

ABSTRACT
African swine fever virus (ASFV) is a major threat for porcine production that has been slowly spreading in Eastern Europe since its first appearance in the Caucasus in 2007. ASFV enters the cell by endocytosis and gains access to the cytosol to start replication from late endosomes and multivesicular bodies. Cholesterol associated with low-density lipoproteins entering the cell by endocytosis also follows a trafficking pathway similar to that of ASFV. Here we show that cholesterol plays an essential role in the establishment of infection as the virus traffics through the endocytic pathway. In contrast to the case for other DNA viruses, such as vaccinia virus or adenovirus 5, cholesterol efflux from endosomes is required for ASFV release/entry to the cytosol. Accumulation of cholesterol in endosomes impairs fusion, resulting in retention of virions inside endosomes. ASFV also remodels intracellular cholesterol by increasing its cellular uptake and redistributes free cholesterol to viral replication sites. Our analysis reveals that ASFV manipulates cholesterol dynamics to ensure an appropriate lipid flux to establish productive infection.
IMPORTANCE Since its appearance in the Caucasus in 2007, African swine fever (ASF) has been spreading westwards to neighboring European countries, threatening porcine production. Due to the lack of an effective vaccine, ASF control relies on early diagnosis and widespread culling of infected animals. We investigated early stages of ASFV infection to identify potential cellular targets for therapeutic intervention against ASF. The virus enters the cell by endocytosis, and soon thereafter, viral decapsidation occurs in the acid pH of late endosomes. We found that ASFV infection requires and reorganizes the cellular lipid cholesterol. ASFV requires cholesterol to exit the endosome to gain access to the cytoplasm to establish productive replication. Our results indicate that there is a differential requirement for cholesterol efflux for vaccinia virus or adenovirus 5 compared to ASFV.
INTRODUCTION
Endocytosis is a common uptake pathway for nutrients, lipids, and receptors that is frequently hijacked by viruses to gain entry into cells (1). Cargoes taken up by endocytosis initially converge on early endosomes (EE), from where they can be recycled back to the plasma membrane (PM) (2). However, more usually, internalized cargo is transported to late endosomes (LE), from where it can be directed to lysosomes (LY) for degradation. Endosomal maturation from EE to lysosomes is a dynamic process that involves a progressive reduction in intraluminal pH (2). At the start of their maturation, invaginations of the limiting membrane into the lumen of the EE give rise to the intraluminal vesicles (ILV), leading to the formation of multivesicular bodies (MVB), in which the pH is proportional to the number of ILV (2). MVB mature to late endosomes ending in the lysosomes (3). Both MVB and LE are late endosomal compartments. The ordered maturation of early endosomes to lysosomes depends on endosomal membrane signaling that is regulated by both proteins and lipids, including cholesterol (CH) (2, 4).
Cholesterol enters the cell by endocytosis in the form of low-density lipoproteins (LDL) and accumulates in the intraluminal vesicles (ILV) in the multivesicular bodies and late endosomes (5). Those ILV that accumulate cholesterol are also enriched in the long-lived lysobisphosphatidic acid (LBPA) (6). Cholesterol must be redistributed to the endosomal limiting membrane so that it becomes available for export to other cellular destinations (7). LBPA regulates cholesterol efflux by a mechanism known as “backfusion,” in which the ILV fuses with the endosomal limiting membrane (6, 8). Backfusion ensures that cholesterol in ILV escapes lysosomal degradation by exiting the LE. This cholesterol efflux is controlled by a number of lipid transporters, including the NPC1 and NPC2 proteins (9). Mutations in these proteins lead to the hereditary Niemann-Pick C (NPC) disease, which is characterized by a severe defect in the export of cholesterol from late endosomes (10). The abnormal accumulation of cholesterol in LE leads to altered protein and lipid trafficking that results in a progressive neurodegenerative disorder (11). This disease phenotype can be mimicked by treating cells with U18666A, an amphipathic steroid that blocks the exit of free cholesterol from the late endosomal compartment, resulting in aggregation of endosomes filled with cholesterol and other lipids (12).
The endocytic pathway ensures a highly dynamic and ordered sorting of cargoes to their correct cellular locations. Not surprisingly, many viruses exploit endocytosis to facilitate their uptake and ensure that they reach the correct site for replication or gain access to the cytoplasm (1, 13, 14). For example, African swine fever virus (ASFV) is a large double-stranded DNA virus that enters the cell by dynamin-, clathrin-, and cholesterol-dependent endocytosis (15, 16). Actin-dependent macropinocytosis also assists ASFV entry into cells (17). Once the virus is inside the endosome, viral decapsidation occurs in the acid pH of LE within a few minutes after entry (18). After uncoating, the virus needs to exit the endosome and gain access to the cytoplasm to start replication in a specialized structure called the viral factory (19). The viral factory is devoid of a limiting membrane and is organized in a single perinuclear site near the microtubule-organizing center (20, 21). After completing morphogenesis in the viral factory, newly assembled virions are moved to the plasma membrane on microtubules by kinesin-1, where they acquire an additional lipid membrane when they exit the cell by budding (22, 23). In this study, we set out to investigate the relevance of cholesterol homeostasis in ASFV entry and replication. Our results clearly demonstrate that cholesterol efflux and endosomal trafficking play essential roles during the establishment of ASFV replication.
MATERIALS AND METHODS
Cells, virus, and infections.
Vero cells were mock infected or infected with ASFV isolate Ba71V as previously described (24, 25) at a multiplicity of infection (MOI) of 1 or 10 (for early-time-point experiments) or 100 (for transmission electron microscopy [TEM]) PFU/cell. When synchronization of infection was required, virus adsorption was performed for 90 min at 4°C, and after a cold phosphate-buffered saline (PBS) wash to remove unbound virus, cells were rapidly shifted to 37°C with fresh prewarmed medium. Virus stocks and/or infective ASFV production yields from infected samples were titrated by plaque assay in Vero cells as previously described (24). Recombinant B54GFP is a recombinant ASFV expressing green fluorescent protein (GFP) as a fusion protein with viral p54 (26). Infections with B54GFP were performed at an MOI of 1 PFU/cell. Vaccinia virus (VV) recombinant vtag2GFP contained the tag2GFP under the control of a strong synthetic VV early/late promoter (27) and was kindly provided by R. Blasco (INIA, Spain). VV infections were performed at an MOI of 1 PFU/cell for 16 h postinfection (hpi). Adenovirus 5/attB (Adv) was provided by Carmen San Martín (CNB, CSIC). This recombinant virus carries a packaging modification that lengthens the viral cycle and GFP to facilitate detection (28, 29). Vero cells were infected with Adv at an MOI of 1 PFU/cell for 16 hpi.
