hugp350 BAC was transfected into the P3HR-1 cell line, and stable BAC-containing clones were selected by growth in hygromycin medium (400 μg/ml). Virus was induced by culturing cells in RPMI containing 20 ng/ml phorbol 12-myristate 13-acetate (TPA) and 3 mM butyrate. The cell supernatants were harvested after 5 to 7 days, filtered through a 0.45-μm filter, and used to infect rhesus PBMC. The infected cells were cultured for 6 to 8 weeks in RPMI with 10% FCS and 0.5 μg/ml cyclosporine and observed for outgrowth of a virus-immortalized LCL. An rhLCV-hugp350 LCL clone was confirmed to be free of P3HR-1 DNA by PCR, and the integrity of the chimeric rhLCV was confirmed by PCR and restriction analysis of BAC DNA rescued from the LCL clone and recovered in bacteria. The BAC vector sequence was removed by expression of Cre recombinase in the LCL, as previously described (22).
72A1 immunoprecipitation.
rhLCV-hugp350 LCLs and WT-rhLCV LCLs were cultured in the presence of 20 ng/ml TPA and 3 mM butyrate for 3 days to stimulate lytic replication and expression of the major membrane glycoprotein. The induced cells were lysed in lysis buffer (50 mM HEPES, pH 7.4, 250 mM NaCl, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.5% NP-40, 1× protease inhibitor mix) on ice for 30 min, and insoluble material was removed by centrifugation. The lysates were precleared by incubation with protein G Sepharose beads (GE Healthcare) for 30 min on ice and removal of the beads by centrifugation. The precleared lysates were incubated with 50 μl 72A1 hybridoma supernatant overnight at 4°C. The next day, samples were incubated with protein G Sepharose beads for 2 h at 4°C. The beads were washed three times with fresh lysis buffer by centrifugation, and bound proteins were analyzed by SDS-PAGE and immunoblotting with EBV-immune human sera diluted 1:1,000.
Large-scale production of virus.
Large numbers (>1 × 109 cells) of WT-rhLCV or rhLCV-hugp350 LCLs were induced by culture with 20 ng/ml TPA and 3 mM butyrate. The following day, the cells were resuspended in fresh medium at a concentration of 3 × 106 cells/ml. The cell supernatants were collected 7 days after induction, filtered through a 0.45-μm filter, and concentrated by centrifugation at 13,000 × g for 2 h at 4°C. Virus was resuspended in RPMI (1/300 to 1/550 of the starting culture volume). The titers of virus stocks were determined for B cell immortalization of rhesus lymphocytes as described below.
Determination of transforming titers and relative DNA contents of virus stocks.
The titers of virus lots were determined for transforming units by making serial 10-fold dilutions of virus and plating multiple replicates of each virus dilution into microtiter wells with 200,000 rhesus lymphocytes/well in RPMI, 10% FCS, and 0.5 μg/ml cyclosporine. Wells were scored positive at 8 weeks postinfection by visual confirmation of cell outgrowth. Transforming units were calculated as the dilution necessary for outgrowth in 50% of the wells based on the Reed-Muench equation.
Virus lots were assayed for relative DNA content by real-time PCR with SYBR green (Life Technologies) detection (35 cycles of 95°C for 15 s, 65°C for 30 s, and 72°C for 40 s) using rhBALF4-specific primers. All virus lots were tested using three independent dilutions (1:4,000), amplified in duplicate, and run simultaneously against a standard curve derived from a plasmid DNA clone containing a portion of rhBALF4. The 6 values obtained for each lot were averaged, and the relative values for each lot were expressed as arbitrary DNA units, resulting in a DNA unit/TU ratio of 10 for WT-rhLCV, as previously described (13).
Reverse transcriptase-mediated PCR detection of rhEBER in PBMC.
For detection of rhLCV-encoded hyperabundant RNAs (rhEBERs), total RNA was isolated from aliquots of 5 × 106 PBMC using RNA-Bee (Tel-Test, Inc.) and converted to cDNA using Moloney murine leukemia virus (M-MuLV) reverse transcriptase (NEB) and primers for rhEBER (173R) and rhGAPDH (glyceraldehyde-3-phosphate dehydrogenase) (R). Ninety percent of the cDNA was tested for rhEBER, and 10% was tested for GAPDH expression by PCR amplification (35 cycles of 95°C for 30 s, 60°C for 30 s, and 68°C for 30 s), as previously described (13). PCR products were separated by agarose gel electrophoresis, transferred to a nitrocellulose membrane by Southern blotting, hybridized with a digoxigenin-labeled rhEBER probe, and detected using an HRP-conjugated anti-digoxigenin (DIG) antibody (Jackson ImmunoResearch).
Frequency analysis for rhLCV-infected cells by limiting dilution.
To determine the frequency of rhLCV-infected cells in the peripheral blood, serial 2-fold dilutions of freshly isolated PBMC were prepared, starting at 2 × 106 cells, and aliquoted into multiple replicates for each dilution. RNA was then isolated from each replicate and used for the detection of rhEBERs, as described above. The frequency of infected cells was calculated by the number of positive and negative replicates in the dilutions using extreme-limiting-dilution analysis (24).
Nucleotide sequence accession numbers.
The nucleotide sequences for the variable regions of H1 and L2 have been submitted to GenBank (accession numbers KT211017 and KT211018, respectively).
RESULTS
Cloning of an EBV-neutralizing monoclonal antibody, 72A1.
Flow cytometric analysis of the 72A1 hybridoma cell line revealed simultaneous expression of at least two different immunoglobulin molecules, one with a kappa light chain and one with a lambda light chain (Fig. 1A). Both kappa- and lambda-containing immunoglobulin molecules could be detected by solid-phase antigen detection in all 72A1 hybridoma supernatants obtained from multiple sources. Subcloning and sequence analysis of the immunoglobulin mRNAs from the 72A1 hybridoma confirmed the expression of two unique IgG1 heavy chains and two unique light chains, one kappa and one lambda.
