

Abstract
Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) is a unique paracaspase protein whose protease activity mediates oncogenic NF-κB signalling in activated B cell-like diffuse large B cell lymphomas (ABC-DLBCLs). ABC-DLBCLs are aggressive lymphomas with high resistance to current chemotherapies. Low survival rate among patients emphasizes the urgent need for alternative treatment options. The characterization of the MALT1 will be an essential tool for developing new target-directed drugs against MALT1 dependent disorders. As the first step in the atomic-level NMR studies of the system, here we report, the 15N/13C/1H backbone assignment of the apo form of the MALT1 paracaspase region together with the third immunoglobulin-like (Ig3) domain, 44 kDa, by high resolution NMR. In addition, the non-uniform sampling (NUS) based targeted acquisition procedure is evaluated as a mean of decreasing acquisition and analysis time for larger proteins.
Introduction
Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) has a central role in transcription factor NF-κB signalling [1, 2]. NF-κB controls the expression of numerous anti-apoptotic and proliferation-promoting genes and has a key role in B-cell activation. Constitutive MALT1 activity is one characteristic of specific types of B-cell lymphomas [3, 4], rendering MALT1 as a potential drug target for these malignancies.
MALT1 exerts its regulating function by two routes. Upon antigen stimulation MALT1 acts as a scaffold for a protein complex formed with CARMA1 and Bcl10, the CBM complex [5]. The complex mediates the further events that lead to the nuclear translocation and activation of NF-κB. In addition, MALT1 functions as a protease that acts on several proteins involved in the pathway leading to NF-κB activation [6, 7].
MALT1 is the first human paracaspase identified [2]. Differently from caspases, reported MALT1 substrates are all cleaved directly on the C-terminal side of an arginine residue [6, 7]. Another difference is that MALT1 is not cleaved upon activation [8]. Similarly to caspases, MALT1 is dependent on dimerization for catalytic activity [8]. In vivo this is accomplished by ubiquitination of a single lysine residue [9]. In biochemical assays it has been found that high concentration of a kosmotropic salt increases MALT1 catalytic activity [7], probably by promoting dimerization.
Full length MALT1 is a 93 kDa protein consisting of 823 amino acids (Uniprot Q9UDY8). The sequence folds into five domains: the N-terminal DEATH domain, two immunoglobulin-like domains (Ig1 and Ig2), the caspase-like domain (Casp) and a third immunoglobulin-like domain (Ig3) followed by an unstructured C-terminal tail. The structures of individual domains and combinations thereof have been solved by X-ray crystallography [8, 10–12]. In recent studies MALT1 truncated to the caspase-like and Ig3 domains, MALT1Casp-Ig3 was used in characterising the structural determinants of activation [8]. The studies revealed that both apo MALT1Casp-Ig3 (substrate/ligand-free) and MALT1Casp-Ig3 in complex with the irreversible substrate mimetic ligand (z-VRPR-mono fluoro-ketone) forms dimers. However, the trigger of proteolytic activity is substrate-induced. Activation involves stabilisation of loop regions in the caspase domain by the ligand, resulting in the alignment of the catalytic machinery and folding of the substrate binding pocket. In absence of ligand, the loops are disordered and the substrate binding pocket is collapsed.
The flexibility of the major loops in the caspase domain and their relationship with the Ig3 domain seems central to MALT1 activation. Solution NMR is the method of choice for studying such flexible parts in proteins. As the first step towards NMR spectroscopy elucidation of the structure and dynamics of MALT1Casp-Ig3, in solution,. we report, the 15N/13C/1H backbone assignment of MALT1Casp-Ig3 in its ligand-free state by high resolution NMR. In addition, we for the first time show that the targeted acquisition (TA) [13–15] procedure, which previously has been used only for intrinsically disordered proteins and small globular proteins, perform just as well also for a relatively large 44 kDa protein.
Materials and Methods
MALT1Casp-Ig3(338–719) expression and purification
The human MALT1Casp-Ig3(338–719) construct, consisting of the catalytic domain and the Ig3 domain, was chosen for NMR studies. DNA encoding this sequence and a C-terminal His6 tag was inserted between the NdeI and XhoI sites in the expression vector pPET21b (Novagen). Expression of the protein was performed in Escherichia coli strain BL21 Star (DE3) (Invitrogen) at 37°C and with 50 μg/ml carbenicillin as the selective antibiotic. For isotope labelling, 15N/13C/2H-labeled minimal medium based on M9 was prepared in D2O. At an OD600 of approximately 1, induction of the protein expression was started by adding IPTG to 0.4 mM. The cultures were incubated at 18°C for an additional 6 hours. The cells were harvested by centrifugation at 6000g for 20 min and subsequently re-suspended and lysed by high pressure (1,7 kbar) in a Cell Disruptor (Constant Systems). The lysate cleared by centrifugation at 48000g.
