

Abstract
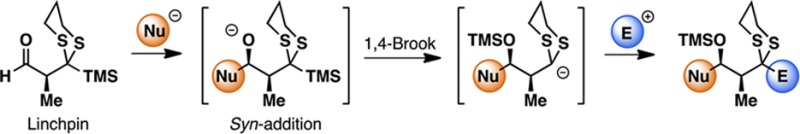
The design, synthesis, and validation of a new bifunctional aldehyde linchpin for Type II anion relay chemistry have been achieved. For this linchpin, the initial nucleophilic addition proceeds under Felkin–Anh control to generate the syn-alkoxide, which undergoes a 1,4-Brook rearrangement to relay the negative charge, thus leading to the formation of a dithiane-stabilized carbanion. Subsequent trapping with an electrophile furnishes a tricomponent adduct with an embedded propionate subunit, a ubiquitous structural motif found in polyketides. The utility of this new linchpin is demonstrated with the construction of a potential C16–C29 fragment for the synthesis of rhizopodin, an actin-binding macrolide.
Propionate and polypropionate subunits are ubiquitous structural motifs found in many polyketide natural products possessing diverse biological properties.1 Stereocontrolled synthesis of such structural motifs has attracted considerable interest in the synthetic community due to the challenges that arise from the inherent architectural complexity of many bioactive natural products.2 Multicomponent anion relay chemistry (ARC), a highly effective, stereocontrolled fragment union process pioneered in our laboratory,3 holds significant promise for the rapid construction of diverse polyketide natural products.
Over the past decade, we have reported extensive studies in the area of fragment union.4 In the area of through-space anion relay chemistry (ARC) (i.e., negative charge migration), we exploited [1,n]-Brook rearrangements that have led to the discovery of Type I and Type II ARC union tactics (Figure 1).5 In Type I ARC, an anion is first generated on linchpin 1 facilitated by an anion-stabilizing group (ASG, e.g., dithiane), which adds to an epoxide to form an alkoxide (2). Upon Brook rearrangement, the negative charge is relayed back to the originating carbon, which is then terminated with either the same electrophile (i.e., homocoupling) or a different electrophile (i.e., heterocoupling) to deliver the three-component adduct 3. In Type II ARC, an external nucleophile is first added to a bifunctional linchpin (4) to generate alkoxide 5, which upon triggering the Brook rearrangement, either by change in solvent polarity, temperature, and/or counterion, the negative charge is then transferred to a new carbon site.5b Subsequent trapping with an electrophile furnishes the three-component adduct 6. Linchpins can also be added in iterative fashion to form multicomponent adducts such as 8 via a process not dissimilar to living polymerization.6
Figure 1.
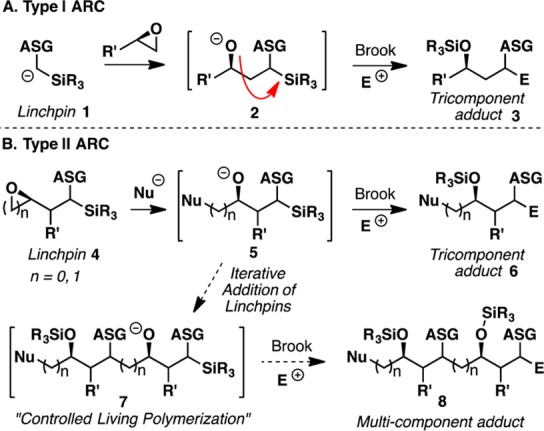
Through-space Type I and Type II ARC tactics. ASG: Anion-Stabilizing Group.
Given the considerable potential of the multicomponent ARC tactic in assembling diverse molecular scaffolds with precise stereocontrol, we initiated a program to focus on the design, synthesis, and validation of a new aldehyde linchpin 9 that would enable the construction of propionate-containing natural products exploiting the Type II ARC tactic. The proposed Type II ARC tactic with linchpin 9 is depicted in Figure 2A. Here, addition of an external nucleophile to the aldehyde (9) would proceed under Felkin–Anh control7 to generate syn-alkoxide 10, which would then undergo 1,4-Brook rearrangement, triggered by the addition of a polar additive (i.e., HMPA), to form anionic dithiane 11. Termination with an electrophile would deliver the three-component adduct 12, which upon reductive dithiane removal would reveal a methylene group (13) or upon dithiane hydrolysis a carbonyl group (14) for further functionalization.