FIG 1.
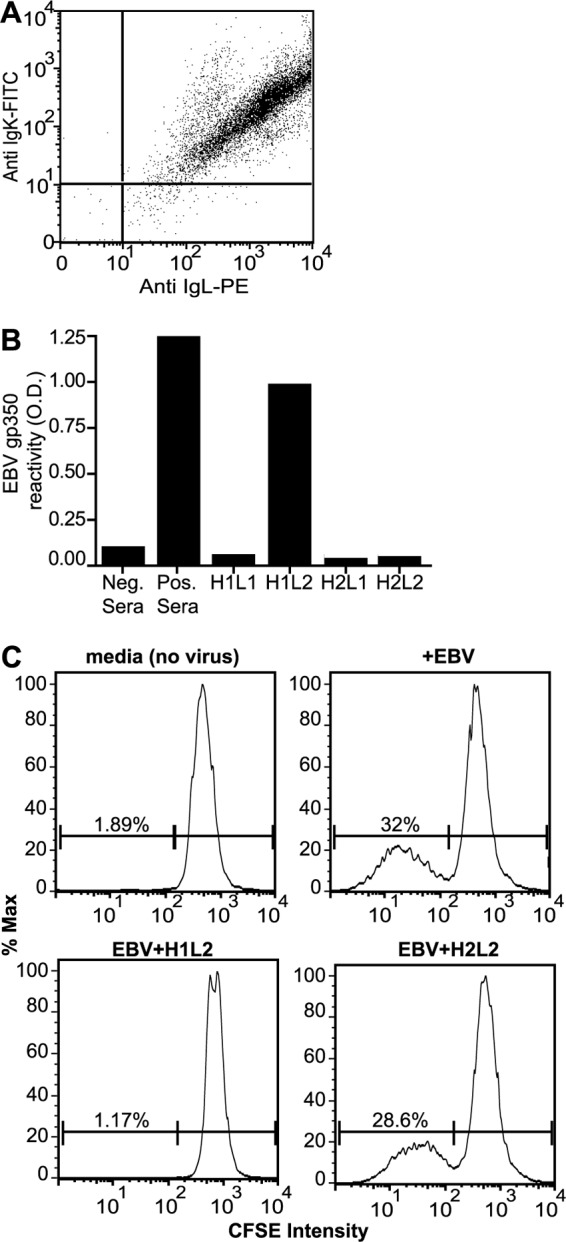
Cloning and expression of recombinant immunoglobulins from the 72A1 hybridoma that bind gp350 and neutralize EBV infection. (A) Simultaneous detection of murine kappa (IgK) and lambda (IgL) light-chain expression on 72A1 hybridoma cells by flow cytometry. (B) Testing recombinant antibodies for EBV gp350 binding by ELISA. 293 cells were transfected with the four different combinations of immunoglobulin expression plasmids [i.e., heavy-chain (H1 and H2) and light-chain (L1 and L2) variable regions of immunoglobulin mRNAs expressed in the 72A1 hybridoma cloned upstream of either the human IgG1 constant region or the human Ig(κ) constant region]. Cell supernatants were harvested 3 days after transfection, and reactivity to recombinant gp350 was measured by ELISA. Human sera from EBV-immune (Pos. Sera) or EBV-nonimmune (Neg. Sera) donors were used as controls. (C) EBV neutralization with recombinant H1L2 72A1. Human PBMC were stained with CFSE and exposed to medium, virus, or virus preincubated with recombinant antibody. Proliferation of human B cells was detected using flow cytometry by gating for live CD20+ cells and monitoring the reduction in CFSE intensity. The percentages of CFSE-low proliferating B cells relative to the percentages of CFSE-high nonproliferating B cells are indicated. The results from preincubation with H2L2 were similar to those with H1L1 and H2L1.
To define the pair that binds EBV gp350 and to confirm that this pair neutralizes EBV infection, we cloned the variable domains of the two heavy chains and two light chains on the constant regions of human IgG1 (H1 and H2) and Ig(κ) (L1 and L2), respectively. All four possible combinations of heavy and light chains were expressed after transfection of 293T cells and could be detected at equal levels by immunoblotting (F. Wang, unpublished data), confirming that each combination of heavy and light chains could interact to produce four different antibody molecules in the original 72A1 hybridoma cell line. All the antibodies were tested for the ability to detect EBV gp350 in an ELISA (Fig. 1B). Of the four combinations, only one (H1L2) bound gp350.
In addition, all four antibody combinations were tested for the ability to neutralize EBV infection of B cells in vitro. EBV infection of PBMC drives B cell proliferation that can be detected by monitoring loss of the cytoplasmic dye CFSE. B cells maintained in medium alone were detected as a single peak of CD20+ CFSE-bright cells (Fig. 1C, top left), and exposure to EBV resulted in B cell proliferation detected as multiple peaks of CD20+ CFSE-low cells (Fig. 1C, top right). Preincubation of EBV with H1L2 (Fig. 1C, bottom left), but not any of the other combinations (H2L2 is shown as a representative example [Fig. 1C, bottom right]), neutralized EBV-driven B cell proliferation and reduced the percentage of proliferated B cells to background levels. These data show that the same unique combination of heavy and light chains (H1L2) was responsible for both gp350 detection and EBV neutralization. Of note, another laboratory recently described a single pair of heavy- and light-chain mRNAs cloned from the 72A1 hybridoma (25). The amino acid sequence of their proposed antibody was identical to that of our H2L1 combination that showed no gp350 reactivity in our studies.
72A1 does not neutralize rhLCV infection of macaque B cells.
We next compared the ability of the recombinant 72A1 (H1L2) to neutralize EBV infection of human PBMC versus rhLCV infection of rhesus PBMC using a CFSE assay. Recombinant 72A1 inhibited EBV infection of human B cells, as expected, with >75% reduction in B cell proliferation when virus was preincubated with 100, 50, or 25 ng/ml of antibody (Fig. 2A). The same concentrations of recombinant 72A1 had no effect on rhLCV infection of rhesus B cells (Fig. 2B).