The protein was subsequently purified by 3 chromatographic steps. First, an Immobilized metal ion affinity chromatography (IMAC) step on a Ni2+ Sepharose 6 Fast Flow column (GE Healthcare) was performed. Second, Ion-exchange chromatography (IEX) on a Q-Sepharose HP column (GE Healthcare) was carried out. Third, a size exclusion chromatography (SEC) step using a HiLoad 16/600 Superdex 200 prep grade column (GE Healthcare) was performed. The buffer was subsequently exchanged to a buffer suitable for NMR experiments on Bio-Spin P-6 gel columns (BioRad). Final yield from a culture of 8 litres was approximately 10 mg of purified protein after concentration to 0.5 mM.
Backbone assignment of MALT1Casp-Ig3(338–719)—the conventional approach
The NMR samples contained approximately 0.5 mM 15N/13C/2H-labeled protein in 10 mM Tris pH 7.5, 50 mM NaCl, 1 mM TCEP-d16, 0.002% NaN3, 10 μM DSS-d6 and 10% D2O. NMR experiments were acquired on a Bruker Avance III spectrometer operating at a frequency of 700 MHz for 1H using a 5 mm cryo-enhanced inverse resonance QCI HFCN probe at 298 K. To enable backbone assignment, transverse relaxation optimized spectroscopy (TROSY)[16–18] versions of HNCO[19], HNCA[19], HN(CO)CA[20], HNCACB[20] and HN(CO)CACB[20], using gradient echo-anti echo TROSY and 2H-decoupling, were acquired together with a 2D 1H-15N TROSY. The total acquisition time sums up to 484 hours (about 3 weeks) and relevant parameters are displayed in Table 1. The backbone assignment was performed manually in CcpNmr Analysis 2.2.2[21].
Table 1. Parameters used for acquisition of 3D NMR spectra using the conventional approach (CA) and Targeted Acquisition (TA).
| Experiment | HNCO | HN(CA)CO | HNCA | HN(CO)CA | HNCACB | HN(CO)CACB | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Approach | CA | TA | CA | TA | CA | TA | CA | TA | CA | TA | CA | TA |
| Number of scans | 4 | 8 | - | 16 | 16 | 8 | 16 | 8 | 64 | 16 | 32 | 16 |
| Sparse, % | 100 | 30 | 100 | 19 | 100 | 19 | 100 | 19 | 100 | 19 | 100 | 19 |
| Maximum evolution time (ms) | ||||||||||||
| F3 (1H) | 91 | 92 | - | 92 | 91 | 92 | 91 | 92 | 91 | 92 | 91 | 92 |
| F1 (13C) | 9 | 25 | - | 25 | 9 | 12 | 9 | 12 | 7 | 12 | 7 | 12 |
| F2 (15N) | 11 | 22 | - | 22 | 11 | 22 | 11 | 22 | 9 | 22 | 11 | 22 |
| Measurement time (hours) | 22 | 13 | - | 17 | 46 | 9 | 46 | 10 | 224 | 44 | 146 | 44 |
The relaxation delay was set to 1 s in all experiments.
Backbone assignment of MALT1Casp-Ig3(338–719) using the Targeted Acquisition (TA) approach
A standard set of 3D TROSY-based triple resonance experiments with deuterium decoupling was acquired using iterative nonuniform sampling (NUS) [14],[22]. The TA procedure was used for automatical processing and analysis of spectra. TA allows a significant reduction in experiment and analysis time for sequential backbone assignment of proteins and has been described in detail previously[13–15]. Essentially, simultaneous co-processing with multi-dimensional decomposition (co-MDD) of all triple-resonance spectra[15] followed by the automated hyper-dimensional spectrum analysis generates clean peak lists with already prebuilt spin systems that can be efficiently used by automated assignment software AutoAssign [23]. The process is repeated (to accumulate more data in the experiments) and iterated until the assignment is sufficiently complete or converges. A 30% sampling schedule was used for the HNCO spectrum and a 19% sampling schedule was used for all other 3D spectra, yielding a total acquisition time of 136 hours (about 1 week). All spectra were acquired at 298 K on a Bruker Avance III HD 800 MHz spectrometer equipped with a 3mm CPTCI cryo-probe. Peak lists generated with the TA procedure were validated by manual analysis of the spectra in SPARKY[24]. Relevant parameters are displayed in Table 1.
