

Abstract
In two-component signal transduction systems (TCSs), responses to stimuli are mediated through phosphotransfer between protein components. Canonical TCSs use His → Asp phosphotransfer in which phosphoryl groups are transferred from a conserved His on a sensory histidine kinase (HK) to a conserved Asp on a response regulator (RR). RRs contain the catalytic core of His → Asp phosphotransfer, evidenced by the ability of RRs to autophosphorylate with small molecule analogs of phospho-His proteins. Phosphorelays are a more complex variation of TCSs that additionally utilize Asp → His phosphotransfer through the use of an additional component, the histidine-containing phosphotransfer domain (Hpt), which reacts with RRs both as phosphodonors and phosphoacceptors. Here we show that imidazole has features of a rudimentary Hpt. Imidazole acted as a nucleophile and attacked phosphorylated RRs (RR-P) to produce monophosphoimidazole (MPI) and unphosphorylated RR. Phosphotransfer from RR-P to imidazole required the intact RR active site, indicating that the RR provided the core catalytic machinery for Asp → His phosphotransfer. Imidazole functioned in an artificial phosphorelay to transfer phosphoryl groups between unrelated RRs. The X-ray crystal structure of an activated RR•imidazole complex showed imidazole oriented in the RR active site similarly to the His of an Hpt. Imidazole interacted with RR non-conserved active site residues, which influenced the relative reactivity of RR-P with imidazole versus water. Rate constants for reaction of imidazole or MPI with chimeric RRs suggested that the RR active site contributes to the kinetic preferences exhibited by the YPD1 Hpt.
Keywords: two-component systems, small molecule analog, response regulators, imidazole, phosphorelays, dephosphorylation, signal transduction, histidine-containing phosphotransfer domain, protein phosphorylation
Microorganisms across all domains of life as well as slime molds and plants use two-component systems (TCSs) to mediate responses to stimuli1, 2. TCSs regulate many different biological functions from development and motility1 to antibiotic resistance3, 4 and virulence5, 6. In TCSs, the “signal” is transmitted amongst protein components through the transfer of phosphoryl groups. The initial phosphotransfer event occurs when the sensory component, the stimulus-regulated histidine kinase (HK), catalyzes phosphotransfer from ATP to its own conserved His. Phosphotransfer proceeds to a conserved Asp on the receiver domain of the response-mediation component, the response regulator (RR). Phosphorylation activates the RR to carry out its output function, e.g. transcription regulation. Finally, termination of the signal results from phosphotransfer from the RR Asp to water. Thus, the typical pathway for phosphotransfer in a canonical TCS is ATP → His → Asp → H2O.
Phosphorelays are a more complex variation of TCSs in which phosphotransfer follows an ATP → His1 → Asp1 → His2 → Asp2 pathway7, 8. Bacteria contain some of the most well characterized phosphorelays9–12 and the vast majority of TCSs found in plants and fungi are phosphorelays7. In phosphorelays, the sensory HK (His1) transfers the phosphoryl group to a RR receiver domain (Asp1) that does not have an output function. The phosphoryl group is then transferred to His2 on a histidine-containing phosphotransfer domain (Hpt) domain, and then to a RR (Asp2), which executes the output response. In phosphorelays, signal termination often occurs by reverse phosphotransfer (Asp2 → His2 → Asp1 → H2O)9, 13. In contrast to canonical TCSs, phosphotransfer from Asp → His is essential for function in phosphorelays.
Optimal functioning of any TCS requires meticulous regulation of the flow of phosphoryl groups on and off of RR(s). In phosphorelays, the varied kinetics and directionality of phosphotransfer for different Hpt/RR pairs14 reflects multiple roles for Hpt domains in directing phosphoryl group flow. In addition to acting as both the source and recipient of RR phosphoryl groups, some Hpts can transfer phosphoryl groups to three RRs, to provide a branch point in a signaling circuit7, 15, or bind to a RR-P to prevent its dephosphorylation16. Bidirectional phosphotransfer to one response regulator and unidirectional phosphotransfer to another response regulator allows the second response regulator to act as a phosphate sink for the first17. The structural determinants, of either Hpts or RRs, that underlie the variation in directionality and kinetics of phosphoryl group flow are not currently known. In particular, protein determinants that favor or disfavor Asp → His phosphotransfer have not been elucidated.
Small molecule analogs of phosphoproteins have been extremely useful in probing the catalytic contributions of protein components in His → Asp phosphotransfer. Two small molecule phosphodonors, monophosphoimidazole (MPI) and phosphoramidate (PAM), represent progressively reduced portions of the His-P side chain. The observation that RRs can autophosphorylate with small molecule phosphodonors demonstrated that RRs contain the core catalytic machinery for His → Asp phosphotransfer18. Small molecule phosphodonors have been used to assign the additional catalytic contributions of HKs19, the RR linker and effector domains20, and the global conformation of the receiver domain21 in His → Asp phosphotransfer.
In light of the significant advances in the dissection of contributions of TCS protein domains to His → Asp phosphotransfer reactions that came about as a result of the discovery of small molecule phosphodonors, we sought a small molecular phosphoacceptor to dissect contributions to Asp → His phosphotransfer. Here we show that imidazole, the functional group of His, acts as a small molecule phosphoacceptor in Asp → His phosphotransfer. Imidazole accelerated the dephosphorylation, when compared to autodephosphorylation with water, of RRs CheY and PhoB by nucleophilic attack on the phospho-Asp. The discovery that imidazole is a recipient in phosphotransfer from RRs suggests that RRs contain the core catalytic machinery for Asp → His phosphotransfer. Furthermore, when imidazole was used in place of an Hpt in an artificial phosphorelay, phosphotransfer between non-partner protein components was achieved, suggesting that imidazole can act as a rudimentary Hpt. Determination of a co-crystal structure showed that imidazole orients in the RR active site similarly to the His of an Hpt poised for attack on the phospho-Asp. The imidazole interacted with nonconserved residues in the RR active site, and altering those amino acids changed the relative reactivities of a phosphorylated RR toward imidazole and water. Quantitative comparison of phosphotransfer reactions between various RRs and either Hpts or imidazole indicated that although the RR is the primary catalyst and dictated at least some of the preference displayed by a particular Hpt for different RRs, the Hpt is essential for high affinity binding between reactants and the primary contributor to overall reaction speed.
EXPERIMENTAL PROCEDURES
Mutagenesis and protein purification
Untagged versions of Escherichia coli wild type CheY (CheYwt), CheY F14E N59M E89R (CheYlarge), and CheY F14Q N59K E89Y (CheYMPI), and CheA were purified as described22, 23. The plasmids encoding His6-tagged CheY N59A E89A (CheYsmall), CheY N59Q E89F (CheYSLN1), CheY N59Q E89S (CheYSSK1), CheY N59V A88G E89N (CheYSKN7) were made using QuikChange (Agilent) with pKC124 as template. CheYsmall, CheYSLN1, CheYSSK1, CheYSKN7 were purified as described25 and removal of the His6-tag by thrombin cleavage left three additional residues (GSH) on the N-terminus. Additional N-terminal residues do not affect CheY autodephosphorylation25 or autophosphorylation24. His6-tagged fusions of E. coli PhoR193-431 and PhoB1-127 (wild type and F20D) were purified as described24. The His6-tag was cleaved from wild type PhoB1-127 leaving an additional (GSH) at the N-terminus and PhoB1-127 F20D was used with the His6-tag intact. The F20D substitution prevents formation of a non-physiological dimer of PhoB26. PhoB F20D exhibits autophosphorylation and autodephosphorylation properties indistinguishable from wild type PhoB24, 27. However, phosphotransfer from PhoR and PhoR-phosphatase susceptibility are diminished27. All proteins were purified using size exclusion chromatography as a final step to remove cleaved peptides, imidazole, and other impurities.
CheY dephosphorylation rate constant measurement by 32P
The effect of imidazole on CheY dephosphorylation was probed with modifications of a previously described method for measuring RR autodephosphorylation by following the loss of 32P 25. Purified [32P]CheA-P (1.5 μM) was mixed with CheY (15 μM) in 35 mM Tris at pH 7.5 and 10 mM MgCl2. After 10 s, to allow for sufficient phosphotransfer, imidazole was added. For determination of pH-dependence of the imidazole-mediated dephosphorylation reaction, 250 mM imidazole was added at pH 4.9 (buffered with 250 mM sodium acetate) or pH 8.2 (buffered with 250 mM Tris). For determination of dephosphorylation rate constants of CheYMPI and CheYSKN7, the ratio of CheY (30 μM) to [32P]CheA-P (0.30 μM) was increased to allow for rapid phosphotransfer. The phosphotransfer buffer was 5 mM Tris at pH 7.5 and 10 mM MgCl2. Next, imidazole (20 μM - 20 mM for CheYMPI, 2.5 mM – 50 mM for CheYSKN7) was added in 100 mM sodium bicarbonate at pH 10.3. Autophosphorylation with MPI is negligible at pH 10.328. For experiments with CheYMPI under conditions that allowed for simultaneous phosphorylation and dephosphorylation, measurements were completed at pH 7.5 with 30 μM CheYMPI, 0.30 μM [32P]CheA-P, and 50 mM imidazole (brought to pH 7.6 using HCl) in 35 mM Tris at pH 7.5 and 10 mM MgCl2. For all reactions, the final pH of mixed components was confirmed. Aliquots were removed at designated time points and quenched with an equal volume of 2X SDS sample buffer. Reaction components were separated by SDS-PAGE, and loss of radiolabel from CheY was detected using a phosphorimager. The signals were quantified using pixel volume analysis in which the background signal was manually subtracted. The amounts of CheY-P were plotted versus time and fit to one phase exponentials, yielding first order rate constants (kobs). Second order rate constants were the slopes calculated by plotting kobs versus imidazole concentration. All 32P experiments were completed at room temperature.
