

Abstract
Purpose
HM781-36B is a novel and irreversible pan-human epidermal growth factor receptor (HER) inhibitor with TEC cytoplasmic kinase inhibition. The aim of this study is to evaluate the antitumor activity and mechanism of action for HM781-36B in CRC cell lines.
Materials and Methods
The CRC cell lines were exposed to HM781-36B and/or oxaliplatin (L-OHP), 5-fluorouracil (5-FU), SN-38. The cell viability was examined by Cell Titer-Glo luminescent cell viability assay kit. Change in the cell cycle and protein expression was determined by flow cytometry and immunoblot analysis, respectively. Synergism between 2 drugs was evaluated by the combination index.
Results
The addition of HM781-36B induced potent growth inhibition in both DiFi cells with EGFR overexpression and SNU-175 cells (IC50 = 0.003 and 0.005 μM, respectively). Furthermore, HM781-36B induced G1 arrest of the cell cycle and apoptosis, and reduced the levels of HER family and downstream signaling molecules, pERK and pAKT, as well as nonreceptor/cytoplasmic tyrosine kinase, BMX. The combination of HM781-36B with 5-FU, L-OHP, or SN-38 showed an additive or synergistic effect in most CRC cells.
Conclusion
These findings suggest the potential roles of HM781-36B as the treatment for EGFR-overexpressing colon cancer, singly or in combination with chemotherapeutic agents. The role of BMX expression as a marker of response to HM781-36B should be further explored.
Keywords: HM781-36B, Colorectal neoplasm, Epidermal growth factor receptor-neu receptor, BMX
Introduction
Colorectal cancer (CRC) is one of the major types of cancer worldwide, in terms of both morbidity and mortality. Despite continuous improvements in adjuvant therapy, the outcomes of treatment for locally advanced and metastatic disease remains disappointing, with 5-year survival rates of lower than 10% in patients with metastatic cancer [1]. Therefore, it is fundamentally important to develop novel agents with reliable biomarkers predicting the responses to such agents.
CRC is frequently associated with a high expression of epidermal growth factor receptors (EGFRs). In some studies, it has been reported that the overexpression of EGFR in colon cancer may be involved in potential metastasis and poor prognosis [2]. EGFR, member of a family of four ErbB receptor tyrosine kinases (EGFR[HER1]/ErbB1, HER2/ErbB2, HER3/ErbB3, and HER4/ErbB4), plays a crucial role in the regulation of cell proliferation, differentiation, angiogenesis, metastasis, and tumor invasiveness. These groups of receptors are activated by the binding of ligands to the external domain of receptors to form homo- or hetero-dimers with each other, which leads to the phosphorylation of the tyrosine kinase domain and the subsequent activation of multiple downstream signaling molecules involved in mitogenic and survival signaling pathways [3]. Therefore, the blockade of EGFR-mediated signaling pathways have been proposed as a specific therapeutic approach for metastatic CRC.
The agents that target EGFR are composed of monoclonal antibodies (mAbs), such as cetuximab and panitumumab, and small molecule tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib [4,5]. Specifically, cetuximab has improved overall survival in patients with KRAS wild-type metastatic CRC and erlotinib has been approved for the treatment of advanced lung cancers but has not been extensively studied in CRC. Also, a subset of patients with colorectal and lung cancer, who initially responded to anti-EGFR agents, develop secondary resistance after the initial benefit [6]. Extensive research based on the mechanisms of resistance to EGFR inhibitors has led to the development of the next-generation EGFR TKIs, more efficient anti-EGFR mAbs, and combination therapy with drugs targeting other ligands and downstream effectors.
The next generation of EGFR TKIs includes EGFR and HER2 reversible dual inhibitor, lapatinib, for the treatment of HER2-positive breast cancer, and a dual irreversible EGFR and HER2 TKI, BIBW-2992, which is capable of overcoming gefitinib resistance via acquired mutation (T790M) of EGFR [5,7]. The other irreversible EGFR TKIs, such as EKB-569 and CI-1033, can also block a gefitinib- and erlotinib-resistant mutant of EGFR (T790M), demonstrating further therapeutic efficacy for the irreversible inhibitors [8]. Currently, several irreversible EGFR TKIs are in clinical development for the remedy of various cancers. However, a previous clinical study reported that CI-1033 is associated with severe toxicity, suggesting that further development of the drug seems unlikely [9].