Figure 2.
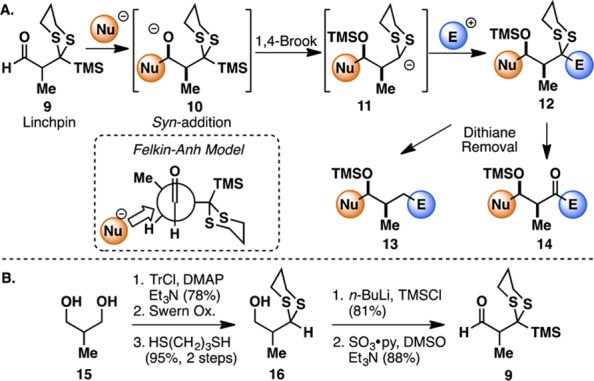
(A) Type II ARC employing aldehyde linchpin 9. (B) Linchpin synthesis in racemic series.
To explore this scenario, we first constructed the prospective racemic aldehyde linchpin 9 from 2-methyl-1,3-propanediol 15 (Figure 2B). Monoprotection of the diol with trityl chloride followed by oxidation of the free alcohol led to the corresponding aldehyde, which upon treatment with 1,3-propanedithiol and BF3·Et2O furnished dithiane 16 with concomitant removal of the trityl group. Subsequent C-silylation with TMSCl/n-BuLi followed by Parikh–Doering oxidation of the free alcohol delivered the desired linchpin 9. This linchpin was also prepared in highly enantiomerically enriched fashion starting from either commercially available Roche ester (vide infra).
With the prospective linchpin in hand, we turned to the proposed Type II ARC reaction. We first investigated n-BuLi and allyl bromide as the initiating nucleophile and terminating electrophile, respectively. After preliminary screenings, we discovered that addition of n-BuLi to the linchpin in Et2O at −78 °C, followed by introduction of allyl bromide and HMPA in Et2O and warming of the reaction mixture to ambient temperature over 4 h, furnished the desired three-component adduct 17 as a single syn-diastereomer (vide infra) in 74% yield after acid-mediated removal of the TMS group (Table 1, entry 1). The use of Et2O as the reaction solvent for the initial nucleophilic addition proved critical, since performing the reaction in THF leads to premature Brook rearrangement, even at low temperature (not shown).
Table 1. Tricomponent Type II ARC with Linchpin 9a.


Reaction conditions: (i) NuLi, Et2O, −78 °C, 30 min; (ii) electrophile, HMPA/Et2O (1/10, v/v), −78 °C to rt, 1 h, then rt, 3 h; and/or (iii) 1.0 N aq HCl, overnight.
Syn/anti ratio was determined by 1H NMR analysis of the crude reaction mixture.
The adduct was obtained as a 1:1 mixture of diastereomers.
Having established the optimal reaction conditions, a brief substrate scope study was performed. As illustrated in Table 1, phenyl-, alkynyl-, and allyllithium, as well as lithiated 2-methyl-1,3-dithiane all proved viable initiating nucleophiles, with the reactions proceeding to deliver the three-component adducts as single syn-diastereomers in moderate to good yields (entries 2–5). A range of electrophiles including benzaldehyde, (R)-1,2-epoxybutane, (S)-epichlorohydrin, and (R)-glycidol benzyl ether (entries 6–9) readily participated as electrophiles in the ARC reactions to furnished the corresponding three-component adducts also in good yield with excellent syn-selectivity as expected. The relative stereochemistry of the three-component adduct 21 (entry 5) was confirmed by single-crystal X-ray diffraction and that of the other congeners was assigned by analogy. Further confirmation was obtained by Mosher ester analysis8 of a derivatized pre-Brook alcohol (see the Supporting Information).
