

ABSTRACT
Measles virus (MeV) and canine distemper virus (CDV) possess tetrameric attachment proteins (H) and trimeric fusion proteins, which cooperate with either SLAM or nectin 4 receptors to trigger membrane fusion for cell entry. While the MeV H-SLAM cocrystal structure revealed the binding interface, two distinct oligomeric H assemblies were also determined. In one of the conformations, two SLAM units were sandwiched between two discrete H head domains, thus spotlighting two binding interfaces (“front” and “back”). Here, we investigated the functional relevance of both interfaces in activating the CDV membrane fusion machinery. While alanine-scanning mutagenesis identified five critical regulatory residues in the front H-binding site of SLAM, the replacement of a conserved glutamate residue (E at position 123, replaced with A [E123A]) led to the most pronounced impact on fusion promotion. Intriguingly, while determination of the interaction of H with the receptor using soluble constructs revealed reduced binding for the identified SLAM mutants, no effect was recorded when physical interaction was investigated with the full-length counterparts of both molecules. Conversely, although mutagenesis of three strategically selected residues within the back H-binding site of SLAM did not substantially affect fusion triggering, nevertheless, the mutants weakened the H-SLAM interaction recorded with the membrane-anchored protein constructs. Collectively, our findings support a mode of binding between the attachment protein and the V domain of SLAM that is common to all morbilliviruses and suggest a major role of the SLAM residue E123, located at the front H-binding site, in triggering the fusion machinery. However, our data additionally support the hypothesis that other microdomain(s) of both glycoproteins (including the back H-binding site) might be required to achieve fully productive H-SLAM interactions.
IMPORTANCE A complete understanding of the measles virus and canine distemper virus (CDV) cell entry molecular framework is still lacking, thus impeding the rational design of antivirals. Both viruses share many biological features that partially rely on the use of analogous Ig-like host cell receptors, namely, SLAM and nectin 4, for entering immune and epithelial cells, respectively. Here, we provide evidence that the mode of binding between the membrane-distal V domain of SLAM and the attachment protein (H) of morbilliviruses is very likely conserved. Moreover, although structural information revealed two discrete conformational states of H, one of the structures displayed two H-SLAM binding interfaces (“front” and “back”). Our data not only spotlight the front H-binding site of SLAM as the main determinant of membrane fusion promotion but suggest that the triggering efficiency of the viral entry machinery may rely on a local conformational change within the front H-SLAM interactive site rather than the binding affinity.
INTRODUCTION
Measles virus (MeV) and canine distemper virus (CDV) belong to the Morbillivirus genus of the Paramyxoviridae family that also includes Rinderpest virus (RPV), peste-de-petits-ruminants virus (PPRV), phocine distemper virus (PDV), and the cetacean dolphin and porpoise morbilliviruses (DMV and PMV, respectively). Among these, CDV exhibits a high potential to cross species barriers, exemplified by major outbreaks in different nonconventional hosts, including nonhuman primates (1–7). Although this might raise concerns for humans, the cross immunity provided by measles virus vaccination is likely to protect against a potential CDV spillover in people (8).
Morbilliviruses share many biological features that partially rely on the use of analogous host cell receptors, namely, SLAM (9, 10) and nectin 4 (11–16), for entering immune and epithelial cells, respectively. The primary replication of MeV and, probably, all morbilliviruses takes place in SLAM-positive immune cells (17, 18). After massive amplification in lymphoid tissues (associated with strong immunosuppression), the virus spreads through the bloodstream to nectin 4-positive epithelial tissues, inducing skin, respiratory, and gastrointestinal symptoms and viral shedding. Hence, the interaction with SLAM receptor is essential to initiate the disease (19). Supporting this view, Leonard and colleagues demonstrated that measles virus particles engineered to lack productive interaction with nectin 4 receptor (nectin 4-blind viruses) were still able to induce immunosuppression, while being defective in replicating in epithelia and, consequently, impaired in shedding (20). Interestingly, both receptors are members of the Ig-like superfamily, which consists of single-pass transmembrane proteins harboring an extracellular region composed of C and V domains. It has been reported that the V domain of both SLAM and nectin 4 is involved in direct physical contacts with the viral receptor-binding protein (13, 16, 21, 22).
Cell entry represents the initial critical step of viral infection and, ultimately, of disease occurrence. Morbilliviruses have evolved finely tuned entry machineries composed of two tightly interacting surface glycoproteins, of which one is a tetrameric attachment protein (H), whose ectodomain is composed of a membrane-proximal helical F-contacting/activating stalk region supporting a membrane-distal receptor-binding head domain, and the other is a trimeric fusion protein (F) (23–26). It is proposed that upon receptor engagement, H proteins undergo sequential conformational changes that activate F trimers (27). In turn, prefusion F structures undergo a series of irreversible structural rearrangements that lead to the merging of the viral envelope with the host cell plasma membrane at neutral pH (24, 28). Our knowledge on the molecular nature of the H-SLAM interaction made a major leap forward based on the recently determined cocrystal structure of the MeV H protein bound to the SLAM receptor (29). Unlike other paramyxovirus attachment proteins, which bind to their cognate receptors at the top of the conserved six-bladed beta-propeller monomeric head domains (30–32), MeV H heads bind to the receptor units in a much more sideways manner (29). More specifically, the crystal structure revealed four main zones of contact in the membrane-distal V domain of SLAM, mediating short-range interactions with the H head's lateral region. On the opposite side, multiple residues in blades 4, 5, and 6 of the H head beta-propeller conformation were located at the SLAM binding site (29).
Remarkably, crystallization of the soluble MeV H ectodomain bound to the SLAM receptor additionally revealed two different oligomeric arrangements (29). While in the first one, four H head domains were assembled in a more planar configuration, with the four receptors protruding at a distance from the tetrameric interface (referred to as the “X shape”), the second conformation displayed a much more staggered overall organization, with two receptors facing externally and the two others sandwiched between two discrete H head domains (referred to as the “V shape”). While an identical interactive site between the V domain of SLAM and the H heads is found in both conformations (referred to as the “front” H-binding site), the V-shaped oligomeric configuration highlighted a supplementary binding interface (referred to as the “back” H-binding site). Although a recent study suggested that both conformational states of MeV H might be relevant for triggering the F protein and subsequent membrane fusion activity (33), further functional and structural analyses must be conducted to validate this important notion.
Interestingly, both MeV H conformations differed significantly from recent crystal structures obtained from a related paramyxovirus attachment protein's soluble ectodomains. Indeed, no tetrameric interface between two dimeric head units was observed in the case of Newcastle disease virus soluble HN. Rather, both dimeric head modules were folded back onto the C-terminal region of the stalk domain (referred to as the “4 heads down” conformation) (34). An intriguing hybrid conformation was also determined for the human parainfluenza virus type 5 (hPIV5) HN protein. In this structure, one head dimer was folded back onto the stalk, whereas the other was stabilized in an upright state (referred to as the “2 heads up and 2 heads down” conformational state) (35). Importantly, these two structures, together with a previously determined alternative hPIV5 HN conformation in which both dimeric head units assembled into tetramers (30) (referred to as the “4 heads up” state), led to a different proposal for the spatiotemporal activation of the F protein. HN and F travel to the cell surface separately and, upon HN receptor engagement, the HN heads may switch position from the down to the up state, thereby exposing the F-binding/activation microdomain of the stalks, leading in turn to productive F triggering (referred to as the “stalk exposure/induced-fit” model) (26, 36).
Although the stalk exposure/induced-fit mechanism has been proposed to act as a general model for paramyxovirus F activation (36), it not only challenged the structural data obtained for MeV H (29) but, unfortunately, could not match some important functional data recorded for MeV and CDV (23, 37, 38). Rather, our recent mechanistic study was suggestive of a model where the H proteins of morbilliviruses may initially assume a conformation (putatively a “4 heads down-like” state) where the head units lock the inherent F-triggering capacity of the stalk domain (referred to as the “H-autorepressed state”) (27). Although we cannot exclude that the V or X shape of MeV H defines the autorepressed conformation, our model rather indicates that the crystal structure(s) determined for MeV H are either not biologically relevant or do not represent the initial pre-F-triggering state. Upon binding to its cognate receptor, the putative autorepressed H structure would experience a first conformational change characterized by movements of the heads (perhaps at this stage reaching the V and/or X shape configuration) that would result in unlocking the stalk domain, which in turn would be freed to undergo a second structural rearrangement strictly required for productive F activation (referred to as the “safety catch” model) (27, 39).
Interestingly, two alternative mechanisms for F activation have been described. According to the first model, the human parainfluenza virus type 3 HN protein may oligomerize into F-triggering-competent structures as a consequence of receptor binding (40). In the second model, it is hypothesized that F trimers would initially interact with the Nipah virus attachment protein (G) head domains to prevent premature F activation of intracellularly preformed G/F complexes. Receptor-induced sequential conformational changes of G (including a stalk exposure step) would then switch this interaction from the heads to the F activation microdomain of the stalks (41). While highlighting clear differences, all of the models are not mutually exclusive and deserve additional work to clarify the mechanism of F activation for each member of the paramyxovirus family. However, how the front and back H-binding sites of the SLAM V domain can initiate the suggested cascade of conformational changes in the morbillivirus H protein remains largely unknown.
While residues regulating receptor-binding efficacy have been identified in both MeV and CDV H proteins (42–44), much less attention has been paid to similar residues in SLAM. The human and mouse SLAM (mSLAM) molecules share only about 60% identity, which precludes MeV entry in mSLAM-expressing cells. However, the replacement of amino acids 60, 61, and 63 in mSLAM with the analogous residues of human SLAM enabled MeV to efficiently infect cells expressing the mouse SLAM receptor (45). Furthermore, bioinformatics-based studies highlighted a putative role of residues 72, 76, 82, and 129 of carnivore SLAM molecules in the affinity and sensitivity of interaction with CDV H and, hence, a possible contribution by these residues to the species barrier and/or the crossing thereof (46). Consistent with this hypothesis, residues 70 and 71 in canine SLAM (cSLAM) were also recently proposed to facilitate CDV-mediated cross-species infections (47).