Previously, it has been reported that HM781-36B, 1-[4-[4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-yloxy]-piperidine-1-yl] propenone hydrochloride, is a novel and irreversible TKI of EGFR, HER2, and HER4, and has more favorable pharmacokinetic properties than that of BIBW-2992, as indicated by a lower effective dosage as previously outlined in an animal model study [10]. Moreover, HM781-36B partially acts as a TEC cytoplasmic kinase inhibitor [11]. At present, HM781-36B is in phase I and II clinical trials for the treatment of various solid tumors and non-small cell lung carcinoma with T790M mutation, refractory to first-line EGFR TKIs, either alone or simultaneously with chemotherapeutic drugs [10,11]. However to date, there have been no studies conducted to investigate the anticancer properties of pan-HER inhibitors in CRC cells, either as a single agent, or in combination with other cytotoxic agents.
In this study, we evaluated the effect of HM781-36B, a small-molecular and quinazoline-based pan-HER inhibitor, in CRC cell lines, with and without other cytotoxic drugs. We also attempted to find the mechanism of response and predictive biomarker of response to HM781-36B.
Materials and Methods
1. Reagents
The irreversible pan-HER inhibitor, HM781-36B, was provided by the Hanmi Pharmaceutical Company. 5-Fluorouracil (5-FU) was purchased from Sigma-Aldrich (St. Louis, MO). Oxaliplatin (L-OHP) and SN-38 were provided by Sanofi-Aventis Korea Co. Ltd. (Seoul, Korea) and CJ Pharmaceutical Company (Seoul, Korea), respectively.
2. Cell lines and culture conditions
The human CRC cell lines Caco-2, COLO-320DM, DLD-1, HCT-8, HCT-15, HT-29, LoVo, SW480, SNU-C2B, SNU-C5, and SNU-175 were purchased from the Korean Cell Line Bank (Seoul, Korea). The DiFi cell line was kindly provided by Dr. J. O. Park (Samsung Medical Center, Seoul, Korea). The KRAS mutation status of all cell lines are summarized in Table 1 [12-15]. All cell lines were cultured in RPMI-1640 medium (Gibco, Grand Island, NY), supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin solution (WelGENE Inc., Daegu, Korea), and were maintained at 37°C in 5% atmospheric CO2.
Table 1.
KRAS mutation status in human colorectal cancer cell lines
| Cell line | KRAS mutation | Reference |
|---|---|---|
| HCT-15 | G13D | [15,17] |
| DiFi | wt | [16,17] |
| HCT-8 | G13D | [17] |
| DLD-1 | G13D | [15-17] |
| SNU-C2B | G12D | [17] |
| LoVo | G13D, A14V | [15-18] |
| Caco-2 | wt | [15,17] |
| SW480 | G12V | [15-17] |
| COLO-320DM | wt | [15,17] |
| SNU-175 | A59T | [16] |
| SNU-C5 | wt | [18] |
| HT-29 | wt | [15-17] |
wt, wild type.
3. Cell growth inhibition assay
Cells were seeded at 3,000-5,000 cells/well in 96-well plates. After an overnight incubation, cells were treated with HM781-36B (0.001-10 μM), 5-FU (1-100 μM), and L-OHP (0.1-50 μM) for 72 hours. The cell viability was determined using the Cell Titer-Glo luminescent cell viability assay kit (Promega, Madison, WI), according to the manufacturer’s instructions. Luminescence was measured using a LMAXII 384 microplate reader (Molecular Devices, Toronto, ON, Canada). The half-maximal inhibitory concentration (IC50) values were calculated using the sigma plot software. The effects of treatments were measured by conducting three independent experiments, each of which was repeated six times.
4. Assessment of combination effects
The simultaneous exposure of cells to HM781-36B, combined with other cytotoxic agents (5-FU, L-OHP, or SN-38), was examined by serial dilutions of each individual drug and combining them at fixed ratios using doses that closely corresponded to the individual IC50 values. After 72 hours of exposure, the viable cell growth was measured using the Cell Titer-Glo luminescent cell viability assay kit (Promega). The synergistic effect was analyzed by the multiple drug-effect equation and quantified by the combination index (CI), using CalcuSyn software ver. 2.1 (Biosoft, Cambridge, UK) [16]. The CI values between 0.9 and 1.1 indicated an additive effect; values between 0.7 and 0.9 indicated moderate synergism; values below 0.7 indicated clear synergism; and antagonism was indicated by CI values above 1.1.
5. Cell cycle analysis
Cells were treated with HM781-36B (0.001, 0.01, and 0.1 μM) for 48 hours. They were harvested and processed by a cell cycle phase determination kit (Abnova Co., Taipei, Taiwan), following the manufacturer’s protocol. In brief, the cell pellet was resuspended at a density of 106 cells/mL in the assay buffer, followed by an addition of 1 mL of fixative. After a minimum of 2 hours, cells were incubated in darkness, in a 0.5 mL of propidium iodide staining solution, containing 10 μL of RNase A, for 30 minutes. Cells were then analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Heidelberg, Germany) with CellQuest-Pro software (10,000 events were recorded for each sample).