PhoB dephosphorylation assayed by 32P
[32P]PhoB-P was monitored to qualitatively determine whether imidazole accelerated dephosphorylation. His6-tagged PhoR (4 μM) was incubated with 0.3 mM [γ-32P]ATP in 35 mM Tris pH 8.0, 3.5 mM MgCl2, and 35 mM KCl for 30 minutes at room temperature. The reaction mix containing [32P]PhoR-P was then pipetted onto ~ 200 μL of a Ni-NTA Agarose (Qiagen) slurry (equilibrated in 35 mM Tris pH 8.0, 3.5 mM MgCl2, and 35 mM KCl buffer) on a 0.22 μm PVDF centrifugal filter column (Millipore). The column was centrifuged, then washed multiple times with PhoR autophosphorylation buffer to remove excess ATP. PhoB (90 μM in 60 μL of PhoR autophosphorylation buffer) was added directly to the beads, mixed, and incubated for 5 min on the column at room temperature to allow for sufficient phosphotransfer. The column was centrifuged to obtain [32P]PhoB-P. 60 μL of [32P]PhoB-P was mixed with either 20 μL of 2 M imidazole or 20 μL of PhoR autophosphorylation buffer. Time courses were completed and analyzed by electrophoresis and phosphorimaging as described above for CheY.
TLC analysis of phosphorylated small molecules
To identify small molecules containing [32P]phosphoryl groups in reaction mixtures, components were separated by thin layer chromatography (TLC). For each time point, a 1 μL aliquot was spotted onto cellulose PEI-F plates (Baker) and the remainder was used to separate proteins by SDS-PAGE. The TLC running buffer was 0.52 M lithium chloride and 1% (v/v) acetic acid29. Plates were air-dried and radiolabel was detected using a phosphorimager. Rf values were calculated and compared to Rf values of non-radiolabeled standards visualized by reacting with an assay spray as described by Bochner et al.30. Rf values for experimental data [0.3 ± 0.001 (SD) for Pi, 0.6 ± 0.02 for MPI] were similar to Rf values for standards [0.3 ± 0.02 for Pi, 0.6 ± 0.05 for MPI]. The MPI used for a non-radiolabeled standard was synthesized as described31.
Artificial phosphorelay
All of the artificial phosphorelay reactions contained 50 mM Tris (pH 7.5), 10 mM MgCl2, 30 μM [γ-32P]ATP, and 3 μM His6-PhoR. In addition, reactions contained different combinations of the following: 25 μM His6-tagged PhoB F20D, 25 μM CheYMPI, and 50 mM imidazole. Reactions were initiated by the addition of the [γ-32P]ATP and incubated for 90 minutes at room temperature before separation by SDS-PAGE. Radiolabel was detected with a phosphorimager. Control experiments were completed using CheYwt instead of CheYMPI.
Crystallization, data collection, structure solution and refinement
Crystals of the CheYMPI • BeF3− • Mn2+ • imidazole complex were made by hanging drop vapor diffusion using crystallization conditions similar to those used for CheYMPI • BeF3− • Mn2+ 32. BeF3− binds CheY tightly and is a stable phosphoryl group analog that is used because of the rapid autodephosphorylation of CheY. Although Mg2+ is the physiologically relevant divalent cation, Mn2+ supports CheY phosphorylation24 and dephosphorylation33. Each drop contained 1.5 μL of a mixture containing: 2.1 mg/mL CheYMPI, 1 mM MnCl2, 1 mM BeCl2, 10 mM NaF, and 200 mM imidazole. Additionally, each drop contained 1.5 μL of well solution: 100 mM Tris – pH 8.4, 2.1–2.4 M ammonium sulfate, and 8% (v/v) glycerol.
Crystals took several days to a week to grow to maximal size. Crystals were moved to cryprotectant as described previously34. The cover slip was moved sequentially to well solutions containing the same initial well solutions but with 11% (v/v) glycerol then 15% (v/v) glycerol. The crystals were then flash cooled in liquid nitrogen.
Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory. Data were collected at a wavelength of 1 Å. Three datasets were processed automatically using the cmdxds tools provided by SER-CAT35. Data collection statistics are shown in Table 1. Phases were determined by molecular replacement using Phaser-MR from the PHENIX suite36 using a single copy of the structure 3FFW32 (the structure of CheYMPI in the absence of imidazole) with all ligands stripped away. Two copies of CheYMPI • BeF3− • Mn2+ • imidazole were found in each asymmetric unit. The best model and electron density maps were examined using Coot37. After manual intervention by stepped real-space refinement with rotamers and visual inspection, computation refinement and automated water additions were performed with phenix.refine36. Following manual intervention with Coot, occupancy refinement was attempted for all ligands with BUSTER38. BUSTER did suggest a small difference in occupancy for the two imidazoles. Following another cycle of manual intervention, the structure was examined by PDB_REDO39. Final refined statistics are shown in Table 1. Ramachandran plots show no dihedral angles in disallowed regions. All three data sets showed nearly identical positioning of the imidazoles and showed no significant differences in the protein or ligands.
Table 1.
Data collection and refinement statistics
| Crystal 1 (5D2C) | Crystal 2 (5DGC) | Crystal 3 (5DKF) | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions | |||
| α, β, γ (Å) | 53.62, 53.72, 162.45 | 53.49, 53.58, 161.98 | 53.58, 53.74, 162.61 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 44.8 – 2.06 (2.11–2.06)* | 26.43 – 1.94 (1.99 – 1.94) | 81.3 – 1.94 (1.99 – 1.94) |
| Rmerge | 0.083 (0.58) | 0.119 (0.65) | 0.073 (0.623) |
| I/σI | 15.1 (3.88) | 10.4 (2.72) | 16.0 (2.84) |
| Completeness (%) | 97.6 (91.7) | 99.7 (99.7) | 100.0 (99.6) |
| Redundancy | 7.2 (7.4) | 7.2 (5.7) | 7.2 (5.8) |
| Refinement (Refmac 5.8.0103) | |||
| Resolution (Å) | 44.8–2.06 (2.11–2.06) | 26.43–1.94 (1.96 – 1.94) | 81.3 – 1.94 (1.96 – 1.94) |
| No. reflections (Rwork/Rfree) | 27677/1953 | 35162/1767 | 35660/1790 |
| Rwork/Rfree | 16.1/18.5 | 16.9/18.1 | 15.7/16.2 |
| Molecules/AU | 2 | 2 | 2 |
| No. atoms | |||
| Protein | 2004 | 2024 | 2088 |
| Ligand/ion | 70 | 53 | 82 |
| Water | 220 | 245 | 304 |
| B-factors | |||
| Protein | 24.1 | 19.2 | 8.9 |
| Ligand/ion | 59.7 | 45.3 | 27.3 |
| Water | 44.1 | 37.8 | 33.1 |
| RMSZ scores# | |||
| Bond lengths | 0.68 | 0.81 | 0.81 |
| Bond angles | 0.78 | 0.83 | 0.85 |
| EDS Fo, Fc correlation | 0.95 | 0.94 | 0.94 |
Values in parentheses are for highest-resolution shell.
RMSZ is the root-mean-square of all Z scores of the bond lengths (or angles) in the structure. A Z score for a bond length (or angle) is the number of standard deviations the observed value is removed from the expected value. A bond length (or angle) with |Z| > 5 is considered an outlier worth inspection.
CheY dephosphorylation rate constant measurement by pH jump/stopped-flow fluorescence
The basic method19, 25 was developed to measure CheY autodephosphorylation kinetics and exploits the strong pH-dependence of RR autophosphorylation with PAM (negligible at pH 10.3) coupled with the near pH-independence of autodephosphorylation over the same pH range28. Thus, a rapid jump in the pH of an equilibrium solution containing CheY-P and excess PAM at neutral pH results in CheY-P autodephosphorylation that can be quantitatively monitored with Trp fluorescence. Here, we measured CheY dephosphorylation rates in the absence and presence of imidazole. The instrument was a Perkin Elmer LS-50B spectrofluorimeter interfaced with an Applied Photophysics RX-2000 stopped-flow accessory. All fluorescence experiments were completed at 25°C. One stopped-flow syringe contained 20 μM wild type or mutant CheY and PAM [450 μM for CheYlarge, 1.4 mM for CheYSLN1, 5 mM for CheYSSK1, 15 mM for CheYwt and CheYsmall] in 5 mM Tris at pH 7.5 and 10 mM MgCl2. PAM is not commercially available and was synthesized as described40. The different PAM concentrations were chosen in light of different rate constants of PAM autophosphorylation25. The other syringe contained 200 mM sodium bicarbonate at pH 10.3 and 0–500 mM imidazole (0–1.0 M imidazole for CheYlarge) or 0–100 mM hydroxylamine. Fluorescence traces were fit to a one-phase exponential. The first order rate constants (kobs) thus obtained were plotted versus imidazole concentration and, in all cases, the relationship approximated linearity. The slopes of the best-fit lines gave the second order rate constants, kdephos,imid. Dephosphorylation data reported for CheYMPI was determined using 32P because CheYMPI exhibited fluorescence characteristics that complicated interpretation of results. Changes in CheYMPI fluorescence upon pH jump were ~30% of the expected change based on autophosphorylation fluorescence traces. The kobs values for CheYMPI measured by pH jump agreed with those determined by 32P at low imidazole concentrations but then appeared to saturate at high imidazole concentration. Therefore, the fluorescence change at high imidazole appeared to be dominated by other changes to the environment of the Trp besides the loss of the phosphoryl group, which was the focus of this study.
Dephosphoryaton rate constants of CheYSKN7 were determined by 32P because autophosphorylation of CheYSKN7 with small molecule phosphodonors was not detectable.
CheY autophosphorylation rate constant measurement using stopped-flow fluorescence
Autophosphorylation rate constants were measured at 25°C for CheYSLN1 and CheYSSK1 using MPI as the phosphodonor as previously described in23. The stopped-flow syringes contained 1) 5 μM CheY, 100 mM Hepes at pH 7.0, 10 mM MgCl2 and 2) varying concentrations of MPI (50 μM – 1.6 mM), 100 mM Hepes at pH 7.0, 10 mM MgCl2, 200 mM KCl. Because salt concentration was much greater than maximal MPI concentration, the ionic strength was approximately the same in each experiment, and varied by < 0.3%.