Estimation of the secondary structure of MALT1Casp-Ig3(338–719)
To estimate the secondary structure in MALT1Casp-Ig3(338–719), random coil chemical shifts corrected for nearest-neighbour effects[25] were subtracted from 13C’, 13Cα and 13Cβ chemical shifts corrected for deuterium isotope shifts[26] and plotted against the amino acid sequence of MALT1Casp-Ig3(338–719). The chemical shift index (CSI) for each carbon type was calculated using a cut-off at ± 0.7 and averaged over all three carbon types to a “consensus” CSI. The secondary structure estimated from the chemical shifts was then compared to the secondary structure of the previously reported X-ray structure of apo MALT1Casp-Ig3[8] (3V55) and of our in house X-ray structure of apo MALT1Casp-Ig3 (data not published).
Results and Discussion
MALT1Casp-Ig3(338–719) assignment by NMR
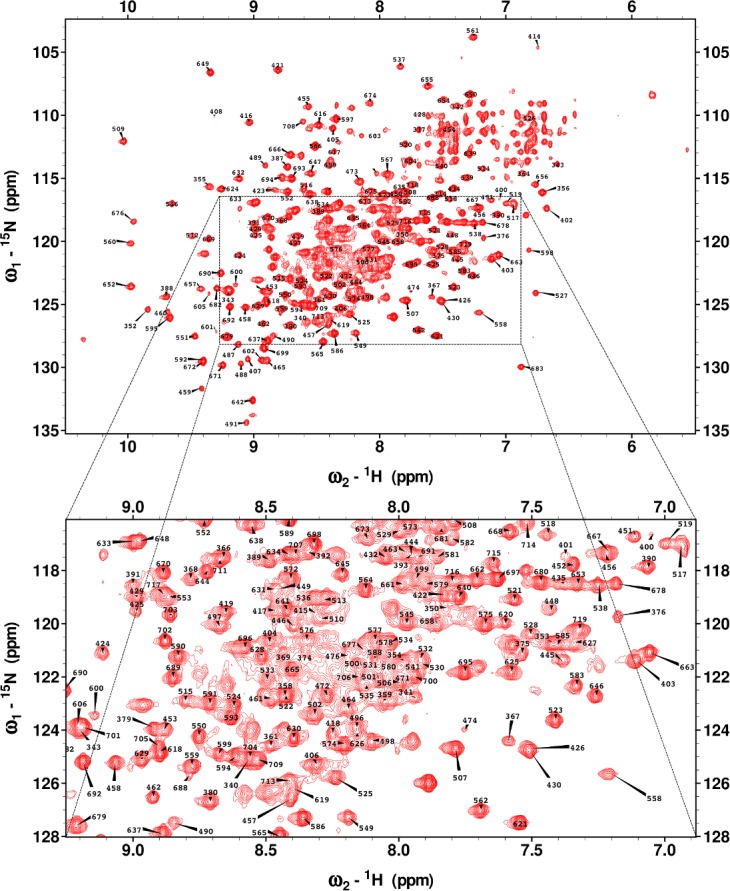
Combining the conventional and TA approaches allowed assignment of 74% of non-proline backbone 15N and 1HN (Fig 1), 80% of 13Cα, 75% of 13Cβ and 76% of 13CO. The 1H, 13C and 15N backbone chemical shifts were referred to DSS-d6, 13C and 15N chemical shifts were referred indirectly, and have been deposited in the Biological Magnetic Resonance Data Bank[27] (http://www.bmrb.wisc.edu/) with the BMRB accession code 25674.
Fig 1. 1H-15N TROSY spectrum of MALT1Casp-Ig3(338–719) with the assigned amino acid residue number annotated.