FIG 2.
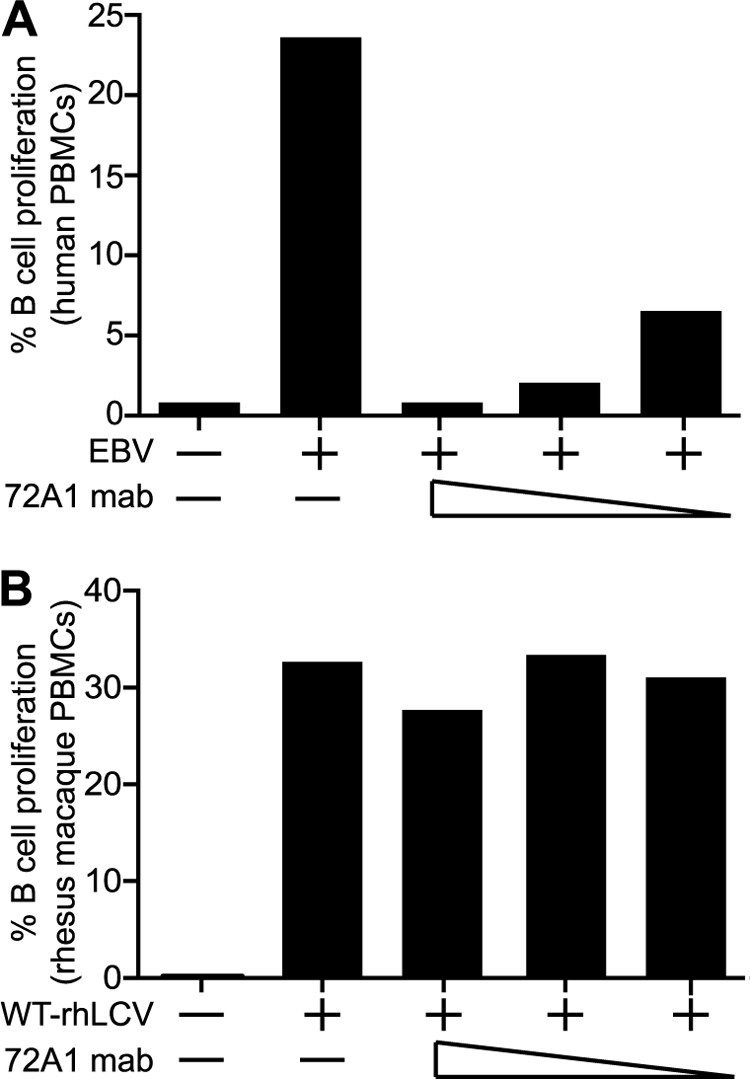
EBV, but not wild-type rhLCV, is sensitive to 72A1 neutralization. (A) CFSE-labeled human PBMC were exposed to medium, EBV, or EBV pretreated with 100, 50, or 25 ng/ml recombinant 72A1. Proliferation was detected using flow cytometry by gating live CD20+ cells and monitoring the reduction in CFSE intensity. The percentages of proliferating (CFSE-low) B cells are indicated. (B) CFSE-labeled rhesus macaque PBMC were similarly assayed following exposure to medium, rhLCV, or rhLCV pretreated with 100, 50, or 25 ng/ml recombinant 72A1.
Generation of a chimeric rhLCV expressing EBV gp350.
Since rhLCV was not neutralized by the prototypic EBV-neutralizing antibody, we tested whether creating a chimeric rhLCV that expressed EBV gp350 in place of rhLCV gp350 would confer sensitivity to 72A1 neutralization while preserving the ability to infect rhesus macaques. We replaced the native rhLCV gp350 (rhgp350) gene with the EBV gp350 (hugp350) gene in the wild-type rhLCV genome cloned as a BAC (WT-rhLCV BAC) (Fig. 3A). Proper insertion of the hugp350 sequence was confirmed by BamHI restriction digestion, Southern blotting, and hybridization with species-specific gp350 oligonucleotide probes. The rhgp350 probe detected the expected 1.4-kb BamHI DNA fragment in the WT-rhLCV BAC, but not in the rhLCV-hugp350 BAC. In contrast, the hugp350 probe did not hybridize to the WT-rhLCV BAC and detected a 10-kb BamHI DNA fragment in the rhLCV-hugp350 BAC representing a fusion of hugp350 with the rhLCV genome flanking the rhgp350 open reading frame (Fig. 3B). The integrity of the hugp350 coding sequence was confirmed by sequencing. Virus derived from the rhLCV-hugp350 BAC was recovered by immortalization of rhesus B cells and generation of a hygromycin-resistant immortalized LCL. PCR analysis confirmed that the viral genome in the LCL was chimeric, i.e., contained hugp350 and rhEBNA2 genes but not other EBV genes or rhgp350 (Fig. 3C).
FIG 3.
