

SUMMARY
Mitral valve prolapse (MVP) is a common cardiac valve disease that affects nearly 1 in 40 individuals1–3. It can manifest as mitral regurgitation and is the leading indication for mitral valve surgery4,5. Despite a clear heritable component, the genetic etiology leading to non-syndromic MVP has remained elusive. Four affected individuals from a large multigenerational family segregating non-syndromic MVP underwent capture sequencing of the linked interval on chromosome 11. We report a missense mutation in the DCHS1 gene, the human homologue of the Drosophila cell polarity gene dachsous (ds) that segregates with MVP in the family. Morpholino knockdown of the zebrafish homolog dachsous1b resulted in a cardiac atrioventricular canal defect that could be rescued by wild-type human DCHS1, but not by DCHS1 mRNA with the familial mutation. Further genetic studies identified two additional families in which a second deleterious DCHS1 mutation segregates with MVP. Both DCHS1 mutations reduce protein stability as demonstrated in zebrafish, cultured cells, and, notably, in mitral valve interstitial cells (MVICs) obtained during mitral valve repair surgery of a proband. Dchs1+/− mice had prolapse of thickened mitral leaflets, which could be traced back to developmental errors in valve morphogenesis. DCHS1 deficiency in MVP patient MVICs as well as in Dchs1+/− mouse MVICs result in altered migration and cellular patterning, supporting these processes as etiological underpinnings for the disease. Understanding the role of DCHS1 in mitral valve development and MVP pathogenesis holds potential for therapeutic insights for this very common disease.
In a previous study, based on specific diagnostic criteria6–9, MMVP2 (Myxomatous Mitral Valve Prolapse-2) was mapped to a 4.3 cM region of chromosome 11p15.4 in a family of Western European descent segregating non-syndromic mitral valve prolapse as an autosomal dominant trait with age-dependent penetrance (Fig. 1A, C)6. We performed tiled capture and high-throughput sequence analysis of genomic DNA from four affected individuals (Fig. 1A), identifying 4891 single nucleotide variants (SNVs) and insertion/deletion polymorphisms in the targeted region (see Methods). After selecting rare protein-coding variants shared among all affected pedigree members, we identified three heterozygous protein-altering variants: two missense SNVs in DCHS1, a member of the cadherin superfamily10, resulting in p.P197L and p.R2513H (Fig. 1B), and a single missense SNV, in APBB1, the amyloid beta (A4) precursor protein-binding family B, member 1 gene resulting in p.R481H. Both DCHS1 mutations, p.P197L and p.R2513H, were rare in the population (the former observed three times in 4300 European-American individuals from the NHLBI Exome Sequencing Project and the latter never observed), and both were predicted to be protein damaging by PolyPhen-211, LRT12, and MutationTaster13. While the APBB1 variant was also rare in population-based data, no cardiac phenotype was observed in apbb1 morphant zebrafish, despite reduction of apbb1 mRNA (Extended Data Fig. 1A, B). Additionally, Apbb1 is not expressed in murine cardiac valves (Extended Data Fig. 2)14, and no cardiac defects have been reported in the Apbb1 knockout mouse15. This suggests that the APBB1 variant is unlikely to be contributing to MVP in this family.
Figure 1. Pedigrees, mutation, and phenotype.

Black symbols = MVP affecteds, gray = unknown, Arrows = probands, If no genotype is shown, the individuals were unavailable for study. (A). Pedigree linked to chromosome 11. #= Individuals under 15 years of age, *- individuals sequenced. Genotypes c. 7538G>A (R2513H) of DCHS1 mutation are shown. (B) DNA sequence of c.7538G>A (p.R2513H). (C) Two-dimensional echocardiographic long-axis view of Family 1 proband. Dashed line marks mitral annulus. (D,E) Family 2 and 3 pedigrees. Genotype c.6988C>T (p.R2330C) shown. (F) DNA sequence c. 6988C>T (p.R2330C) (G) Two-dimensional echocardiographic long-axis view of Family 2 proband.