RESULTS
Imidazole stimulates dephosphorylation of the RR CheY
Because imidazole is the functional group of His, there was reason to speculate that imidazole would react with phosphorylated RRs. To determine whether imidazole affects dephosphorylation of CheYwt-P, loss of [32P]CheY-P was followed (Fig. 1A). In the absence of imidazole, dephosphorylation occurred with a half-life of ~15 s, consistent with CheY autodephosphorylation41. In the presence of 100 mM imidazole, loss of 32P was considerably faster (half-life < 2 s) than the autodephosphorylation of CheY. These results were confirmed by monitoring a unique Trp in the CheY active site18. Fluorescence time courses that monitored CheY-P dephosphorylation showed that the presence of imidazole enhanced the rate relative to CheY-P autodephosphorylation (Fig. 1B).
Figure 1.

CheY-P dephosphorylation kinetics in the absence and presence of imidazole. (A) Phosphorimaging scans of SDS-PAGE gels showing the time-dependent loss of 32P from [32P]CheY-P in the absence (top panel) or presence (bottom panel) of 100 mM imidazole at pH 7.5. (B) Trp fluorescence time traces showing loss of phosphoryl groups with 0 (black), 50 mM (medium gray), or 500 mM (light gray) imidazole at pH 10.3.
Imidazole enhances dephosphorylation by acting as a nucleophile
Two possible mechanisms were considered for the observed acceleration of CheY-P dephosphorylation with imidazole. First, imidazole could act to enhance the known hydrolysis reaction, producing Pi – the same product as autodephosphorylation. Precedence for this mechanism is provided by RR phosphatases, which enhance hydrolysis by orienting the attacking water molecule42–46. Alternatively, imidazole could act as the nucleophile in the reaction, producing MPI. To determine the mechanism of acceleration of CheY-P dephosphorylation, [32P]-small molecule reaction products were analyzed by thin-layer chromatography (TLC). [32P]MPI was produced in the presence of imidazole (Fig. 2A), consistent with imidazole acting as the nucleophile.
Figure 2.

Nucleophilic activity of imidazole in the dephosphorylation of CheY-P. Phosphorimaging scans of TLC plate showing products of [32P]CheY-P dephosphorylation reactions (A) in the absence or presence of 250 mM imidazole at pH 7.5 and (B) in the presence of 250 mM imidazole at pH 4.9 (buffered with 250 mM sodium acetate) or pH 8.2 (buffered with 250 mM Tris). (C) Phosphorimaging scans of SDS-PAGE gels showing the loss of 32P from [32P]CheY-P in the presence of 250 mM imidazole at pH 4.9 (top panel) or pH 8.2 (bottom panel).
Nucleophilic activity of imidazole is pH-dependent47, 48. At a pH below the pKa of 6.0, imidazole is mostly protonated and unable to act as a nucleophile. Above the pKa, imidazole is mostly deprotonated and able to act as a nucleophile. Imidazole was added to [32P]CheY-P either at pH 4.9 or at pH 8.2. The samples were separated using SDS-PAGE (to follow [32P]CheY-P) or by TLC (to follow 32P-labeled small molecules). At pH 4.9, there was no enhancement of CheY dephosphorylation by 250 mM imidazole (half life ~ 20 s−1; similar to autodephosphorylation), and the amount of [32P]MPI produced was barely detectable above background (Fig. 2B–C). In contrast, at pH 8.2, dephosphorylation was enhanced by imidazole (half life < 2 s) and [32P]MPI was detected.
To test whether another nucleophile could enhance dephosphorylation of CheYwt to the same extent as imidazole, dephosphorylation was measured in the presence of hydroxylamine. Hydroxylamine is a highly reactive nucleophile towards acyl phosphates49 and could potentially rapidly dephosphorylate CheY-P. Monitoring CheY-P dephosphorylation by pH-jump fluorescence, hydroxylamine did not accelerate dephosphorylation of CheY-P to the same extent as imidazole (Fig. 3A).
Figure 3.

CheY-P dephosphorylation in the presence of imidazole or hydroxylamine. (A) Plots of kdephos versus small molecule concentration showing the enhancement of CheY-P dephosphorylation with hydroxylamine (open squares and diamonds) compared to imidazole (closed squares and diamonds). CheY dephosphorylation was measured by stopped-flow fluorescence as previously described. The y-intercepts of each fit are approximately the rate constant for water-mediated dephosphorylation. Plots show two individual repeats each for CheY-P measurement with hydroxylamine or imidazole. (B) Phosphorimaging scan showing the loss of 32P from denatured [32P]CheY-P. CheY was phosphorylated using [32P]CheA-P. After a 10 second incubation, SDS was added to denature the proteins. The reaction was separated into three aliquots to which buffer with no nucleophile (lane 1), 250 mM imidazole (lane 2), or 250 mM hydroxylamine (lane 3) were added. After incubating for one hour at room temperature, reaction components were separated by SDS-PAGE, and loss of radiolabel from CheY was detected using a phosphorimager.
Imidazole-mediated dephosphorylation requires the intact RR active site
To further characterize the reaction of CheY-P with imidazole and probe its generality to other RRs, multiple experiments were completed.
Imidazole can act as a nucleophile against acyl phosphates in the absence of protein or other catalyst49. To test whether the dephosphorylation reaction with imidazole was dependent on catalysis by the folded CheY protein, the dephosphorylation reaction of denatured CheY-P with imidazole was assessed. Imidazole (250 mM) was incubated for 40 minutes with [32P]CheY-P that had been denatured in 2% SDS. Subsequent analysis by phosphorimaging (Fig. 3B) showed no detectable loss of [32P]CheY-P relative to a control that did not contain imidazole. Thus, the reaction between CheY-P and imidazole required folded CheY-P protein. In contrast, hydroxylamine (250 mM), which reacted only weakly with folded CheY-P (Fig. 3A) completely dephosphorylated denatured [32P]CheY-P (Fig. 3B), likely due to increased solvent exposure of the acyl phosphate group.
The requirement for folded protein for the reaction of CheY-P with imidazole suggested that additional features of the RR active site might be required. RR dephosphorylation by hydrolysis requires an active site divalent metal ion such as Mg2+ 33, 41. Removal of the metal inhibits both autophosphorylation and autodephosphorylation. To determine whether the metal is required for dephosphorylation with imidazole, the Trp fluorescence of CheY was monitored during addition of various chemicals. CheY-P was generated by incubating 12 μM CheY with 20 mM PAM in 50 mM Tris 7.5 and 14 mM MgCl2. The metal was then removed from the active site of CheY-P by addition of 250 mM EDTA. Next, 1.1 M imidazole was added, which resulted in only minimal dephosphorylation (data not shown). The little dephosphorylation that did occur was likely due to some small portion of CheY-P remaining metal bound. Rapid dephosphorylation (~2 s half life, faster than the reaction with water) occurred when the system was replenished with 440 mM Mg2+. Thus, Mg2+ is also required for catalysis of the reaction between imidazole and CheY-P.
As the functional group of His, imidazole could be a moiety recognized by RRs in general. To probe for generality, another RR, PhoB-P, was reacted with imidazole. 500 mM imidazole enhanced [32P]PhoB-P dephosphorylation by at least 10-fold (Fig. 4A) suggesting that imidazole can react with RRs in general.
Figure 4.

Sensitivity of PhoB-P and CheA-P to imidazole. (A) Phosphorimaging scans showing the loss of 32P from [32P]PhoBN-P in the absence (top panel) or presence (bottom panel) of 500 mM imidazole at pH 7.5. (B) Phosphorimaging scan showing [32P]CheA-P after a 1-hour incubation with different concentrations of imidazole at pH 7.5.
Finally, because imidazole undergoes nucleophilic attack on phosphoramidates31, 48, we tested whether imidazole reacted with the His-P of an HK. The HK [32P]CheA-P (1 μM) was incubated with 0–500 mM imidazole in 5 mM Tris pH 7.5 and 10 mM MgCl2. After an hour-long incubation with imidazole, loss of [32P]CheA-P was not detectable (Fig. 4B). The data described above indicate that the intact RR active site is required for dephosphorylation with imidazole.
Phosphoryl groups reversibly transfer between CheYMPI and imidazole
In the experiments described so far, imidazole-mediated RR-P dephosphorylation was observed under essentially irreversible reaction conditions that (by choice of pH and/or CheY variant) precluded significant re-phosphorylation of CheY. To further characterize the relationship between RRs and imidazole, reactions were carried out under conditions that did not inhibit CheY re-phosphorylation with the MPI product. CheYMPI was chosen for this analysis because it is a variant that previously exhibited enhanced ability to autophosphorylate with MPI23.
Paradoxically, [32P]CheYMPI-P persisted longer in the presence than in the absence of imidazole; 50 mM imidazole increased the apparent half-life by nearly three fold (~21 minutes) (Fig. 5) compared to the half-life in the absence of imidazole [~8 minutes22]. Conversely, the apparent half-life of [32P]CheYwt-P was decreased when imidazole was added under similar conditions (Fig. 1A). TLC analysis of the reaction of CheYMPI-P with imidazole showed a rapid burst of [32P]MPI followed by the gradual consumption of [32P]MPI and accumulation of [32P]Pi (Fig. 5)-consistent with three reactions proceeding simultaneously. Starting with [32P]CheYMPI-P, imidazole rapidly dephosphorylated [32P]CheYMPI-P, producing a burst of [32P]MPI (Fig. 5). CheYMPI was able to then re-phosphorylate to form [32P]CheYMPI-P, consuming the [32P]MPI. Subsequently, some [32P]CheYMPI-P was able to autodephosphorylate resulting in the accumulation of [32P]Pi. Because CheYMPI autophosphorylates with MPI23 much faster than it dephosphorylates with imidazole or water, the [32P]CheYMPI-P persisted longer than in the absence of imidazole.
Figure 5.