6. Western blot analysis
Cells were incubated with HM781-36B, at concentrations of 0.001, 0.01, and 0.1 μM, in 10% FBS media. After 48 hours, cells were washed rapidly with ice-cold phosphate buffered saline (PBS) and lysed in a 1× lysis buffer (Cell Signaling Technology, Danvers, MA) containing 1 mM of the protease inhibitor PMSF (Amresco, Solon, OH). The cell extracts were centrifuged at 13,000 rpm for 20 minutes. Protein concentration of the supernatant was measured using a BCA reagent (Pierce, Rockford, IL). For each sample, equal amounts of protein were denatured and fractionated by 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis, and then transferred onto a nitrocellulose membrane. Membranes were blocked by placing them in a 5% skimmed milk powder solution made up in TBS with 0.1% Tween (TBST) for 1 hour. Membranes were then incubated overnight at 4°C with primary antibodies against p-HER2 (pY1221/1222), p-AKT (pS473), p-ERK (p44/p42), EGFR, HER2, ERK, AKT, Bcl-2, cleaved caspase-3, cleaved caspase-9, poly(ADP-ribose) polymerase (PARP), β-actin (Cell Signaling Technology), BMX (Santa Cruz Biotechnology, Santa Cruz, CA), and p-EGFR (pY1092) (Abcam, Cambridge, MA). After three washes with TBST, membranes were incubated with the appropriate HRP-conjugated secondary antibodies for 1 hour (Santa Cruz Biotechnology). Detection of bound antibodies was carried out using ECL reagents (Amersham Pharmacia Biotech, Buckinghamshire, UK) and an X-ray film (AGFA, Mortsel, Belgium).
7. Fluorescence in situ hybridization analysis
Each cell line was detached and washed in PBS. After centrifugation, the sediment was fixed in 95% ethyl alcohol, followed by a second centrifugation. The packed sediment was then wrapped in lens paper and embedded in paraffin in accordance to conventional histological techniques. Five consecutive 4-μm-thick sections were cut from the cell block of all six lines and processed by fluorescence in situ hybridization (FISH) analysis to analyze gene amplification. FISH analysis was performed using the Vysis EGFR/CEP 7 FISH probe kit (Abbott Molecular, Downers Grove, IL) and the PathVysion assay (HER2 Spectrum Orange/CEP17 Spectrum Green, Abbott Molecular) in accordance with the manufacturers’ instructions. Shortly after deparaffinization, the FFPE specimens were incubated in the pre-treatment solution (Abbott Molecular) at 80°C for 30 minutes, and then digested with a protease solution (Abbott Molecular) for 20 minutes at 37°C. The slides were incubated at 80°C for 10 minutes for co-denaturation of the chromosomal and DNA probes, followed by a hybridization for 24 hours at 37°C. After post-hybridization washing, the slides were then counterstained with 4′,6-diamidino-2 phenylindole (DAPI) and analyzed using a fluorescent microscope. An average of 100 nuclei was counted for each sample. The EGFR gene was considered to be amplified if the EGFR/CEP 7 ratio was ≥ 2.0. An average of 20 nuclei was counted for FISH analysis of HER2, and the HER2 gene was considered to be amplified if the HER2/CEP 17 ratio was ≥ 2.0.
8. Statistical analysis
All experiments were repeated in duplicate or triplicate. All data were presented as the mean±standard error of the mean. A statistical significance was determined by a Student’s t test, and the differences with p-values of < 0.05 were accepted as statistically significant.
Results
1. The effect of HM781-36B on the viability of CRC cell lines
A total of 12 CRC cell lines were used to determine the growth inhibitory ability of HM781-36B. The addition of HM781-36B inhibited the growth activity of all cell types in a dose-dependent manner (Fig. 1). In particular, DiFi and SNU-175 cells had a highly sensitive response to the addition of HM781-36B. The IC50 levels of HM781-36B for DiFi and SNU-175 cells were 0.003 and 0.005 μM, respectively, which was about 500-7,000 fold lower than the IC50 levels of the other CRC cells (Table 2).
Fig. 1.