The functional impact of the DCHS1 variants was evaluated in the zebrafish Danio rerio, as this model system lends itself to functional annotation of mutations implicated in human disease16–18. Zebrafish have two DCHS1 homologues, dachsous1a and dachsous1b. Dachsous1b is located in a region of Danio rerio chromosome 10 that is syntenic to the DCHS1 region of human chromosome 11. Knockdown of dachsous1a did not result in a cardiac phenotype despite reduction in mRNA levels (Extended Data Fig. 1A–B); however, knockdown of dachsous1b (dchs1b) led to significant changes in cardiac morphology (Fig. 2A; Extended Data Fig. 1A). Control zebrafish hearts undergo looping and develop an atrioventricular (AV) constriction by 48 hours post-fertilization (hpf), whereas dchs1b knockdown disrupts this process resulting in impaired formation of the atrioventricular constriction (Fig. 2A–B). While control embryos have unidirectional blood flow between the atrium and ventricle at 72 hpf (Supplemental Video 4), dchs1b knockdown causes regurgitation of blood from the ventricle into the atrium (Supplemental Video 5). An AV canal defect was defined as failure of cardiac looping combined with any AV regurgitation at 72 hpf. Using a high morpholino dose (1.5 ng) to establish the phenotype, the prevalence of AV canal defects was 76% (n=170), whereas spontaneous cardiac defects were rarely observed in controls (0.5%, n=205) (Fig. 2B). Whole-mount in situ hybridization of dchs1b confirmed predominant expression at the AV junction at 54 and 72hpf, corresponding to the temporal defects observed in the morphants (Extended Data Fig. 3A–C). We evaluated gene expression patterns in the developing AV ring, and observed that bmp4 expression is expanded into the ventricle at 48 hpf in dchs1b knockdown embryos while it is restricted to the AV ring in controls (Extended Data Fig. 4A–B). Additionally, has2 expression was not detectable at 48 hpf, and only faintly at 72 hpf in the dchs1b knockdown (Extended Data Fig. 4I–L). To test mutation pathogenicity in this model, rescue experiments were performed using both wild-type human DCHS1 and P197L/R2513H mutant mRNA, which were injected into dchs1b knockdown zebrafish with a lower dose of morpholino (0.75 ng) to minimize combined morpholino/mRNA toxicity. Human wild-type DCHS1 mRNA rescued the AV canal defect observed upon dchs1b knockdown, whereas injection of an equimolar amount of mutant DCHS1 mRNA failed to rescue (Figure 2C). Injection of the mutant DCHS1 mRNA alone did not cause AV canal defects, supporting a loss of function mechanism for the DCHS1 mutation.
Figure 2. Zebrafish Dchs1b Is Required For AV Canal Development.

(A) By 72 hpf, zebrafish hearts develop a constriction in the atrioventricular canal (AVC) that separates the atrium (a) from the ventricle (v). (B) Knockdown of dchs1b results in absence of the AV constriction (bracket). (C) ~75% of Dchs1b morphants exhibit AVC defects (*p= 1×10−62). DCHS1 human mRNA rescues the dchs1b morpholino AVC phenotype whereas human mutant DCHS1 mRNA (P197H/R2513H) fails to rescue (**p=.009). Total number of fish analyzed was 611 and statistical values were obtained using Fisher’s Exact test.
Having demonstrated segregation of a loss-of-function DCHS1 mutation with MVP in our large pedigree, we sought to determine if genetic variation in DCHS1 plays a role in MVP beyond the linked family. By evaluating a cohort of MVP patients, we identified 2 additional families in which MVP segregated with the novel DCHS1 protein variant p.R2330C (Fig. 1D–G). The proband of family 2 underwent surgical mitral valve repair for severe MVP and mitral regurgitation at age 21. Valve tissue was resected to repair the posterior leaflet and examination of the tissue showed classic myxomatous degeneration19,20 (Extended Data Fig. 5). Mitral valve interstitial cells were isolated from a portion of the posterior leaflet resected during surgery, providing a unique resource for the functional studies described below. The proband’s sister, evaluated at age 27 and also heterozygous for the p.R2330C mutation, demonstrated classical MVP with thickened leaflets and moderate regurgitation. The father and maternal grandmother were unaffected and do not carry the mutation. The mother (age 49) and maternal grandfather (age 76) are both affected with mild MVP and both carry p.R2330C. In family 3, the proband (Fig. 1E) had moderate to severe heart failure due to severe mitral regurgitation with posterior leaflet prolapse requiring surgery at age 72. The proband’s sister, age 69, is unaffected and negative for the p.R2330C mutation. The son (age 52) and the daughter (age 53) are both affected with MVP and both carry p.R2330C. The second son (age 55) also carries p.R2330C, however his MVP status is indeterminate due to due to mild LV inferior wall hypokinesis that tethers the leaflets down into the LV cavity21, masking leaflet prolapse motion toward the left atrium.