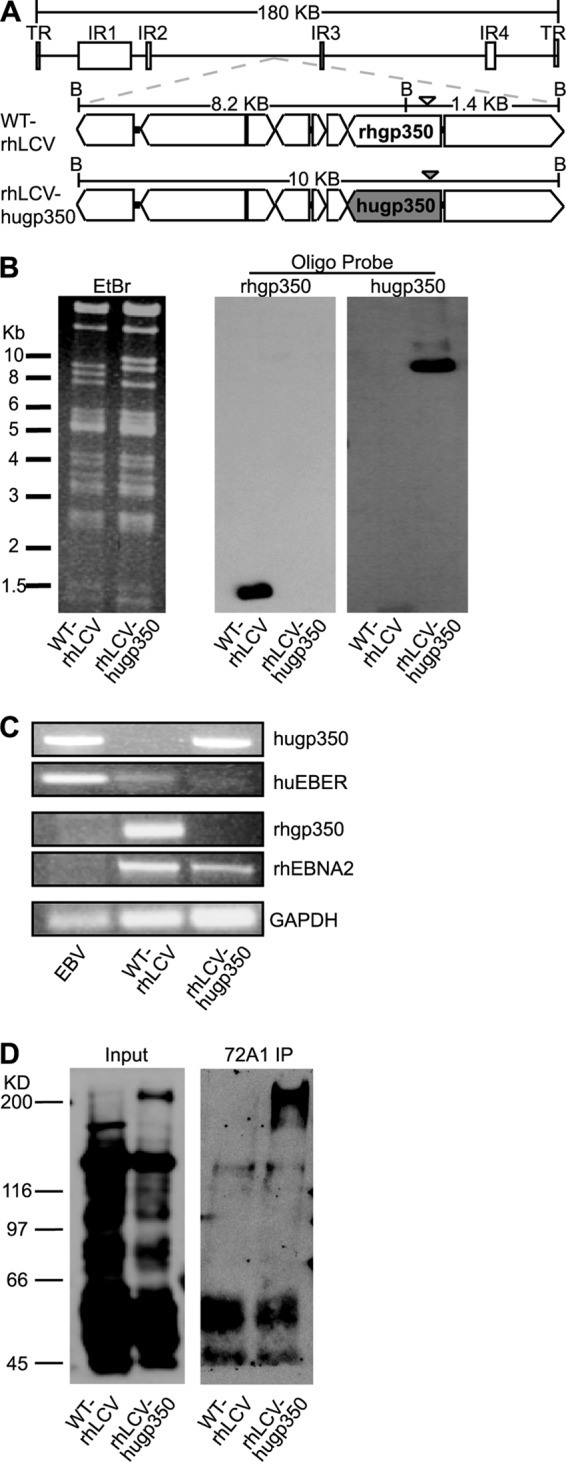
Generation of chimeric rhLCV-hugp350. (A) Schematic representation of the 10-kb region of the WT-rhLCV BAC (top) containing the gp350 open reading frame and surrounding genes. The native rhgp350 of the WT-rhLCV BAC is shown in the middle structure, and the replacement with the EBV gp350 in the chimeric rhLCV-hugp350 BAC is shaded in gray in the bottom structure. BamHI restriction sites are indicated by a B, and the distance between the restriction sites is noted. DNA probes used for detection of rhgp350 or hugp350 fragments after Southern blotting are shown as an open or filled arrowhead, respectively. (B) Validation of rhgp350 replacement with hugp350 in the rhLCV BAC. BamHI digests of WT-rhLCV BAC and chimeric rhLCV-hugp350 BAC recovered from the rhLCV-hugp350 LCL were separated by agarose gel electrophoresis. The ethidium bromide (EtBr)-stained gel is shown on the left. After Southern blotting, the membrane was hybridized with rhgp350- or hugp350-specific probes, as shown on the right. (C) The rhLCV-hugp350 LCL was infected with the BAC-derived chimeric rhLCV-hugp350. DNA from LCLs stably transformed with EBV, WT-rhLCV, and the chimeric rhLCV-hugp350 was assayed for hugp350, huEBER, rhgp350, and rhEBNA2 genes by PCR. GAPDH was amplified as a control. (D) The rhLCV-hugp350 LCL expresses EBV gp350. WT-rhLCV and rhLCV-hugp350 LCLs were induced for lytic replication, and EBV gp350 was precipitated from the cell lysates by incubation with 72A1. Whole-cell lysates are shown on the left, and 72A1 immunoprecipitation (IP) is shown on the right. EBV-immune human serum was used for immunoblotting.
Expression of the EBV gp350 protein in the rhLCV-hugp350 LCL was detected by immunoprecipitation with 72A1, followed by immunoblotting with EBV-immune human serum. After induction of lytic replication, a high-molecular-weight protein consistent with EBV gp350 was readily immunoprecipitated from the rhLCV-hugp350 LCL, but not the WT-rhLCV LCL (Fig. 3D, right). Thus, a recombinant rhLCV with the native rhgp350 deleted and expressing the EBV major membrane glycoprotein was successfully recovered.
rhLCV-hugp350 infects macaque B cells comparably to WT-rhLCV.
To determine whether use of EBV gp350 compromised the ability of rhLCV to enter macaque B cells, we assessed the in vitro transformation efficiency of rhLCV-hugp350 compared to that of WT-rhLCV. Any differences in the entry properties of the two gp350s should be detected as a difference in transforming activity, since the chimeric rhLCV-hugp350 and WT-rhLCV are otherwise identical, i.e., both are BAC derived and have identical latent proteins (EBNAs and LMPs) necessary for B cell immortalization.
First, multiple lots of cell-free virus were generated from WT-rhLCV and rhLCV-hugp350 LCLs, and the relative amount of virus in each lot was determined by real-time PCR. All the viral lots were assayed simultaneously to allow direct comparison between the wild-type and chimeric viruses, and values were expressed as arbitrary DNA units (Fig. 4A). A wide range of viral DNAs was expected, since each lot was independently derived from varying numbers of cells in which the induction of lytic replication and virus production might differ. Next, the number of transforming units per milliliter for each virus lot was determined by infecting multiple replicates of rhesus lymphocytes with serial dilutions of virus to derive the titer required to induce LCL outgrowth in 50% of the replicates (Fig. 4A).
FIG 4.