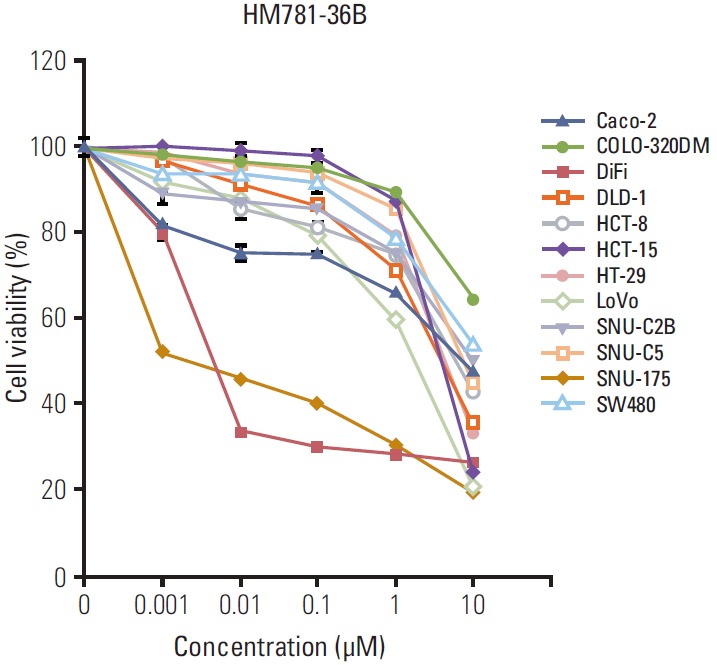
The growth inhibitory effect of HM781-36B on a panel of human colorectal carcinoma cell lines. The cells were treated with increasing doses of HM781-36B (0.001-10 μM) for 72 hours. The number of viable cells after the treatment was measured using the luminescent Cell Titer-Glo assay and expressed as percentage viable cells. Data represents the mean±standard error of the mean of three independent experiments (n=3), each of which was replicated six times.
Table 2.
Comparison of cell growth inhibition activity of HM781-36B
| Cell line | IC50 (μM) |
|---|---|
| HCT-15 | 5.28 |
| DiFi | 0.003 |
| HCT-8 | 7.57 |
| DLD-1 | 6.48 |
| SNU-C2B | 8.48 |
| LoVo | 2.54 |
| Caco-2 | 14.85 |
| SW480 | 12.16 |
| COLO-320DM | 21.58 |
| SNU-175 | 0.005 |
| SNU-C5 | 7.11 |
| HT-29 | 5.21 |
2. The effect of HM781-36B on the cell cycle and apoptosis of colorectal tumor cell lines
We examined the changes in distribution of the cell cycle in six CRC cell lines treated with HM781-36B at different doses (0.001, 0.01, and 0.1 μM), using fluorescence-activated cell sorting analysis (Fig. 2A). As expected, there was a considerable dose-dependent increase in the sub G1 phase and G1 phase fraction, and a decrease in the S phase fraction for drug-sensitive DiFi cells. Furthermore, we observed a slight increase in the G1 fraction and a reduced S phase fraction in SNU-175 cells; however, an increase in the sub G1 phase was not detected. In the case of drug-insensitive COLO-320DM cells, there were no statistically significant differences in the proportion of cells in all phases of the cell cycle.
Fig. 2.
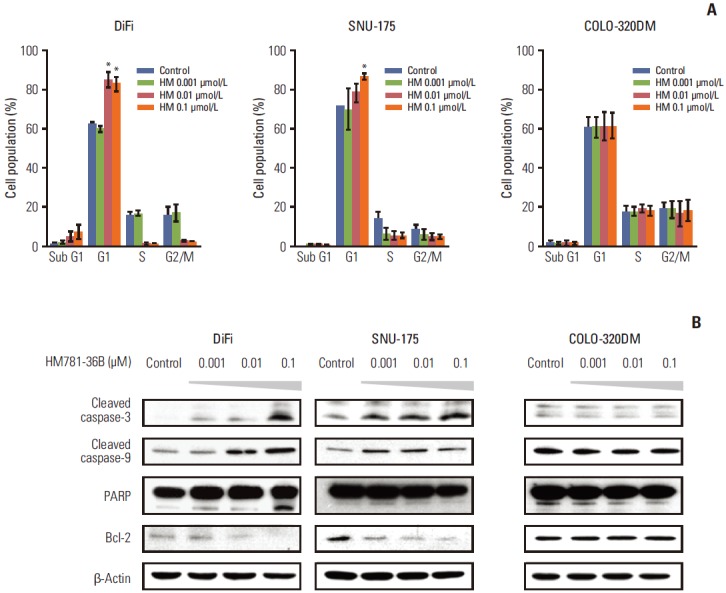
Analysis of the cell cycle and apoptosis in colorectal carcinoma cell lines after HM781-36B treatment. The cells were treated with the indicated concentrations of HM781-36B for 48 hours. (A) Distribution of the cell cycle was examined by propidium iodide staining and analyzed using fluorescence-activated cell sorting. The mean percentage of cells in the sub G1, G1, S, and G2/M phases of the cell cycle for duplicate independent experiments were plotted. Data represents the mean±standard error of the mean. Sample means were compared using a Student’s t test. *p < 0.01 compared with the control. (B) Apoptosis-related proteins were visualized by Western blotting using anti–cleaved caspase-3, anti–cleaved caspse-9, anti–poly(ADP-ribose) polymerase (PARP), and anti–Bcl-2 antibodies. Equal loading was identified by showing the total β-actin levels. The results are representative of two independent experiments (n=2).