To evaluate the functional consequence of the DCHS1 mutations, we quantified protein levels in cells transfected with either wild-type or variant (p.P197L/p.R2513H) DCHS1 cDNA constructs. Expression of the DCHS1 mutant protein was ~60% less than wild-type with no significant change in mRNA levels, suggesting that the DCHS1 variants reduce protein stability (Fig. 3A–C). Cycloheximide treatment revealed that wild-type DCHS1 protein in transfected cells had a half-life of 5.8 hours while the mutant DCHS1 protein (p.P197L/p.R2513H) had a half-life of 1.6 hours (Fig. 3D, E). DCHS1 constructs harboring either p.P197L or p.R2513H were evaluated showing that p.R2513H markedly reduced protein levels, implicating this variant as pathogenic in the family (Fig. 1, Extended Data Fig. 6). A similar analysis of DCHS1 protein half-life was conducted using p.R2330C MVICs from the proband of family 2. Consistent with the data obtained from the p.R2513H transfectants, these studies showed significant reduction in protein half-life compared to control MVICs (t1/2 = 0.46 hours vs. 1.73 hours, Fig. 3D,E). Together, our studies show that p.R2513H and p.R2330C result in DCHS1 loss of function. In order to evaluate DCHS1 loss of function in a mammalian model, we analyzed Dchs1 deficient mice for phenotypic similarities to human MVP.
Figure 3. DCHS1 Mutations Result In Diminished Protein Levels.

(A–C) Western blot: (p.P197L/p.R2513H) mutant DCHS1 results in a 60% decrease in protein with no change in RNA expression. (D) Left Panel: DCHS1 wild-type (WT) or mutant (p.P197L/p.R2513H) transfectants treated with cycloheximide for specified times followed by Western analyses. Right Panel: Cycloheximide on Control (DCHS1 WT) or MVP patient (p.R2330C) MVICs. Tubulin=loading control (E) Graphical depiction of calculated protein half-lives. WT and mutant transfectants half-life = 5.8 hours versus 1.6, respectively (left graph). Control and mutant DCHS1 half-life is 1.73 hours versus 0.46 hours, respectively (right graph). Analyses performed in triplicate and repeated four times. Error bars = standard deviations; p-values calculated using two-tailed Students t-test.
Homozygous knockout of Dchs1 in mice results in neonatal lethality and multi-organ impairment22; however, the relevant genetic model for human MVP is the heterozygous Dchs1 mouse. Dchs1+/− mice exhibit mitral valve prolapse with pronounced involvement of the posterior leaflet, which is elongated and shifts the leaflet coaptation anteriorly (Fig. 4) as in the proband from Family 1 (Fig. 1C, Supplementary Videos 1–3, 6, 7)23. Micro-MRI analyses and 3D reconstructions of adult Dchs1+/− mice reveal prominent posterior leaflet thickening with quantitative increases in valve volume (Fig. 4 and Supplementary Video 8). All other echocardiographic measurements were unchanged (Extended Data Fig. 7). Histological and molecular characterization of Dchs1+/− mice confirmed leaflet thickening and showed myxomatous degeneration with increased proteoglycan accumulation in both mitral leaflets (Fig. 4). These results clearly show that Dchs1 heterozygosity results in mitral valve prolapse in mice.
Figure 4. Dchs1 Deficiency Causes MVP and Myxomatous Degeneration in the Adult Mouse.

Echocardiography (Echo), MRI, Histopathology, and 3D reconstructions performed on 9-month old male Dchs1+/+ and Dchs1+/− mouse hearts. Echo: posterior leaflet prolapse in Dchs1+/− (green arrow) (N=6/genotype). Immunohistochemistry (IHC): Dchs1+/− (N=5) anterior, posterior leaflets (AL, PL) exhibit myxomatous degeneration and expansion of proteoglycan expression compared to Dchs1+/+ (N=7), collagen (red), proteoglycans (green). MRI show posterior leaflet (PL) thickening in Dchs1+/− (arrow-inset) compared to control littermates. 3D reconstructions of MRI: Dchs1+/− mice exhibit thickened and elongated leaflets compared to Dchs1+/+. (Two-tailed Student t-test was used to calculate p-values; p=.01, N=4/genotype).