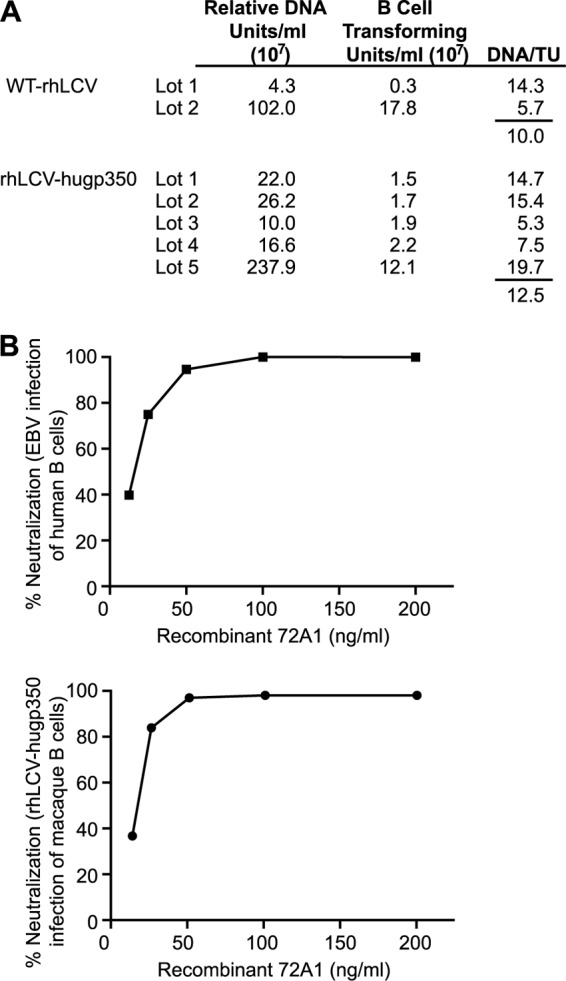
rhLCV-hugp350 immortalizes rhesus B cells similarly to the wild type and is sensitive to 72A1 neutralization. (A) The transformation efficiencies of WT-rhLCV and rhLCV-hugp350 were compared by calculating the DNA unit/TU ratio. Multiple large-scale virus preparations (lots 1 to 5) were assayed for B cell-transforming activity, and transforming units per milliliter were calculated as the dilution necessary for 50% outgrowth. The relative DNA content was assayed by real-time PCR, and all the samples were amplified in the same PCR run to allow comparison of DNA units. (B) rhLCV-hugp350 is as sensitive to 72A1 neutralization as EBV. Shown are CFSE neutralization assay results for rhLCV-hugp350 infection of rhesus PBMC (bottom) and EBV infection of human PBMC (top) when preincubated with 2-fold dilutions of recombinant 72A1. For both EBV and rhLCV infections, virus alone induced proliferation in approximately 25% of human and rhesus macaque B cells, respectively. Neutralization was calculated as follows: (percent CFSE-low cells with virus alone − percent CFSE-low cells of virus preincubated with antibody)/percent CFSE-low cells with virus alone.
The transformation efficiency was represented by the number of relative DNA units required for each transforming unit (Fig. 4A). The transformation efficiency for rhLCV-hugp350 was 12.5 DNA units per transforming unit and was not significantly different from the 10 DNA units/TU for WT-rhLCV. Thus, rhLCV infections of macaque B cells in vitro were comparable regardless of whether the EBV or rhLCV gp350 was used for attachment, i.e., EBV gp350 does not restrict rhLCV infection in vitro.
rhLCV-hugp350 is sensitive to 72A1 neutralization.
To determine whether the chimeric rhLCV-hugp350 had become sensitive to neutralization by 72A1, virus was incubated with recombinant 72A1 or control antibody before infection of CFSE-labeled rhesus macaque PBMC (Fig. 4B). Infection of macaque B cells with rhLCV-hugp350 was now readily neutralized by 72A1, and dilution studies showed that chimeric rhLCV-hugp350 infection of macaque B cells was as sensitive to 72A1 neutralization as EBV infection of human B cells (50% inhibitory concentrations, 14.5 ng/ml and 15.8 ng/ml, respectively). rhLCV-hugp350 did not induce proliferation or immortalize human PBMC.
rhLCV-hugp350 infects and persists in rhesus macaques.
To test whether the chimeric rhLCV-hugp350 could successfully infect a primate host, two rhLCV-naive rhesus macaques were orally inoculated with 106 TU of virus. In order to detect virus penetration of the oral mucosa and infection of peripheral blood B cells during acute infection (0 to 16 weeks), we assayed for the presence of the hyperabundant small RNAs (rhEBERs) in PBMC after oral virus inoculation. Multiple aliquots of 5 × 106 PBMC were tested for rhEBER expression as a semiquantitative measure of the acute viral load. Since the total amount of blood available over a given period was limited, this approach allowed more frequent sampling during the acute phase, when the number of virus-infected cells is likely to be changing more rapidly over time. Virus-infected B cells were first detected in at least one PBMC aliquot starting at week 1 postinoculation for both animals and remained detectable throughout the time course, consistent with previous reports of WT-rhLCV infection (Fig. 5A) (13). Overall, 18/48 (38%) and 31/48 (65%) aliquots were rhEBER positive during the acute phase for Mm218-05 and Mm143-97, respectively.
FIG 5.
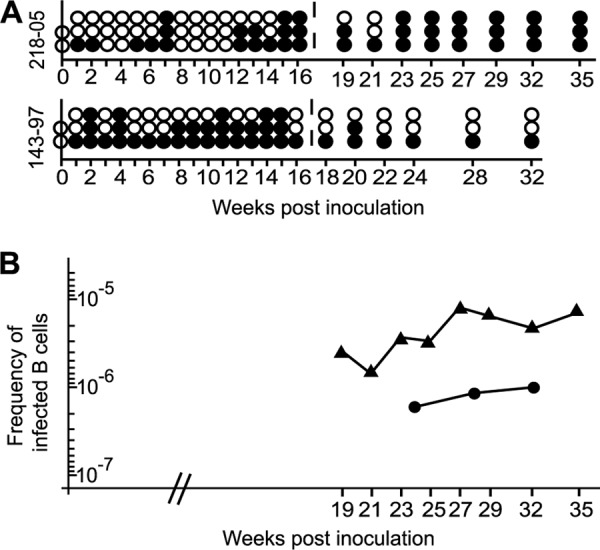
Chimeric rhLCV-hugp350 infects rhesus macaques following oral inoculation. (A) Detection of rhLCV-hugp350 infection in circulating PBMC of experimentally infected macaques during acute (0 to 16 weeks) and persistent (>16 weeks) infection. Shown is a schematic representation of rhEBER reverse transcription (RT)-PCR-positive (filled circles) and -negative (open circles) PBMC aliquots for two macaques (Mm218-05 and Mm143-97) after oral inoculation with rhLCV-hugp350. (B) rhLCV-hugp350 establishes a persistent infection in rhesus macaques. The frequency of rhLCV-hugp350-infected B cells was determined by limiting dilution of PBMC, rhEBER RT-PCR, and Poisson distribution (24). The frequency of infected B cells during persistent infection in Mm218-05 is represented by triangles and that in Mm143-97 by circles.