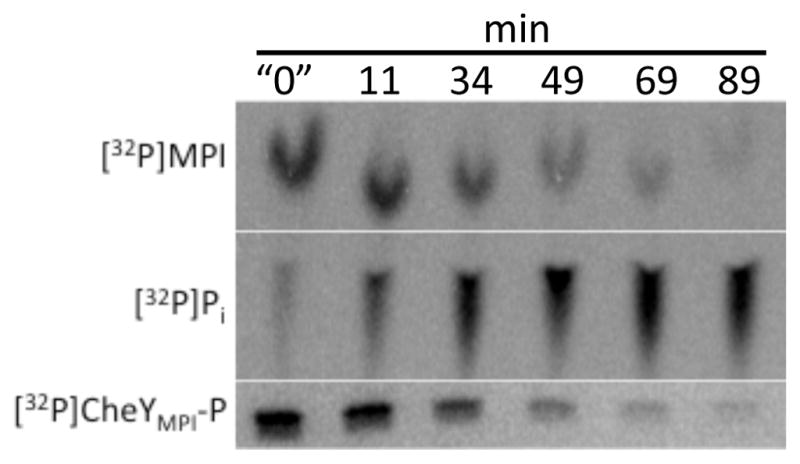
[32P]CheYMPI-P reaction with imidazole. Phosphorimaging scans showing changes in [32P]CheYMPI-P (SDS-PAGE), [32P]MPI (TLC), and [32P]Pi (TLC) over time upon reaction of CheYMPI-P with 50 mM imidazole at pH 7.5. The “0” time point was quenched less than 15 seconds after addition of imidazole to [32P]CheYMPI-P.
Imidazole can act as a rudimentary Hpt in an artificial phosphorelay
The reversible phosphotransfer between CheYMPI and imidazole is analogous to reversible transfer between RRs and Hpts, and suggests that imidazole may be able to function in a manner reminiscent of an Hpt in a phosphorelay. An artificial phosphorelay was designed to test whether imidazole could transfer phosphoryl groups between different response regulators. To probe the robustness of such an ability, instead of just replacing the Hpt of a known phosphorelay with imidazole, the artificial phosphorelay was assembled from components that are not part of natural phosphorelays. Following progressive inclusion of phosphorelay components, transfer of phosphoryl groups was tracked (Fig. 6A). The PhoR193-431 HK cytoplasmic fragment was incubated with [32P]ATP, resulting in [32P]PhoR-P (lane 1). When PhoR partner RR PhoB1-127 F20D was added, phosphorylation of PhoB F20D was observed as expected (lane 2). His6-tagged PhoB1-127 F20D was used for clean separation from PhoR193-431 and CheYMPI on SDS-PAGE. When imidazole was added, there was a loss of [32P]PhoB F20D-P (lane 3), consistent with phosphotransfer from PhoB F20D to imidazole. If non-partner RR CheYMPI was added in the absence of imidazole, then the [32P]PhoB F20D-P remained and accumulation of [32P]CheYMPI-P was not detected (lane 4). However, if CheYMPI was added in the presence of imidazole, both loss of [32P]PhoB F20D-P and accumulation of [32P]CheYMPI-P were observed (lane 5). Thus, in the presence of imidazole, CheYMPI becomes phosphorylated though sequential phosphotransfer from ATP → PhoR → PhoB → imidazole → CheYMPI.
Figure 6.
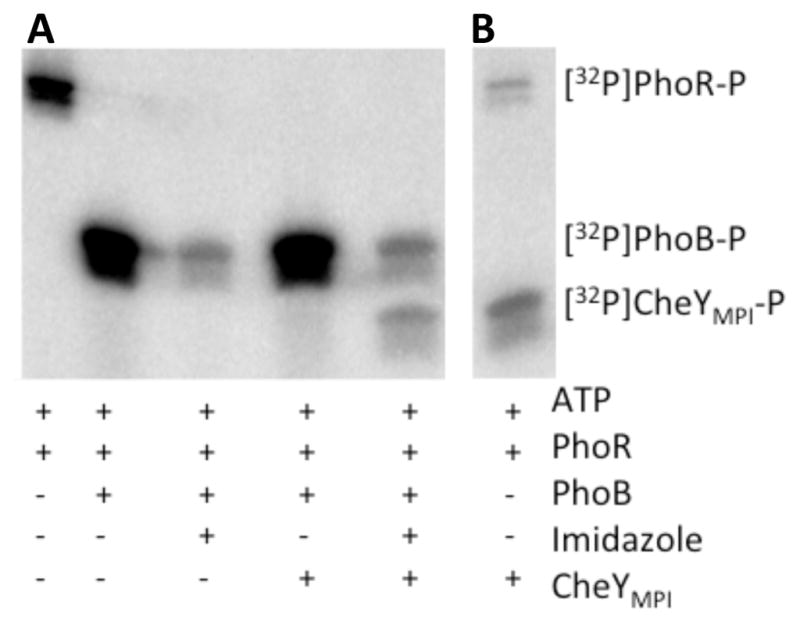
The role of imidazole in an artificial phosphorelay. (A) Phosphorimaging scan of SDS-PAGE gel showing the presence of 32P on protein components in an artificial phosphorelay at pH 7.5. The PhoBN F20D variant was used for this analysis. (B) Phosphorimaging scan showing transfer of [32P] from PhoR to CheYMPI at pH 7.5. Reaction components in (A) and (B) were incubated for 30 minutes at room temperature.
Control experiments for the artificial phosphorelay revealed that, under normal experimental conditions, CheYMPI was directly phosphorylated by PhoR (Fig. 6B). PhoB F20D had to be absent in order for crosstalk between PhoR and CheYMPI to occur (cf. lane 4 of Fig. 6A and Fig. 6B). Additionally, crosstalk was not observed between PhoR and CheYwt under the same conditions (data not shown). The ability of CheYMPI to accept phosphoryl groups from a nonpartner HK is consistent with CheYMPI having an enhanced affinity for the phosphorylated imidazole (MPI) moiety23.
Nonconserved residues D+2, T+1, and T+2 interact with imidazole ring in the response regulator active site
To further explore the basis by which imidazole could act similarly to an Hpt in reactions with CheYMPI-P, three crystal structures of CheYMPI in complex with Mn2+, the stable phosphoryl group analog BeF3−, and imidazole were determined to a resolution of 1.9–2.1Å (Table 1). To our knowledge, these represent the first structures of an activated response regulator complexed with a small molecule phosphoacceptor. Two imidazoles were found complexed with each of the two CheYMPI per asymmetric unit (Fig. 7A). However, the linear dependence of CheY-P dephosphorylation on imidazole concentration (Fig. 3A for wildtype CheY and data not shown for other CheY variants) strongly suggests that only one of the two imidazoles is involved in the dephosphorylation reaction. One imidazole, at full occupancy but likely irrelevant to the dephosphorylation reaction, was coordinated with the Mn2+, consistent with the affinity between imidazoles and transition metals exploited in the use of His-tagged proteins and Ni-affinity column50. The other imidazole, at partial occupancy, was positioned over the phosphomimic oriented for direct in-line attack of the phosphoryl group (Fig. 7). For six individual CheYMPI monomers, the closest atom from the imidazole was 3.4 ± 0.2 Å from the beryllium, consistent with the ability reported here of imidazole to dephosphorylate response regulators. Despite the lower occupancy of the imidazole poised for nucleophilic attack, clear electron density for the plane of the imidazole was observed in all three independent structures (Fig. 7B). Furthermore, when the CheYMPI • BeF3− • Mn2+• imidazole structure was overlaid (Fig. 8) with two activated RR/Hpt complexes – SLN1 • BeF3− • Mg2+ • YPD1 (PDB 2R25)51 and Spo0F • BeF3− • Mg2+• Spo0B (PDB 2FTK)52 – the imidazole in the CheYMPI active site was oriented similarly to the imidazole moieties of the Hpt (YPD1 and Spo0B) histidines; all were positioned for in-line attack.
Figure 7.

Structure showing imidazole poised for nucleophilic attack in CheYMPI•BeF3-. (A) Two imidazoles (purple) were complexed with CheY (PDB 5D2C). One imidazole is oriented over the BeF3- (phosphomimic) in a fashion consistent with nucleophilic attack. Another imidazole was associated with the metal (light teal). (B) Electron density map (2Fo-Fc) shows the presence of the imidazole over the Be. In both panels, the divalent metal is light teal, BeF3- is green and light cyan, and the imidazoles are purple.
Figure 8.

Structures showing imidazole oriented in the active site similarly to Hpt His. (A) The co-crystal structures of CheYMPI•BeF3-•imidazole (PDB 5D2C), Spo0F•BeF3-•Spo0B (PDB 2FTK)52, SLN1•BeF3-•YPD1 (PDB 2R25)51 showing interactions between D+2, T+1, and/or T+2 side chains from each RR and the imidazole rings. CheYMPI/imidazole residues are represented by purple, Spo0F/Spo0B by cyan, SLN1/YPD1 by slate, and the phosphomimic BeF3- is green and light cyan in all three. Purple (CheY) and slate (SLN1) dots and cyan (Spo0F) spheres represent space-filling models of non-conserved active site residues. (B) Using the same coloring as panel (A) the imidazole rings from all three structures are oriented over the Be within appropriate distance for nucleophilic attack.
For the CheYMPI • BeF3− • Mn2+ • imidazole structures (PDB 5D2C, 5DGC, 5DKF), residues at positions D+2 (two positions C-terminal to the site of phosphorylation, K in CheYMPI), T+1 (one position C-terminal to the conserved Thr/Ser, A in CheYMPI), and T+2 (two positions C-terminal to the conserved Thr/Ser, Y in CheYMPI) were in direct contact with the imidazole through van der Waal’s interactions. Similar interactions were observed between residues at D+2, T+1, and T+2 on the SLN1 receiver domain and the histidine of YPD1. In both cases, non-conserved active site residues at D+2, T+1, and T+2 formed a discrete pocket around the imidazole (Fig. 8A). These observations suggest a role for non-conserved active site residues in RRs in the reaction with imidazole, and, potentially, with Hpts. This is direct evidence that imidazole is able to fulfill some of the function of an Hpt in a similar fashion to the Hpt.