Detection and quantitation of the ASFV genome.
The quantitation of the number of copies of the ASFV genome was achieved by quantitative real-time PCR (qPCR) as described previously (15). The qPCR assay used fluorescent hybridization probes to amplify a region of the p72 viral gene as described previously (30). Each sample was included in triplicates, and values were normalized to the standard positive controls. Reactions were performed using the ABI 7500 Fast real-time PCR system (Applied Biosystems).
Antibodies.
Monoclonal antibodies against the virus major capsid protein p72 (Ingenasa) and early protein p30 were used at 1:1,000 and 1:100 dilutions, respectively (31). ASFV p72 is a very abundant late protein in infection and is accumulated in the viral factory (32).
EE were labeled with anti-mouse EEA1 antibody (BD Biosciences Pharmingen), and LE with anti-rabbit Rab7 (Cell Signaling), both at a 1:50 dilution. MVB were labeled with anti-CD63 (clone H5C6; Developmental Studies Hybridoma Bank, University of Iowa), a tetraspanin characteristic of this compartment, at a 1:200 dilution. LY were labeled with anti-Lamp1 (Abcam) at a 1:50 dilution. Antibody 6C4 against lysobisphosphatidic acid (LBPA), which is enriched in intraluminal vesicles of multivesicular bodies, was used at a concentration of 50 μg/ml (kindly provided by Jean Gruenberg). Anti-mouse immunoglobulin G (IgG) antibody conjugated to Alexa Fluor 594 and anti-rabbit IgG antibody conjugated to Alexa Fluor 488 (Molecular Probes) were used as secondary antibodies and diluted 1:200.
Protein detection by WB.
Protein extracts were electrophoresed in 12% acrylamide-bisacrylamide gels. Separated proteins were transferred to nitrocellulose membranes and detected with corresponding antibodies. As a secondary antibody, anti-mouse IgG (GE Healthcare) or anti-rabbit IgG (Bio-Rad) conjugated to horseradish peroxidase was used at a 1:5,000 dilution. β-Tubulin (Sigma) was used as a loading control in Western blotting (WB) analysis. Finally, bands obtained after development with ECL reagent were detected on a Molecular Imager Chemidoc XRSplus imaging system. Bands were quantified by densitometry and data normalized to control values using Image lab software (Bio-Rad).
Indirect immunofluorescence and conventional and confocal microscopy.
Immunofluorescence experiments were performed as previously described (18). Vero cells were fixed in PBS–4% paraformaldehyde (PFA) for 15 min and permeabilized with PBS–0.1% Triton X-100 or saponin (Sigma) for 10 min. Cells were then incubated with 50 mM NH4Cl in PBS for 10 min. After blocking with bovine serum albumin (BSA) (Sigma) or normal goat serum (Sigma), cells were incubated with corresponding antibodies and nuclei were stained with Topro3 (Molecular Probes) before mounting.
Confocal microscopy was carried out in a Leica TCS SPE confocal microscope. Conventional fluorescence microscopy to analyze cholesterol staining with filipin was carried out using a Leica DM RB microscope. Image analyses were performed with Leica Application Suite advanced fluorescence software (LAS AF) and ImageJ software.
TEM.
Vero cells were pretreated with U18666A for 16 h or with dimethyl sulfoxide (DMSO). Cells were then infected with Ba71V at an MOI of 100 PFU/cell. Virus adsorption was carried out by spinoculation as previously described (33), and cells were then placed at 37°C for 30 or 45 min postinfection (mpi). Cells were then washed with PBS and fixed with a mixture of 2% PFA and 2.5% glutaraldehyde in PBS for 1 h. The cell monolayer on coverslips was postfixed with 1% osmium tetroxide in PBS (45 min), treated with 1% aqueous uranyl acetate (45 min), dehydrated with ethanol, and embedded in epoxy resin 812 (TAAB) for 2 days at room temperature as described previously (34). Ultrathin, 70-nm-thick sections were obtained with the Ultracut UCT ultramicrotome (Leica Microsystems), transferred to nickel EM grids (Gilder), and stained with 3% aqueous uranyl acetate for 20 min and lead citrate for 2 min. Sections were examined on a JEOL JEM 1200 EXII electron microscope (operating at 100 kV).
Flow cytometry.
At 6 hpi, cells were washed with PBS and harvested by trypsinization. Flow cytometry experiments were performed by detection of infected cells with an anti-p30 monoclonal antibody as described elsewhere (18) or by GFP expression at 16 hpi when using recombinant viruses; 104 cells per tube in triplicates were scored and analyzed in a FACSCalibur flow cytometer (BD Sciences) to determine the percentage of infected cells under these conditions. The obtained infection rates were normalized to the corresponding controls.
Cell treatment with U18666A.
U18666A (Sigma) is an amphipathic steroid which is widely used to block the intracellular trafficking of cholesterol and mimic the Niemann-Pick type C disease phenotype (12, 35). Cytotoxicity was tested using the CellTiter 96 nonradioactive cell proliferation assay (Promega) following the manufacturer′s instructions. Vero cells were pretreated for 16 h with DMSO or 5 or 10 μM U18666A and then infected with the different viruses tested at an MOI of 1 PFU/cell without washing.
Cholesterol quantitation and determination.
Cholesterol was determined and quantified enzymatically using the Amplex Red cholesterol assay kit, following the manufacturer's instructions (Invitrogen). Fluorescence values at an excitation wavelength of 532 nm and an emission wavelength of 590 nm were counted in a Genios Spectrafluor microplate reader (Tecan), normalized to control sample values, and expressed in percentages.
CHDM, CHEM, and cell treatment.