The apoptotic effect of HM781-36B was also assessed by a Western blot analysis in CRC cells (Fig. 2B). The addition of HM781-36B to drug-sensitive DiFi and SNU-175 cells upregulated the expression of pro-apoptotic proteins, i.e., cleaved caspase-3, -9, and PARP, and down-regulated the expression of the pro-survival protein Bcl-2. Taken together, these observations suggest that HM781-36B induces G1 cell cycle arrest and apoptosis in pan-HER inhibitor-sensitive CRC cell lines.
3. The effect of HM781-36B on the HER family and its downstream signaling molecules
Prior to establishing the underlying mechanism of HM781-36B action, we confirmed the basic protein expression level of EGFR, HER2, and BMX in six colon cancer cell lines using a Western blot and amplification of EGFR and HER2 by FISH analysis. The phosphorylation of the HER family and BMX was induced in all cell lines. In particular, pEGFR was overexpressed in DiFi cells, and BMX was highly expressed in SNU-175 and COLO-320DM cells (Fig. 3A). Moreover, only DiFi cells showed EGFR amplification with a copy number ratio of 12, while none of the other CRC cell lines had a copy number ratio > 1.8 (Table 3, Fig. 3B). The amplification of the HER2 gene was not detected in any of the CRC cell lines (data not shown).
Fig. 3.
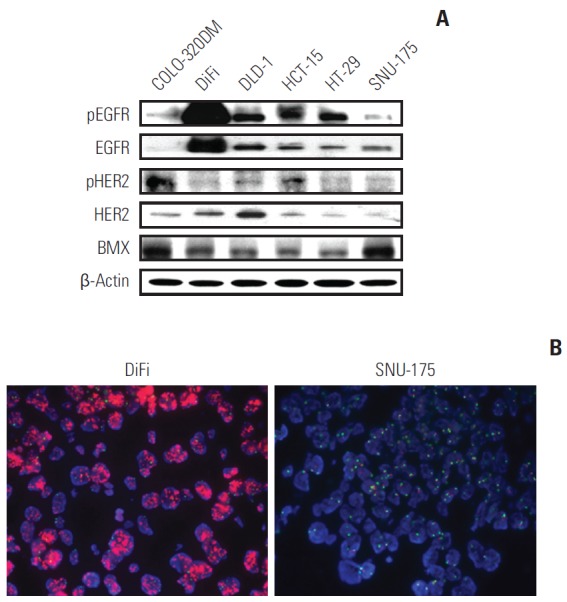
Basal level expression of the HER family and BMX, and fluorescence in situ hybridization (FISH) analysis in CRC cell lines. (A) The basal protein levels of epidermal growth factor receptor (EGFR), HER2, and BMX were evaluated in six colorectal cancer cell lines by Western blotting. (B) Hybridization of prepared cell lines with Vysis EGFR/CEP 7 FISH probe kit was performed, as described in the Materials and Methods section. The amplification of EGFR is positive in DiFi cells (left) and negative in the SNU-175 cells (right) (×100).
Table 3.
EGFR and HER2 gene copy number by FISH analysis in colorectal cancer cell lines
| Cell line | EGFR/CEP 7 ratio | HER2/CEP 17 ratio |
|---|---|---|
| DLD-1 | 0.91 | 0.96 |
| COLO-320DM | 0.99 | 1.29 |
| HT-29 | 1.69 | 1.78 |
| SNU-175 | 1.02 | 1.02 |
| DiFi | 12 | 1.32 |
EGFR, epidermal growth factor receptor; FISH, fluorescence in situ hybridization.
Subsequently, we examined the changes in protein expressions of the HER family and in the downstream signaling pathways, after treatment with HM781-36B. As predicted, the expression of pEGFR and pHER2 was decreased by HM781-36B in EGFR-overexpressing DiFi cells in a dose-dependent manner. In SNU-175 cells, reduced expression of pEGFR and pHER2 was also observed (Fig. 4).
Fig. 4.

Protein expression of the HER family and downstream signaling molecules in colorectal cancer cells following treatment with HM781-36B. Cells were treated with increasing concentrations of HM781-36B (0.001, 0.01, and 0.1 μM) for 48 hours. Cells were lysed, and proteins were analyzed by Western blotting with the indicated antibodies. Results are representative of two independent experiments (n=2). β-Actin was used as a loading control.
Treatment with HM781-36B inhibited downstream signaling molecules in DiFi and SNU-175 cells, as shown by decreased protein levels of pERK, pAKT, and BMX, which is a member of the TEC family nonreceptor/cytoplasmic tyrosine kinases. In contrast, in COLO-320DM and HCT-15 cells, which are relatively resistant to HM781-36B, the protein levels of pEGFR, pERK, pAKT, and BMX were not changed in response to HM781-36B treatment, and only the level of pHER2 was down-regulated (Fig. 4).