Further validation of the new protocol as a viable synthetic tactic logically required the synthesis of both enantiomers of 9. To this end, (+)-9 was prepared first from known compound (−)-16, the latter constructed from commercially available (R)-Roche ester (−)-26 in four steps (Figure 3).9 Silylation at the dithiane 2-position followed by Parikh–Doering oxidation furnished aldehyde linchpin (+)-9 in 66% yield over two steps. Importantly, following the oxidation step, removal of excess triethylamine from the crude reaction mixture employing satd aq CuSO4 proved critical in preserving the optical purity of the linchpin. The enantiomer (−)-9 was then prepared in a similar fashion. Pleasingly, X-ray quality crystals were obtained in this case. Both series were carried out on multigram scale; chiral phase chromatography indicated high levels of enantiomeric excess (>98%, see the Supporting Information).
Figure 3.

Synthesis of (+)- and (−)-9.
With an effective bifunctional linchpin for Type II ARC in hand, we turned to demonstrate the synthetic utility in polyketide synthesis by employing linchpin (+)-9 in a convergent synthesis of a potential C16–C29 fragment for rhizopodin (27, Figure 4), a C2-symmetric, 38-membered macrodiolide isolated from the myxobacterium Myxococcus stipitatus(10) that displays impressive biological properties, including selective potent cytotoxicity against a range of human cancer cell lines,11 with the enamide side chain, a highly conserved moiety, responsible for protein recognition.12 As such, 27 has been the object of numerous synthetic efforts.13 Our retrosynthetic analysis of the C16–C29 fragment is depicted in Figure 4. Here, we envisioned a highly convergent assembly of the full C16–C29 carbon segment 28 via a single-flask Type II ARC tactic, involving lithiation of vinyl iodide 29, Felkin-controlled addition to aldehyde (+)-9, [1,4]-Brook rearrangement, and termination with epoxide 30.
Figure 4.
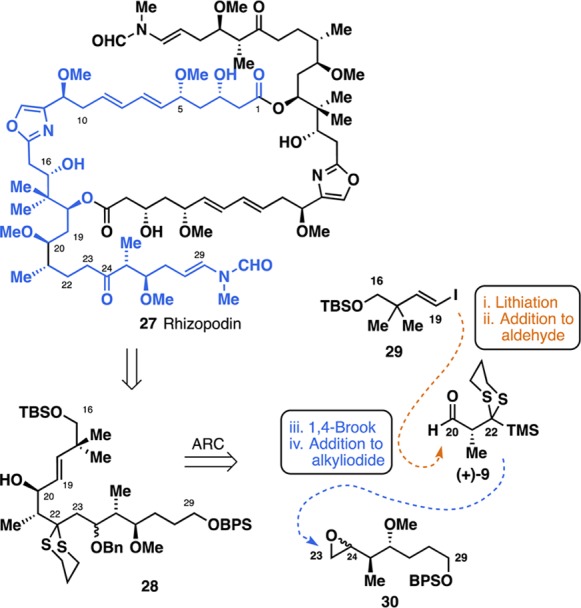
Retrosynthetic analysis of the C16–C29 side chain of rhizopodin.
The synthesis of 28 thus began with the preparation of the requisite ARC fragments, 29 and 30 (Figure 5). Vinyl iodide 29 was constructed from diol 31 as follows: monoprotection with TBSCl followed by Parikh–Doering oxidation14 afforded aldehyde 32; Takai olefination15 then delivered 29 in 77% yield (Figure 5A). In turn, elaboration of 30 entailed the asymmetric crotylation of aldehyde 33(16) to furnish homoallylic alcohol (−)-34 in 93% yield. O-Methylation followed by epoxidation led to 30, obtained as a 1:1 mixture of diastereomers at C24, and then employed as such in preliminary studies of the ARC key step; in fact, the stereoconfiguration at C24 ultimately becomes irrelevant as it comprises a carbonyl in the targeted natural product (see Figure 4). Having demonstrated the viability of 30 as a terminating electrophile for the key fragment union (not shown), we decided to move forward in our rhizopodin synthetic studies with a single diastereomer of 30 in order to facilitate both reaction monitoring and chromatographic separation. We thus turned to the iodo carbonate cyclization of (−)-34 to install the requisite epoxide functionality, which generally favors formation of 1,3-syn stereoarrays with good selectivities.17 To our surprise, the well-established protocols employing N-iodosuccinimide17b,17c resulted in diastereomeric mixtures when applied to the Boc carbonate of (−)-34, while iodine17a led to loss of the terminal BPS protection. Pleasingly, use of iodine monobromide, a protocol developed in our laboratory,17d cleanly furnished the desired stereoisomer (dr 11:1; the minor diastereomer was removed by column chromatography). The synthesis of (−)-30 was thus completed by removal of the Boc-carbonate under basic conditions with concomitant closure of the oxirane ring, followed by O-methylation (Figure 5B).