In the present study, we investigated the relevance of the front and back H-binding sites of cSLAM with regard to productive triggering of morbillivirus membrane fusion machineries. Based on structure-based alanine-scanning mutagenesis, a panel of canine SLAM mutants was generated and their phenotypes analyzed with regard to cell surface expression, cell-cell fusion, and the efficiency of the interaction with the CDV H protein. Collectively, our data strongly support the notion that the front H-binding site of the V domain of canine SLAM is the major molecular determinant in the activation of the CDV membrane fusion machinery. Furthermore, beyond mediating the efficiency of binding to CDV H, the conserved glutamate residue of SLAM (E at position 123 [E123]) in the front H-binding site may convey an essential fusion-triggering signal to the opposite conserved arginine residue (R529) that resides at the top of the lateral side of the attachment protein head domain.
MATERIALS AND METHODS
Cell cultures.
293T cells (ATCC CRL-11268), Vero cells (ATCC CCL-81), and derivative Vero cells expressing the canine SLAM receptor (Vero-cSLAM, kindly provided by Yusuke Yanagi, Kyushu University, Japan) were grown in Dulbecco's modified Eagle's medium (Gibco; Invitrogen) with 10% fetal calf serum at 37°C in the presence of 5% CO2.
Construction of expression plasmids and transfections.
All single (and multiple) substitutions performed in the pCI-HA-cSLAM construct were obtained using the QuikChange lightning site-directed mutagenesis kit (Stratagene). For the cysteine-based approach, we used the previously described pCI-sH-expressing strain (derived from CDV strain A75/17, residues 59 to 607). The construct additionally carries an N-terminal GCN4 motif, a C-terminal FLAG tag (48), and pCI-sSLAM plasmids (43) as a backbone for mutagenesis. Both proteins lack the transmembrane and cytosolic tail regions, thus representing soluble forms of the protein (sH and sSLAM, respectively). For transfection experiments, Vero cells at 90% confluence were transfected with the various expression vectors (as indicated in the figure legends) using TransIT-LT1 (Mirus) according to the manufacturer's instructions. All primers are available upon request.
Semiquantitative SLAM-H binding assay.
The interaction between either the canine SLAM (cSLAM) wild-type (wt) or mutant protein and a recombinant soluble H protein (sH) was assessed through a semiquantitative assay, as previously described by Ader and colleagues (48). Briefly, the soluble form of FLAG-tagged H protein (sHFLAG) was expressed in 293T cells. The sHFLAG-containing supernatant was harvested 3 day posttransfection and further concentrated using 30-kDa-cutoff filtration columns (Millipore). sHFLAG was then added for 1 h at 4°C to Vero cells that had previously been transfected with either hemagglutinin (HA)-tagged cSLAM or plasmids expressing cSLAM-derived mutants. Receptor-bound soluble H proteins were stained with a mouse anti-FLAG monoclonal antibody (MAb) (1:500; Sigma, Switzerland) and then with a goat anti-mouse Alexa Fluor 488-conjugated antibody (1:500; Invitrogen). The occurrence of sHFLAG binding to the HA-cSLAM proteins was then monitored by flow cytometry analysis (BD LSRII; Becton Dickinson). The semiquantitative assessment of the sH-cSLAM binding activity was calculated as follows: all FLAG values were normalized according to the surface expression of the given cSLAM protein (recorded by flow cytometry analysis using an anti-HA monoclonal antibody 16B12 [1:500; Covance]). Finally, all ratios were standardized to the one obtained for the combination of wt sHFLAG and wt cSLAM, which was arbitrarily set at 100%. The experiments were repeated three times in triplicate.
Luciferase reporter gene-based cell content mixture assay.
The quantitative fusion assay was performed as described previously (49, 50), with slight modifications. Briefly, one population of Vero cells were cotransfected with the F (1.9 μg) and H (1 μg) expression plasmids and 0.1 μg of pTM-Luc (kindly provided by Laurent Roux, University of Geneva). In parallel, separate six-well plates containing another population of Vero cells were infected with modified vaccinia virus Ankara expressing T7 polymerase (MVA-T7) at a multiplicity of infection (MOI) of 1 and, 2 h later, were transfected with SLAM expression plasmids (2 μg). Four hours posttransfection, the cells were detached and plated (1:2 dilution) into new six-well plates. After overnight incubation, both cell populations were mixed and incubated at 37°C. Five hours later, the cells were lysed using Bright-Glo lysis buffer (Promega), and the luciferase activity was determined using a luminescence counter (PerkinElmer Life Sciences) and the Britelite reporter gene assay system (PerkinElmer Life Sciences).
Cell surface expression of SLAM proteins.
Vero cells were transfected with 1-μg amounts of various HA-tagged cSLAM-expressing plasmids. One day posttransfection, unfixed and unpermeabilized cells were washed twice with cold phosphate-buffered saline (PBS) and then stained with the anti-HA MAb (1:500) (Covance) at 4°C. This was followed by washes with cold PBS and incubation of the cells with Alexa Fluor 488-conjugated secondary antibody (1:500) for 1 h at 4°C. The cells were subsequently washed 2 times with cold PBS and detached from the wells by adding PBS-EDTA (50 μM) for 20 min at 37°C. The mean fluorescence intensity (MFI) of 10,000 cells was then measured by using a BD LSRII flow cytometer (Becton Dickinson).
Western blotting.
Western blot analyses were performed as previously described (49, 50). Briefly, the cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1% deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS]) containing protease inhibitor (cOmplete mixture; Roche) and were cleared by centrifugation for 20 min at 4°C. The supernatant was mixed with an equal amount of 2× Laemmli sample buffer (Bio-Rad) containing 5% β-mercaptoethanol, boiled at 95°C for 5 min, and fractionated on SDS-PAGE or 3 to 8% Tris-acetate gels (Invitrogen) under reducing or nonreducing conditions. The separated proteins were transferred to nitrocellulose membranes by electroblotting. The membranes were then incubated with the anti-CDV H polyclonal antibody (PAb) (1:1,000) (51) or an anti-HA PAb (1:1,000) (Covance). Following incubation with a peroxidase-conjugated secondary antibody, the membranes were subjected to enhanced chemiluminescence (ECL) using an ECL kit (Amersham Pharmacia Biotech) according to the manufacturer's instructions.
SLAM-H coimmunoprecipitation (co-IP) assay.
Vero cells in a six-well-plate format were transfected with 2-μg amounts of canine SLAM-expressing plasmids (wt or derived mutants) and 2 μg of wt pCI-H. At 24 h posttransfection, cells were lysed in RIPA buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1% deoxycholate, 1% Triton X-100, 0.1% SDS) containing protease inhibitor (cOmplete mix; Roche). Lysates cleared by centrifugation (20,000 × g for 20 min at 4°C) were incubated for 2 h with anti-HA monoclonal antibody 16B12 (1:1,000) (Covance), followed by overnight incubation with immunoglobulin G-coupled Sepharose beads (GE Healthcare) and then by 3 washes with RIPA buffer. The samples were then subjected to Western blot analysis as described above using either an anti-HA polyclonal antibody (Covance) or a rabbit anti-CDV H polyclonal antibody.
Immunoprecipitation of covalent SLAM/H complexes.
293T cells in 6-well plates were transfected with 2-μg amounts of plasmids encoding cysteine variants of the soluble versions of SLAM and H (both constructs additionally carried a hexahistidine tag). At 72 h posttransfection, the supernatant was harvested, and soluble proteins were immunoprecipitated for 2 h with an anti-histidine MAb (AbD Serotec) (1:1,000). This was followed by adding protein G-Sepharose beads overnight (GE Healthcare) and then by fractionation in 10% SDS-PAGE or 3 to 8% Tris-acetate gels under regular reducing or nonreducing conditions. Immunoprecipitated SLAM/H complexes were finally revealed by Western blotting, as described above, using an anti-H polyclonal antibody or the anti-SLAM antibody (1:1,000).
Surface plasmon resonance (SPR).
Binding analysis was performed on a Biacore X100 instrument (GE Healthcare, Chalfont St. Giles, United Kingdom) with HBS-EP+ running buffer (GE Healthcare) at a flow rate of 20 μl/min. CMD500m chips (XanTec Bioanalytics GmBH, Düsseldorf, Germany) were used. The CDV H protein was immobilized on the chip using a standard amine-coupling protocol, resulting in a coupling level of about 2,000 response units. The biosensor surface was regenerated after each cycle by injecting 25 mM NaOH. To measure the affinities of cSLAM mutants, cSLAM G71A (mutated from G to A at position 71), N72A, and E123A mutants and a quintuple mutant (H61A G71A N72A E123A H130A; referred to as “5A”) in a 2-fold serial-dilution series (5 concentrations, starting at 2,000 nM) were injected over the CDV H-coupled surface for 120 s, followed by a dissociation time of 400 s. The binding affinities of the different cSLAMs at a concentration of 1,000 nM were compared. Kinetic analyses were performed using the BIAevalution software (GE Healthcare) by applying a 1:1 Langmuir binding model.
Purification of soluble proteins.
The soluble CDV H construct carrying an N-terminal HA epitope tag (sH-HA) was expressed in 293T cells. After 3 days, 1 ml of Pierce HA epitope-tagged antibody-agarose conjugate (Pierce) was loaded into an Econo-Column chromatography column (1.0 by 5 cm; Bio-Rad). The column was packed and thoroughly rinsed with equilibration buffer (20 mM Tris, 0.1 M NaCl, 0.1 mM EDTA, pH 7.5), and then the supernatant was loaded on the column at a flow rate of 0.7 ml/h. After complete loading of the supernatant, the column was rinsed with 20 ml of wash buffer (20 mM Tris, 0.1 M NaCl, 0.1 mM EDTA, pH 7.5, 0.05% Tween). The column was then detached from the pump, and all the solution to the very top of the resin was run out by gravity flow. Three subsequent elutions were done by adding 1 ml of elution buffer (HA peptide reconstituted to 1 mg/ml in equilibration buffer) to the resin. The properly closed column was then incubated for 15 min at 37°C, and the eluate was collected by gravity flow. To purify soluble SLAM molecules, the proteins were first expressed in 293T cells. After 3 days, the cell culture supernatant was harvested and filtered with a 0.22-μm filter. Then, 10× binding buffer (500 mM NaH2PO4, 1.5 M NaCl, 100 mM imidazole, pH 8.0) and 400 μl of the cOmplete His tag purification resin (Roche) were added. The mixture was incubated for 20 h at 4°C on a shaking platform. To pellet the beads, the tubes were centrifuged at 2,100 × g for 15 min. The supernatant was then carefully removed, and the beads containing the bound proteins were resuspended in 1 ml wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 0.05% Tween 20, pH 8.0) and then transferred to an Eppendorf tube. The beads were washed four more times with 1 ml wash buffer. The proteins were eluted by the addition of 500 μl elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 0.05% Tween 20, pH 8.0) and incubation on the rotor for 15 min at room temperature. The agarose beads were then pelleted at 13,000 × g in the centrifuge, and the resulting supernatant was used for further analysis.