4. The effect of the co-administration of HM781-36B with clinically relevant chemotherapeutic drugs in CRC cell lines
We investigated the effect of combining HM781-36B with cytotoxic drugs (L-OHP, 5-FU, and SN-38) on six CRC cell lines. We established the IC50 values of CRC cells for cytotoxic drugs, and determined the fixed dose ratios of drug combinations based on these IC50 values (Table 4). Synergistic or additive effects between the two drugs were displayed, in accordance with the CI values at the 50% fraction affected (Fa; the range of cell kill level).
Table 4.
The IC50 values for chemotherapeutic drugs against human colorectal cancer cells
| Cell line | IC50
(μM) |
||
|---|---|---|---|
| L-OHP | 5-FU | SN-38 | |
| HCT-15 | 8.64 | 75.76 | 0.006 |
| DiFi | 10.95 | 92.41 | 0.39 |
| DLD-1 | 8.65 | 4.53 | 0.09 |
| COLO-320DM | 5.38 | 38.84 | 0.10 |
| SNU-175 | 1.51 | 5.81 | 0.004 |
| HT-29 | 5.22 | 29.51 | 0.08 |
5-FU, 5-fluorouracil.
The simultaneous exposure to HM781-36B with chemotherapeutic agents produced an additive or synergistic effect in most CRC cells (Fig. 5). The antagonistic activity was only observed where COLO-320DM cells were treated with a combination of HM781-36B and L-OHP. In particular, EGFR-overexpressing DiFi cells had the highest concentration of IC50 values for all the chemotherapeutic agents studied. Furthermore, a strong synergistic effect was observed in these cells when HM781-36B was combined with other agents.
Fig. 5.

The synergistic effect of colorectal cancer cells treated with HM781-36B in combination with chemotherapeutic drugs (oxaliplatin [L-OHP], 5-fluorouracil [5-FU], and SN-38). Synergism was assessed by the Chou and Talalay equation and the CalcuSyn software ver. 2.1 (Biosoft). Each column shows a combination index (CI) value at the 50% fraction affected. Data represents the means±standard error of the mean.
Discussion
Our study provides the first data available that demonstrates the growth inhibitory effect of the irreversible pan-HER inhibitor, HM781-36B, and its mechanism of action in CRC cell lines. The HM781-36B inhibitor had potent antitumor activity in DiFi and SNU-175 cells (IC50, 0.003 μM and 0.005 μM, respectively) (Table 2, Fig. 1). We observed the suppression of Bcl-2 and the induction of the cleaved forms of caspase-3, -9, and PARP, and an increase in the G1 phase fraction after the treatment of HM781-36B in two drug-sensitive cell lines (Fig. 2A and B). Therefore, we propose that HM781-36B induced G1 arrest of the cell cycle and that this resulted in apoptosis. HM781-36B also down-regulated the level of pEGFR, pHER2, pERK, pAKT, and BMX in EGFR-overexpressing DiFi and SNU-175 cells (Fig. 4). Moreover, HM781-36B, in combination with chemotherapeutic agents such as L-OHP, 5-FU, or SN-38, exerted a synergistic or an additive effect in almost all CRC cells studied (Fig. 5).
During the last two decades, novel strategies that target EGFR have been evaluated in CRC. The monoclonal antibodies directed against EGFR leads to antitumor activity in CRCs. The ATP-site-directed, small molecule TKIs are distinct advantages over monoclonal antibodies by the lower cost of care, oral bioavailability, and the ability to target multiple signaling pathways. However, manifold early-phase trials of EGFR TKIs failed to prove antitumor efficacy in CRC [17]. The TKIs in combination with standard chemotherapy also did not show a significant benefit and were associated with increased toxicities [18]. Other EGFR TKIs have been developed, including lapatinib, EKB-569, CI-1033, AEE788, and ZD6474. These TKIs are multi-targeted agents that act through the inhibition of ErbB 1/2 and vascular endothelial growth factor receptors. Although these have been examined in phase I/II trials for patients with various solid tumors, these data regarding efficacy in patients with CRC have not yet been determined [19].
Many studies have reported that DiFi cells are EGFR-overexpressing cell lines, and we have also confirmed the levels of EGFR expression in DiFi cells using Western blot and FISH analyses (Table 3, Fig. 3). Whether EGFR expression can be used as a predictive marker of response to anti-EGFR mAbs has been a matter of controversy. In earlier studies, the addition of cetuximab in CRC patients with EGFR overexpression was significantly correlated with survival. However, other studies found no relationship between EGFR expression and cetuximab response [20]. Some studies have suggested that the expression of other growth factor receptors, like HER2, HER3 and IGF-IR, EGFR gene amplification, mutations in exons 18 to 21 of the kinase domain of EGFR, and mutations of KRAS or PTEN might be associated with the response/resistance to therapy with the EGFR inhibitors [21,22].