Vero cells were grown in cholesterol-depleted medium (CHDM) containing Dulbecco modified Eagle medium containing 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine [DMEM (P/S+G)], 5% lipoprotein deficient serum (LPDS) (Sigma), 10 μM mevastatin (Santa Cruz Biotechnology), 50 μM mevalonate (Sigma), and 5 mM methyl-β-cyclodextrin (MβCDX) (Sigma). Chemicals were previously tested for cytotoxicity at the concentrations used. Cells were incubated in CHDM for 2 h before infection, at the time of infection (0 hpi), or 2 and 4 hpi. To avoid cytotoxicity of MβCDX (36), after 2 h, CHDM was shifted to CHDM lacking MβCDX (CHDM−) and maintained up to 6 or 16 hpi. Cell extracts were analyzed for early p30 expression at 6 hpi and for viral replication using qPCR at 16 hpi. Viral infectivity was evaluated by GFP expression of recombinant ASFV, VV, and Adv at 16 hpi in cells incubated in CHDM for 2 h before infection.
Cholesterol-enriched medium (CHEM) was prepared as described previously (37, 38). Briefly, 10 mM cholesterol (CH) (Santa Cruz Biotechnology) stock was prepared in stirred and warmed DMSO at 80°C on a heating block. The clarified solution was filtered and added in increasing concentrations (0, 100, and 500 μM) to DMEM (P/S+G) containing 5 mM MβCDX to form CH/MβCDX complexes. Vero cells were incubated with CHEM for 2 h, medium was removed, and cells were washed for 6 times to clear remnant MβCDX. After washing, cells were infected at an MOI of 1 PFU/cell and maintained in DMEM (P/S+G) containing 2% LPDS. At 2 hpi, the medium was replaced with DMEM–2% fetal calf serum (FCS) (normal infection medium), and infection progressed for 16 hpi. Finally, cell extracts were processed for qPCR.
Cholesterol staining.
To detect free intracellular cholesterol, we used fluorescent filipin (Sigma) as previously described (39–42). The filipin signal was recorded using a 390- to 415-nm-wavelength excitation filter and a 450- to 470-nm-wavelength emission filter. For living cells, we used fluorescently labeled bodipy-cholesterol (TopFluor-Cholesterol; Avanti Polar Lipids), a cholesterol live-cell mimic. Vero cells were loaded with 5 μg/ml of bodipy-cholesterol (bodipy-CH) in DMEM–2% FBS for 30 min at 37°C, and then cells were infected at an MOI of 5 PFU/cell for 1 or 3 h in order to analyze its uptake by endocytosis. Cells were then quickly rinsed with PBS and fixed with 4% PFA before confocal imaging. Intracellular fluorescence measurement of bodipy-CH from mock- or ASFV-infected cells was performed using ImageJ software.
Statistical analysis.
Differences between groups were analyzed by the Bonferroni test with GraphPad Prism software and Instat software. All experiments were performed more than two times, and data are presented as mean values and standard deviations (SD) from independent experiments. Metrics were normalized to control values. Asterisks denote statistically significant differences (***, P < 0.001; **, P < 0.01; and *, P < 0.05).
RESULTS
We set out to investigate the role of cholesterol at different stages of the ASFV infection and replication cycle (Fig. 1A). To examine the impact of cholesterol depletion on viral infectivity, we infected cells previously incubated in cholesterol-depleted medium (CHDM) for 2 h. This treatment resulted in a significant reduction of cellular cholesterol pools as revealed by filipin staining (Fig. 1B). To assess whether cholesterol is required for ASFV entry and/or replication, we infected cells that were grown in cholesterol-depleted medium at various times before and after virus binding at 4°C. Lowering cholesterol pools 2 h before infection significantly impaired ASFV early protein expression and viral replication (Fig. 1C). However, the impact was lower after virus binding (0 to 4 hpi), suggesting that cholesterol was required at early time points. Consistent with this, incubating cells grown in CHDM with cholesterol-enriched medium at increasing concentrations recovered viral replication to control levels (Fig. 1D). In these experiments, cells were shifted to normal infection medium at 2 hpi (baseline) or to medium enriched with 100 or 500 μM. Next, we analyzed the effect of cholesterol-depleted medium on ASFV infectivity in comparison with that of other DNA viruses, such as vaccinia virus (VV) and adenovirus 5 (Adv). To evaluate infectivity, we used recombinant viruses expressing GFP and found that CHDM impaired infectivity in all viruses tested, as expected from a general requirement of membrane cholesterol for endocytic uptake.
FIG 1.

Effect of low cholesterol concentrations on ASFV infection. (A) ASFV infectious cycle milestones. (B) Intracellular cholesterol before (Ctrl) and after incubation of cells in cholesterol-depleted medium (CHDM). (C) Graphical representation of viral p30 protein expression at 6 hpi and of viral replication at 16 hpi in ASFV-infected cells incubated with CHDM added at the indicated times. (D) Viral replication in cells shifted at 2 hpi to cholesterol-enriched medium at the indicated cholesterol concentration (100 or 500 μM) or DMEM with 2% fetal calf serum (not lipoprotein depleted). (E) Variations in ASFV, VV, and Adv infectivity in Vero cells incubated in control medium or CHDM. Infected cells were detected by fluorescence-activated cell sorter (FACS) analysis and percentages normalized to controls. Statistically significant differences are indicated by asterisks (***, P < 0.001; **, P < 0.01). Representative FACS profiles of control, ASFV-infected, or ASFV-infected cells in CHDM are shown.
We next analyzed whether ASFV infection affects the levels of free, esterified, and total cholesterol during its replication cycle (Fig. 2A). Cholesterol content values were normalized to the same values obtained in mock-infected controls at each time point. Early in infection (1 to 3 hpi), total cholesterol levels increased, before returning to noninfected levels at late viral replication times. Free cholesterol increased from 70 to 90% at these early times. A similar increase was also seen for both free and esterified cholesterol before a return to normal levels at late infection times (Fig. 2A). As a result of the stimulation of cholesterol storage and following the rise in free cholesterol, esterified cholesterol increased from 30 to 60% at 3 hpi, and then the relative amounts of free and esterified cholesterol reached similar net increase at this time point. High cholesterol levels were maintained by peak replication, returning back to levels for uninfected cells at 16 hpi. These results suggest that cholesterol may play an important role in early ASFV infection after initial entry.