Non-conserved active site residues influence dephosphorylation with imidazole
Active sites of RRs consist of five conserved residues and several residues that vary amongst different RRs53. Non-conserved active site residues modulate autophosphorylation23 and autodephosphorylation22, 32 of RRs. Because structural analysis of CheYMPI complexed with imidazole, as well as of the SLN1 receiver domain complexed with the YPD1 Hpt protein, showed that non-conserved residues at D+2, T+1, and T+2 could facilitate appropriate orientation of the imidazole in the active site, it seemed logical that these three residues might influence the rates of dephosphorylation with imidazole. Ala occupies position T+1 in about half of response regulators, so dephosphorylation rate constants with imidazole were measured for a set of CheY mutants containing Ala at T+1 and various substitutions at D+2 and T+2. The initial set of mutants explored four distinct active site environments: CheYwt contains the polar amino acids Asn at D+2 and Glu at T+2. CheYsmall represents a reduced active site in which the D+2 and T+2 residues are both Ala. With CheYlarge, a previously solved crystal structure indicates that the large Met and Arg side chains (D+2 and T+2, respectively) could potentially occlude the active site, hindering interaction between the nucleophile and the phospho-Asp32. CheYMPI (Lys at D+2 and Tyr at T+2) is highly reactive with MPI [103-fold faster autophosphorylation than CheYwt 23], possibly reflective of enhanced affinity for the imidazole moiety. Rate constants for imidazole-mediated dephosphorylation of these four CheY variants varied over a ~20-fold range (Table 2). Similarly, autodephosphorylation rate constants of this mutant set span a ~30-fold range; however, the rank order from fastest to slowest varied considerably for imidazole versus water-mediated dephosphorylation. For the mutants studied here, the relative sensitivity to imidazole, defined by the ratio of imidazole-mediated to water-mediated dephosphorylation rate constants, varied over a 100-fold range. All of the CheY-P variants were at least 1000-fold more reactive with imidazole than with water. The differences in reactivity were potentially due to the chemical properties of imidazole and/or due to features of the RR active site.
Table 2.
Rate constants for dephosphorylation of CheY variants with imidazole and water
| Receiver domaina | kdephos,imida(M−1s−1) | kdephos,waterb ×10−5 (M−1s−1) | Relative sensitivity to imidazolec |
|---|---|---|---|
| CheYwt | 1.1 ± 0.1 | 75.0 ± 9.0d | 1,500 |
| CheYsmall | 1.2 ± 0.04 | 9.0 ± 0.6e | 14,000 |
| CheYlarge | 0.25 ± 0.005 | 3.3 ± 0.0d | 8,200 |
| CheYMPI | 4.3 ± 0.05 | 2.6 ± 0.3e | 170,000 |
Values are averages of second order rate constants from 2–3 measurements (completed at pH 10.3) ± SD and are determined by pH-jump fluorescence except CheYMPI was measured using 32P.
Calculated by dividing first order water-mediated dephosphorylation rate constants by the concentration of water (55.5 M).
Relative sensitivity to imidazole was calculated by dividing kdephos,imid by kdephos,water.
First order water-mediated dephosphorylation rate constants from 22.
First order water-mediated dephosphorylation rate constant determined using 32P as previously described in 22.
Non-conserved active site residues influence phosphorylation reactions in CheY variants that mimic phosphorelay receiver domains
Considering the previous results that imidazole could function in some ways similarly to an Hpt, we next used imidazole as a tool to dissect the contributions of response regulator and Hpt proteins to Asp → His phosphotransfer reactions. To our knowledge, the yeast osmosensing phosphorelay is the only phosphorelay system for which rate constants have been measured for His → Asp and Asp → His phosphotransfer events14. The system contains one Hpt, YPD1, which interacts with three different receiver domains: SLN1, SSK1, and SKN7. YPD1 reacts differentially with each receiver domain (Tables 3 & 4) such that phosphotransfer is: rapid in both directions with SLN1, rapid to SSK1 and does not occur from SSK1 to YPD1, and slow in both directions with SKN7. Although phosphotransfer rate constants varied between YPD1 and each of the receiver domains, binding constants were similar (1.4–7.8 μM14), suggesting that the variation in phosphotransfer arises from the reaction chemistry. Because the three receiver domains all react with the same Hpt, it is logical to hypothesize that the receiver domains, and not the Hpt, contain the determinants of the relative reactivities. Alternatively, the differential reactivities may arise due to differences in interactions between the Hpt and each of the receiver domains; for example, binding may not align reactants optimally. We predicted that, based on the first hypothesis, reactions between RR-P and imidazole (analogous to Asp → His phosphotransfer) or between MPI and RR (His → Asp) would reflect the relative reactivities observed in the yeast system. Based on the CheY. • BeF3−. • Mn2+.• imidazole structure, variants of CheY were made that matched the three yeast receiver domains at the three non-conserved residues at D+2, T+1, and T+2, which interacted with the imidazole ring in both the CheY and SLN1 structures. Additionally, the CheY variants match the yeast receiver domain active sites at conserved residues D (site of phosphorylation), T, K, and the two acids that coordinate the divalent metal (DD in CheY, ED in the yeast receiver domains).
Table 3.
Rate constants for “His → Asp” phosphotransfer
| Receiver domain | Amino acid at
|
YPD1-P ➝ yeast RR
|
MPI ➝ CheY
|
||||
|---|---|---|---|---|---|---|---|
| D+2 | T+1 | T+2 | kmaxa (s−1) | Kda (μM) | kmax/Kd (× 106 M−1s−1) | kphos/KS (M−1s−1) | |
| SLN1/CheYSLN1 | Q | A | F | 230 ± 130 | 7.8 ± 5.7 | 29 | 1500 ± 76 |
| SSK1/CheYSSK1 | Q | A | S | 160 ± 70 | 2.4 ± 1.9 | 67 | 68 ± 12 |
| SKN7/CheYSKN7 | V | G | N | 1.4 ± 0.2 | 1.5 ± 0.5 | 0.9 | < 0.1b |
Values are from 14.
Phosphorylation with MPI was below the limit of detection by fluorescence quenching. The observed rate of approach to equilibrium is given by kobs = (kphos/Ks)[phosphodonor] + kdephos,water. To detect phosphorylation, the term describing the phosphorylation rate (which changes with phosphodonor concentration) must be at least as large as the term for dephosphorylation (which is invariant). With [MPI] = 25 mM and kdephos,water for CheYSKN7 = 0.002 s−1, setting the phosphorylation and dephosphorylation terms equal gives kphos/KS = 0.08 M−1 s−1.
Table 4.
Rate constants for “Asp → His” phosphotransfer
Relative to each other, the yeast receiver domains SLN1 and SSK1 were faster than SKN7 for His → Asp phosphotransfer14. Similarly, CheYSLN1 and CheYSSK1 were faster than CheYSKN7 for autophosphorylation with MPI (Table 3). In fact, autophosphorylation of CheYSKN7 with MPI (or two other small molecule phosphodonors, PAM and acetyl phosphate) was not detectable. To determine whether the failure of CheYSKN7 to autophosphorylate was a meaningful result or the trivial consequence of a nonfunctional protein, other activities of CheYSKN7 were assayed. Binding of Mg2+ (required for catalysis33) to CheYSKN7 was detected using fluorescence quenching (data not shown) and CheYSKN7 was phosphorylated in the presence of CheA and [32P]ATP (data not shown), indicating that CheYSKN7 is properly folded.
For Asp → His phosphotransfer, SLN1 transfers more rapidly than SKN7 to YPD1 (Table 4)14. Asp → His phosphotransfer was not observed for SSK1-P and YPD114. This is likely due to formation of an inhibitory YPD1•SSK1-P complex that results in a ~200× reduction in autodephosphorylation of SSK1-P (i.e. reaction with water), presumably by steric occlusion of the active site. Consistent with comparisons between SLN1 and SKN7, CheYSLN1 was faster than CheYSKN7 for dephosphorylation with imidazole – though the difference was modest (Table 4).
Taken together, the results reported in Tables 3 and 4 indicate that the receiver domain active site provides at least some of the determinants for the relative preferences in phosphotransfer reported between the YPD1 Hpt protein and the SLN1, SSK1, and SKN7 receiver domains.
DISCUSSION
Phosphotransfer reactions in TCSs: Hpt domains allow bidirectional His/Asp phosphotransfer
To provide context for thinking about potential implications of the reactions of imidazole characterized here, we first summarize the phosphotransfer reactions known to occur in TCS proteins. Recall that HK DHp (dimerization and histidine phosphotransfer) domains and Hpt domains are phosphorylated on His residues, whereas RR proteins are phosphorylated on Asp residues. Therefore, in the ATP →HK DHp →RR →H2O phosphotransfer pathway of canonical TCSs, the phosphotransfer between HKs and RRs is His →Asp. Although phosphotransfer from RR →HK DHp has been observed in vitro54, 55, the physiologically relevant reaction between most HKs and RRs is believed to be unidirectional from HK →RR (see exception below). The inclusion of Hpt domains, however, which participate in bidirectional phosphotransfer, allows more complex phosphorelays. The typical phosphotransfer pathway in a phosphorelay is ATP →HK →RR1 →Hpt →RR2 →Hpt →RR1 →H2O11, 56. Thus, in addition to the HK DHp→RR phosphotransfer utilized in canonical TCSs, phosphorelays require a second type of His →Asp phosphotransfer (Hpt →RR), as well as Asp →His (RR →Hpt) phosphotransfer for function. In CheA-type HKs, the DHp domain of a canonical HK is replaced by an Hpt domain57. The ability of Hpt domains to support bidirectional phosphotransfer can then allow reverse phosphotransfer from one RR through CheA to another RR that acts as a phosphate sink17: ATP →Hpt →RR1 →Hpt →RR2 →H2O.