RESULTS
Identification of a critical residue at the front H-binding site of the canine SLAM V domain.
The cocrystal structure of MeV H bound to the SLAM receptor revealed four major sites within the V domain that are responsible for physical interactions with the attachment protein. To investigate whether canine SLAM (cSLAM) may interact with CDV H in a similar manner, we initially targeted SLAM residues located at the putative front cSLAM-H binding interface. Forty-three residues located at (or nearby) the putative four analogous binding sites in the cSLAM V domain were replaced with alanine, and the mutant proteins were subjected to a previously reported semiquantitative SLAM-H binding assay (Fig. 1A and B) (48). While most of the cSLAM mutants returned wild-type-like avidities of interaction, the cSLAM H61A, G71A, N72A, E123A, E124A, and H130A mutants exhibited values below 50% for interaction with soluble CDV H compared to the value for wt cSLAM (Fig. 1C). When mapped in our 3-dimensional (3-D) structural homology model (52), residues H61, E123, and E124 cluster in one side of SLAM, while amino acids G71 and N72 are located on the opposite side. Residue H130 maps more centrally (Fig. 1D). Because E124 is adjacent to E123 and shows only partial reduction in CDV H-binding activity when replaced by alanine, the following five residues were subjected to further analysis: H61A, G71A, N72A, E123A, and H130A. The fusion-triggering ability of the five selected mutants was next examined qualitatively (cell-cell fusion assay) and quantitatively (reporter gene-based cell content mixture assay). Interestingly, although some mutants displayed reduced membrane fusion triggering, bioactivity was never entirely inhibited (Fig. 1E and F). Furthermore, while the cSLAM N72A mutant exhibited a slight reduction in intracellular transport competence, all of the others were properly surface expressed (Fig. 1F). Of note, the cSLAM E123A mutant was characterized by a clear increase in surface expression, which interestingly correlated with the most dramatic loss-of-function phenotype (Fig. 1E and F).
FIG 1.

Identification of key regulatory residues in the front H-binding site of the V domain of canine SLAM. (A, B, and D) Structural homology model of the canine SLAM V domain in complex with one CDV H head or alone (52). (A) The canine SLAM (cSLAM) V domain is color coded in green, and the H protein in red. (B) All residues targeted for alanine-scanning mutagenesis are highlighted in blue. (D) Color coding identifies the residues whose mutation to alanine had a moderate (yellow) or strong (red) effect on the binding activity to CDV H. (C) Results of the semiquantitative CDV H-cSLAM binding assay. Vero cells were transfected with wt SLAM or one of its mutants (HA tagged) and treated 24 h later with the soluble form of CDV H (FLAG tagged). The cells were then stained with an anti-HA MAb and subjected to flow cytometry to record quantitative values. The CDV H-cSLAM binding activities were calculated as the ratios of mean fluorescence intensities obtained with the anti-FLAG MAb (reporting the binding activity of soluble H molecules to each of the membrane-anchored cSLAM variants) normalized to the levels obtained with the anti-HA MAb (reporting the cell surface expression of each of the cSLAM variants). The ratio obtained for the unmodified CDV H-cSLAM combination was arbitrarily set at 100%. Orange and red bars indicate interactions between CDV H and the selected cSLAM variant of less than 50% or less than 20%, respectively. (E) Results of qualitative syncytium formation assays in Vero cells triggered by coexpression of CDV H (strain A75/17), CDV F (strain A75/17), and cSLAM or a cSLAM mutant. To improve the sensitivity of the assay, the cells were additionally transfected with a plasmid encoding the red fluorescent protein. Images of fluorescence emissions from induced cell-cell fusion in representative fields are shown. The pictures were taken 24 h posttransfection with a confocal microscope (Fluoroview FV1000; Olympus). (F) Dark-gray bars show the results for cell surface expression of cSLAM and its mutants as determined by treating Vero cells at 24 h posttransfection with an anti-HA MAb. After the addition of the secondary antibody, MFI values were recorded by flow cytometry. All values were normalized to the one recorded with the unmodified cSLAM molecule. Means ± standard deviations (SD) of data from three independent experiments performed in triplicate are shown. Light-gray bars show the results of the quantitative cell-cell fusion assay. One population of Vero cells (target cells) were infected with MVA-T7 (MOI of 1) and transfected with DNA plasmids encoding the various SLAM mutants. In parallel, another population of Vero cells (effector cells) was transfected with vectors expressing wt F and wt H and a plasmid containing the luciferase reporter gene under the control of the T7 promoter. At 4 h posttransfection, the effector cells were split (1:2) and seeded into new wells. Fifteen hours later, the target cells were detached and mixed with the effector cells. After 5 h at 37°C, fusion was quantified indirectly by using a commercial luciferase-measuring kit. For each experiment, the value obtained for the unmodified F and H combination was set to 100%. Means ± SD of data from three independent experiments performed in duplicate are shown.
Overall, our alanine-based mutagenesis screen identified five residues in cSLAM that affected (i) binding activity to a soluble form of CDV H and (ii) fusion promotion through contacts with standard H tetramers. Because analogous SLAM regions have previously been reported to contact MeV H, these results further indicate a common mode of binding to the immune cell receptor SLAM by different members of the Morbillivirus genus.
Cysteine-based substitution led to successful engineering of covalent soluble cSLAM/H complexes.
To further validate the presumptive mode of binding between CDV H and cSLAM, we selected residues G71 and P541 in soluble constructs of cSLAM and CDV H, respectively, for mutagenesis. Indeed, our homology model indicates a close spatial positioning between these two amino acids, with a putative distance between their alpha carbons of less than 7.5 Å (Fig. 2A). We thus speculated that replacing these two residues with cysteine might lead to the generation of covalent CDV H/cSLAM complexes.
FIG 2.

Successful covalent CDV H-SLAM complex engineering. (A) Structural homology model of the cSLAM V domain in complex with one CDV H head. The inset highlights the putative distances between residue P541of CDV H and amino acids G71 and N72 of cSLAM. (B) Atomic structure of the human SLAM V domain homodimer. The two critical E71 residues are shown. (C and D) Biochemical assessment of the disulfide bond formation linking CDV H to cSLAM. His-tagged soluble versions of CDV H and cSLAM were coexpressed in 293T cells, and the protein complex immunoprecipitated (IP) with an anti-His MAb. Proteins were then run in Tris-acetate gels (nonreducing conditions) (C) or SDS-PAGE gels (reducing conditions) (D) and detected by immunoblotting (IB) using either anti-H (top) or anti-SLAM (bottom) polyclonal antibodies. H, H monomers; H2, H dimers; H4, H tetramers; H2-SLAM (1×), H dimers bound to one SLAM unit; H2-SLAM (2×), H dimers bound to two SLAM units.
Unmodified or mutant soluble engineered constructs (additionally carrying hexahistidine tags) were expressed alone or in different combinations for 3 days in 293T cells. Upon harvesting of the cell supernatants, the soluble constructs were subjected to immunoprecipitation (IP) using an anti-His monoclonal antibody (MAb) and fractionated on SDS-PAGE gels (reducing conditions) or Tris-acetate gels (nonreducing conditions). Antigenic materials were then detected using either an anti-CDV H (51) or anti-cSLAM (53) polyclonal antibody (PAb). As expected from our previous study, when membrane-anchored and soluble forms of CDV H (expressed alone) were separated under nonreducing conditions, a prominent band representing dimers was detected, although a small proportion of resisting tetramers were also systematically observed (Fig. 2C, top) (48). In contrast, while unmodified soluble cSLAM essentially ran as monomers under nonreducing conditions, the G71C mutant generated one population of monomers and a second one very likely representing dimers (Fig. 2C, bottom). This finding is not completely unexpected, since residues 71 oppose each other in the SLAM-SLAM homodimeric interface (Fig. 2B), as observed in a previously generated structural homology model (29). Consistent with a noncovalent CDV H-cSLAM interaction, coexpression of both unmodified soluble constructs resulted in running profiles that were very similar to those detected when expressed individually. In sharp contrast, however, when both cysteine mutants were expressed together, a single band of intermediate molecular weight was recognized by the anti-CDV H PAb (Fig. 2C, top). The latter most likely represented H dimers covalently bound to two SLAM units, since a band of similar size was also detected using the anti-cSLAM PAb (Fig. 2C, bottom). All of the proteins' migration profiles were identical when they were run under reducing conditions, thereby confirming the validity of the assay (Fig. 2D).
Overall, the successful engineering of covalent soluble CDV H/cSLAM complexes strongly argues in favor of an overall similar mode of binding between morbillivirus attachment proteins and their respective immune cell-specific receptor SLAMs.
Combined mutations at the front H-binding interface are required to fully abrogate fusion triggering.
Having demonstrated that mutants with single alanine mutations at the front H-binding interface of cSLAM nevertheless remained partially functional, we next examined whether other types of substitutions or combined mutations might lead to complete membrane fusion-triggering inhibition. Because it was recently reported that both glycoproteins of CDV strain A75/17 induced membrane fusion in Vero cells expressing the lion SLAM receptor inefficiently (54), we mutated residues G71, N72, and H130 (H61 and E123 are conserved) into the corresponding amino acids found in the lion SLAM predicted amino acid sequence. Interestingly, the derived mutants (G71Q, N72K, G71Q N72K, and H130Q) were not deficient in either intracellular transport competence or cell-cell fusion triggering, although bioactivity was slightly reduced for the G71Q N72K and G71Q cSLAM mutants (Fig. 3A and D).