To determine if MVP has a developmental origin, we performed expression and functional analyses during embryonic and fetal timepoints. RNA in situ hybridization and immunohistochemistry showed expression of Dchs1 in endocardial and mesenchymal cells of atrioventricular valve leaflets at all timepoints examined (Extended Data Fig. 8). While no morphological defects were observed in the Dchs1+/− mice during early embryonic development (E11.5–E13.5), at later timepoints (E15.5–E17.5) Dchs1+/− mice displayed changes in mitral-valve shape (Fig. 5A–C), which were more severe in Dchs1−/− animals. Histology and three-dimensional reconstructions of anterior and posterior mitral leaflets at E17.5 of Dchs1+/+, Dchs1+/−, and Dchs1−/− mice showed comparable leaflet volumes. However, Dchs1+/− and Dchs1−/− animals exhibited statistically significant changes in valve length and width (Supplemental Video 9 and Fig. 5A–C). In most leaflet regions measured, Dchs1+/− animals displayed an intermediate phenotype, demonstrating a gene dosage effect. These shape changes implicate Dchs1 as critical for proper anatomical patterning of the valve, consistent with previous reports of dachsous function in the Drosophila wing24. Thus, in vivo lineage-tracing studies were performed on Dchs1+/+ and Dchs1+/− mice. Crossing the WT1-Cre/ROSA-eGFP25 line onto both Dchs1+/+ and Dchs1+/− backgrounds allowed visualization of patterning defects of epicardial-derived cells (EPDCs) during migration into the posterior leaflet. This EPDC population initially migrates into the posterior leaflet as a sheet of cells. However, in the Dchs1+/− mice this sheet-like appearance is disrupted and an increase in EPDCs infiltrating diffusely throughout the valve tissue is observed (Extended Data Fig. 9A–C, Supplementary Video 10), concomitant with altered cellular patterning and alignment (Extended Data Fig. 9D, E). In vitro studies of MVICs from Dchs1+/− and from the Family 2 MVP proband (p.R2330C) also show this increased migration phenotype (Extended Data Fig. 10A, B). Taken together, the mouse and human studies support a developmental etiology for MVP, and invoke a model for MVP in which cell migration and patterning defects mediated by DCHS1 contribute to disease pathogenesis.
Figure 5. Developmental Etiology for MVP.

(A,B) H&E and 3D reconstructions of E17.5 Dchs1+/+, Dchs1+/−, and Dchs1−/− mouse hearts showing thickening of anterior and posterior leaflets (AL, PL) in Dchs1−/− mice compared to Dchs1+/+. Dchs1+/− valves display an intermediate phenotype. (C) Quantification of valve dimensions showing Dchs1−/− (green bars) and Dchs1+/− (orange bars) anterior and posterior lengths were significantly reduced compared to Dchs1+/+ (blue bars) leaflets. Dchs1−/− and Dchs1+/− valves displayed increased thickness throughout the leaflets compared to Dchs1+/+. Scale Bars= 100μm. N=5/genotype and two-tailed Student’s t-test was used to calculate p-values; *p<.01
MVP is one of the most common cardiovascular diseases, affecting nearly 1 in 40 people world-wide1–3. Although its heritability and variable expression in large pedigrees has been known for decades, its genetic underpinnings have remained elusive23,26. We report the discovery of two loss-of-function mutations in DCHS1 that segregate with MVP in three families. Our mouse models exhibit classical MVP, a phenotype that was traced back to developmental errors during valve morphogenesis. These findings provide a model for understanding inherited non-syndromic MVP as a developmentally based disease that progresses over the lifespan of affected individuals, consistent with previous reports on the natural history of MVP27. A robust estimate of the total contribution of rare DCHS1 genetic variation to sporadic MVP has yet to be determined and will require sequencing of large cohorts of MVP patients. Nonetheless, discovery of this novel mechanistic pathway elucidated by intensively studying rare familial mutations will facilitate the identification of additional MVP genes and reveal pathogenic mechanisms that hold the potential for pre-surgical therapy for this very common cardiac disease.