Small molecule analogs as kinetic probes of TCSs
Autophosphorylation of RRs with PAM and MPI, small molecule analogs of phospho-His, has long been informative for dissecting catalytic contributions to His →Asp (HK DHp →RR or Hpt →RR) phosphotransfer reactions20, 23, 28. The imidazole-mediated dephosphorylation of RR-P reported here (RR-P + imidazole →RR + MPI) is the reverse of the autophosphorylation reaction with MPI. Analogously, this reaction is informative for the kinetic dissection of Asp →His phosphotransfer, which is characteristic of phosphorelays.
The reaction rates of a single Hpt protein for both His →Asp and Asp →His phosphotransfer can differ dramatically with different RRs14. The determinants contained in either Hpts or RRs that underlie the variation in kinetics, specificity, and directionality of Hpt ←→RR phosphotransfer reactions are not known. Here, we explore the use of imidazole to probe Asp →His phosphotransfer. Additionally, we used MPI to probe His →Asp phosphotransfer in the context of phosphorelays.
Receiver domain active sites account for some kinetic preference in phosphotransfer reactions involving Hpts
Comparing the properties of a phosphotransfer reaction between two proteins with a reaction in which one protein is replaced with a small molecule allows us to ask what the remaining protein can contribute to the reaction. Thus, in principle, replacement of an Hpt with imidazole could be used to assess the contributions of a RR to a RR-P →Hpt reaction. The fact that the reaction with imidazole occurred at all is evidence that the RR provides the core catalytic machinery for the chemistry (see section below). But the availability of co-crystal structures between CheY, BeF3−, and imidazole (Fig. 7) allowed us to go further in dissecting contributors to the RR-P →Hpt reaction. Because the structure of CheYMPI complexed with imidazole revealed interactions between imidazole and non-conserved active site residues at D+2, T+1, and T+2, we could assess the contributions of the active site portion of the RR in regulating the degree of catalysis. We constructed three chimeric proteins, in which the active sites of three yeast response regulators were spliced onto the same RR backbone, CheY. The CheYSLN1, CheYSSK1, and CheYSKN7 chimeras matched the wild type yeast receiver domains at four out of five conserved active site residues (one of the conserved metal-coordinating acid residues was a D in CheY instead of the E found in the yeast RRs) as well as the three non-conserved positions. If the relative kinetics of the chimeras correlated with the relative kinetics of the yeast proteins, it could be concluded that differences in the RR active sites contribute to modulation of the variable kinetics in the yeast system.
The kinetics of phosphotransfer between the YPD1 Hpt and the three yeast receiver domains are saturable, allowing the measurement of binding constants (Kd) and first-order kinetic constants for catalysis (kmax)14. In contrast, reactions between CheY and small molecules are not saturable at experimentally accessible concentrations58, so only bimolecular rate constants incorporating both binding and catalysis were obtained. For “Asp →His” phosphotransfer, CheYSLN1-P →imidazole was faster than CheYSKN7-P →imidazole (Table 4), just as SLN1-P →YPD1 is faster than SKN7-P →YPD114. However, the difference in rate constants for the model reactions of CheY chimeras with imidazole was a modest four-fold, whereas the difference in kmax for the phosphotransfer reactions between yeast RR-P and Hpt proteins was 70-fold. This suggests that the non-conserved active site residues in the receiver domains contribute to, but do not entirely account for, the differential reactivities for Asp →His phosphotransfer observed in the yeast phosphorelay.
For “His →Asp” phosphotransfer, rate constants for transfer from MPI to CheYSLN1, CheYSSK1, or CheYSKN7 resulted in the same rank order as rate constants (kmsx) for phosphotransfer from YPD1-P to SLN1, SSK1, or SKN7 (Table 3). In contrast to the Asp →His results, in this case, the rate constants (kphos/Ks) for the CheY chimeras spanned a range of more than four orders of magnitude, whereas the rate constants for the yeast receiver domains span two orders of magnitude. The correlation is consistent with the notion that the receiver domain non-conserved active site residues strongly contribute to the differential activities of multiple RRs with a single Hpt in His →Asp transfer.
RRs contain the primary catalytic machinery for Asp to His phosphotransfer
In Asp →His phosphotransfer, the His residue on the Hpt acts as a nucleophile to attack the phospho-Asp on the RR-P, resulting in unphosphorylated RR and Hpt-P12, 14. In analogous fashion, imidazole was able to accept phosphoryl groups from both CheY-P and PhoB-P, resulting in unphosphorylated RR and MPI (Figs. 2, 4, 5 & 6). Analogous to an Hpt, imidazole was able to reversibly transfer phosphoryl groups with CheYMPI (Fig. 5) and acted in place of a His2 in an artificial phosphorelay (Fig. 6). Dephosphorylation of RRs with imidazole requires the intact RR active site, but does not require input from other proteins. Previous work established that RRs contain the catalytic core of His →Asp18, 28 and Asp →H2O41, 45 phosphotransfer reactions. Zhao, et al. proposed that, in order to catalyze the phosphotransfer chemistry between Hpts and RRs, the RR conserved active site residues and divalent active site metal interact with the equatorial oxygens of the phosphoryl group, stabilizing the proposed trigonal bipyramidal transition state [Fig. 7 of 51]. The ability of RRs studied here to achieve phosphotransfer to imidazole in a fashion that required the intact metal-bound RR active site provides functional evidence that RRs are sufficient for catalysis of Asp →His phosphotransfer.
The ability of RRs to support reactions with imidazole and MPI, i.e. support phosphotransfer reactions in the absence of a protein partner, is consistent with the assertion59 that Hpt active sites have minimal known catalytic features beyond the phosphorylatable His. For Hpt domains that constitute the phosphorylation site of CheA-type HKs, several amino acids that form a hydrogen bonding network with the His are critical for phosphorylation with ATP60. However, alteration of similar residues in YPD1 had only modest effects on phosphotransfer reactions with SLN17, 14, 61. Replacement of a particular Gly with a larger residue had a substantial effect on the ability of YPD1 to support phosphotransfer, presumably due to blocking access of the receiver domain to the YPD1 His7. In summary, there does not appear to be compelling evidence that specific Hpt residues other than the His make important contributions to catalysis of phosphotransfer with RRs.
Although not a primary contributor of catalytic machinery, Hpt proteins clearly do make important contributions to phosphotransfer reactions with RRs. The bimolecular rate constants for phosphotransfer between CheY and imidazole/MPI are many orders of magnitude slower than the corresponding rate constants for phosphotransfer between the yeast RRs and YPD1 (Tables 3 & 4). One means by which Hpts enhance phosphotransfer is through binding between phosphodonor and recipient through protein-protein interactions. Additionally, the Hpt domain could possibly enhance phosphotransfer by (i) orienting the His in the active site and/or (ii) acting as a cap and excluding water from the RR active site.
Imidazole orients similarly to Hpts in Asp →His phosphotransfer
Biochemical analysis of reactions between CheY-P (and PhoB-P) and imidazole showed the capability of the receiver domains to support Asp → His phosphotransfer. To explore the model reaction further, we determined the structure of CheYMPI complexed with imidazole and BeF3- (Fig. 7) and compared it to the few reported structures of HKs or Hpts complexed with RRs and a phosphoryl group or phosphomimic. In the available structures of HK-P•RR (PDB 3KYI) or HK•BeF3-•RR (PDB 4JAV) complexes, the His and Asp residues are not oriented to react with one another. We are aware of two co-crystal structures of RRs complexed with Hpts in the presence of a phosphomimic – Spo0F•BeF3-•Mg2+•Spo0B (PDB 2FTK)52 and SLN1•BeF3-•Mg2+•YPD1 (PDB 2R25)51. Note that these structures represent the two known classes of Hpt domains. Both classes form a four-helix bundle, but the Spo0B class is dimeric and apparently arose from degenerate HKs62, whereas more typical Hpt proteins are monomeric63. When the structure of CheYMPI•BeF3-• Mn2+•imidazole was aligned with each RR•BeF3-•Hpt co-crystal structure, the imidazole was oriented similarly to the His residues from both types of Hpts and positioned appropriately for nucleophilic attack (Fig. 8). In particular, the distances from the imidazole ring to the Be atom (P analog) were 3.4 ± 0.2 Å for imidazole alone, 3.6 ± 0.8 Å (from four separate protamers) for Spo0B, and 3.2 Å for YPD1.
Non-conserved active site residues differentiate between nucleophiles
In addition to similar orientation of the imidazole ring, alignment of the CheYMPI•BeF3-•imidazole structure with two RR•BeF3-•Hpt structures showed that non-conserved active site residues in each RR (D+2, T+1, and T+2 in CheY and SLN1; D+2 and T+1 in Spo0F) interacted with the imidazole ring and formed a pocket directly over the phosphomimic. The overall suggestion was that non-conserved active site residues at D+2, T+1 and T+2 functioned to orient the imidazole ring in the active site and could potentially influence catalysis. Residues at these positions influence autodephosphorylation reactions in receiver domains23, so it seemed reasonable to predict that residues at D+2, T+1, and T+2 would influence dephosphorylation reactions with imidazole. In this work, imidazole-mediated dephosphorylation of seven CheY variants was assessed (Tables 2 & 4). Rate constants for water and imidazole-mediated dephosphorylation spanned similar ranges – 29 and 33-fold, respectively (Table 5). However, rank orders from fastest to slowest were different for water versus imidazole. As a result, the relative sensitivities of the CheY variants to the two nucleophiles spanned a 110-fold range. The large range in relative sensitivities suggests that the non-conserved active site residues discriminate between water and imidazole as nucleophiles.
Table 5.
Relative sensitivity of CheY variants for imidazole versus water
| Receiver domain | kdephos,imida (M−1s−1) | kdephos,waterb ×10−5 (M−1s−1) | Relative sensitivity to imidazolec |
|---|---|---|---|
| CheYMPI | 4.3 ± 0.05 | 2.6 ± 0.3d | 170,000 |
| CheYSKN7 | 1.9 ± 0.4 | 3.9 ± 0.4d | 48,000 |
| CheYSLN1 | 8.2 ± 0.4 | 18.0 ± 4.2e | 45,000 |
| CheYsmall | 1.2 ± 0.04 | 9.0 ± 0.6d | 14,000 |
| CheYSSK1 | 3.1 ± 0.1 | 34.0 ± 7.2e | 9,000 |
| CheYlarge | 0.25± 0.005 | 3.3 ± 0.0f | 8,200 |
| CheYwt | 1.1 ± 0.1 | 75.0 ± 9.0f | 1,500 |
Values are averages of second order rate constants from 2–3 measurements (completed at pH 10.3) ± SD and are determined by pH-jump fluorescence except CheYMPI and CheYSKN7 were measured using 32P.