It has been shown that HM781-36B is both a receptor and a nonreceptor/cytoplasmic TKI. The irreversible HER TKIs are capable of potently inhibiting the TEC family of nonreceptor/cytoplasmic tyrosine kinases, including BMX [11,23]. Other members of the TEC family includes Btk, Tec, Txk, and Itk. Unlike other TEC family kinases that are predominantly expressed in hematopoietic cells, BMX is expressed in various cell types, such as endothelial, epithelial, and importantly, metastatic carcinoma cells. BMX mediates various signaling pathways and plays a critical role in several cellular functions [23]. In our study, the basal level of BMX was up-regulated in COLO-320DM and SNU-175 cells (Fig. 3A). Moreover, HM781-36B inhibited the phosphorylation of BMX in EGFR-overexpressing DiFi and SNU-175 cells (Fig. 4). Interestingly, although SNU-175 is an EGFR non-amplified cell line with KRAS mutation, the growth of SNU-175 was inhibited by the irreversible EGFR inhibitor, HM781-36B, which may likely be a result of the inhibition of cytoplasmic kinase, BMX.
HM781-36B, a quinazoline-based irreversible pan-HER TKI, has already shown potent antitumor activity in EGFR-and HER2-amplified cancer cell lines [10,11,24]. Furthermore, it exerted synergistic effects with chemotherapeutic agents on the HER2-amplified and some non-amplified cancer cell lines [10,24]. Our study shows that HM781-36B exerted a synergistic, or an additive effect, in almost all CRC cells studied when combined with a clinically relevant cytotoxic agent, such as L-OHP, 5-FU, or SN-38 (Fig. 5). In particular, although DiFi cells were the most resistant cell line to all of the chemotherapeutic drugs studied, these cells showed a potent synergistic effect in combination therapy with HM781-36B. In addition, HM781-36B alone did not inhibit the growth of non-EGFR–amplified cells, with the exception of SNU-175 cells. However, HM781-36B, in combination with chemotherapeutic agents, resulted in synergistic effects in both EGFR amplified and non-amplified cells, including two cell lines with KRAS mutations (DLD-1, HCT-15, SNU-175) and one cell line with BRAF mutations (HT-29).
Limitations of our study include few numbers of cell lines used in the experiment, especially only one CRC cell line with EGFR overexpression was used. Also, we cannot conclude on the role of BMX in the response of CRC cells to HM781-36B because we did not perform various functional experiments for BMX inhibition. However, there is a possibility that phosphoinositide 3-kinase (PI3K)/AKT or STAT can be one of the mechanisms to explain the role of BMX in response to HM781-36B because BMX was associated with survival signaling pathways, such as PI3K/AKT or STAT, which are well-established targets for anti-cancer therapy [25].
Conclusion
To summarize, we showed that HM781-36B is a potential upcoming anticancer drug that acts through the irreversible inhibition of both the EGFR family of tyrosine kinases and the TEC family of nonreceptor/cytoplasmic tyrosine kinases for the treatment of CRC. In addition, our findings suggest that the administration of HM781-36B, when combined with chemotherapeutic drugs, may be effective in EGFR-overexpressing colorectal cancer. Further studies are needed to elucidate the role of BMX expression as a marker of response to HM781-36B in colon cancer.
Footnotes
Conflict of interest relevant to this article was not reported.