FIG 2.

Increased uptake and free cholesterol pools upon ASFV infection. (A) Quantitation of total (dark bars), free (gray bars), and esterified (white bars) cholesterol pools in mock- or ASFV-infected cells at several time points. Values were normalized to mock-infected control values at each time point. (B) Bodipy-cholesterol uptake after ASFV infection at 1 and 3 hpi compared to that in control cells in the graph. representative images are shown. **, P < 0.01; *, P < 0.05. Bar, 10 μm.
Bodipy-cholesterol (bodipy-CH) is a fluorescent mimic of free cholesterol that partitions into the plasma membrane (PM) when added exogenously to live cells (43). Taking advantage of this, we investigated the uptake of fluorescent bodipy-CH in control and ASFV-infected cells. Calculating the integrated density of endocytosed bodipy-CH using the confocal microscope, we found that ASFV infection stimulated cholesterol uptake at 1 and 3 hpi (Fig. 2B). This would increase the availability of free cholesterol levels in early infection.
ASFV infection is dependent on cholesterol endosomal traffic.
Cholesterol is diffusely distributed in the plasma membrane, cytoplasm, and endosomes in control cells (Fig. 3). The chemical U18666A is an amphipathic steroid which is widely used to block the intracellular trafficking of cholesterol (12, 45). In contrast to the diffuse distribution of cholesterol in control cells, in the presence of U18666A, cholesterol accumulates in perinuclear swollen endosomes. These endosomes label with CD63, LBPA, and Rab7, markers of multivesicular bodies (MVB) and late endosomes (LE). LE clusters exhibited a dilated lumen morphology characteristic of U18666A treatment (Fig. 3).
FIG 3.
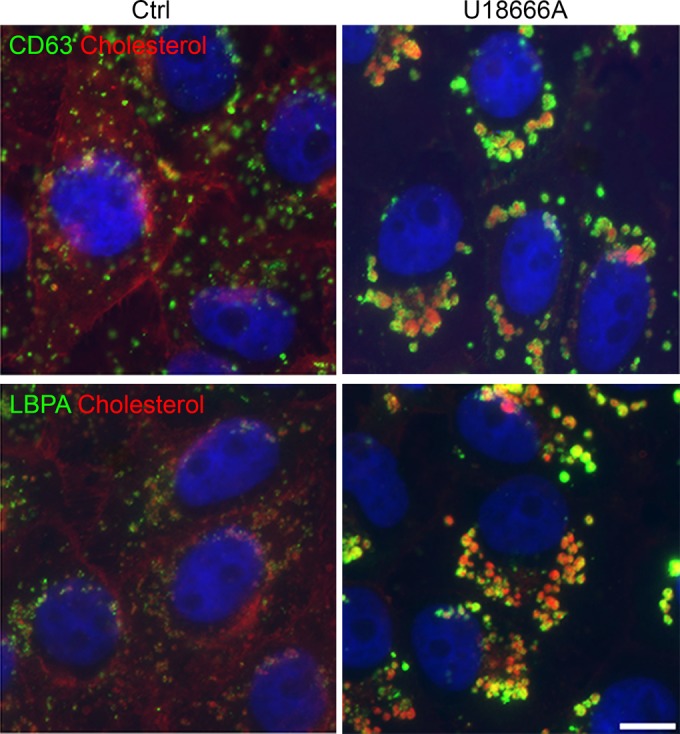
Alterations of cholesterol distribution and endosomal traffic with U18666A. Ctrl, cholesterol distribution in control cells with normal cholesterol efflux. CD63 and LBPA are stained in green and cholesterol in red. Upon U18666A treatment, inhibition of cholesterol efflux from endosomes caused cholesterol-laden vesicular clustering at the perinuclear area, while free cholesterol was reduced in other cellular locations such as the plasma membrane. Bar, 10 μm.
In infected, vehicle-treated cells, aggregated endosomes and cholesterol were found at the viral factory together with viral proteins at 16 hpi (Fig. 4A). In U18666A-treated and infected cells, very few viral factories were found. Consistent with this, treatment of cells with U18666A resulted in a potent dose-dependent reduction of ASFV infectivity by as much as 80% (Fig. 4C). Early p30 protein expression was also decreased by up to ∼50% in U18666A-treated cells compared to controls (Fig. 4D). However, in the few viral factories, it was possible to identify a direct contact with some LE devoid of cholesterol (Fig. 4A, arrows). Endosomes close to the viral factory also lacked the dilated appearance of endosomes filled with cholesterol. The reduction in ASFV infectivity due to U1866A is comparable to the impact of inhibiting endosomal acidification with bafilomycin A (BAF) (15, 18).
FIG 4.

Cholesterol efflux is necessary for viral infectivity. (A) Distribution of endosomes and cholesterol at the viral factory in ASFV-infected untreated cells at 16 hpi. Endosomes stained with Rab 7 are shown in green, cholesterol in blue, and viral protein p72 accumulated in the viral replication site in red. N, nucleus. High magnifications of the merged boxed area are shown. The color channels are shown individually in black-and-white pictures. Bar, 10 μm. Bottom left, general view of cholesterol-laden endosomes in uninfected control cells treated with U18666A. Bottom right, in treated and infected cells at 16 hpi, LE devoid of cholesterol were found in close proximity to viral factories (arrows). Bar, 25 μm. (B) Cellular viability at increasing concentrations of U18666A. (C) Infectivity at 6 hpi with U18666A, analyzed by flow cytometry by p30 expression (***, P < 0.001; *, P < 0.05). (D) Early p30 viral protein expression at 6 hpi by WB after U18666A treatment.