FIG 3.

Investigation of the ability of the identified SLAM mutants to trigger the CDV membrane fusion machinery. (A to C) Cell-cell fusion activity in Vero cells triggered by coexpression of CDV H, CDV F, and cSLAM (or cSLAM mutants). To improve the sensitivity of the assay, the cells were additionally transfected with a plasmid encoding the red fluorescent protein. Images of fluorescence emissions from induced cell-cell fusion in representative fields are shown. The pictures were taken 24 h posttransfection with a confocal microscope (Fluoroview FV1000; Olympus). (D and E) Dark-gray bars show the results for cell surface expression of the wt SLAM and SLAM mutants, determined by treating Vero cells 24 h posttransfection with an anti-HA MAb. After the addition of the secondary antibody, MFI values were recorded by flow cytometry. All values were normalized to the one recorded with the unmodified cSLAM molecule. Means ± SD of data from three independent experiments performed in triplicates are shown. Light-gray bars show the results for quantitative cell-cell fusion assay performed as described in the legend to Fig. 1F.
Thus, because very similar results were obtained regardless of the nature of the side chains of the selected SLAM positions, we generated mutants with sets of combinatorial alanine mutations as follows: double mutations G71A N72A, H61A E123A, and E123A H130A, a triple mutation (H61A E123A H130A; referred to as “3A”), a quadruple mutation (G71A N72A E123A H130A; referred to as “4A”), and a quintuple mutation (H61A G71A N72A E123A H130A; referred to as “5A”). The effects of the latter SLAM mutants on membrane fusion triggering were next assessed in Vero cells expressing CDV F and H. Strikingly, while the G71A N72A double mutant preserved substantial fusion-triggering competency (Fig. 3B and E), the other two double mutants and the further combinatorial variants (3A, 4A, and 5A) exhibited strong inhibition of bioactivity (Fig. 3B, C, and E). Importantly, the latter loss-of-function results were not due to gross protein misfolding, since all mutants were properly surface expressed (Fig. 3E). Of note, as observed in the case of the E123A single alanine mutant, all combinatorial variants carrying the E123A substitution exhibited a clear enhancement of cell surface transport efficacy (Fig. 3E).
Taken together, these data suggest that, although single alanine mutations of the five critical residues identified in the front H-binding site of SLAM lead to severe impairments in physical interaction, a combination of mutations is strictly required to achieve complete lack of bioactivity. The results thus indicate that only subtle binding affinity between CDV H and cSLAM is sufficient to trigger plasma membrane fusion activity.
Residue E123A in the front H-binding site of SLAM is essential in supporting CDV H-F fusion activity.
Among the panel of single alanine mutants that we generated, the replacement of residue E123 led to the most significant reduction of membrane fusion triggering. To determine the effect of residue E123 in the context of the combinatorial alanine mutations, we mutated that position back to the parental residue (E) or to a nonconserved amino acid (R or S). The mutations were introduced in the genetic background of the SLAM mutants 3A, 4A, and 5A, thereby producing three additional variants, referred to as 2-A′+E123, 3-A′+E123, and 4-A′+E123, respectively. For controls, SLAM E123R and E123S single mutants were also generated.
Remarkably, regardless of the number of combinatorial alanine mutations, mutating the cSLAM alanine residue at position 123 back to the original amino acid (A123E) led to substantial restoration of membrane fusion triggering (Fig. 4A to F), whereas the E123R- and E123S-carrying combinatorial mutants remained fully inactive (Fig. 4A to F). In addition, while the cSLAM mutant harboring the single E123S substitution was characterized by an intermediate triggering efficacy of the CDV membrane fusion machinery, SLAM E123R exhibited strong inhibition of bioactivity (Fig. 4G and H). Cell surface expression determination revealed that all of the mutants generated were intracellular transport competent. As described above, all SLAM variants carrying a mutation at position 123 exhibited higher cell surface expression (Fig. 4B, D, F, and H). It should be emphasized that the ability of SLAM E123S to substantially trigger the CDV fusion machinery, while being expressed at the cell surface with improved efficiency compared to that of wt SLAM, argued against the possibility that the fusion-triggering deficiency exhibited by some specific SLAM mutants resulted from an increased amount of molecules at the plasma membrane. Consistent with this idea, reducing the amounts of wt and mutant SLAM proteins expressed at the cell surface by decreasing the amount of DNA transfected invariably led to a downregulation of the bioactivity (data not shown).
FIG 4.

Investigation of the impact of residue E123 of SLAM in triggering fusion. (A, C, E, and G) Cell-cell fusion activity in Vero cells triggered by coexpression of CDV H, CDV F, and cSLAM or a cSLAM mutant. To improve the sensitivity of the assay, the cells were additionally transfected with a plasmid encoding the red fluorescent protein. Images of fluorescence emissions from induced cell-cell fusion in representative fields are shown. The pictures were taken 24 h posttransfection with a confocal microscope (Fluoroview FV1000; Olympus). (B, D, F, and H) Dark-gray bars show the results for cell surface expression of the wt SLAM and SLAM mutants as determined by treating Vero cells 24 h posttransfection with an anti-HA MAb. After the addition of the secondary antibody, MFI values were recorded by flow cytometry. All values were normalized to the one recorded with the unmodified cSLAM molecule. Means ± SD of data from three independent experiments performed in triplicate are shown. Light-gray bars show the results for quantitative cell-cell fusion assay performed as described in the legend to Fig. 1F.
Altogether, these findings underline the critical effect of the SLAM residue E123 in supporting the membrane fusion process.
Membrane-anchored SLAM mutants and CDV H proteins exhibit physical interactions.
We next conducted coimmunoprecipitation (co-IP) experiments to biochemically confirm the notion that fusion modulation by mutations residing in the front H-binding site of SLAM resulted from impaired physical interactions. Toward this aim, we coexpressed the full-length versions of cSLAM and CDV H proteins in Vero cells and pulled down the potential complexes using an anti-HA MAb that targeted cSLAM. Finally, bound H proteins were detected using an anti-H PAb. Unexpectedly, as shown by the results in Fig. 5A, wt cSLAM, together with all of the cSLAM single mutants tested, could very efficiently bind to full-length CDV H protein. Likewise, all of the combined alanine mutants, as well as the variants with selected residues mutated into their lion SLAM amino acid counterparts, also exhibited significant physical interaction with CDV H (Fig. 5B and C). Conversely, the lion SLAM molecule appeared to bind to CDV H with very weak affinity. Indeed, only trace amounts of CDV H were detected by co-IP (Fig. 5B and C), despite efficient intracellular expression and cell surface targeting of the lion SLAM molecule (Fig. 3D). Hence, this finding argued against the hypothesis that the CDV H-SLAM interaction being monitored resulted from the experimental settings of the co-IP assay. As expected from the increased intracellular and surface expression profiles exhibited by SLAM variants harboring the E123A substitution, enhanced amounts of co-IP H materials were detected from coexpressing Vero cells (Fig. 5A and B). Interestingly, the SLAM variants carrying E123A also generated a second band with slower gel mobility, which may correspond to SDS-PAGE-resistant noncovalent homodimers.
FIG 5.

Biochemical assessment of the avidity of CDV H-cSLAM interactions. (A to C) Coimmunoprecipitation assays. CDV HFLAG and HA-tagged wt cSLAM, mutant cSLAM, or lion SLAM were coexpressed in Vero cells and subsequently lysed with RIPA buffer 24 h posttransfection. The CDV H-SLAM complexes were then immunoprecipitated (IP) with an anti-HA MAb and protein G-Sepharose bead treatment. Proteins were boiled and subjected to immunoblotting (IB) using an anti-CDV H polyclonal antibody to detect H antigenic materials (co-IP). Co-IP CDV H proteins were detected in comparison with CDV H proteins present in cell lysates prior to IP by immunoblotting using the same anti-H antibody. Gels illustrating total expression and immunoprecipitation of SLAM molecules are also shown (detected using an anti-HA polyclonal antibody). The specific MAbs used for the immunoprecipitation (IP) or immunoblotting (IB) steps are indicated on the left of the gels. (B) Of note, the white line on the right side of the gels shows where we cropped the gels for easier comparisons between the canine SLAM mutants and the lion SLAM molecule.
Overall, with the exception of the lion SLAM receptor, our co-IP assays revealed unperturbed CDV H-SLAM interactions when both molecules were coexpressed as full-length constructs in the identical cells. Furthermore, SLAM variants carrying the E123A substitution may also display stronger SLAM-SLAM homophilic interactions.
Binding kinetics and affinities of soluble CDV H and cSLAM constructs.
The discrepancies in the CDV H-SLAM interactions recorded using our flow cytometry-based cellular assay or our co-IP experiments prompted us to employ surface plasmon resonance (SPR) analyses to further investigate in detail the binding kinetics and affinities of the attachment protein with its receptor and mutants of the receptor. In this experimental setting (where the soluble forms of both CDV H and cSLAM are required), wt CDV H and cSLAM exhibited a substantial equilibrium of the dissociation constant (KD), although it was relatively weaker than the one previously recorded for the related MeV H-human SLAM (hSLAM) interaction (Fig. 6A) (29, 55). Variations of the experimental settings and/or specific values monitored with the H protein of the neurovirulent CDV strain A75/17 may explain the recorded differences in receptor-binding affinities. Interestingly, under these conditions, all cSLAM variants tested (G71A, N72A, E123A, and 5A) returned values translating to substantially reduced binding affinities (Fig. 6B and Table 1). Although they defined very low binding affinities (at the limit of the SPR sensitivity), these values nevertheless may still represent some relevant physical interactions between the two molecules, since they are within the range of values previously determined for the productive homophilic SLAM-SLAM binding affinity (29).
FIG 6.