Evaluation of the TA approach
Apart from obtaining the backbone assignment of MALT1, the secondary goal of this project was to evaluate the performance of the TA approach for a large triple labelled protein. Fig 2 shows the build-up of the peaks with the measurement time, i.e. with increasing number of data points in individual backbone experiments. The peak counts do not level-off at the end of the TA procedure, suggesting that further measurements might improve the result of the assignment. However, the sample stability proved to be the limiting factor. Since the assignment was already obtained using the conventional approach, we decided that the TA data set accumulated during one week was sufficient for the purpose of evaluation of the TA procedure. To make a fair comparison, it should be noted that the TA procedure in this work included modifications that could have benefited the conventional approach as well. These are running the experiment at higher magnetic field (800 MHz), which improves sensitivity and resolution, and use of an additional HN(CA)CO experiment that increases the success and reliability of assignment, albeit in expense of extending the total measurement time.
Fig 2. Peak appearance progress during the course of the TA procedure for the MALT1 sample.

The horizontal axis shows the total measurement time excluding the HNCO experiment, which was recorded prior to the TA. The spectral processing and analysis was done automatically during the course of the data acquisition.
To evaluate the automated peak picking algorithm used in the TA procedure, all of the spectra were manually carefully analysed in SPARKY[24]. In Table 2 it is clearly seen that the TA approach was successful. Specifically, almost as many peaks were picked when using the TA approach as when using the conventional approach, using only one third of the acquisition time and only one NMR sample instead of two samples. Furthermore, during this measurement time, it was possible to include an additional experiment, the HN(CA)CO, which was essential to disambiguate assignments for several residues and correct a few errors in the assignment produced by the traditional approach.
Table 2. Comparison of automated and manual peak picking of the spectra in the TA approach.
| SPECTRA | HNCO | HN(CA)CO | HNCA | HN(CO)CA | HNCACB | HN(CO)CACB | |
|---|---|---|---|---|---|---|---|
| MANUAL | Peaksa | 358 | 280 | 492 | 268 | 726 | 291 |
| AUTOMATIC | Peaksb | 387 | 224 | 459 | 232 | 619 | 290 |
| False peaksc | 14 | 2 | 2 | 3 | 1 | 3 | |
| Peaks picked twiced | 15 | 4 | - | - | - | - | |
| Correct not visiblee | - | - | 2 | 1 | - | 2 | |
| New peaksf | - | 3 | 3 | - | — | 5 | |
| Missingg | - | 65 | 40 | 40 | 108 | 11 |
a Peaks picked manually from data acquired by targeted acquisition.
b Peaks picked automatically.
c Peaks picked automatically which were not visible in TA or in the conventional spectra.
d Peaks that were picked twice by the automatic method.
e Peaks picked automatically that were correct (visible in conventional spectra) while not visible in TA spectra.
f Automatically picked peaks that could not be verified.
g Additional peaks that were found in TA spectra upon manual inspection.
It should be noted that peak picking in the HNCO spectrum is semi-automated and its correctness, which is insured by manual verification of the peaks in this spectrum, is crucial for the automated peak detection in all other spectra. Thus, the statistics on the quality of the peak lists refer to all spectra except for the HNCO. The number of false peaks in these spectra is lower than 0.8% in total. For an additional 0.6% of the peaks the obtained data does not allow discrimination between true or false. In addition to the automated peak list, careful manual peak picking allows finding approximately 5% to 20% more peaks and in case of the weakest spectrum, HN(CA)CO, almost 30% more peaks. This was expected, since the TA peak counts did not level-off in our data set (Fig 2).
Comparison of the backbone assignment obtained using the conventional and TA approaches is presented in Table 3. Overall the presented results clearly show that TA can be reliably used for in backbone assignment of large proteins. We clearly show that the method can be used to speed up data acquisition and analysis for backbone assignment of proteins with size of over 40 kDa.
Table 3. Summary of the total chemical shifts assigned using the conventional approach and picked (automatically and/or manually) using the TA approach.