METHODS
Study Participants
Family 1 was originally recruited through the Echocardiography Laboratory at Massachusetts General Hospital as part of a phenotype-driven genetic study of MVP. MVP was diagnosed by specific criteria (>2 mm atrial leaflet displacement in a parasternal long-axis view)6–9. The study was approved by the Institutional Review Board of Partners Healthcare, Boston, MA, and all participants provided written informed consent. Complete details of the linkage analysis on this large, multigenerational family have been previously published6. In brief, the family contains 41 individuals in five generations. Echocardiograms and DNA were obtained on 28 subjects, of whom 12 were diagnosed with MVP, three were classified as having nondiagnostic minimal leaflet displacement, and 13 were unaffected6. Three patients had non-diagnostic valve leaflet displacement and were considered unknown for the original linkage analysis. The proband had prominent MVP with thickened leaflets, severe MR, and heart failure ultimately requiring surgical valve repair (Fig. 1C, Supplemental Videos 1–3). Other affected members also showed diffuse leaflet thickening, prolapse, and MR of varying severity; one required surgical repair. No extracardiac manifestations of connective-tissue abnormalities or Marfan syndrome were present in any family members. Following a complete genome scan, parametric and non-parametric analyses confirmed linkage of this family to a 4.3 cM region of chromosome 11p15.4. Consistent with the model of sex- and age-dependent penetrance, several of the unaffected members who carried the MVP allele were less than 15 years old at the time of evaluation (Fig. 1A). Importantly, an analysis utilizing only affected individuals confirmed the linkage result.
DNA Sequencing and Variant Calling in Family 1
In order to identify the mutation, four affected individuals who shared the disease haplotype were chosen for sequencing (Fig. 1A). To reduce the likelihood of random haplotype sharing, we selected individuals with four distinct haplotypes on the non-MVP allele. A 2.1 Mb region of human chromosome 11 (5094774–7248926; NCBI36 coordinates) was targeted and screened for repetitive regions using the SureSelect system (Agilent). DNA extraction was performed using the AutoGenFlex STAR automated system (Autogen) and FlexiGen DNA purification reagents (Qiagen) according to the manufacturers’ instructions. Bait oligonucleotides were designed to the non-repetitive regions of the targeted linkage peak, resulting in 1.03 Mb of target sequence using the SureSelect in-solution long RNA baits (Agilent). Captured DNA was amplified and quantified using the Agilent High Sensitivity DNA Kit for the Agilent 2100 Bioanalyzer, and sequenced using Illumina sequencing chemistry (paired-end, 100 cycles) at the Venter Institute supported by the NHLBI Resequencing and Genotyping Program. One hundred bases were sequenced from each end of the captured DNA fragments. Image analysis and base calling were performed using Illumina’s GA Pipeline version 1.6.0. Sequence reads were mapped to the human genome (ncbi36) and variants identified using clc-ngs-cell-2.0.5-linux_64 (clc_ref_assemble_long -q -p fb ss 180 360 –I –r, and find_variations -c 8 -v -f 0.2). Variants were classified using VariantClassifier28. 4891 SNVs were identified in the four subjects, 1951 were shared by all four subjects. We classified all rare SNVs or those of unknown frequency based on conservation data, population genetic data, and predictive functional impact from public resources. We performed analyses of conservation of the variant locus using PhyloP29, PhastCONS30 and GERP31, and assessed the population frequency initially using the Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/) as an initial filter and the 1,000 Genomes Project32 as a secondary filter, cognizant of the limitations of the low-depth coverage of the 1,000 Genomes Project to characterize rare mutations, which were available during our initial analyses.
Sporadic MVP Patient Cohort
As part of an international consortium on mitral valve disease, we initiated collection of MVP patients with the eventual goal of performing GWAS studies in MVP. To date, 1896 patients have been collected in the United States, France, and Spain. MVP was defined as systolic displacement of one or both mitral leaflets ≥2mm beyond the annulus in parasternal or apical long-axis views, asymmetric posterior leaflet prolapse was also included in any view, including apical 4-chamber, when confirmed by side-to-side long-axis scanning 7,8. Patients were required to have no evidence by history, physical examination, or imaging for Marfan syndrome or other connective tissue disorders associated with MVP.