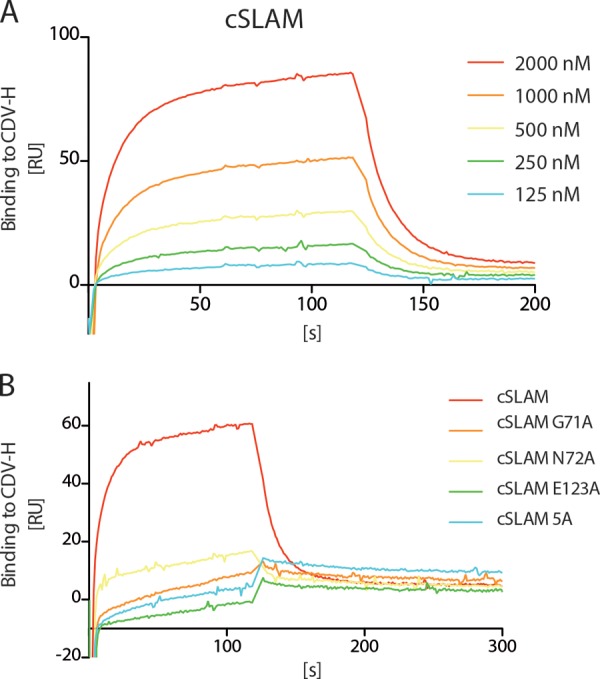
Lack of binding of the soluble cSLAM mutants to CDV H. (A) Surface plasmon resonance was employed to determine the binding kinetics of soluble CDV H to different concentrations of unmodified soluble cSLAM molecules. (B) Surface plasmon resonance was employed to determine the binding affinity of soluble CDV H to soluble cSLAM and cSLAM mutants (at the same concentration, 1,000 nM).
TABLE 1.
Binding kinetics of soluble CDV H ectodomain to soluble SLAM ectodomains
| Ligand | ka (1/s) | kd (1/s) | KD (μM) |
|---|---|---|---|
| cSLAM | 2.5 × 104 | 2.0 × 10−1 | 8.0 |
| cSLAM G71A | 6.3 × 103 | 5.9 × 10−1 | 93.7 |
| cSLAM N72A | 2.1 × 104 | 5.7 × 10−1 | 27.1 |
| cSLAM E123A | 2.6 × 104 | 6.4 × 10−1 | 24.6 |
| cSLAM 5A | 1.2 × 104 | 5.7 × 10−1 | 47.5 |
aka, association constant (on rate); kd, dissociation constant (off rate); KD, equilibrium dissociation constant.
Taken together, when using at least one soluble molecule (of SLAM or CDV H) in the experimental setting, both glycoproteins exhibited limited (wt CDV H and wt SLAM; SPR) or very weak (wt CDV H and SLAM mutants; flow cytometry based and SPR) binding affinities. Conversely, when using the full-length, membrane-anchored versions of the SLAM and H proteins (which were additionally coexpressed in the same cell), strong physical interaction could be demonstrated regardless of the mutation introduced into the V domain of SLAM.
Investigation of the putative back H-binding site of the SLAM V domain.
A high-resolution structure of MeV H in complex with the receptor SLAM revealed two tetrameric organizations (29). Interestingly, in one of these conformational states (the V shape), the V domain of two SLAM units was found to be sandwiched between two globular heads of two different H dimers, thereby illustrating a front and a back H-binding site (Fig. 7A and B). To investigate the functional relevance of the back H-binding site of cSLAM, we selected three critical residues (P33, P36, and K38) that map to this region and are potentially involved in direct interaction with the H head (Fig. 7C). Three SLAM variants with nonconservative substitutions were generated, bE (carrying all three residues mutated into E), bN (carrying all three residues mutated into N), and bRKN (carrying the three amino acids mutated into R, K, and N, respectively). The results in Fig. 7D and F illustrate that none of the three mutations had a substantial effect on the fusion-triggering ability of SLAM, while the mutants were expressed at the cell surface in a wild-type-like manner (Fig. 7F). In addition, all three mutants retained wild-type-like binding efficacy to CDV H, as determined by semiquantitative SLAM-H binding and co-IP assays (Fig. 7E and G).
FIG 7.

Determination of the functional impacts of SLAM mutants with mutations of residues in the back H-binding site. (A to C) Structural homology model of the CDV H tetrameric head domains (V shape) in complex with four V domains of cSLAM. (B) Image focusing on head 1 (light gray) of the CDV H tetramer bound to SLAM1 (red) and SLAM4 (dark gray). (C) The inset represents a zoomed depiction of the binding of cSLAM to the back of H. Key residues selected for mutagenesis are highlighted in magenta (P33, P36, and K38). (D) Cell-cell fusion activities in Vero cells triggered by coexpression of CDV H, CDV F, and cSLAM or a cSLAM mutant. To improve the sensitivity of the assay, the cells were additionally transfected with a plasmid encoding the red fluorescent protein. Images of fluorescence emissions from induced cell-cell fusion in representative fields are shown. The pictures were taken 24 h posttransfection with a confocal microscope (Fluoroview FV1000; Olympus). (E) Semiquantitative CDV H-cSLAM binding assay performed as described in the legend to Fig. 1C. (F) Dark-gray bars show the results for cell surface expression of the wt SLAM and cSLAM mutants as determined by treating Vero cells 24 h posttransfection with an anti-HA MAb. After the addition of the secondary antibody, MFI values were recorded by flow cytometry. All values were normalized to the one recorded with the unmodified cSLAM molecule. Means ± SD of data from three independent experiments performed in triplicates are shown. Light-gray bars show the results for quantitative cell-cell fusion assay performed as described in the legend to Fig. 1F. (G) Co-IP assays were performed as described in the legend to Fig. 5.
Overall, these results suggest that either the V-shaped conformation of H tetramers is not biologically relevant or the putative back H-binding site of SLAM is not required to control the triggering of the CDV fusion machinery. Alternatively, we cannot rule out the hypothesis that additional mutations should be introduced at (or near) the back binding site to achieve a recordable loss-of-function phenotype.
Combined front and back mutations in SLAM did not significantly alter the activation of the CDV fusion machinery.
Although introducing mutations at three critical positions in the putative back H-binding site of cSLAM did not significantly modulate its capacity to trigger the CDV membrane fusion machinery, we thought to further investigate the effects of these substitutions in SLAM variants characterized by partial impairments in fusion triggering. To this aim, we replaced the three key residues in the genetic background of the SLAM mutants 4-A′+E123, 4-A′+E123R, and 4-A′+E123S. As expected from the results of our previous experiments, qualitative and quantitative cell-cell fusion assays revealed that only the variant bE+4-A′+E123 exhibited residual bioactivity (Fig. 8A and B). Importantly, compared to the 4-A′+E123 variant, the SLAM mutant additionally harboring the back-site mutations (bE+4-A′+E123) did not display a further reduction in fusion triggering (Fig. 4E and 8A and B). Consistent with our previous results, co-IP assays performed with full-length versions of CDV H and the selected SLAM mutants invariably returned efficient physical interactions (Fig. 8C and D). Correlating with increased intracellular (Fig. 8C and D) and cell surface expression (Fig. 8B, E, and F), SLAM mutants carrying a substitution at position 123 displayed larger amounts of co-IP H materials, regardless of the presence or absence of the back mutations (Fig. 8C and D). Very similar results were obtained independently of whether the back H-binding site was mutated into glutamic acid (E) or asparagine (N) (Fig. 8D). An exception was that enhanced amounts of H proteins were copurified from SLAM 4-A′+E123 pulldown experiments, while this mutant exhibited wild-type-like intracellular and cell surface expression (Fig. 8C to F), thereby suggesting an increased binding affinity of this specific SLAM mutant with CDV H. Strikingly, additional substitutions of either three E or three N residues in the putative back H-binding site of SLAM 4-A′+E123 generated variants (bE+4-A′+E123 and bN+4-A′+E123) that lost the enhanced binding affinity that was observed with the parental version (4-A′+E123) (Fig. 8D).
FIG 8.

Determination of the functional impact of SLAM variants harboring front and back mutations. (A) Cell-cell fusion activity in Vero cells triggered by coexpression of CDV H, CDV F, and cSLAM or a cSLAM mutant. To improve the sensitivity of the assay, the cells were additionally transfected with a plasmid encoding the red fluorescent protein. Images of fluorescence emissions from induced cell-cell fusion in representative fields are shown. The pictures were taken 24 h posttransfection with a confocal microscope (Fluoroview FV1000; Olympus). (B, E, and F) Dark-gray bars show the results for cell surface expression of the wt SLAM and SLAM mutants, determined by treating Vero cells 24 h posttransfection with an anti-HA MAb. After the addition of the secondary antibody, MFI values were recorded by flow cytometry. All values were normalized to the one recorded with the unmodified cSLAM molecule. Means ± SD of data from three independent experiments performed in triplicates are shown. (B) Light-gray bars show the results for quantitative cell-cell fusion assay performed as described in the legend to Fig. 1F. (C and D) Co-IPs were performed as described in the legend to Fig. 5.
Collectively, while these results support the notion that the back H-binding site of SLAM does not significantly affect its bioactivity, physical interactions with CDV H might, however, be influenced.
The back H-binding site of SLAM interferes slightly with CDV H binding.
The above-described findings tend to argue for a putative effect of the back H-binding site of SLAM in modulating physical interactions with the membrane-anchored version of CDV H. It is important, however, to note that the latter phenotype was recorded in a SLAM mutant (4-A′+E123) that initially exhibited an increased interaction with CDV H, which therefore complicated the interpretation of the results. To overcome this problem, we introduced the back-site mutations (E, N, or RKN) in the genetic background of the G71A N72A SLAM variant. Indeed, the latter mutant was selected for three main reasons: (i) it displays near wild-type-like ability to trigger the CDV membrane fusion machinery, (ii) it interacts with CDV H in a manner similar to that of the original canine SLAM molecule, and (iii) it has good surface expression. The results in Fig. 9B show that the three newly generated SLAM variants did not exhibit any significant reduction in cell surface expression. However, co-IP experiments using these SLAM variants indicated slightly reduced interaction affinities to CDV H compared to those detected for the SLAM G71A N72A and 3-A′ front-only H-binding site mutants. Furthermore, the efficiency of binding exhibited by the double binding-site mutants might be even more pronounced, since the intracellular expression rate of the SLAM G71A N72A variant was limited in this experiment (Fig. 9C). While any differences in bioactivity between the three mutants could hardly be detected based on the qualitative assay (Fig. 9A), slight reductions compared to the results for the parental G71A N72A SLAM variant were observed when assessed using the quantitative fusion assay (Fig. 9B). Although this discrepancy may rely on different sensitivities for the two assays, they could also emerge from the different times of incubation used between the qualitative and quantitative fusion assays (overnight versus 5 h, respectively).