Figure 5.

ARC: synthesis of pronucleophile and electrophile.
With the three molecular fragments in hand [29, (+)-9 and (−)-30], we turned to the central Type II ARC tricomponent fragment union employing the new aldehyde linchpin (Figure 6). To our delight, addition of the vinyl lithium species derived from 29 to linchpin (+)-9, followed by triggering of the [1,4]-Brook rearrangement with HMPA and trapping of epoxide (−)-30 delivered the desired adduct (+)-28 in 69% yield, thus permitting a highly convergent construction of the C16–C29 carbon framework of rhizopodin.
Figure 6.

Fragment assembly via Type II ARC.
Having validated the key step, we focused on the further elaboration of the ARC adduct [(+)-28] with the goal of accessing aldehyde 37, a potentially suitable partner for the union with the C1–C15 fragment developed by Nicolaou et al. in their synthesis of related natural product monorhizopodin12d (Figure 6). The synthesis of such aldehyde would require, notably (i) reductive removal of the C22-dithiane and (ii) stereoselective installation of the C18-acetate, the latter planned via Sharpless asymmetric epoxidation18 followed by reductive epoxide opening.19 Prior protection of the C24-hydroxyl was achieved employing KHMDS and excess BnBr in up to 91% yield. Of note, these conditions proved critical to suppress the byproduct arising from undesired migration of the neighboring TMS with concomitant benzylation of the C20-hydroxyl (see the Supporting Information). Subsequent dithiane removal mediated by Raney nickel under a hydrogen atmosphere, followed by a mild acidic workup (0.2 N aq HCl), provided the free allylic alcohol without loss of the terminal BPS ether (69% yield). Next, Sharpless asymmetric epoxidation was accomplished utilizing (−)-diethyl-d-tartrate [(−)-DET] as ligand to furnish the single diastereomer (−)-36 in 73% yield. Disappointingly, all attempts to date to effect the desired epoxide opening directed by the C20 hydroxyl, utilizing hydrides at various temperatures (e.g., Red-Al, LiAlH4)19a,19b or protocols relying on single-electron transfer,19c,19d have proved unfruitful, presumably due to the significant steric hindrance at the projected reactive site. Alternative pathways are currently being investigated in our laboratory.
In summary, we have achieved the design, synthesis and validation of a new effective aldehyde bifunctional linchpin (9) for Type II ARC fragment unions. Importantly, this linchpin enables the rapid construction of propionate-containing polyketide fragments for natural product total synthesis, exploiting highly efficient multicomponent ARC tactics. The utility of this new protocol is demonstrated in ongoing synthetic studies toward the C16–C29 fragment of rhizopodin.
Acknowledgments
Financial support was provided by the NIH through Grant No. R01-GM-29028 and a NIH postdoctoral fellowship (1F32CA171736) to M.Z.C. We thank Dr. Patrick J. Carroll at the University of Pennsylvania for obtaining X-ray crystal structures. We are also grateful to Drs. George Furst and Jun Gu and Dr. Rakesh K. Kohli for assistance obtaining NMR and high-resolution mass spectra, respectively.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.5b03235.
Author Present Address
† Pfizer Inc., 445 Eastern Point Road, Groton, CT 06340.