References
- 1.Venook A. Critical evaluation of current treatments in metastatic colorectal cancer. Oncologist. 2005;10:250–61. doi: 10.1634/theoncologist.10-4-250. [DOI] [PubMed] [Google Scholar]
- 2.McKay JA, Murray LJ, Curran S, Ross VG, Clark C, Murray GI, et al. Evaluation of the epidermal growth factor receptor (EGFR) in colorectal tumours and lymph node metastases. Eur J Cancer. 2002;38:2258–64. doi: 10.1016/s0959-8049(02)00234-4. [DOI] [PubMed] [Google Scholar]
- 3.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–54. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 4.Wong SF. Cetuximab: an epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin Ther. 2005;27:684–94. doi: 10.1016/j.clinthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 6.Dienstmann R, De Dosso S, Felip E, Tabernero J. Drug development to overcome resistance to EGFR inhibitors in lung and colorectal cancer. Mol Oncol. 2012;6:15–26. doi: 10.1016/j.molonc.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cameron D, Casey M, Oliva C, Newstat B, Imwalle B, Geyer CE. Lapatinib plus capecitabine in women with HER-2-positive advanced breast cancer: final survival analysis of a phase III randomized trial. Oncologist. 2010;15:924–34. doi: 10.1634/theoncologist.2009-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U S A. 2005;102:11011–6. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marrero-Ponce Y, Khan MT, Casanola Martin GM, Ather A, Sultankhodzhaev MN, Torrens F, et al. Prediction of tyrosinase inhibition activity using atom-based bilinear indices. ChemMedChem. 2007;2:449–78. doi: 10.1002/cmdc.200600186. [DOI] [PubMed] [Google Scholar]
- 10.Nam HJ, Kim HP, Yoon YK, Hur HS, Song SH, Kim MS, et al. Antitumor activity of HM781-36B, an irreversible Pan-HER inhibitor, alone or in combination with cytotoxic chemotherapeutic agents in gastric cancer. Cancer Lett. 2011;302:155–65. doi: 10.1016/j.canlet.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Cha MY, Lee KO, Kim M, Song JY, Lee KH, Park J, et al. Antitumor activity of HM781-36B, a highly effective pan-HER inhibitor in erlotinib-resistant NSCLC and other EGFR-dependent cancer models. Int J Cancer. 2012;130:2445–54. doi: 10.1002/ijc.26276. [DOI] [PubMed] [Google Scholar]
- 12.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknaes M, Hektoen M, et al. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013;2: doi: 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee J, Lee I, Han B, Park JO, Jang J, Park C, et al. Effect of simvastatin on cetuximab resistance in human colorectal cancer with KRAS mutations. J Natl Cancer Inst. 2011;103:674–88. doi: 10.1093/jnci/djr070. [DOI] [PubMed] [Google Scholar]
- 14.Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70:5901–11. doi: 10.1158/0008-5472.CAN-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Achmad A, Hanaoka H, Yoshioka H, Yamamoto S, Tominaga H, Araki T, et al. Predicting cetuximab accumulation in KRAS wild-type and KRAS mutant colorectal cancer using 64Cu-labeled cetuximab positron emission tomography. Cancer Sci. 2012;103:600–5. doi: 10.1111/j.1349-7006.2011.02166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 17.Rothenberg ML, LaFleur B, Levy DE, Washington MK, Morgan-Meadows SL, Ramanathan RK, et al. Randomized phase II trial of the clinical and biological effects of two dose levels of gefitinib in patients with recurrent colorectal adenocarcinoma. J Clin Oncol. 2005;23:9265–74. doi: 10.1200/JCO.2005.03.0536. [DOI] [PubMed] [Google Scholar]
- 18.Fisher GA, Kuo T, Ramsey M, Schwartz E, Rouse RV, Cho CD, et al. A phase II study of gefitinib, 5-fluorouracil, leucovorin, and oxaliplatin in previously untreated patients with metastatic colorectal cancer. Clin Cancer Res. 2008;14:7074–9. doi: 10.1158/1078-0432.CCR-08-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuo T, Fisher GA. Current status of small-molecule tyrosine kinase inhibitors targeting epidermal growth factor receptor in colorectal cancer. Clin Colorectal Cancer. 2005;5 Suppl 2:S62–70. doi: 10.3816/ccc.2005.s.009. [DOI] [PubMed] [Google Scholar]
- 20.Lenz HJ, Van Cutsem E, Khambata-Ford S, Mayer RJ, Gold P, Stella P, et al. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J Clin Oncol. 2006;24:4914–21. doi: 10.1200/JCO.2006.06.7595. [DOI] [PubMed] [Google Scholar]
- 21.Modjtahedi H, Essapen S. Epidermal growth factor receptor inhibitors in cancer treatment: advances, challenges and opportunities. Anticancer Drugs. 2009;20:851–5. doi: 10.1097/CAD.0b013e3283330590. [DOI] [PubMed] [Google Scholar]
- 22.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–74. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 23.Hur W, Velentza A, Kim S, Flatauer L, Jiang X, Valente D, et al. Clinical stage EGFR inhibitors irreversibly alkylate Bmx kinase. Bioorg Med Chem Lett. 2008;18:5916–9. doi: 10.1016/j.bmcl.2008.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HJ, Kim HP, Yoon YK, Kim MS, Lee GS, Han SW, et al. Antitumor activity of HM781-36B, a pan-HER tyrosine kinase inhibitor, in HER2-amplified breast cancer cells. Anticancer Drugs. 2012;23:288–97. doi: 10.1097/CAD.0b013e32834e7d9b. [DOI] [PubMed] [Google Scholar]
- 25.Jarboe JS, Dutta S, Velu SE, Willey CD. Mini-review: bmx kinase inhibitors for cancer therapy. Recent Pat Anticancer Drug Discov. 2013;8:228–38. doi: 10.2174/15748928113089990043. [DOI] [PubMed] [Google Scholar]