FIG 9.
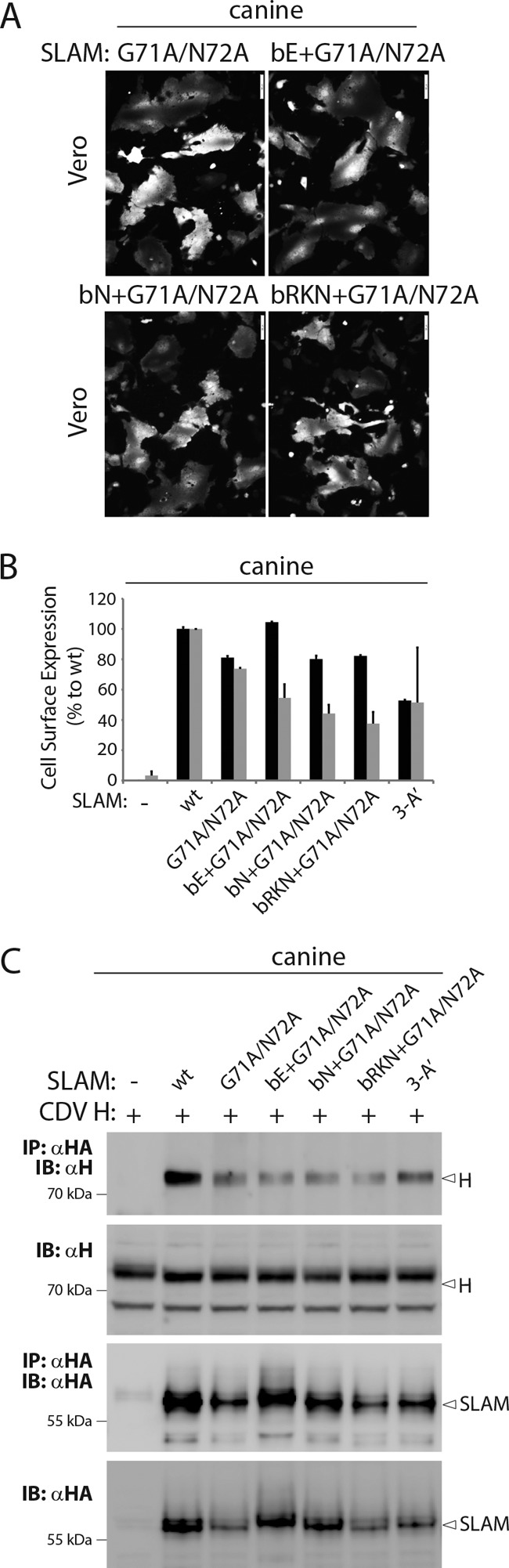
Determination of the functional impact of back substitutions in the background of the front SLAM mutant G71A/N72A. (A) Cell-cell fusion activity in Vero cells triggered by coexpression of CDV H, CDV F and cSLAM (or derivative mutants). To improve the sensitivity of the assay, the cells were additionally transfected with a plasmid encoding the red fluorescent protein. Images of fluorescence emissions from induced cell-cell fusion representative fields are shown. The pictures were taken 24 h posttransfection with a confocal microscope (Fluoroview FV1000; Olympus). (B) Dark-gray bars show the results for cell surface expression of the wt SLAM- and SLAM mutants, determined by treating Vero cells 24 h posttransfection with an anti-HA MAb. After the addition of the secondary antibody, MFI values were recorded by flow cytometry. All values were normalized to the one recorded with the unmodified cSLAM molecule. Means ± SD of data from three independent experiments performed in triplicates are shown. Light-gray bars show the results for quantitative cell-cell fusion assay performed as described in the legend to Fig. 1F. (C) Co-IPs were performed as described in the legend to Fig. 5.
DISCUSSION
Cell entry of morbilliviruses relies on the concerted action of two surface glycoproteins (H and F) that undergo conformational changes upon the binding of H to a host cell receptor, resulting in membrane fusion (26, 27, 48, 56–59). In this study, we aimed at improving our fundamental understanding of the first step that triggers the CDV membrane fusion machinery. The atomic structure of MeV H in complex with its immune cell receptor SLAM not only revealed the precise binding interface but, interestingly, indicated two discrete tetrameric conformations assumed by H (29). Importantly, in one of these, the V shape, it appeared that two regions of the V domain of SLAM (referred to as the front and back H-binding sites) were involved in short-range interactions with two monomeric H head domains of two discrete H dimers. Pending the determination of the crystal structure of CDV H in complex with the canine SLAM receptor to definitely spotlight the binding interface, we used alanine-scanning and structure-guided mutagenesis to shed light on the initial cSLAM and CDV H interaction in the work presented here.
Using a previously described semiquantitative flow cytometry-based cellular assay and surface plasmon resonance experiments, we could identify five critical residues clustering at the front H-binding site of the SLAM V domain that are very likely involved in short-range interaction with CDV H. Indeed, the replacement of SLAM residues H61, G71, N72, E123, and H130 with alanine led to severe impairments in these physical interactions. Since all of the corresponding five amino acids are located at, or in the case of G71, adjacent to, the binding interface of the MeV H/SLAM complex, our findings strongly suggest that the overall mode of binding between the attachment protein and SLAM is conserved among all members of the Morbillivirus genus. Consistent with this idea, using bioinformatics analyses of 3-D homology models of SLAM, Ohishi and colleagues recently hypothesized that these amino acids (among others) could be involved in productive interaction between the morbillivirus H protein and the immune receptor SLAM (46).
Interestingly, although even a single alanine substitution at one of these five critical positions located at the front H-binding site of cSLAM led to severe impairment in physical interaction with CDV H (as observed using flow cytometry-based and surface plasmon resonance assays), the cell-cell fusion-triggering capacity nevertheless remained substantial, being most affected by mutation of residue E123. When this residue was replaced with alanine, the resulting SLAM E123A variant required at least two additional mutations (among the five regulatory residues identified) to achieve complete inhibition of its fusion promotion ability. These findings led to the assumptions that (i) some kind of interaction must have nevertheless occurred between membrane-anchored versions of CDV H and the selected individual cSLAM variants and (ii) weak interaction between CDV H and cSLAM is sufficient to retain, at least partially, the biological function. In support of the first hypothesis, we could indeed readily detect physical interaction between CDV H and the SLAM variants using our co-IP assay, which was based on the coexpression of full-length versions of CDV H and cSLAM in the same cells. It is also noteworthy that we could record by SPR slight interactions between CDV H and the SLAM single mutants. Moreover, consistent with the second hypothesis, our data illustrated efficient triggering of the CDV membrane fusion machinery by the lion SLAM molecule, although the binding activity between CDV H and lion SLAM remained almost undetectable biochemically.
It is thus tempting to speculate that productive interactions between CDV H and cSLAM may depend on a more complex mode of binding, perhaps involving additional domains of the receptor and/or the attachment protein, as suggested in the V shape conformation of the MeV H and SLAM cocrystal structure (29). Therefore, investigation of the interaction based on purified truncated constructs of H and/or SLAM, while being relevant to study the front binding interface, may lack the necessary domains and/or specific conformational states that might be required to mimic physical binding of full-length, membrane-anchored forms of the two molecules. Alternatively, it cannot be excluded that (i) the experimental settings of our biochemical CDV H-cSLAM co-IP assay generated unspecific interactions or (ii) an unknown cellular factor assembled with the full-length H and SLAM proteins to form a tripartite functional complex. Future studies are therefore necessary to discriminate between these different possibilities.
If interactions between the front H-binding site of SLAM and H are nonetheless considered to be (at least partially) maintained even with SLAM variants harboring combined mutations at the interactive site, then the regulatory residues identified may not merely affect fusion triggering by modulating the binding affinity with CDV H. In this case, we can speculate that, beyond regulating the SLAM-H affinity of interaction, some of these key residues may additionally contribute to the transmission of a critical fusion-triggering signal to the CDV H head domains. Strikingly, alignment of various species-specific SLAM sequences illustrates that the glutamate residue at position 123 of SLAM (E123) is the only amino acid of the front H-binding site (among the five identified positions) that is completely conserved (not shown). Furthermore, as stated above, the replacement of residue E123 led to the most pronounced detrimental effect on fusion-triggering activity, thereby strengthening its potentially essential role in regulating SLAM's bioactivity. Interestingly, based on the MeV H-SLAM atomic structure, E123 is involved in short-range contacts with residue R533 of MeV H (29). Strikingly, recombinant measles viruses harboring the R533A mutation in their receptor-binding protein exhibit a selective deficiency of cell entry in SLAM-expressing cells (19, 42). Likewise, and consistent with our hypothesis, Navaratnarajah and colleagues proposed a putative role of residue R533 of MeV H in transmitting an essential signal necessary to support the membrane fusion process, rather than merely engaging physical interactions with SLAM (55). We thus hypothesize that, besides the essential proper positioning of the H head's lateral region with the front H-binding site of the V domain of SLAM, productive triggering of the morbillivirus membrane fusion machinery may rely not only on proper affinity and binding between the two partners but also on a local conformational change. With regard to the recently proposed safety catch model of morbillivirus F activation (27, 39), this local structural modification may involve a repositioning of the side chains of residues SLAM E123 and H R533 (or the analogous R529 residue of CDV H) that will translate into the initiation of the large-scale movements of the heads (putatively reaching conformational intermediates, such as the X and/or V shapes), ultimately leading to structural rearrangements of the H stalk. Although structural information argues against large-scale conformational changes occurring within the paramyxovirus attachment protein monomeric head unit as a consequence of receptor binding (22, 29–31, 60–64), subtle local modifications cannot be excluded in all models proposed for F activation (26, 27, 36, 40). In fact, recent functional data obtained with henipaviruses are consistent with this idea (41). However, although the latest mechanistic data obtained with morbilliviruses support the safety catch model for F activation (39, 65, 66), we cannot formally exclude at this stage that the proposed productive SLAM-H interactions translate into membrane fusion triggering by one of the alternative mechanisms (stalk exposure/induced fit [36], attachment protein oligomerization [40], and bidentate attachment protein-F interaction [41] models).