To obtain additional insights into the stage at which virus infection is inhibited, we next analyzed the impact of impaired cholesterol efflux using electron microscopy. In controls, remnants of decapsidated viral cores together with intraluminal vesicles typical of MVB were observed, as expected from infection progression with uncoating (Fig. 5, DMSO). Treatment of cells with the endosomal acidification inhibitor bafilomycin A (BAF) led to virions with icosahedral capsids being retained in endosomes (arrowhead). Ultrastructural analyses of cells infected with ASFV in the presence of U18666A revealed dilated endosomes in aggregates close to the nucleus (N), containing “onion-skin” membranes, which are characteristic of sphingolipid-rich membranes. Such “onion-skin” structures were never observed in DMSO controls or bafilomycin A-treated cells. In ASFV-infected cells with U18666A, virions with relatively intact capsids (arrowhead) were evident inside dilated endosomes in close association with the nucleus. The presence of fully encapsidated virions in the lumen of membrane compartments close to the nucleus suggests that virus uncoating is inhibited. This suggests that cholesterol endosomal flux is required for ASFV to gain access to the cytoplasm from the endosome.
FIG 5.

A cholesterol efflux block impairs viral exit from endosomes. TEM of control or ASFV-infected cells at 45 mpi is shown. DMSO-treated control cells with remnants of viral cores (arrowheads) are found in endosomes with intraluminal vesicles (arrow). In BAF-treated cells, virions with intact capsids were found in endosomes (arrowhead), as BAF-induced endosomal acidification inhibition impairs virus decapsidation. Scale bars, 500 nm. Bottom panels, TEM images of U18666A-treated and ASFV-infected cells with vesicular aggregates and “onion-leaf” images characteristic of lipids at 45 mpi. Virions are shown inside vesicles close to the nucleus (arrowheads).Scale bars, 500 nm. N, nucleus.
In parallel, we found that in contrast to controls, in cells treated with U18666A, encapsidated virions were trapped inside perinuclear lipid-laden dilated endosomes (Fig. 6A). The numbers of virions colocalizing with the late endosomal marker Rab 7 were significantly higher in drug-treated cells than in controls (Fig. 6B).
FIG 6.

A cholesterol efflux block impairs viral exit from endosomes. (A) Top panels, Rab7-labeled endosomes in control untreated, uninfected cells (DMSO), aggregates of dilated endosomes in uninfected cells treated with U18666A, and control infected, untreated cells. Virions are labeled with anti-p72 antibody, and endosomes labeled with Rab7 marker are shown in green. Bottom panels, U18666A-pretreated cells infected with ASFV at 1 hpi. Dilated perinuclear endosomes filled with cholesterol shown in black and white in an independent channel. Arrowheads and zoom images show viral capsid staining of intact virions retained at the periphery of dilated endosomes. Scale bars, 10 μm. (B) Number of virions found inside endosomes in control and U18666A-treated cells (***, P < 0.001). (C) Effect of cholesterol flux inhibition on ASFV, VV, and Adv infectivity evaluated by flow cytometry (***, P < 0.001; *, P < 0.05).
We also tested the impact of U18666A on adenovirus 5 and vaccinia virus infectivity (Fig. 6C). A cholesterol efflux block using U18666A resulted in a statistically significant inhibition of ASFV infectivity (P < 0.001). However, this treatment did not affect adenovirus infectivity and slightly reduced vaccinia infectivity, suggesting the existence of other mechanisms unrelated to cholesterol efflux for cytoplasmic entry of these viruses.
DISCUSSION
Cellular cholesterol is a crucial host component in a number of viral infections. In general, the amount of cellular cholesterol is controlled at two different levels (7). The first checkpoint is the entry of cholesterol by endocytosis through specific receptors on the plasma membrane (46). The second checkpoint is at the endosomal membrane by controlled efflux to the cytosol. Lowering cellular cholesterol pools specifically at early infection severely affected ASFV infectivity and replication, indicating that the virus needs to ensure a constant supply of this lipid to achieve a successful infection. Depletion of extracellular cholesterol in the medium resulted in a substantial inhibition of ASFV infection even though there are other sources of intracellular cholesterol, such as de novo cholesterol biosynthesis. In fact, inhibition of cholesterol biosynthesis with statins negatively impacts several virus infections, including human immunodeficiency virus (HIV), influenza virus, and hepatitis C virus (HCV) infections (47). In the case of ASFV, it also impairs virion morphogenesis, maturation, and exit (48). Moreover, in order to deplete cholesterol from the medium, it is necessary to include chemicals such as mevastatin and methyl-β-cyclodextrin that would affect the cholesterol membrane content. This would affect the entry of a number of viruses using different entry mechanisms, as was the case for vaccinia virus and adenovirus 5.
ASFV infection also reorganized the cholesterol cellular landscape. ASFV infection increased free cholesterol levels, ensuring exogenous supplies by augmented uptake. A similar mechanism has recently been reported for enterovirus to increase cellular cholesterol levels (49). Cholesterol is enriched at the PM, at the endocytic recycling pathway (it is recycled back to the PM), and at the trans-Golgi compartment, while the endoplasmic reticulum (ER) has a low free cholesterol content. When free cholesterol is very abundant, it traffics directly to the ER, where it becomes esterified and stored in lipid droplets to maintain homeostasis (50). Free cholesterol increased from 70 to 90% at early times after infection, and the relative amounts of free and esterified cholesterol reached similar net increase at this time point. High cholesterol levels were maintained by peak replication, returning back to uninfected cell rates at later time points.
More importantly, we found that cholesterol entering the cell by endocytosis was important for ASFV uncoating and penetration at early infection stages. Cholesterol is stored in multivesicular bodies and late endosomes, from where it should efflux to the cytosol. This efflux is controlled by cholesterol transporter proteins, such as NPC1 and -2, vesicle membrane protein-associated protein A (VAPA), and others (45). In fact, cholesterol endosomal traffic integrity is necessary for a number of virus infections (51–53). Endosomes acquire cholesterol by endocytosis as cholesteryl esters. In acidic compartments, acid lipase hydrolyzes cholesteryl esters, and the resulting free cholesterol partitions into neighboring membranes (7, 54). Lipids can diffuse rapidly within membranes that are connected by membrane flow during transport, and this could facilitate viral fusion and exit by mechanisms still partially unexplored. It is conceivable that taking advantage of this process and under physiological conditions of cholesterol homeostasis, endocytosed viral particles could fuse with the limiting membrane of the endosome in order to release the viral genome to start replication. In fact, in cells where cholesterol efflux has been biochemically disrupted (by U18666A treatment), ASFV infectivity was impaired at late entry in a postbinding stage. Encapsidated ASF viral particles remained trapped in endosomes, suggesting an alteration in viral uncoating and penetration that ultimately inhibits infection progression.