| H | N | CO | CA | CB | |
|---|---|---|---|---|---|
| Total chemical shifts assigned | 272 | 272 | 289 | 304 | 271 |
| Chemical shifts assigned by conventional NMR | 264 | 264 | 255 | 283 | 256 |
| Chemical shifts confirmed by TA | 256 | 256 | 275 | 288 | 239 |
The secondary structure of MALT1Casp-Ig3(338–719)
The secondary chemical shifts of 13C’, 13Cα and 13Cβ were calculated and is shown in Fig 3 together with the “consensus” chemical shift index CSI. The CSI derived secondary structure was compared to the secondary structure of the apo MALT1Casp-Ig3 crystal structures (unpublished in-house structure and published, PDB ID: 3V55), to validate the assignment. As can been seen in Fig 4 the secondary structure estimated from chemical shifts is close to identical to that of the X-ray structures. However some small differences are seen. Most of these differences, i.e. β1, gap in αC, gap in β3AB, gap between β2* and β3* can be explained by gaps in the assignment that creates gaps in the secondary structure estimation. These differences do not imply any real difference between the X-ray structure and the NMR assignment, merely that we do not have enough information to estimate the secondary structure from chemical shifts in these parts of the protein. Further β5* was not identified as a β-strand based on the strict criteria used for the “consensus” CSI. However, if the threshold is lowered slightly also this secondary structure element would be identified. All secondary structure elements except β1 and β5Ig3 (denoted β5* in Fig 4) could be assigned based on the data, i.e. αB, β2, αC, β3, β3A, β3B, αD, β4, loop 2 (L2), β5, loop 3 (L3), αE, αF, β6, α1Ig3, β1Ig3, β2Ig3, β3Ig3, β4Ig3, β6Ig3, β7Ig3. The different oligomerisation states of apo MALT1Casp-Ig3 in solution (monomer) and crystals (dimer) are not reflected by great differences in secondary structure, suggesting that the secondary structure elements necessary for oligomerisation are pre-formed in solution.
Fig 3. Estimate of the secondary structure in MALT1Casp-Ig3(338–719).
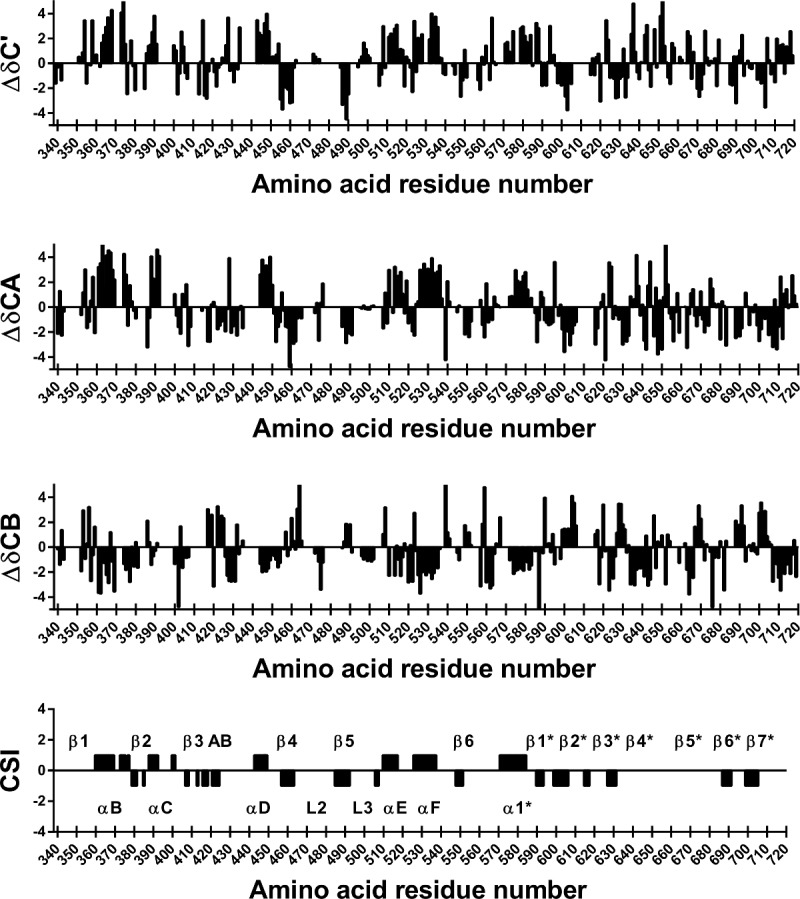
Secondary chemical shifts (Δδ) were calculated by subtracting random coil chemical shifts corrected for nearest-neighbour effects from 13C’, 13Cα and 13Cβ chemical shifts corrected for deuterium isotope shifts. Consecutive values above 0.7 indicates alpha helix, while consecutive values below -0.7 indicates beta strand for Δδ13C’ and Δδ13Cα. The opposite is true for Δδ13Cβ. The CSI for the three nuclei were averaged and reported as a “consensus” CSI. β3, β3A and β3B are denoted β3 AB in the Fig. The star (*) indicates that the secondary structure is part of the Ig3 domain.