However, as stated above, H-SLAM interactions may be more complex than just relying on the front H-binding interface. Indeed, the tetrameric V shape conformation of MeV H in complex with four units of SLAM revealed a second binding interface for two receptor molecules in the back. Since Hashiguchi and colleagues proposed that fusion triggering might rely on a conformational shift between the X- and V-shaped tetramers (thus potentially involving the back H-binding site in fusion triggering) (29), we mutated three selected residues that mapped to the putative back H-binding site of cSLAM to evaluate their effects on the activation of the CDV membrane fusion machinery. Regardless of whether the mutations were introduced into SLAM molecules additionally harboring substitutions in the front H-binding site or not, no substantial functional effect could be recorded. Rather, the back-site SLAM variants bE+G71A N72A and bE+4A-A′+E123 were the only mutants generated in this study that were characterized by weakened interaction with membrane-anchored CDV H proteins, as determined in our co-IP assays. It should be noted, however, that the recorded weaker H-binding activity of these mutants nevertheless remained stronger than that recorded with the lion SLAM receptor (essentially not detectable), which could nevertheless trigger membrane fusion to a substantial extent. Therefore, although selective deletions and/or additional substitutions at (or near) the back H-binding site might be required to achieve a noticeable functional impact, the finding that mutations in the back H-binding site weakened the cSLAM-H interaction but did not substantially weaken fusion efficacy supports a major role of the front H-binding site in triggering fusion. Taken together, while the back H-binding site of cSLAM might indeed contribute to short-range (or long-range) interaction with a specific structural state of CDV H, further experiments are required in order to determine the biological relevance of the back H-binding site with regard to the activation of the CDV cell entry machinery. Alternatively, we cannot yet formally exclude the hypothesis that, when studied in the context of membrane-anchored proteins, another domain of SLAM may contribute to physical interaction with a discrete H conformation.
Collectively, our findings advanced our general mechanistic understanding of the initial steps that are required to activate the membrane fusion complex of CDV. First, they highlight a mode of binding between the morbillivirus attachment protein and the immune receptor SLAM which may be common to all members of the genus, whereby functional interactions rely on the proper alignment of the H head's lateral region (blades 4, 5, and 6 of the beta-propeller) with the front site of the V domain of SLAM. Second, they spotlight a putative key role of the highly conserved residue E123, which is located within the front site of SLAM and may transmit a productive fusion-triggering signal to the H head domains. Third, they support the hypothesis that the entire molecular framework supporting functional interactions between membrane-anchored versions of CDV H and canine SLAM may depend either on additional microdomains of one (or both) protein(s) or on putative, yet-to-be-determined supplementary host cellular factor(s), which may assemble with the H/receptor complex. Finally, they provide further evidence that membrane fusion activity in morbillivirus infections appears to be finely tuned by a range of subtle molecular interactions between cell-specific receptors and the viral glycoproteins. This may translate into important variations in viral transmission and the ensuing cellular pathology, as well as immune responses, both of which determine the clinical course and outcome of the infection.
ACKNOWLEDGMENTS
We are grateful to Yusuke Yanagi for having provided the Vero-cSLAM cells.
This work was supported by the Swiss National Science Foundation (ref. no. 310030_153281 to P.P.) and the Berne University Research Foundation (ref. no. 14/2014 to P.P.).
REFERENCES
- 1.Roelke-Parker ME, Munson L, Packer C, Kock R, Cleaveland S, Carpenter M, O'Brien SJ, Pospischil A, Hofmann-Lehmann R, Lutz H, Mwamengele GL, Mgasa MN, Machange GA, Summers BA, Appel MJ. 1996. A canine distemper virus epidemic in Serengeti lions (Panthera leo). Nature 379:441–445. doi: 10.1038/379441a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakai K, Nagata N, Ami Y, Seki F, Suzaki Y, Iwata-Yoshikawa N, Suzuki T, Fukushi S, Mizutani T, Yoshikawa T, Otsuki N, Kurane I, Komase K, Yamaguchi R, Hasegawa H, Saijo M, Takeda M, Morikawa S. 2013. Lethal canine distemper virus outbreak in cynomolgus monkeys in Japan in 2008. J Virol 87:1105–1114. doi: 10.1128/JVI.02419-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osterhaus AD, Vedder EJ. 1988. Identification of virus causing recent seal deaths. Nature 335:20. doi: 10.1038/335020a0. [DOI] [PubMed] [Google Scholar]
- 4.Hall AJ. 1995. Morbilliviruses in marine mammals. Trends Microbiol 3:4–9. doi: 10.1016/S0966-842X(00)88861-7. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy S. 1998. Morbillivirus infections in aquatic mammals. J Comp Pathol 119:201–225. doi: 10.1016/S0021-9975(98)80045-5. [DOI] [PubMed] [Google Scholar]
- 6.Origgi FC, Plattet P, Sattler U, Robert N, Casaubon J, Mavrot F, Pewsner M, Wu N, Giovannini S, Oevermann A, Stoffel MH, Gaschen V, Segner H, Ryser-Degiorgis MP. 2012. Emergence of canine distemper virus strains with modified molecular signature and enhanced neuronal tropism leading to high mortality in wild carnivores. Vet Pathol 49:913–929. doi: 10.1177/0300985812436743. [DOI] [PubMed] [Google Scholar]
- 7.Origgi FC, Sattler U, Pilo P, Waldvogel AS. 2013. Fatal combined infection with canine distemper virus and orthopoxvirus in a group of Asian marmots (Marmota caudata). Vet Pathol 50:914–920. doi: 10.1177/0300985813476060. [DOI] [PubMed] [Google Scholar]
- 8.de Vries RD, Ludlow M, Verburgh RJ, van Amerongen G, Yuksel S, Nguyen DT, McQuaid S, Osterhaus AD, Duprex WP, de Swart RL. 2014. Measles vaccination of nonhuman primates provides partial protection against infection with canine distemper virus. J Virol 88:4423–4433. doi: 10.1128/JVI.03676-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 10.Tatsuo H, Ono N, Yanagi Y. 2001. Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J Virol 75:5842–5850. doi: 10.1128/JVI.75.13.5842-5850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noyce RS, Richardson CD. 2012. Nectin 4 is the epithelial cell receptor for measles virus. Trends Microbiol 20:429–439. doi: 10.1016/j.tim.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pratakpiriya W, Seki F, Otsuki N, Sakai K, Fukuhara H, Katamoto H, Hirai T, Maenaka K, Techangamsuwan S, Lan NT, Takeda M, Yamaguchi R. 2012. Nectin4 is an epithelial cell receptor for canine distemper virus and involved in neurovirulence. J Virol 86:10207–10210. doi: 10.1128/JVI.00824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noyce RS, Delpeut S, Richardson CD. 2013. Dog nectin-4 is an epithelial cell receptor for canine distemper virus that facilitates virus entry and syncytia formation. Virology 436:210–220. doi: 10.1016/j.virol.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 16.Delpeut S, Noyce RS, Richardson CD. 2014. The V domain of dog PVRL4 (nectin-4) mediates canine distemper virus entry and virus cell-to-cell spread. Virology 454–455:109–117. doi: 10.1016/j.virol.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 17.de Swart RL, Ludlow M, de Witte L, Yanagi Y, van Amerongen G, McQuaid S, Yuksel S, Geijtenbeek TB, Duprex WP, Osterhaus AD. 2007. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog 3:e178. doi: 10.1371/journal.ppat.0030178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yuksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog 7:e1001263. doi: 10.1371/journal.ppat.1001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leonard VH, Hodge G, Reyes-Del Valle J, McChesney MB, Cattaneo R. 2010. Measles virus selectively blind to signaling lymphocytic activation molecule (SLAM; CD150) is attenuated and induces strong adaptive immune responses in rhesus monkeys. J Virol 84:3413–3420. doi: 10.1128/JVI.02304-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leonard VH, Sinn PL, Hodge G, Miest T, Devaux P, Oezguen N, Braun W, McCray PB Jr, McChesney MB, Cattaneo R. 2008. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J Clin Invest 118:2448–2458. doi: 10.1172/JCI35454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ono N, Tatsuo H, Tanaka K, Minagawa H, Yanagi Y. 2001. V domain of human SLAM (CDw150) is essential for its function as a measles virus receptor. J Virol 75:1594–1600. doi: 10.1128/JVI.75.4.1594-1600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Lu G, Qi J, Li Y, He Y, Xu X, Shi J, Zhang CW, Yan J, Gao GF. 2013. Structure of measles virus hemagglutinin bound to its epithelial receptor nectin-4. Nat Struct Mol Biol 20:67–72. doi: 10.1038/nsmb.2432. [DOI] [PubMed] [Google Scholar]
- 23.Plattet P, Plemper RK. 2013. Envelope protein dynamics in paramyxovirus entry. mBio 4(4):e00413–13. doi: 10.1128/mBio.00413-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1449–1496. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 25.Mateo M, Navaratnarajah CK, Cattaneo R. 2014. Structural basis of efficient contagion: measles variations on a theme by parainfluenza viruses. Curr Opin Virol 5:16–23. doi: 10.1016/j.coviro.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bose S, Jardetzky TS, Lamb RA. 2015. Timing is everything: fine-tuned molecular machines orchestrate paramyxovirus entry. Virology 479–480:518–531. doi: 10.1016/j.virol.2015.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ader-Ebert N, Khosravi M, Herren M, Avila M, Alves L, Bringolf F, Orvell C, Langedijk JP, Zurbriggen A, Plemper RK, Plattet P. 2015. Sequential conformational changes in the morbillivirus attachment protein initiate the membrane fusion process. PLoS Pathog 11:e1004880. doi: 10.1371/journal.ppat.1004880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russell CJ, Jardetzky TS, Lamb RA. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J 20:4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hashiguchi T, Ose T, Kubota M, Maita N, Kamishikiryo J, Maenaka K, Yanagi Y. 2011. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol 18:135–141. doi: 10.1038/nsmb.1969. [DOI] [PubMed] [Google Scholar]