At this level, we were able to find significant differences between ASFV and other viruses. Successful ASFV infectivity was strongly dependent on the integrity of cholesterol endosomal efflux. Infectious entry of the flaviviruses hepatitis C virus (HCV) and dengue virus (DV) into host cells is also dependent on cholesterol efflux, and the cholesterol efflux inhibitor U18666A affected HCV and DV infection (51, 52). Under U18666A treatment, Dengue viral particles remained trapped in the late endosome/lysosome compartment. Similar to the case for ASFV, viral traffic and cytoplasmic release were impaired in cholesterol-laden endosomes (52). U18666A blocks the release of HCV particles that accumulate in late endosomes (51). Moreover, the cellular cholesterol-sensing receptor Niemann-Pick C1 (NPC1)-like 1 is an important HCV entry factor. The FDA-approved antagonist of this receptor, Ezetimibe, inhibits virion-cell membrane fusion before cytoplasmic penetration (55).
U18666A and other cationic amphiphiles inhibit Ebola virus (EBOV) entry and infection at the step of endosomal membrane fusion. These chemical compounds have the ability to accumulate cholesterol in late endosomes and inhibit EBOV entry through an NPC1-dependent pathway (53). Filovirus entry seems to require the interaction of GP with NPC1 protein as an intracellular receptor (44, 56). EBOV, DV, and HCV uncoating and cytoplasmic penetration occur at the late endosomal level as a multistep process that could be similar for ASFV and other enveloped viruses. In this process, the acid pH environment could act as a “primer” for later membrane fusion, in which cholesterol transport acts by mechanisms not fully understood.
Adenovirus (Adv) infectivity was independent of cholesterol endosomal efflux. In contrast to ASFV and VV, Adv is a nonenveloped virus that relies mainly on mechanical cues for uncoating (57). With the Adv 2 and 5 serotypes, uncoating starts at receptor binding. Integrins and coxsackievirus adenovirus receptor (CAR) cooperate to induce conformational changes, untwisting penton base, resulting in fiber loss, and exposing membrane lytic protein VI (58). This protein induces mechanical ruptures at the endosomal membrane to allow virus endosomal escape to the cytosol (59). Like other enveloped viruses, poxviruses respond to uncoating cues immediately after fusion, and viral cores undergo dramatic morphological changes (60). Vaccinia virus genome uncoating follows a multistep process. First, the viral core becomes acid activated, probably in macropinosomes, and early genes are transcribed within the intact viral core (61). This mode of entry, related to endocytosis, could be somewhat inhibited by blocking cholesterol efflux. Next, early gene transcription and mRNA export would be initiated via the action of viral RNA polymerases and transcription factors inside the core (62). The activation of the transcription machinery is considered a priming step, as it is a prerequisite for the next phase of uncoating. The proteasome activity (63) and the recently characterized uncoating factor D5 ATPase then are required for VV cytoplasmic genome release (64).
In conclusion, we have used biochemical assays, fluorescent imaging, and electron microscopy to investigate the impact of cholesterol in ASFV infection and the regulation of cholesterol levels induced by this virus. Cholesterol-depleted medium early in infection severely inhibits ASFV infectivity and replication. ASFV infection causes an increase in free cholesterol levels, remodels intracellular cholesterol, and redistributes free cholesterol to the viral replication site. Inhibition of cholesterol efflux severely impairs ASFV infectivity, as occurs with HCV. Other DNA viruses, such as adenovirus and vaccinia virus, are also sensitive to cholesterol-depleted medium, but the main difference is in ASFV sensitivity to cholesterol efflux inhibition. Hence, cholesterol homeostasis at endosomes appears to be a selective target for some viruses. Endosomal cholesterol might be essential for ASFV at a postbinding step, at the viral membrane fusion with the endosomal membrane and cytoplasmic exit. Together, our results provide new avenues for research focused on early targets at the molecular signaling of endosomal traffic for DNA viruses.
ACKNOWLEDGMENTS
This work was supported by grants AGL2012-34533, AGL2009-09209, and BFU2014-54181 (to J.L.C.) from the Ministerio de Economía y Competitividad of Spain.
We thank Michael Way of the Francis Crick Institute of London for his critical comments and suggestions on the manuscript and Jean Gruenberg of the University of Geneva for the generous gift of the anti-LBPA antibody.