Fig 4. Estimated secondary structure from NMR experiments (in black) compared to secondary structure from the in-house X-ray structure (in dark grey) and from the published X-ray structure of apo MALT1Casp-Ig3, PDB ID: 3V55 (in light grey).

Alpha helices are indicated with a greater symbol size than the beta-sheets. β3, β3A and β3B are denoted β3 AB in the Fig. The star (*) indicates that the secondary structure is part of the Ig3 domain.
Conclusions
In this work we present the NMR backbone assignment of apo MALT1Casp-Ig3 in solution (monomer) and show that the secondary structure deduced from the chemical shifts resemble the secondary structure observed in the apo X-ray structures (dimer). In addition, we tested the performance of the TA assignment procedure for a large protein system and demonstrated for the first time clear benefit of the approach in reducing measurement and analysis time.
The nature of MALT1 activation and regulation suggests a highly dynamic system in solution. Key to understanding such system is to acquire knowledge on the conformational ensembles and their equilibrium to help elucidate the structure recognised by substrate. For ligand free MALT1Casp-Ig3 this includes retrieving structure-dynamics information on the major flexible loops in the caspase domain, and their structural relationship with the C-terminal Ig3 domain. This necessitates solution-based techniques as a complement to X-ray crystallography. The 15N/13C/1H backbone assignment of MALT1Casp-Ig3 presented in this report forms a spring board to such future studies aimed at clarifying the role of MALT1 structure-dynamics in substrate recognition and processing.
Acknowledgments
We thank Esmeralda Woestenenk for help with protein production.
Data Availability
The 1H, 13C and 15N backbone chemical shifts have been deposited in the Biological Magnetic Resonance Data Bank (http://www.bmrb.wisc.edu/) with the BMRB accession code 25674.
Funding Statement
M.N would like to thank the National Centre of Science for support with Sonata Bis 2 Grant No. DEC-2012/07/E/ST4/01386. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Medivir AB provided support in the form of salaries for authors SU, VB, GS, JL and TA, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section.
References
- 1.Lenz G. Insights into the Molecular Pathogenesis of Activated B-Cell-like Diffuse Large B-Cell Lymphoma and Its Therapeutic Implications. Cancers. 2015;7(2):811–22. 10.3390/cancers7020812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uren AG, O'Rourke K, Aravind L, Pisabarro MT, Seshagiri S, Koonin EV, et al. Identification of Paracaspases and Metacaspases: Two Ancient Families of Caspase-like Proteins, One of which Plays a Key Role in MALT Lymphoma. Molecular Cell. 2000;6(4):961–7. [DOI] [PubMed] [Google Scholar]
- 3.Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M, et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(47):19946–51. 10.1073/pnas.0907511106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunleavy K, Wilson WH. Appropriate management of molecular subtypes of diffuse large B-cell lymphoma. Oncology (Williston Park, NY). 2014;28(4):326–34. [PMC free article] [PubMed] [Google Scholar]
- 5.Che T, You Y, Wang D, Tanner MJ, Dixit VM, Lin X. MALT1/paracaspase is a signaling component downstream of CARMA1 and mediates T cell receptor-induced NF-κB activation. Journal of Biological Chemistry. 2004;279(16):15870–6. [DOI] [PubMed] [Google Scholar]
- 6.Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D, et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nature immunology. 2008;9(3):272–81. 10.1038/ni1568 [DOI] [PubMed] [Google Scholar]
- 7.Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, et al. T cell antigen receptor stimulation induces MALT1 paracaspase–mediated cleavage of the NF-κB inhibitor A20. Nature immunology. 2008;9(3):263–71. 10.1038/ni1561 [DOI] [PubMed] [Google Scholar]
- 8.Wiesmann C, Leder L, Blank J, Bernardi A, Melkko S, Decock A, et al. Structural Determinants of MALT1 Protease Activity. Journal of Molecular Biology. 2012;419(1–2):4–21. 10.1016/j.jmb.2012.02.018 [DOI] [PubMed] [Google Scholar]