Author Contributions
‡ B.M. and M.Z.C. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- For reviews, see:; a O’Hagan D.The Polyketide Metabolites; Ellis Horwood: Chichester, 1991. [Google Scholar]; b O’Hagan D. Nat. Prod. Rep. 1995, 12, 1. 10.1039/np9951200001. [DOI] [Google Scholar]; c Davies-Coleman M. T.; Garson M. J. Nat. Prod. Rep. 1998, 15, 477. 10.1039/a815477y. [DOI] [PubMed] [Google Scholar]; d Menche D. Nat. Prod. Rep. 2008, 25, 905. 10.1039/b707989n. [DOI] [PubMed] [Google Scholar]
- For reviews on polyketide syntheses, see:; a Paterson I. Pure Appl. Chem. 1992, 64, 1821. 10.1351/pac199264121821. [DOI] [Google Scholar]; b Faul M. M.; Huff B. E. Chem. Rev. 2000, 100, 2407. 10.1021/cr940210s. [DOI] [PubMed] [Google Scholar]; c Yeung K.-S.; Paterson I. Chem. Rev. 2005, 105, 4237. 10.1021/cr040614c. [DOI] [PubMed] [Google Scholar]; d Koskinen A. M. P.; Karisalmi K. Chem. Soc. Rev. 2005, 34, 677. 10.1039/b417466f. [DOI] [PubMed] [Google Scholar]; e Schetter B.; Mahrwald R. Angew. Chem., Int. Ed. 2006, 45, 7506. 10.1002/anie.200602780. [DOI] [PubMed] [Google Scholar]
- For reviews on anion relay chemistry, see:; a Smith A. B. III; Adams C. M. Acc. Chem. Res. 2004, 37, 365. 10.1021/ar030245r. [DOI] [PubMed] [Google Scholar]; b Smith A. B. III; Wuest W. M. Chem. Commun. 2008, 5883. 10.1039/b810394a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. B. III; Fox R. J.; Razler T. M. Acc. Chem. Res. 2008, 41, 675. 10.1021/ar700234r. [DOI] [PubMed] [Google Scholar]
- Early ARC studies: Type I:; a Smith A. B. III; Boldi A. M. J. Am. Chem. Soc. 1997, 119, 6925. 10.1021/ja970371o. [DOI] [Google Scholar]; Type II:; b Smith A. B. III; Xian M. J. Am. Chem. Soc. 2006, 128, 66. 10.1021/ja057059w. [DOI] [PubMed] [Google Scholar]; Relevant work in the area of Type II ARC:; c Smith A. B. III; Kim W.-S.; Tong R. Org. Lett. 2010, 12, 588. 10.1021/ol902784q. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Smith A. B. III; Tong R. Org. Lett. 2010, 12, 1260. 10.1021/ol100130x. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Smith A. B. III; Smits H.; Kim D.-S. Tetrahedron 2010, 66, 6597. 10.1016/j.tet.2010.01.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A.; Nakahama S. Acta Polym. 1998, 49, 133. 10.1002/(SICI)1521-4044(199804)49:4<133::AID-APOL133>3.0.CO;2-P. [DOI] [Google Scholar]
- Anh N. T.; Eisenstein O. Tetrahedron Lett. 1976, 17, 155. 10.1016/0040-4039(76)80002-0. [DOI] [Google Scholar]; Studies to develop anti Felkin–Anh addition are underway in our laboratory. For an enantioselective, stepwise alternative, see:Mukherjee S.; Lee D. Org. Lett. 2009, 11, 2916. 10.1021/ol900923c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoye T. R.; Jeffrey C. S.; Shao F. Nat. Protoc. 2007, 2, 2451. 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- Ley S. V.; Tackett M. N.; Maddess M. L.; Anderson J. C.; Brennan P. E.; Cappi M. W.; Heer J. P.; Helgen C.; Kori M.; Kouklovsky C.; Marsden S. P.; Norman J.; Osborn D. P.; Palomero M. A.; Pavey J. B. J.; Pinel C.; Robinson L. A.; Schnaubelt J.; Scott J. S.; Spilling C. D.; Watanabe H.; Wesson K. E.; Willis M. C. Chem. - Eur. J. 2009, 15, 2874. 10.1002/chem.200801656. [DOI] [PubMed] [Google Scholar]
- Sasse F.; Steinmetz H.; Hofle G.; Reichenbach H. J. Antibiot. 1993, 46, 741. 10.7164/antibiotics.46.741. [DOI] [PubMed] [Google Scholar]