REFERENCES
- 1.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu Rev Biochem 79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 2.Huotari J, Helenius A. 2011. Endosome maturation. EMBO J 30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gruenberg J. 2001. The endocytic pathway: a mosaic of domains. Nat Rev Mol Cell Biol 2:721–730. doi: 10.1038/35096054. [DOI] [PubMed] [Google Scholar]
- 4.Bissig C, Lenoir M, Velluz MC, Kufareva I, Abagyan R, Overduin M, Gruenberg J. 2013. Viral infection controlled by a calcium-dependent lipid-binding Module in ALIX. Dev Cell 25:364–373. doi: 10.1016/j.devcel.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitt L, Tampe R. 2002. Structure and mechanism of ABC transporters. Curr Opin Struct Biol 12:754–760. doi: 10.1016/S0959-440X(02)00399-8. [DOI] [PubMed] [Google Scholar]
- 6.Chevallier J, Chamoun Z, Jiang G, Prestwich G, Sakai N, Matile S, Parton RG, Gruenberg J. 2008. Lysobisphosphatidic acid controls endosomal cholesterol levels. J Biol Chem 283:27871–27880. doi: 10.1074/jbc.M801463200. [DOI] [PubMed] [Google Scholar]
- 7.Ikonen E. 2008. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- 8.Le Blanc I, Luyet PP, Pons V, Ferguson C, Emans N, Petiot A, Mayran N, Demaurex N, Faure J, Sadoul R, Parton RG, Gruenberg J. 2005. Endosome-to-cytosol transport of viral nucleocapsids. Nat Cell Biol 7:653–664. doi: 10.1038/ncb1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Infante RE, Radhakrishnan A, Abi-Mosleh L, Kinch LN, Wang ML, Grishin NV, Goldstein JL, Brown MS. 2008. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem 283:1064–1075. [DOI] [PubMed] [Google Scholar]
- 10.Rosenbaum AI, Maxfield FR. 2011. Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J Neurochem 116:789–795. doi: 10.1111/j.1471-4159.2010.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karten B, Peake KB, Vance JE. 2009. Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim Biophys Acta 1791:659–670. doi: 10.1016/j.bbalip.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 12.Liscum L, Faust JR. 1989. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J Biol Chem 264:11796–11806. [PubMed] [Google Scholar]
- 13.Greber UF. 2002. Signalling in viral entry. Cell Mol Life Sci 59:608–626. doi: 10.1007/s00018-002-8453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marsh M, Helenius A. 2006. Virus entry: open sesame. Cell 124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galindo I, Cuesta-Geijo MA, Hlavova K, Munoz-Moreno R, Barrado-Gil L, Dominguez J, Alonso C. 2015. African swine fever virus infects macrophages, the natural host cells, via clathrin- and cholesterol-dependent endocytosis. Virus Res 200:45–55. doi: 10.1016/j.virusres.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 16.Hernaez B, Tarrago T, Giralt E, Escribano JM, Alonso C. 2010. Small peptide inhibitors disrupt a high-affinity interaction between cytoplasmic dynein and a viral cargo protein. J Virol 84:10792–10801. doi: 10.1128/JVI.01168-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanchez EG, Quintas A, Perez-Nunez D, Nogal M, Barroso S, Carrascosa AL, Revilla Y. 2012. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog 8:e1002754. doi: 10.1371/journal.ppat.1002754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuesta-Geijo MA, Galindo I, Hernaez B, Quetglas JI, Dalmau-Mena I, Alonso C. 2012. Endosomal maturation, Rab7 GTPase and phosphoinositides in African swine fever virus entry. PLoS One 7:e48853. doi: 10.1371/journal.pone.0048853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Netherton CL, Wileman TE. 2013. African swine fever virus organelle rearrangements. Virus Res 173:76–86. doi: 10.1016/j.virusres.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 20.Alonso C, Galindo I, Cuesta-Geijo MA, Cabezas M, Hernaez B, Munoz-Moreno R. 2013. African swine fever virus-cell interactions: from virus entry to cell survival. Virus Res 173:42–57. doi: 10.1016/j.virusres.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munoz-Moreno R, Barrado-Gil L, Galindo I, Alonso C. 2015. Analysis of HDAC6 and BAG3-aggresome pathways in African swine fever viral factory formation. Viruses 7:1823–1831. doi: 10.3390/v7041823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jouvenet N, Monaghan P, Way M, Wileman T. 2004. Transport of African swine fever virus from assembly sites to the plasma membrane is dependent on microtubules and conventional kinesin. J Virol 78:7990–8001. doi: 10.1128/JVI.78.15.7990-8001.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salas ML, Andres G. 2013. African swine fever virus morphogenesis. Virus Res 173:29–41. doi: 10.1016/j.virusres.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 24.Enjuanes L, Carrascosa AL, Moreno MA, Vinuela E. 1976. Titration of African swine fever (ASF) virus. J Gen Virol 32:471–477. doi: 10.1099/0022-1317-32-3-471. [DOI] [PubMed] [Google Scholar]
- 25.Yanez RJ, Rodriguez JM, Nogal ML, Yuste L, Enriquez C, Rodriguez JF, Vinuela E. 1995. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 208:249–278. doi: 10.1006/viro.1995.1149. [DOI] [PubMed] [Google Scholar]
- 26.Hernaez B, Escribano JM, Alonso C. 2006. Visualization of the African swine fever virus infection in living cells by incorporation into the virus particle of green fluorescent protein-p54 membrane protein chimera. Virology 350:1–14. doi: 10.1016/j.virol.2006.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blasco R, Moss B. 1995. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 158:157–162. doi: 10.1016/0378-1119(95)00149-Z. [DOI] [PubMed] [Google Scholar]
- 28.Alba R, Cots D, Ostapchuk P, Bosch A, Hearing P, Chillon M. 2011. Altering the Ad5 packaging domain affects the maturation of the Ad particles. PLoS One 6:e19564. doi: 10.1371/journal.pone.0019564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alba R, Hearing P, Bosch A, Chillon M. 2007. Differential amplification of adenovirus vectors by flanking the packaging signal with attB/attP-PhiC31 sequences: implications for helper-dependent adenovirus production. Virology 367:51–58. doi: 10.1016/j.virol.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 30.King DP, Reid SM, Hutchings GH, Grierson SS, Wilkinson PJ, Dixon LK, Bastos AD, Drew TW. 2003. Development of a TaqMan PCR assay with internal amplification control for the detection of African swine fever virus. J Virol Methods 107:53–61. doi: 10.1016/S0166-0934(02)00189-1. [DOI] [PubMed] [Google Scholar]