Virus-infected B cells were also detectable at all time points assayed during the persistent phase (>16 weeks), with 21/24 (88%) and 7/18 (39%) aliquots scoring rhEBER positive (Fig. 5A). As a more quantitative measure of persistent viral infection, we analyzed the frequency of infected B cells in the blood by limiting-dilution analysis at these later time points when larger blood volumes were available. The frequency of LCV-infected B cells in the peripheral blood at 6 different time points between weeks 19 and 35 postinoculation ranged from 1 infected cell in 707,276 B cells to 1 in 127,735 B cells for Mm218-05 (Fig. 5B). Mm143-97 had a range of 1 infected cell in 1,002,143 B cells to 1 in 1,668,387 B cells when assayed at three different time points from weeks 24 to 32 (Fig. 5B). Together, these results show that rhLCV-hugp350 can successfully cross the oral mucosa and establish a stable, persistent infection in macaques. Thus, EBV gp350 can substitute for the native rhLCV major membrane glycoprotein to confer susceptibility to 72A1 neutralization and still support acute and persistent infection of the natural host upon oral challenge.
DISCUSSION
EBV-related herpesviruses, or LCVs, naturally infect most, if not all, primates, i.e., hominoids, Old World nonhuman primates (NHP), and New World NHP. The ability of LCV to cross species, i.e., to immortalize B cells or infect hosts from other primate species, is becoming better defined and has important implications for EBV animal models. It is well established that EBV can immortalize B cells from New World NHP and can be used to infect New World monkeys (26, 27). However, the New World NHP animal model does not accurately reproduce many important aspects of EBV biology, since it requires parenteral injection of virus that bypasses the oral mucosa, and infection is either cleared or results in lethal lymphomas, i.e., there is no establishment of asymptomatic, lifelong persistent infection.
Experimental infection of rhesus macaques accurately reproduces the biology of EBV infection in humans, but it is unlikely that EBV could be successfully used as a challenge virus in this model. Experiments using EBV to inoculate rhesus macaques are dated and confounded by conflicting results. In two reports from the 1970s, seroconversion to viral capsid antigens was construed as evidence of successful infection after experimental inoculation of rhesus macaques (28, 29). However, the P3HR-1 and Raji EBV strains used for inoculation are now well recognized to have genomic deletions rendering them incapable of immortalizing human B cells and thus are likely to be defective in their ability to infect even their native human hosts (30). In many of these experiments, natural rhLCV infection among the test animals was not well recognized. Thus, the apparent “seroconversions” may have been recall responses of LCV cross-reactive antibodies induced by the antigenic stimulus of parenterally inoculated EBV. In contrast, a study that controlled for the transformation efficiency of EBV by using the same stocks of B95-8 EBV that had previously induced lymphomas in New World NHP was unable to detect seroconversion or illness in newborn or juvenile rhesus monkeys after experimental inoculation (31).
Indeed, successful experimental infection of rhesus macaques with EBV is highly unlikely, given that EBV is unable to immortalize rhesus macaque B cells in tissue culture. The published literature on LCV immortalization of B cells across hominoid and Old World NHP species was similarly dated and often used LCV isolates that were poorly characterized and are no longer available (for a review, see reference 32). We recently used molecularly characterized isolates with internal controls for virus titers and B cells from multiple species to reproducibly demonstrate a host range restriction between hominoids and Old World NHP that prevents EBV from immortalizing rhesus macaque B cells (33).
The fact that EBV cannot immortalize rhesus macaque B cells or establish successful infection in rhesus macaques is curious, since EBV and rhLCV share identical repertoires of viral genes. One might predict the host range restriction would be linked to the viral genes most divergent between EBV and rhLCV, i.e., those encoding gp350 (49% amino acid similarity) and the latent infection proteins essential for B cell immortalization (29 to 57% amino acid similarity) (34). Our current experiments formally demonstrate that EBV gp350 and rhLCV gp350 are functionally equivalent for infection of rhesus macaque B cells in vitro and that rhLCV can use EBV gp350 to penetrate the oral mucosa and establish persistent infection in rhesus macaques. Thus, suspicion for the mechanism underlying the host range restriction shifts to the latent infection proteins associated with the B cell immortalization process itself. Formal proof that EBV gp350 does not restrict infection of rhesus macaques is also important, because it allows the use of EBV-specific agents against gp350, e.g., the 72A1 MAb, in the more biologically authentic model of rhesus macaques.
The finding that the 72A1 hybridoma does not produce a single antibody was surprising, since this antibody has been used in EBV research for decades (4). It is not clear why the functional mRNA sequences we identified for 72A1 (H1L2) differ from those recently reported by another laboratory (25). The group did not describe the presence of two different heavy- and light-chain mRNAs in the 72A1 hybridoma, and their putative 72A1 sequences were identical to our H2L1 sequences, which did not react with EBV gp350 in our studies. The H2L1 sequences are highly similar to immunoglobulin molecules produced from the mineral oil-induced plasmacytoma (MOPC) fusion partner originally used to create the 72A1 hybridoma and are unlikely to encode EBV-specific antibodies (4).
