

The US Food and Drug Administration (FDA) notified Congress July 31, 2014,1 of its intent to regulate laboratory developed tests. These encompass thousands of clinical assays currently used in medical practice including most pharmacogenetic tests. This guidance has the potential to impact the innovation and sustainability of pharmacogenetic research and its clinical implementation.
We anticipate that several requirements could have substantial implications for both clinical research and the clinical implementation of pharmacogenetics. Input from federal funding agencies (eg, National Institutes of Health, Agency for Health Research Quality, Patient-Centered Outcomes Research Institute) will be needed to clarify who will bear the financial burden of some of the increased requirements. Further, key clarifications will be required from the FDA to understand how implementation will occur over the next decade (Figure 1). Clarity now will allow the research and clinical communities to effectively plan and budget for these changes. Below, we identify and describe the key issues.
Figure 1.
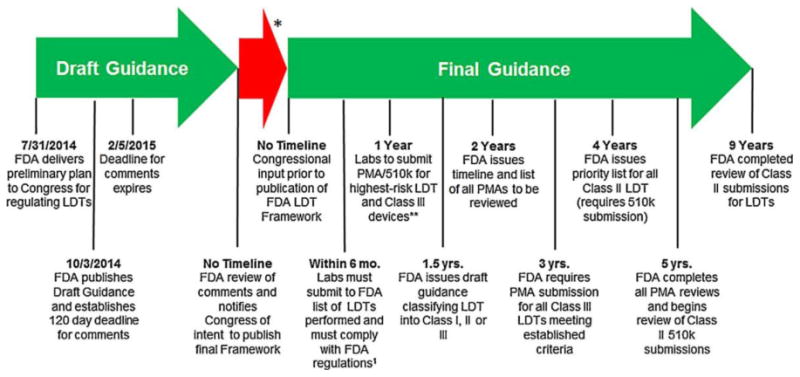
FDA timeline for implementation of oversight of laboratory-developed tests. *Time from FDA comment review and congressional input to the release of the final guidance is unknown. **PMA process is based on a per se demonstration of safety and effectiveness through “adequate and well-controlled” clinical trials. A successful PMA submission results in approval of the new device. The 510(k) process is somewhat analogous to the “generic” drug concept in that Premarket Notification is used to obtain marketing clearance for a device that is “substantially equivalent” in safety and effectiveness to another lawfully marketed device or to a standard recognized by the FDA when used for the same intended purpose(s).1 Federal regulations include medical device reporting as defined in the Code of Federal Register (CFR §803) and implementation of quality systems regulation (21 CFR §820).
Over the past decades, many pharmacogenomics biomarkers that are responsible for clinical variability in the pharmacokinetics and pharmacodynamics of drug therapy have been identified. These studies have mostly been conducted using laboratory-developed procedures or tests (LDTs).2 According to the published guidance, it appears that these clinical discovery and validation studies will now need to obtain investigational device exemptions (IDEs) from the FDA. For example, conducting a simple clinical study to determine if genetic variants in a drug-metabolizing enzyme are associated with the pharmacokinetics of a drug would require that the genotyping assay laboratory obtain an IDE before conducting the study. IDE submissions require substantial expertise and resources that are not typically common in academic research centers or Clinical Laboratory Improvement Amendment (CLIA)–certified clinical laboratories. This may even be new for drug companies that may have expertise in new drug application submissions, but not IDEs. Therefore, it will substantially increase the cost and resources needed to conduct these studies. It is not clear to us what harm has been done that will be solved by the IDE requirement that justifies such a change. The question of funding is important. Will funding agencies be willing to support these additional costs, or will it just reduce the scope or number of studies that will be able to be conducted? We suggest that the FDA use its enforcement discretion and not require IDEs for these types of translational studies.