Calculated by dividing first order water-mediated dephosphorylation rate constants by the concentration of water (55.5 M). Multiply column values by 10−5.
Relative sensitivity was calculated by dividing kdephos,imid by kdephos,water.
First order water-mediated dephosphorylation rate constant determined using 32P as previously described in 22.
First order water-mediated dephosphorylation rate constant determined using pH-jump fluorescence described in 25.
First order water-mediated dephosphorylation rate constants from 22.
Considering the roles of RRs and Hpts in phosphotransfer reactions, the variation in relative sensitivities between CheY variants reported here may reflect some aspect of RR/Hpt function in phosphorelays. From the perspective of Hpts, one function in phosphorelays is to react differentially with multiple receiver domains14 (which we set out to probe with this work) while maintaining specificity to protect against cross reactivity with RRs from other TCSs in the cell10 (which we do not address experimentally with this work). From the perspective of RRs, one function in phosphorelays may be to distinguish between phosphotransfer to His or to water. The slowest rate constant measured here for phosphotransfer from a CheY variant to imidazole was 1,500 times greater than the rate constant for transfer to water, but some CheY variants achieved even greater discrimination between nucleophiles. When considering ability of non-conserved active site residues to influence both water and imidazole-mediated dephosphorylation, the CheYsmall variant represents a reduced active site, which we used as a convenient reference point for purposes of interpretation. Conceivably, phosphorelay RRs that undergo rapid phosphotransfer to Hpts may represent cases in which reaction with imidazole is preferred over autodephosphorylation. CheY variants that mimic faster phosphorelay RRs - CheYMPI (contains same D+2, T+2 residues as phosphorelay RR Spo0F), CheYSLN1, CheYSKN7 – exhibit relative sensitivities to imidazole larger than that of CheYsmall. Consistent with predicted behavior, these variants were better at differentiating between water and imidazole. For CheY variants that did not mimic known phosphorelay RRs, relative sensitivities to imidazole were smaller. CheYSSK1 had a relative sensitivity to imidazole that was lower than that of CheYsmall, consistent with the observation that SSK1 does not transfer phosphoryl groups to YPD1. Notably, CheYwt had the lowest relative sensitivity to imidazole.
Practical applications of imidazole
In addition to the use of imidazole as an experimental tool in the study of TCSs, there are several potential practical applications. One use of imidazole could be ‘partial phosphorelays’ to generate [32P]MPI or [32P]RR-P. Radiolabeled MPI is not commercially available and, currently, making [32P]MPI requires synthesis of [32P]PAM first. Using imidazole and RRs could simplify the protocol for the generation of radiolabeled MPI. Currently, radiolabeling RRs typically utilizes a partner HK and commercially available [γ-32P]ATP as the phosphodonor. However, HKs are often transmembrane proteins, and isolation of catalytically active soluble fragments of HKs can be difficult and time consuming. Using an artificial phosphorelay to radiolabel RRs could eliminate the need for isolation and use of the partner HK. Finally, imidazole could be used to rapidly dephosphorylate RRs in vitro. As a closing note, given the data reported here, it is important to eliminate imidazole from His-tagged TCS proteins eluted off of a Ni-column for use in phosphorylation studies.
Acknowledgments
Funding
This work was supported by the National Institutes of Health Grant RO1 GM050860 to RBB and by the National Institutes of Health Training Grant T32 GM008570 to SCP. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or National Institutes of Health.
We thank Stephanie Thomas for providing purified CheYlarge and CheYMPI, Rachel Creager-Allen for providing PhoB F20D, and Robert Immormino for PhoR. We also thank Stephanie Thomas, Rachel Creager-Allen, and Robert Immormino for helpful discussions regarding this work.
ABBREVIATIONS
- HK
histidine kinase
- RR
response regulator and/or receiver domain
- Hpt
histidine-containing phosphotransfer domain
- -P
phosphorylated molecule
- MPI
monophosphoimidazole
- PAM
phosphoramidate
- Ni-NTA
nickel-nitrilotriacetic acid
- CheYsmall
CheY N59A E89A
- CheYMPI
CheY F14Q N59K E89Y
- CheYlarge
CheY F14E N59M E89R
- CheYSLN1
CheY N59Q E89F
- CheYSSK1
CheY N59Q E89S
- CheYSKN7
CheY N59V A88G E89N
- D+2
two positions C-terminal to the site of phosphorylation Asp
- T+1
one position C-terminal to the conserved Thr/Ser
- T+2
two positions C-terminal to the conserved Thr/Ser
- Dhp
dimerization and histidine phosphotransfer domain
References
- 1.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 2.Podgornaia AI, Laub MT. Determinants of specificity in two-component signal transduction. Curr Opin Microbiol. 2013;16:156–162. doi: 10.1016/j.mib.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Qureshi NK, Yin S, Boyle-Vavra S. The role of the Staphylococcal VraTSR regulatory system on vancomycin resistance and vanA operon expression in vancomycin-resistant Staphylococcus aureus. PLoS One. 2014;9:e85873. doi: 10.1371/journal.pone.0085873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gebhard S, Fang C, Shaaly A, Leslie DJ, Weimar MR, Kalamorz F, Carne A, Cook GM. Identification and characterization of a bacitracin resistance network in Enterococcus faecalis. Antimicrob Agents Chemother. 2014;58:1425–1433. doi: 10.1128/AAC.02111-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung JK, Awad MM, McGowan S, Rood JI. Functional analysis of the VirSR phosphorelay from Clostridium perfringens. PLoS One. 2009;4:e5849. doi: 10.1371/journal.pone.0005849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Gouw D, Hermans PW, Bootsma HJ, Zomer A, Heuvelman K, Diavatopoulos DA, Mooi FR. Differentially expressed genes in Bordetella pertussis strains belonging to a lineage which recently spread globally. PLoS One. 2014;9:e84523. doi: 10.1371/journal.pone.0084523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fassler JS, West AH. Histidine phosphotransfer proteins in fungal two-component signal transduction pathways, Eukaryot. Cell. 2013;12:1052–1060. doi: 10.1128/EC.00083-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Appleby JL, Parkinson JS, Bourret RB. Signal transduction via the multi-step phosphorelay: not necessarily a road less traveled. Cell. 1996;86:845–848. doi: 10.1016/s0092-8674(00)80158-0. [DOI] [PubMed] [Google Scholar]
- 9.Freeman JA, Bassler BL. Sequence and function of LuxU: a two-component phosphorelay protein that regulates quorum sensing in Vibrio harveyi. J Bacteriol. 1999;181:899–906. doi: 10.1128/jb.181.3.899-906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perraud AL, Kimmel B, Weiss V, Gross R. Specificity of the BvgAS and EvgAS phosphorelay is mediated by the C-terminal HPt domains of the sensor proteins. Mol Microbiol. 1998;27:875–887. doi: 10.1046/j.1365-2958.1998.00716.x. [DOI] [PubMed] [Google Scholar]
- 11.Ansaldi M, Jourlin-Castelli C, Lepelletier M, Theraulaz L, Mejean V. Rapid dephosphorylation of the TorR response regulator by the TorS unorthodox sensor in Escherichia coli. J Bacteriol. 2001;183:2691–2695. doi: 10.1128/JB.183.8.2691-2695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Georgellis D, Kwon O, De Wulf P, Lin EC. Signal decay through a reverse phosphorelay in the Arc two-component signal transduction system. J Biol Chem. 1998;273:32864–32869. doi: 10.1074/jbc.273.49.32864. [DOI] [PubMed] [Google Scholar]
- 13.Georgellis D, Kwon O, De Wulf P, Lin EC. Signal decay through a reverse phosphorelay in the Arc two-component signal transduction system. J Biol Chem. 1998;273:32864–32869. doi: 10.1074/jbc.273.49.32864. [DOI] [PubMed] [Google Scholar]
- 14.Janiak-Spens F, Cook PF, West AH. Kinetic analysis of YPD1-dependent phosphotransfer reactions in the yeast osmoregulatory phosphorelay system. Biochemistry. 2005;44:377–386. doi: 10.1021/bi048433s. [DOI] [PubMed] [Google Scholar]
- 15.Perraud AL, Weiss V, Gross R. Signalling pathways in two-component phosphorelay systems. Trends Microbiol. 1999;7:115–120. doi: 10.1016/s0966-842x(99)01458-4. [DOI] [PubMed] [Google Scholar]
- 16.Janiak-Spens F, Sparling DP, West AH. Novel role for an HPt domain in stabilizing the phosphorylated state of a response regulator domain. J Bacteriol. 2000;182:6673–6678. doi: 10.1128/jb.182.23.6673-6678.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sourjik V, Schmitt R. Phosphotransfer between CheA, CheY1, and CheY2 in the chemotaxis signal transduction chain of Rhizobium meliloti. Biochemistry. 1998;37:2327–2335. doi: 10.1021/bi972330a. [DOI] [PubMed] [Google Scholar]
- 18.Lukat GS, McCleary WR, Stock AM, Stock JB. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc Natl Acad Sci U S A. 1992;89:718–722. doi: 10.1073/pnas.89.2.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayover TL, Halkides CJ, Stewart RC. Kinetic characterization of CheY phosphorylation reactions: comparison of P-CheA and small-molecule phosphodonors. Biochemistry. 1999;38:2259–2271. doi: 10.1021/bi981707p. [DOI] [PubMed] [Google Scholar]