- 9.Pelzer C, Cabalzar K, Wolf A, Gonzalez M, Lenz G, Thome M. The protease activity of the paracaspase MALT1 is controlled by monoubiquitination. Nature immunology. 2013;14(4):337–45. 10.1038/ni.2540 [DOI] [PubMed] [Google Scholar]
- 10.Eitelhuber Andrea C, Vosyka O, Nagel D, Bognar M, Lenze D, Lammens K, et al. Activity-Based Probes for Detection of Active MALT1 Paracaspase in Immune Cells and Lymphomas. Chemistry & Biology. 2015;22(1):129–38. [DOI] [PubMed] [Google Scholar]
- 11.Schlauderer F, Lammens K, Nagel D, Vincendeau M, Eitelhuber AC, Verhelst SH, et al. Structural analysis of phenothiazine derivatives as allosteric inhibitors of the MALT1 paracaspase. Angewandte Chemie International Edition. 2013;52(39):10384–7. [DOI] [PubMed] [Google Scholar]
- 12.Jong WY, Jeffrey PD, Ha JY, Yang X, Shi Y. Crystal structure of the mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) paracaspase region. Proceedings of the National Academy of Sciences. 2011;108(52):21004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isaksson L, Mayzel M, Saline M, Pedersen A, Rosenlöw J, Brutscher B, et al. Highly efficient NMR assignment of intrinsically disordered proteins: application to B-and T cell receptor domains. 2013. 10.1371/journal.pone.0062947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaravine VA, Orekhov VY. Targeted acquisition for real-time NMR spectroscopy. Journal of the American Chemical Society. 2006;128(41):13421–6. [DOI] [PubMed] [Google Scholar]
- 15.Jaravine VA, Zhuravleva AV, Permi P, Ibraghimov I, Orekhov VY. Hyperdimensional NMR spectroscopy with nonlinear sampling. Journal of the American Chemical Society. 2008;130(12):3927–36. 10.1021/ja077282o [DOI] [PubMed] [Google Scholar]
- 16.Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proceedings of the National Academy of Sciences. 1997;94(23):12366–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulte-Herbrüggen T, Sørensen OW. Clean TROSY: compensation for relaxation-induced artifacts. Journal of Magnetic Resonance. 2000;144(1):123–8. [DOI] [PubMed] [Google Scholar]
- 18.Eletsky A, Kienhöfer A, Pervushin K. TROSY NMR with partially deuterated proteins. Journal of biomolecular NMR. 2001;20(2):177–80. [DOI] [PubMed] [Google Scholar]
- 19.Salzmann M, Pervushin K, Wider G, Senn H, Wüthrich K. TROSY in triple-resonance experiments: New perspectives for sequential NMR assignment of large proteins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(23):13585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salzmann M, Wider G, Pervushin K, Senn H, Wüthrich K. TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins. Journal of the American Chemical Society. 1999;121(4):844–8. [Google Scholar]
- 21.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, et al. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins: Structure, Function, and Bioinformatics. 2005;59(4):687–96. [DOI] [PubMed] [Google Scholar]
- 22.Orekhov VY, Jaravine VA. Analysis of non-uniformly sampled spectra with multi-dimensional decomposition. Progress in nuclear magnetic resonance spectroscopy. 2011;59(3):271–92. 10.1016/j.pnmrs.2011.02.002 [DOI] [PubMed] [Google Scholar]
- 23.Moseley HN, Sahota G, Montelione GT. Assignment validation software suite for the evaluation and presentation of protein resonance assignment data. Journal of biomolecular NMR. 2004;28(4):341–55. [DOI] [PubMed] [Google Scholar]
- 24.Goddard T, Kneller D. SPARKY 3. University of California, San Francisco: 2004;14:15. [Google Scholar]
- 25.Wishart DS. Interpreting protein chemical shift data. Progress in nuclear magnetic resonance spectroscopy. 2011;58(1):62–87. [DOI] [PubMed] [Google Scholar]
- 26.Niklasson M, Ahlner A, Andrésen C, Marsh JA, Lundström P. Fast and Accurate Resonance Assignment of Small-to-Large Proteins by Combining Automated and Manual Approaches. PLoS computational biology. 2015;11(1):e1004022 10.1371/journal.pcbi.1004022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, et al. BioMagResBank. Nucleic Acids Research. 2008;36(Database issue):D402–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The 1H, 13C and 15N backbone chemical shifts have been deposited in the Biological Magnetic Resonance Data Bank (http://www.bmrb.wisc.edu/) with the BMRB accession code 25674.