Based on the language in the guidance document, clinical (ie, CLIA) laboratories providing pharmacogenetic genotyping using an LDT will be required to obtain premarket approvals (PMAs). As there are very few FDA-approved pharmacogenetic in vitro diagnostic devices, most clinical testing using LDTs is performed in CLIA laboratories, often with College of American Pathologists accreditation. The time and costs involved in obtaining PMAs are high. In a 2010 survey and analysis conducted by AdvaMed,3 US patients waited an average of 2 years longer than those in Europe to gain access to new medical technologies, which was attributable to the FDA device approval. In addition, the average total cost to bring a low to moderate in vitro diagnostic through the FDA 510(k) process was $31 million, with $24 million spent on FDA-dependent or -related activities. For higher-risk products submitted for approval, the average cost increased to $94 million, with $75 million linked to FDA requirements. In addition, the industry reported an average time from first communication with the FDA to clearance or approval ranging from 31 to 54 months. The FDA claims that CLIA certification of a laboratory performing an LDT does not provide the safety, oversight, and adverse event reporting requirements that exist with devices cleared or approved by the agency; however, this has not been supported by statistically valid evidence that this has caused harm to patients. At least publicly, they have only cited antidotal cases where a patient received inappropriate therapy as a result of a faulty LDT. In the FDA's notice to Congress of its intent to initiate oversight of LDTs (Federal Register/Vol. 79, No. 192/Friday, October 3, 2014), there was no data submitted that supports the claim that the status quo condition was causing harm. Despite this lack of evidence, it does express a concern about the potential risk to patients if LDTs do not receive oversight. The FDA in their October 2014 filing in the Federal Register noted that the complexity and expanded use of LDTs today are significantly different from 1976 when the Congress enacted Medical Device Amendment (MDA) and expanded the FDA oversight of medical devices intended for human use. To support the oversight of LDTs, the FDA is using the 1976 MDA in attempt to redefine laboratories that perform LDTs as manufacturers of medical devices intended for human use. There are potentially many implications, including financial and health economics, if the FDA is allowed to categorize these laboratories as manufacturers. During the September 9, 2014 House Committee on Energy and Commerce Hearing on the Proposed FDA Laboratory Developed Test Policy, Dr. Jeff Shuren, the Director of the Center for Devices and Radiological Health at the FDA, stated that the “FDA has not conducted a formal economic analysis” to justify the increased time and cost required to implement the proposed framework. In this context, it would be valuable to consider a “fast track” approval process or other creative approaches for new tests that have evidence of life-saving benefits (i.e., HLA-B*57:01 and abacavir), in the same way as drugs has occurred for drug approvals.
Currently, many laboratories decide on a genotyping platform that fits the needs of their laboratory and then design LDTs for multiple assays on a single platform. However, with companion diagnostics in which the drug and device are approved together by the FDA, drug and device manufacturers essentially have 1 instrument per drug, making it necessary for the laboratories to obtain multiple different instruments to provide pharmacogenomic services for multiple drugs. The initial capital investment and routine running costs (eg, maintenance contracts, training, and competency) will thus be much higher for laboratories. Depending on the device labeling, it could even require different instruments for the same genetic test when used for different drugs (eg, CYP2D6 testing for codeine and antidepressants), even though they test the exact same genetic variants. If the device (ie, instrument and test kit) is used for a different intended use (ie, same gene, different drug), then the laboratory will need to submit to the FDA to demonstrate cross-platform equivalency.
Last, input from payers will be needed to understand how reimbursement for clinical testing will be handled. For example, once a test is FDA-approved or -cleared, will the payers reimburse those tests at a significantly higher rate, so the clinical laboratory can recoup its cost for the FDA submission? The current procedural terminology coding system for reimbursement is platform agnostic. Though Centers for Medicare and Medicaid Service has a pilot program to differentiate reimbursement based on platform,4 will the payers even allow reimbursement for non-FDA-approved tests when there are FDA-approved tests available?
In conclusion, many questions remain to be answered related to the new FDA guidance on LDTs with respect to clinical research and implementation of pharmacogenetic testing. The FDA should provide specific evidence about the harm caused by LDTs, so a less obstructive solution could be identified. Although it may be true that there are some CLIA-accredited laboratories that have developed their own methods and tests that do not meet acceptable quality standards, why not selectively pursue these entities through existing or enhanced CLIA regulations rather than imposing FDA-directed oppressive and costly guidance across the entire spectrum of laboratory service providers and researchers?
Acknowledgments
Research reported in this publication was supported by National Human Genome Institute of the National Institutes of Health under award number 1U01HG007762-01 Embedding Pharmacogenotyping in an Integrated Health System for the Under-served. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Draft Guidance for Industry; Food and Drug Administration Staff and Clinical Laboratories. Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs) 2014 Jul 31; [Google Scholar]
- 2.Ferreira-Gonzalez A, Emmadi R, Day SP, et al. Revisiting oversight and regulation of molecular-based laboratory-developed tests. J Mol Diag. 2014;16(1):3–6. doi: 10.1016/j.jmoldx.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 3.FDA Impact on U.S. Medical Technology Innovation: A Survey of Over 200 Medical Technology Companies. 2010 Nov; [Google Scholar]
- 4.Molecular Diagnostic Program (MolDX1) Version 3.0. PALMETTO GPA, A Celerian Group Company. [computer program] http://www.palmettogba.com/palmetto/moldx.nsf/MolDX_Manual.pdf.