Identifying and cloning the functional EBV-neutralizing MAb from the 72A1 hybridoma has translational implications. If recombinant 72A1 can effectively block viral amplification during acute primary LCV infection in the rhesus macaque model, then passive transfer of recombinant 72A1 may provide effective prophylaxis in clinical situations where patients are at high risk for complications associated with primary EBV infection. Among these high-risk patients are pediatric transplant recipients, who are frequently EBV negative prior to transplantation and who have an extremely high risk of posttransplantation lymphoproliferative disorder (PTLD) if they acquire primary EBV infection while immunosuppressed (35). Additionally, boys with the congenital immunodeficiency X-linked lymphoproliferative syndrome (XLP) are uniquely sensitive to primary EBV infection, with severe and often fatal outcomes (36). Antibody from the 72A1 hybridoma has been tested for safety in a limited number of pediatric liver transplant patients, but we now know that only a fraction of the hybridoma-produced antibody is truly EBV neutralizing (37). Thus, use of a recombinant, humanized 72A1 MAb in human studies will minimize the hypersensitivity reactions seen in the clinical trial and be more meaningful than polyclonal hybridoma antibody for testing efficacy of an EBV-neutralizing antibody.
The studies described here provide both technical and conceptual advancements with important translational implications. We have created the first chimeric LCV in which viral genes from different LCV species were exchanged, and we used this recombinant genetic approach to better understand the evolutionary differences between and mechanisms of host range restriction in LCV species. The chimeric virus provides a novel modification to the rhesus macaque animal model by conferring sensitivity to a potent, now well-defined EBV-neutralizing MAb targeting gp350. Thus, use of the rhLCV-hugp350 virus in the rhesus macaque model will allow testing whether serum neutralizing antibodies to B cell or epithelial cell infection are most effective in blocking viral amplification during acute infection after oral virus transmission. Direct testing of EBV-neutralizing MAbs in this “humanized” rhesus macaque animal model will also provide important preclinical data for their potential therapeutic use in humans.
ACKNOWLEDGMENTS
We thank Lisa Cavacini for providing the pEIG and pCIRN plasmids, Elliott Kieff for reagents, and Nina Orlova for technical assistance.
Funding Statement
This work was also supported by generous philanthropic donations to the Wang laboratory at Brigham and Women's Hospital. Services at the New England Primate Research Center (NEPRC) were supported in part by NEPRC base grant OD0111103.
REFERENCES
- 1.Cohen JI, Fauci AS, Varmus H, Nabel GJ. 2011. Epstein-Barr virus: an important vaccine target for cancer prevention. Sci Transl Med 3:107fs7. doi: 10.1126/scitranslmed.3002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nemerow GR, Mold C, Schwend VK, Tollefson V, Cooper NR. 1987. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J Virol 61:1416–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanner J, Weis J, Fearon D, Whang Y, Kieff E. 1987. Epstein-Barr Virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 50:203–213. doi: 10.1016/0092-8674(87)90216-9. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman GJ, Lazarowitz SG, Hayward SD. 1980. Monoclonal antibody against a 250,000-dalton glycoprotein of Epstein-Barr virus identifies a membrane antigen and a neutralizing antigen. Proc Natl Acad Sci U S A 77:2979–2983. doi: 10.1073/pnas.77.5.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorley-Lawson DA, Geilinger K. 1980. Monoclonal antibodies against the major glycoprotein (gp350/220) of Epstein-Barr virus neutralize infectivity. Proc Natl Acad Sci U S A 77:5307–5311. doi: 10.1073/pnas.77.9.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorley-Lawson D, Poodry CA. 1982. Identification and isolation of the main component (gp350-gp220) of Epstein-Barr virus responsible for generating neutralizing antibodies in vivo. J Virol 43:730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao J, Palefsky JM, Herrera R, Berline J, Tugizov SM. 2008. The Epstein-Barr virus BMRF-2 protein facilitates virus attachment to oral epithelial cells. Virology 370:430–442. doi: 10.1016/j.virol.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Kenyon WJ, Li Q, Mullberg J, Hutt-Fletcher LM. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J Virol 72:5552–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franken M, Devergne O, Rosenzweig M, Annis B, Kieff E, Wang F. 1996. Comparative analysis identifies conserved tumor necrosis factor receptor-associated factor 3 binding sites in the human and simian Epstein-Barr virus oncogene LMP1. J Virol 70:7819–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blake N, Moghaddam A, Rao P, Kaur A, Glickman R, Cho Y, Marchini A, Haigh T, Johnson R, Rickinson A, Wang F. 1999. Inhibition of antigen presentation by the glycine:alanine repeat domain is not conserved in simian homologues of Epstein-Barr virus nuclear antigen 1. J Virol 73:7381–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng R, Gordadze A, Panana EF, Wang F, Zong J, Hayward G, Tan J, Ling P. 2000. Sequence and functional analysis of EBNA-LP and EBNA2 proteins from nonhuman primate lymphocryptoviruses. J Virol 74:379–389. doi: 10.1128/JVI.74.1.379-389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moghaddam A, Rosenzweig M, Lee-Parritz D, Annis B, Johnson RP, Wang F. 1997. An animal model for acute and persistent Epstein-Barr virus infection. Science 276:2030–2033. doi: 10.1126/science.276.5321.2030. [DOI] [PubMed] [Google Scholar]