- 30.Yuan P, Thompson TB, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS. 2005. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13:803–815. doi: 10.1016/j.str.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 31.Xu K, Rajashankar KR, Chan YP, Himanen JP, Broder CC, Nikolov DB. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc Natl Acad Sci U S A 105:9953–9958. doi: 10.1073/pnas.0804797105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowden TA, Crispin M, Harvey DJ, Aricescu AR, Grimes JM, Jones EY, Stuart DI. 2008. Crystal structure and carbohydrate analysis of Nipah virus attachment glycoprotein: a template for antiviral and vaccine design. J Virol 82:11628–11636. doi: 10.1128/JVI.01344-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakashima M, Shirogane Y, Hashiguchi T, Yanagi Y. 2013. Mutations in the putative dimer-dimer interfaces of the measles virus hemagglutinin head domain affect membrane fusion triggering. J Biol Chem 288:8085–8091. doi: 10.1074/jbc.M112.427609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc Natl Acad Sci U S A 108:14920–14925. doi: 10.1073/pnas.1111691108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welch BD, Yuan P, Bose S, Kors CA, Lamb RA, Jardetzky TS. 2013. Structure of the parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) ectodomain. PLoS Pathog 9:e1003534. doi: 10.1371/journal.ppat.1003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bose S, Song AS, Jardetzky TS, Lamb RA. 2014. Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J Virol 88:3925–3941. doi: 10.1128/JVI.03741-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plattet P, Langedijk JP, Zipperle L, Vandevelde M, Orvell C, Zurbriggen A. 2009. Conserved leucine residue in the head region of morbillivirus fusion protein regulates the large conformational change during fusion activity. Biochemistry 48:9112–9121. doi: 10.1021/bi9008566. [DOI] [PubMed] [Google Scholar]
- 38.Plemper RK, Hammond AL, Cattaneo R. 2001. Measles virus envelope glycoproteins hetero-oligomerize in the endoplasmic reticulum. J Biol Chem 276:44239–44246. doi: 10.1074/jbc.M105967200. [DOI] [PubMed] [Google Scholar]
- 39.Brindley MA, Chaudhury S, Plemper RK. 2015. Measles virus glycoprotein complexes preassemble intracellularly and relax during transport to the cell surface in preparation for fusion. J Virol 89:1230–1241. doi: 10.1128/JVI.02754-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gui L, Jurgens EM, Ebner JL, Porotto M, Moscona A, Lee KK. 2015. Electron tomography imaging of surface glycoproteins on human parainfluenza virus 3: association of receptor binding and fusion proteins before receptor engagement. mBio 6:e02393–14. doi: 10.1128/mBio.02393-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Q, Stone JA, Bradel-Tretheway B, Dabundo J, Benavides Montano JA, Santos-Montanez J, Biering SB, Nicola AV, Iorio RM, Lu X, Aguilar HC. 2013. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog 9:e1003770. doi: 10.1371/journal.ppat.1003770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vongpunsawad S, Oezgun N, Braun W, Cattaneo R. 2004. Selectively receptor-blind measles viruses: identification of residues necessary for SLAM- or CD46-induced fusion and their localization on a new hemagglutinin structural model. J Virol 78:302–313. doi: 10.1128/JVI.78.1.302-313.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zipperle L, Langedijk JP, Orvell C, Vandevelde M, Zurbriggen A, Plattet P. 2010. Identification of key residues in virulent canine distemper virus hemagglutinin that control CD150/SLAM-binding activity. J Virol 84:9618–9624. doi: 10.1128/JVI.01077-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Messling V, Oezguen N, Zheng Q, Vongpunsawad S, Braun W, Cattaneo R. 2005. Nearby clusters of hemagglutinin residues sustain SLAM-dependent canine distemper virus entry in peripheral blood mononuclear cells. J Virol 79:5857–5862. doi: 10.1128/JVI.79.9.5857-5862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohno S, Seki F, Ono N, Yanagi Y. 2003. Histidine at position 61 and its adjacent amino acid residues are critical for the ability of SLAM (CD150) to act as a cellular receptor for measles virus. J Gen Virol 84:2381–2388. doi: 10.1099/vir.0.19248-0. [DOI] [PubMed] [Google Scholar]
- 46.Ohishi K, Suzuki R, Maeda T, Tsuda M, Abe E, Yoshida T, Endo Y, Okamura M, Nagamine T, Yamamoto H, Ueda M, Maruyama T. 2014. Recent host range expansion of canine distemper virus and variation in its receptor, the signaling lymphocyte activation molecule, in carnivores. J Wildl Dis 50:596–606. doi: 10.7589/2013-09-228. [DOI] [PubMed] [Google Scholar]
- 47.Bieringer M, Han JW, Kendl S, Khosravi M, Plattet P, Schneider-Schaulies J. 2013. Experimental adaptation of wild-type canine distemper virus (CDV) to the human entry receptor CD150. PLoS One 8:e57488. doi: 10.1371/journal.pone.0057488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ader N, Brindley MA, Avila M, Origgi FC, Langedijk JP, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J Biol Chem 287:16324–16334. doi: 10.1074/jbc.M112.342493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiener D, Plattet P, Cherpillod P, Zipperle L, Doherr MG, Vandevelde M, Zurbriggen A. 2007. Synergistic inhibition in cell-cell fusion mediated by the matrix and nucleocapsid protein of canine distemper virus. Virus Res 129:145–154. doi: 10.1016/j.virusres.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 50.Plattet P, Cherpillod P, Wiener D, Zipperle L, Vandevelde M, Wittek R, Zurbriggen A. 2007. Signal peptide and helical bundle domains of virulent canine distemper virus fusion protein restrict fusogenicity. J Virol 81:11413–11425. doi: 10.1128/JVI.01287-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cherpillod P, Beck K, Zurbriggen A, Wittek R. 1999. Sequence analysis and expression of the attachment and fusion proteins of canine distemper virus wild-type strain A75/17. J Virol 73:2263–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langedijk JP, Janda J, Origgi FC, Orvell C, Vandevelde M, Zurbriggen A, Plattet P. 2011. Canine distemper virus infects canine keratinocytes and immune cells by using overlapping and distinct regions located on one side of the attachment protein. J Virol 85:11242–11254. doi: 10.1128/JVI.05340-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wenzlow N, Plattet P, Wittek R, Zurbriggen A, Grone A. 2007. Immunohistochemical demonstration of the putative canine distemper virus receptor CD150 in dogs with and without distemper. Vet Pathol 44:943–948. doi: 10.1354/vp.44-6-943. [DOI] [PubMed] [Google Scholar]
- 54.Nikolin VM, Osterrieder K, von Messling V, Hofer H, Anderson D, Dubovi E, Brunner E, East ML. 2012. Antagonistic pleiotropy and fitness trade-offs reveal specialist and generalist traits in strains of canine distemper virus. PLoS One 7:e50955. doi: 10.1371/journal.pone.0050955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navaratnarajah CK, Vongpunsawad S, Oezguen N, Stehle T, Braun W, Hashiguchi T, Maenaka K, Yanagi Y, Cattaneo R. 2008. Dynamic interaction of the measles virus hemagglutinin with its receptor signaling lymphocytic activation molecule (SLAM, CD150). J Biol Chem 283:11763–11771. doi: 10.1074/jbc.M800896200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ader N, Brindley M, Avila M, Orvell C, Horvat B, Hiltensperger G, Schneider-Schaulies J, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2013. Mechanism for active membrane fusion triggering by morbillivirus attachment protein. J Virol 87:314–326. doi: 10.1128/JVI.01826-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brindley MA, Takeda M, Plattet P, Plemper RK. 2012. Triggering the measles virus membrane fusion machinery. Proc Natl Acad Sci U S A 109:E3018–E3027. doi: 10.1073/pnas.1210925109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Navaratnarajah CK, Negi S, Braun W, Cattaneo R. 2012. Membrane fusion triggering: three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J Biol Chem 287:38543–38551. doi: 10.1074/jbc.M112.410563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navaratnarajah CK, Kumar S, Generous A, Apte-Sengupta S, Mateo M, Cattaneo R. 2014. The measles virus hemagglutinin stalk: structures and functions of the central fusion activation and membrane-proximal segments. J Virol 88:6158–6167. doi: 10.1128/JVI.02846-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lawrence MC, Borg NA, Streltsov VA, Pilling PA, Epa VC, Varghese JN, McKimm-Breschkin JL, Colman PM. 2004. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J Mol Biol 335:1343–1357. doi: 10.1016/j.jmb.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 61.Bowden TA, Aricescu AR, Gilbert RJ, Grimes JM, Jones EY, Stuart DI. 2008. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat Struct Mol Biol 15:567–572. doi: 10.1038/nsmb.1435. [DOI] [PubMed] [Google Scholar]
- 62.Colf LA, Juo ZS, Garcia KC. 2007. Structure of the measles virus hemagglutinin. Nat Struct Mol Biol 14:1227–1228. doi: 10.1038/nsmb1342. [DOI] [PubMed] [Google Scholar]
- 63.Hashiguchi T, Kajikawa M, Maita N, Takeda M, Kuroki K, Sasaki K, Kohda D, Yanagi Y, Maenaka K. 2007. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc Natl Acad Sci U S A 104:19535–19540. doi: 10.1073/pnas.0707830104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santiago C, Celma ML, Stehle T, Casasnovas JM. 2010. Structure of the measles virus hemagglutinin bound to the CD46 receptor. Nat Struct Mol Biol 17:124–129. doi: 10.1038/nsmb.1726. [DOI] [PubMed] [Google Scholar]
- 65.Brindley MA, Suter R, Schestak I, Kiss G, Wright ER, Plemper RK. 2013. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J Virol 87:11693–11703. doi: 10.1128/JVI.01945-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Avila M, Khosravi M, Alves L, Ader-Ebert N, Bringolf F, Zurbriggen A, Plemper RK, Plattet P. 2015. Canine distemper virus envelope protein interactions modulated by hydrophobic residues in the fusion protein globular head. J Virol 89:1445–1451. doi: 10.1128/JVI.01828-14. [DOI] [PMC free article] [PubMed] [Google Scholar]