- 31.Hernaez B, Escribano JM, Alonso C. 2008. African swine fever virus protein p30 interaction with heterogeneous nuclear ribonucleoprotein K (hnRNP-K) during infection. FEBS Lett 582:3275–3280. doi: 10.1016/j.febslet.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cobbold C, Wileman T. 1998. The major structural protein of African swine fever virus, p73, is packaged into large structures, indicative of viral capsid or matrix precursors, on the endoplasmic reticulum. J Virol 72:5215–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter GC, Law M, Hollinshead M, Smith GL. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol 86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- 34.Calero M, Chiappi M, Lazaro-Carrillo A, Rodriguez MJ, Chichon FJ, Crosbie-Staunton K, Prina-Mello A, Volkov Y, Villanueva A, Carrascosa JL. 2015. Characterization of interaction of magnetic nanoparticles with breast cancer cells. J Nanobiotechnol 13:16. doi: 10.1186/s12951-015-0073-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobayashi T, Beuchat MH, Lindsay M, Frias S, Palmiter RD, Sakuraba H, Parton RG, Gruenberg J. 1999. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat Cell Biol 1:113–118. doi: 10.1038/10084. [DOI] [PubMed] [Google Scholar]
- 36.Hernaez B, Alonso C. 2010. Dynamin- and clathrin-dependent endocytosis in African swine fever virus entry. J Virol 84:2100–2109. doi: 10.1128/JVI.01557-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danthi P, Chow M. 2004. Cholesterol removal by methyl-beta-cyclodextrin inhibits poliovirus entry. J Virol 78:33–41. doi: 10.1128/JVI.78.1.33-41.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin JJ, Holguera J, Sanchez-Felipe L, Villar E, Munoz-Barroso I. 2012. Cholesterol dependence of Newcastle disease virus entry. Biochim Biophys Acta 1818:753–761. doi: 10.1016/j.bbamem.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Appelqvist H, Nilsson C, Garner B, Brown AJ, Kagedal K, Ollinger K. 2011. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am J Pathol 178:629–639. doi: 10.1016/j.ajpath.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamada H, Sato K, Iwai M, Ohta K, Nagano I, Shoji M, Abe K. 2003. Changes of free cholesterol and neutral lipids after transient focal brain ischemia in rats. Acta Neurochir Suppl 86:177–180. [DOI] [PubMed] [Google Scholar]
- 41.Kruth HS. 1984. Localization of unesterified cholesterol in human atherosclerotic lesions. Demonstration of filipin-positive, oil-red-O-negative particles. Am J Pathol 114:201–208. [PMC free article] [PubMed] [Google Scholar]
- 42.Kruth HS, Comly ME, Butler JD, Vanier MT, Fink JK, Wenger DA, Patel S, Pentchev PG. 1986. Type C Niemann-Pick disease. Abnormal metabolism of low density lipoprotein in homozygous and heterozygous fibroblasts. J Biol Chem 261:16769–16774. [PubMed] [Google Scholar]
- 43.Holtta-Vuori M, Uronen RL, Repakova J, Salonen E, Vattulainen I, Panula P, Li Z, Bittman R, Ikonen E. 2008. BODIPY-cholesterol: a new tool to visualize sterol trafficking in living cells and organisms. Traffic 9:1839–1849. doi: 10.1111/j.1600-0854.2008.00801.x. [DOI] [PubMed] [Google Scholar]
- 44.Cote M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. 2011. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liscum L, Sturley SL. 2004. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim Biophys Acta 1685:22–27. doi: 10.1016/j.bbalip.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 46.Goldstein JL, Brown MS. 1984. Progress in understanding the LDL receptor and HMG-CoA reductase, two membrane proteins that regulate the plasma cholesterol. J Lipid Res 25:1450–1461. [PubMed] [Google Scholar]
- 47.Bader T, Fazili J, Madhoun M, Aston C, Hughes D, Rizvi S, Seres K, Hasan M. 2008. Fluvastatin inhibits hepatitis C replication in humans. Am J Gastroenterol 103:1383–1389. doi: 10.1111/j.1572-0241.2008.01876.x. [DOI] [PubMed] [Google Scholar]
- 48.Munoz-Moreno R, Galindo I, Cuesta-Geijo MA, Barrado-Gil L, Alonso C. 2015. Host cell targets for African swine fever virus. Virus Res 209:118–127. doi: 10.1016/j.virusres.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 49.Ilnytska O, Santiana M, Hsu NY, Du WL, Chen YH, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, Dixon JL, Altan-Bonnet N. 2013. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 14:281–293. doi: 10.1016/j.chom.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.English AR, Voeltz GK. 2013. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb Perspect Biol 5:a013227. doi: 10.1101/cshperspect.a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lai CK, Jeng KS, Machida K, Lai MM. 2010. Hepatitis C virus egress and release depend on endosomal trafficking of core protein. J Virol 84:11590–11598. doi: 10.1128/JVI.00587-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poh MK, Shui G, Xie X, Shi PY, Wenk MR, Gu F. 2012. U18666A, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antiviral Res 93:191–198. doi: 10.1016/j.antiviral.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 53.Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, White JM. 2013. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 8:e56265. doi: 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maxfield FR, van Meer G. 2010. Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol 22:422–429. doi: 10.1016/j.ceb.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suomalainen M, Greber UF. 2013. Uncoating of non-enveloped viruses. Curr Opin Virol 3:27–33. doi: 10.1016/j.coviro.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 58.Lindert S, Silvestry M, Mullen TM, Nemerow GR, Stewart PL. 2009. Cryo-electron microscopy structure of an adenovirus-integrin complex indicates conformational changes in both penton base and integrin. J Virol 83:11491–11501. doi: 10.1128/JVI.01214-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luisoni S, Suomalainen M, Boucke K, Tanner LB, Wenk MR, Guan XL, Grzybek M, Coskun U, Greber UF. 2015. Co-option of membrane wounding enables virus penetration into cells. Cell Host Microbe 18:75–85. doi: 10.1016/j.chom.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt FI, Bleck CK, Reh L, Novy K, Wollscheid B, Helenius A, Stahlberg H, Mercer J. 2013. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep 4:464–476. doi: 10.1016/j.celrep.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 61.De Silva FS, Lewis W, Berglund P, Koonin EV, Moss B. 2007. Poxvirus DNA primase. Proc Natl Acad Sci U S A 104:18724–18729. doi: 10.1073/pnas.0709276104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Z, Moss B. 2009. Interaction of the vaccinia virus RNA polymerase-associated 94-kilodalton protein with the early transcription factor. J Virol 83:12018–12026. doi: 10.1128/JVI.01653-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mercer J, Snijder B, Sacher R, Burkard C, Bleck CK, Stahlberg H, Pelkmans L, Helenius A. 2012. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep 2:1036–1047. doi: 10.1016/j.celrep.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 64.Kilcher S, Schmidt FI, Schneider C, Kopf M, Helenius A, Mercer J. 2014. siRNA screen of early poxvirus genes identifies the AAA+ ATPase D5 as the virus genome-uncoating factor. Cell Host Microbe 15:103–112. doi: 10.1016/j.chom.2013.12.008. [DOI] [PubMed] [Google Scholar]