- 13.Ohashi M, Fogg MH, Orlova N, Quink C, Wang F. 2012. An Epstein-Barr virus encoded inhibitor of Colony Stimulating Factor-1 signaling is an important determinant for acute and persistent EBV infection. PLoS Pathog 8:e1003095. doi: 10.1371/journal.ppat.1003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fogg MH, Kaur A, Cho YG, Wang F. 2005. The CD8+ T-cell response to an Epstein-Barr virus-related gammaherpesvirus infecting rhesus macaques provides evidence for immune evasion by the EBNA-1 homologue. J Virol 79:12681–12691. doi: 10.1128/JVI.79.20.12681-12691.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fogg MH, Garry D, Awad A, Wang F, Kaur A. 2006. The BZLF1 homolog of an Epstein-Barr-related-herpesvirus is a frequent target of the CTL response in persistently infected rhesus macaques. J Immunol 176:3391–3401. doi: 10.4049/jimmunol.176.6.3391. [DOI] [PubMed] [Google Scholar]
- 16.Orlova N, Fogg MH, Carville A, Wang F. 2011. Antibodies to lytic infection proteins in lymphocryptovirus-infected rhesus macaques: a model for humoral immune responses to Epstein-Barr virus infection. Clin Vaccine Immunol 18:1427–1434. doi: 10.1128/CVI.05126-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orlova N, Wang F, Fogg MH. 2011. Persistent infection drives the development of CD8+ T cells specific for late lytic infection antigens in lymphocryptovirus-infected macaques and Epstein-Barr virus-infected humans. J Virol 85:12821–12824. doi: 10.1128/JVI.05742-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kutok JL, Klumpp S, Simon M, MacKey JJ, Nguyen V, Middeldorp JM, Aster JC, Wang F. 2004. Molecular evidence for rhesus lymphocryptovirus infection of epithelial cells in immunosuppressed rhesus macaques. J Virol 78:3455–3461. doi: 10.1128/JVI.78.7.3455-3461.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rivailler P, Carville A, Kaur A, Rao P, Quink C, Kutok JL, Westmoreland S, Klumpp S, Simon M, Aster JC, Wang F. 2004. Experimental rhesus lymphocryptovirus infection in immunosuppressed macaques: an animal model for Epstein-Barr virus pathogenesis in the immunosuppressed host. Blood 104:1482–1489. doi: 10.1182/blood-2004-01-0342. [DOI] [PubMed] [Google Scholar]
- 20.Wu L, Hutt-Fletcher LM. 2007. Compatibility of the gH homologues of Epstein-Barr virus and related lymphocryptoviruses. J Gen Virol 88:2129–2136. doi: 10.1099/vir.0.82949-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whang Y, Silberklang M, Morgan A, Munshi S, Lenny AB, Ellis RW, Kieff E. 1987. Expression of the Epstein-Barr virus gp350/220 gene in rodent and primate cells. J Virol 61:1796–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohashi M, Orlova N, Quink C, Wang F. 2011. Cloning of the Epstein-Barr virus-related rhesus lymphocryptovirus as a bacterial artificial chromosome: a loss-of-function mutation of the rhBARF1 immune evasion gene. J Virol 85:1330–1339. doi: 10.1128/JVI.01411-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y, Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 25.Tanner JE, Coincon M, Leblond V, Hu J, Fang JM, Sygusch J, Alfieri C. 2015. Peptides designed to spatially depict the Epstein-Barr virus major virion glycoprotein gp350 neutralization epitope elicit antibodies that block virus-neutralizing antibody 72A1 interaction with the native gp350 molecule. J Virol 89:4932–4941. doi: 10.1128/JVI.03269-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falk L, Wolfe L, Deinhardt F, Paciga J, Dombos L, Klein G, Henle W, Henle G. 1974. Epstein-Barr virus: transformation of non-human primate lymphocytes in vitro. Int J Cancer 13:363–376. doi: 10.1002/ijc.2910130312. [DOI] [PubMed] [Google Scholar]
- 27.Miller G, Shope T, Coope D, Waters L, Pagano J, Bornkamn G, Henle W. 1977. Lymphoma in cotton-top marmosets after inoculation with Epstein-Barr virus: tumor incidence, histologic spectrum antibody responses, demonstration of viral DNA, and characterization of viruses. J Exp Med 145:948–967. doi: 10.1084/jem.145.4.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen MH, Bernstein AD, Levine PH. 1974. Hematological and serological effects of Rauscher leukemia virus and Epstein-Barr virus on immunosuppressed newborn subhuman primates. Oncology 29:253–263. [PubMed] [Google Scholar]
- 29.Levine PH, Leiseca SA, Hewetson JF, Traul KA, Andrese AP, Granlund DJ, Fabrizio P, Stevens DA. 1980. Infection of rhesus monkeys and chimpanzees with Epstein-Barr virus. Arch Virol 66:341–351. doi: 10.1007/BF01320630. [DOI] [PubMed] [Google Scholar]
- 30.Heller M, Dambaugh T, Kieff E. 1981. Epstein-Barr virus DNA. IX. Variation among viral DNAs from producer and nonproducer infected cells. J Virol 38:632–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shope T, Dechairo D, Miller G. 1973. Malignant lymphoma in cottontop marmosets after inoculation with Epstein-Barr virus. Proc Natl Acad Sci U S A 70:2487–2491. doi: 10.1073/pnas.70.9.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mühe J, Wang F. 2015. Non-human primate lymphocryptoviruses: past, present, and future. Curr Top Microbiol Immunol 391:385–405. doi: 10.1007/978-3-319-22834-1_13. [DOI] [PubMed] [Google Scholar]
- 33.Mühe J, Wang F. 2015. Host range restriction of Epstein-Barr virus and related lymphocryptoviruses. J Virol 89:9133–9136. doi: 10.1128/JVI.01235-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rivailler P, Jiang H, Cho Y, Quink C, Wang F. 2002. Complete nucleotide sequence of the rhesus lymphocryptovirus: genetic validation for an Epstein-Barr virus animal model. J Virol 76:421–426. doi: 10.1128/JVI.76.1.421-426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Green M, Michaels MG. 2013. Epstein-Barr virus infection and posttransplant lymphoproliferative disorder. Am J Transplant 13(Suppl 3):41–54. doi: 10.1111/ajt.12004. [DOI] [PubMed] [Google Scholar]
- 36.Purtilo D, Cassel C, Yang J, Harper R. 1975. X-linked recessive progressive combined variable immunodeficiency (Duncan's disease). Lancet 26:935–940. [DOI] [PubMed] [Google Scholar]
- 37.Haque T, Johannessen I, Dombagoda D, Sengupta C, Burns D, Bird P, Hale G, Mieli-Vergani G, Crawford D. 2006. A mouse monoclonal antibody against Epstein-Barr virus envelope glycoprotein 350 prevents infection both in vitro and in vivo. J Infect Dis 194:584–587. doi: 10.1086/505912. [DOI] [PubMed] [Google Scholar]