- Gronewold T. M. A.; Sasse F.; Lunsdorf H.; Reichenbach H. Cell Tissue Res. 1999, 295, 121. 10.1007/s004410051218. [DOI] [PubMed] [Google Scholar]
- a Hagelueken G.; Albrecht S. C.; Steinmetz H.; Jansen R.; Heinz D. W.; Kalesse M.; Schubert W. D. Angew. Chem., Int. Ed. 2009, 48, 595. 10.1002/anie.200802915. [DOI] [PubMed] [Google Scholar]; b Jansen R.; Steinmetz H.; Sasse F.; Schubert W. D.; Hagelueken G.; Albrecht S. C.; Mueller R. Tetrahedron Lett. 2008, 49, 5796. 10.1016/j.tetlet.2008.07.132. [DOI] [Google Scholar]; c Horstmann N.; Menche D. Chem. Commun. 2008, 5173. 10.1039/b810405k. [DOI] [PubMed] [Google Scholar]; d Nicolaou K. C.; Jiang X.; Lindsay-Scott P. J.; Corbu A.; Yamashiro S.; Bacconi A.; Fowler V. M. Angew. Chem., Int. Ed. 2011, 50, 1139. 10.1002/anie.201006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Total syntheses of 27:; a Kretschmer M.; Dieckmann M.; Li P.; Rudolph S.; Herkommer D.; Troendlin J.; Menche D. Chem. - Eur. J. 2013, 19, 15993.and references cited therein 10.1002/chem.201302197. [DOI] [PubMed] [Google Scholar]; b Dalby S. M.; Goodwin-Tindall J.; Paterson I. Angew. Chem., Int. Ed. 2013, 52, 6517. 10.1002/anie.201301978. [DOI] [PubMed] [Google Scholar]; Total synthesis of congener monorhizopodin: see ref (12d). Selected fragment syntheses:; c Chen Z.; Song L.; Xu Z.; Ye T. Org. Lett. 2010, 12, 2036. 10.1021/ol100515m. [DOI] [PubMed] [Google Scholar]; d Kretschmer M.; Menche D. Org. Lett. 2012, 14, 382. 10.1021/ol203130b. [DOI] [PubMed] [Google Scholar]; e Pulukuri K. K.; Chakraborty T. K. Org. Lett. 2012, 14, 2858.and references cited therein 10.1021/ol301103d. [DOI] [PubMed] [Google Scholar]; f Bender T.; Loits D.; White J. M.; Rizzacasa M. A. Org. Lett. 2014, 16, 1450. 10.1021/ol500261n. [DOI] [PubMed] [Google Scholar]
- Parikh J. R.; Doering W. v. E. J. Am. Chem. Soc. 1967, 89, 5505. 10.1021/ja00997a067. [DOI] [Google Scholar]
- Takai K.; Nitta K.; Utimoto K. J. Am. Chem. Soc. 1986, 108, 7408. 10.1021/ja00283a046. [DOI] [PubMed] [Google Scholar]
- Luo J.; Li H.; Wu J.; Xing X.; Dai W.-M. Tetrahedron 2009, 65, 6828. 10.1016/j.tet.2009.06.070. [DOI] [Google Scholar]
- a Bartlett P. A.; Meadows J. D.; Brown E. G.; Morimoto A.; Jernstedt K. K. J. Org. Chem. 1982, 47, 4013. 10.1021/jo00142a002. [DOI] [Google Scholar]; b Taylor R. E.; Jin M. Org. Lett. 2003, 5, 4959. 10.1021/ol0358814. [DOI] [PubMed] [Google Scholar]; c Kumar D.; Reddy C.; Das B. Synthesis 2011, 2011, 3190. 10.1055/s-0030-1260177. [DOI] [Google Scholar]; d Duan J. J. W.; Smith A. B. III J. Org. Chem. 1993, 58, 3703. 10.1021/jo00066a024. [DOI] [Google Scholar]
- Katsuki T.; Sharpless K. B. J. Am. Chem. Soc. 1980, 102, 5974. 10.1021/ja00538a077. [DOI] [Google Scholar]
- a Yamazaki T.; Ichige T.; Kitazume T. Org. Lett. 2004, 6, 4073. 10.1021/ol048229x. [DOI] [PubMed] [Google Scholar]; b Abe H.; Aoyagi S.; Kibayashi C. J. Am. Chem. Soc. 2005, 127, 1473. 10.1021/ja040213e. [DOI] [PubMed] [Google Scholar]; c RajanBabu T. V.; Nugent W. A. J. Am. Chem. Soc. 1994, 116, 986. 10.1021/ja00082a021. [DOI] [Google Scholar]; d Jorgensen K. B.; Suenaga T.; Nakata T. Tetrahedron Lett. 1999, 40, 8855. 10.1016/S0040-4039(99)01860-2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.