- 20.Barbieri CM, Mack TR, Robinson VL, Miller MT, Stock AM. Regulation of response regulator autophosphorylation through interdomain contacts. J Biol Chem. 2010;285:32325–32335. doi: 10.1074/jbc.M110.157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuster M, Silversmith RE, Bourret RB. Conformational coupling in the chemotaxis response regulator CheY. Proc Natl Acad Sci U S A. 2001;98:6003–6008. doi: 10.1073/pnas.101571298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas SA, Brewster JA, Bourret RB. Two variable active site residues modulate response regulator phosphoryl group stability. Mol Microbiol. 2008;69:453–465. doi: 10.1111/j.1365-2958.2008.06296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas SA, Immormino RM, Bourret RB, Silversmith RE. Nonconserved active site residues modulate CheY autophosphorylation kinetics and phosphodonor preference. Biochemistry. 2013;52:2262–2273. doi: 10.1021/bi301654m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Creager-Allen RL, Silversmith RE, Bourret RB. A link between dimerization and autophosphorylation of the response regulator PhoB. J Biol Chem. 2013;288:21755–21769. doi: 10.1074/jbc.M113.471763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourret RB, Thomas SA, Page SC, Creager-Allen RL, Moore AM, Silversmith RE. Measurement of response regulator autodephosphorylation rates spanning six orders of magnitude. Methods Enzymol. 2010;471:89–114. doi: 10.1016/S0076-6879(10)71006-5. [DOI] [PubMed] [Google Scholar]
- 26.Mack TR, Gao R, Stock AM. Probing the roles of the two different dimers mediated by the receiver domain of the response regulator PhoB. J Mol Biol. 2009;389:349–364. doi: 10.1016/j.jmb.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao R, Stock AM. Probing kinase and phosphatase activities of two-component systems in vivo with concentration-dependent phosphorylation profiling. Proc Natl Acad Sci U S A. 2013;110:672–677. doi: 10.1073/pnas.1214587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silversmith RE, Appleby JL, Bourret RB. Catalytic mechanism of phosphorylation and dephosphorylation of CheY: kinetic characterization of imidazole phosphates as phosphodonors and the role of acid catalysis. Biochemistry. 1997;36:14965–14974. doi: 10.1021/bi9715573. [DOI] [PubMed] [Google Scholar]
- 29.Klein AH, Shulla A, Reimann SA, Keating DH, Wolfe AJ. The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J Bacteriol. 2007;189:5574–5581. doi: 10.1128/JB.00564-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bochner BR, Maron DM, Ames BN. Detection of phosphate esters on chromatograms: an improved reagent. Anal Biochem. 1981;117:81–83. doi: 10.1016/0003-2697(81)90695-3. [DOI] [PubMed] [Google Scholar]
- 31.Rathlev T, Rosenberg T. Non-enzymic formation and rupture of phosphorus to nitrogen linkages in phosphoramido derivatives. Arch Biochem Biophys. 1956;65:319–339. doi: 10.1016/0003-9861(56)90197-7. [DOI] [PubMed] [Google Scholar]
- 32.Pazy Y, Wollish AC, Thomas SA, Miller PJ, Collins EJ, Bourret RB, Silversmith RE. Matching biochemical reaction kinetics to the timescales of life: structural determinants that influence the autodephosphorylation rate of response regulator proteins. J Mol Biol. 2009;392:1205–1220. doi: 10.1016/j.jmb.2009.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukat GS, Stock AM, Stock JB. Divalent metal ion binding to the CheY protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry. 1990;29:5436–5442. doi: 10.1021/bi00475a004. [DOI] [PubMed] [Google Scholar]
- 34.Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE. Crystal structure of activated CheY. Comparison with other activated receiver domains. J Biol Chem. 2001;276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 35.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joosten RP, Long F, Murshudov GN, Perrakis A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 2014;1:213–220. doi: 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheridan RC, McCullough JF, Wakefield ZT, Allcock HR, Walsh EJ. Phosphoramidic Acid and its Salts. In: Cotton FA, editor. Inorganic Syntheses. John Wiley & Sons, Inc; 1972. pp. 23–26. [Google Scholar]
- 41.Hess JF, Bourret RB, Oosawa K, Matsumura P, Simon MI. Protein phosphorylation and bacterial chemotaxis. Cold Spring Harb Symp Quant Biol. 1988;53(Pt 1):41–48. doi: 10.1101/sqb.1988.053.01.008. [DOI] [PubMed] [Google Scholar]
- 42.Zhao R, Collins EJ, Bourret RB, Silversmith RE. Structure and catalytic mechanism of the E. coli chemotaxis phosphatase CheZ. Nat Struct Biol. 2002;9:570–575. doi: 10.1038/nsb816. [DOI] [PubMed] [Google Scholar]
- 43.Pazy Y, Motaleb MA, Guarnieri MT, Charon NW, Zhao R, Silversmith RE. Identical phosphatase mechanisms achieved through distinct modes of binding phosphoprotein substrate. Proc Natl Acad Sci U S A. 2010;107:1924–1929. doi: 10.1073/pnas.0911185107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parashar V, Mirouze N, Dubnau DA, Neiditch MB. Structural basis of response regulator dephosphorylation by Rap phosphatases. PLoS Biol. 2011;9:e1000589. doi: 10.1371/journal.pbio.1000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silversmith RE. Auxiliary phosphatases in two-component signal transduction. Curr Opin Microbiol. 2010;13:177–183. doi: 10.1016/j.mib.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huynh TN, Stewart V. Negative control in two-component signal transduction by transmitter phosphatase activity. Mol Microbiol. 2011;82:275–286. doi: 10.1111/j.1365-2958.2011.07829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conley MP, Berg HC, Tawa P, Stewart RC, Ellefson DD, Wolfe AJ. pH dependence of CheA autophosphorylation in Escherichia coli. J Bacteriol. 1994;176:3870–3877. doi: 10.1128/jb.176.13.3870-3877.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jencks WP, Gilchris M. Reactions of nucleophilic reagents with phosphoramidate. J Am Chem Soc. 1965;87:3199–3209. [Google Scholar]
- 49.Disabato G, Jencks WP. Mechanism and catalysis of reactions of acyl phosphates. 1. Nucleophilic reactions. J Am Chem Soc. 1961;83:4393–4400. [Google Scholar]
- 50.Schmitt J, Hess H, Stunnenberg HG. Affinity purification of histidine-tagged proteins. Mol Biol Rep. 1993;18:223–230. doi: 10.1007/BF01674434. [DOI] [PubMed] [Google Scholar]
- 51.Zhao X, Copeland DM, Soares AS, West AH. Crystal structure of a complex between the phosphorelay protein YPD1 and the response regulator domain of SLN1 bound to a phosphoryl analog. J Mol Biol. 2008;375:1141–1151. doi: 10.1016/j.jmb.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varughese KI, Tsigelny I, Zhao H. The crystal structure of beryllofluoride Spo0F in complex with the phosphotransferase Spo0B represents a phosphotransfer pretransition state. J Bacteriol. 2006;188:4970–4977. doi: 10.1128/JB.00160-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bourret RB. Receiver domain structure and function in response regulator proteins. Curr Opin Microbiol. 2010;13:142–149. doi: 10.1016/j.mib.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dutta R, Inouye M. Reverse phosphotransfer from OmpR to EnvZ in a kinase−/phosphatase+ mutant of EnvZ (EnvZ.N347D), a bifunctional signal transducer of Escherichia coli. J Biol Chem. 1996;271:1424–1429. doi: 10.1074/jbc.271.3.1424. [DOI] [PubMed] [Google Scholar]
- 55.Shi L, Liu W, Hulett FM. Decay of activated Bacillus subtilis pho response regulator, PhoP approximately P, involves the PhoR approximately P intermediate. Biochemistry. 1999;38:10119–10125. doi: 10.1021/bi990658t. [DOI] [PubMed] [Google Scholar]
- 56.Pena-Sandoval GR, Kwon O, Georgellis D. Requirement of the receiver and phosphotransfer domains of ArcB for efficient dephosphorylation of phosphorylated ArcA in vivo. J Bacteriol. 2005;187:3267–3272. doi: 10.1128/JB.187.9.3267-3272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dutta R, Qin L, Inouye M. Histidine kinases: diversity of domain organization. Mol Microbiol. 1999;34:633–640. doi: 10.1046/j.1365-2958.1999.01646.x. [DOI] [PubMed] [Google Scholar]
- 58.Da Re SS, Deville-Bonne D, Tolstykh T, MVr, Stock JB. Kinetics of CheY phosphorylation by small molecule phosphodonors. FEBS Lett. 1999;457:323–326. doi: 10.1016/s0014-5793(99)01057-1. [DOI] [PubMed] [Google Scholar]
- 59.Capra EJ, Laub MT. Evolution of two-component signal transduction systems. Annu Rev Microbiol. 2012;66:325–347. doi: 10.1146/annurev-micro-092611-150039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quezada CM, Hamel DJ, Gradinaru C, Bilwes AM, Dahlquist FW, Crane BR, Simon MI. Structural and chemical requirements for histidine phosphorylation by the chemotaxis kinase CheA. J Biol Chem. 2005;280:30581–30585. doi: 10.1074/jbc.M505316200. [DOI] [PubMed] [Google Scholar]
- 61.Janiak-Spens F, West AH. Functional roles of conserved amino acid residues surrounding the phosphorylatable histidine of the yeast phosphorelay protein YPD1. Mol Microbiol. 2000;37:136–144. doi: 10.1046/j.1365-2958.2000.01973.x. [DOI] [PubMed] [Google Scholar]
- 62.Salazar ME, Laub MT. Temporal and evolutionary dynamics of twocomponent signaling pathways. Curr Opin Microbiol. 2015;24:7–14. doi: 10.1016/j.mib.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Varughese KI, Madhusudan Zhou XZ, Whiteley JM, Hoch JA. Formation of a novel four-helix bundle and molecular recognition sites by dimerization of a response regulator phosphotransferase. Mol Cell. 1998;2:485–493. doi: 10.1016/s1097-2765(00)80148-3. [DOI] [PubMed] [Google Scholar]