Exome Sequencing, Genotyping, and Variant Evaluation
As part of an MVP exome sequencing pilot project conducted by the Leducq Mitral Network, exome data were generated on twenty-one severe, early onset MVP patients and made available to identify variants in DCHS1. Fifteen patients were collected in Paris and had severe bileaflet mitral valve prolapse with myxomatous leaflets and an average age of onset of 15 years. Six patients were collected in Nantes, France, and had similar clinical characteristics with an average age at onset of 42. Exome capture was carried out using the SureSelect Human All Exon System using the manufacturer’s protocol version 1.0 that is compatible with Illumina paired-end sequencing. Exome-enriched genomes were multiplexed by flow cell for 101-bp paired-end read sequencing according to the protocol for the Hiseq 2000 sequencer (version 1.7.0; Illumina) to allow a minimum coverage of 30x. Reads were aligned to the human reference genome (UCSC NCBI36/hg19) using the Burrows-Wheeler Aligner (version 0.5.9). Evaluation of the DCHS1 gene yielded 4 novel coding sequence variants that confirmed following repeat Sanger sequencing: 6646587 G/A (p.R2330C) 6646709 G/A (p.A2289V), 6648584 C/T (p.A1896T), 6648820 A/G (p.V1817A), base pair positions are NCBI36 coordinates. These four variants, in addition to the variants identified in Family 1, were genotyped using Sequenom technology in the sporadic cohort. The major steps included primer and multiplex assay design using Sequenom’s MassARRAY® Designer software, DNA amplification by PCR, post-PCR nucleotide deactivation using shrimp alkaline phosphatase (SAP) to remove phosphate groups from unincorporated dNTPs, single-base extension reaction for allele differentiation, salt removal using ion-exchange resin, and mass correlated genotype calling using SpectroCHIP® array and MALDI-TOF mass spectrometry. Quality control to determine sample and genotyping quality and to potentially remove poor SNPs and/or samples was performed in PLINK, a whole genome association analysis toolset. We predicted the impact on gene function using PolyPhen211, Mutation Taster13 and LRT12.
Identification of Family 2 and 3
In order to identify other mutations in DCHS1, we first evaluated DCHS1 in the exome sequence data described above, reasoning that early onset forms may be more likely to have strong genetic etiologies. Rare variants causing amino acid substitutions in DCHS1 were identified in four individuals (p.V1817A, p.A1896T, p.A2289V, and p.R2330C) and genotyped in a cohort of 1864 sporadic MVP patients that included the 21 individuals with exome data; two of these variants, both localized to exon 19, were observed in the MVP cohort (p.A2289V in two cases and p.R2330C in three cases). The proband in family 2 carried the p.R2330C variant and underwent surgery for MVP in Paris. We were able to collect DNA and echocardiograms on first-degree relatives at that time. Additional clinical characteristics of the proband in family 2 included congestive heart failure (NYHA II/III) with left ventricular [LV] dilatation (70/50 mm end-diastolic/end-systolic dimensions), impaired LV systolic function (ejection fraction 53%, low for this volume overload), recurrent symptomatic atrial fibrillation, non-sustained ventricular tachycardia, and exercise-induced pulmonary hypertension (70 mmHg systolic). The proband in family 3 was originally collected in Amiens, France. All echocardiograms were read in both Boston and Paris and readers were blind to genotype data.
Danio Rerio Studies
Husbandry, knockdown and expression analyses were performed in the wild-type Danio rerio (zebrafish) strain Tubingen AB. Morpholinos were injected at a dose of 1.5 ng (after dose optimization) into single-cell embryos to achieve gene knockdown, and phenotypes were examined at 48 and 72 hours post-fertilization in three separate experiments of 50–75 embryos and compared to controls using the Fisher’s Exact test. Morpholino GeneTools LLC (Philomath, OR) sequences were as follows: apbb1 AACAAAGCGTACCACTCAGATTAGC, dchs1a TAAAGAAATGACAGTCCTACCTCCA, and dchs1b CATAACTGTTAAGAGTTCCGCTACA. Knockdown was confirmed by quantitative polymerase chain reaction. qPCR was performed as previously described 33. In brief, 20–30 morpholino-injected embryos were collected at 72 hpf, and snap frozen in liquid nitrogen. Trizol (Sigma) was added, RNA was purified according to the manufacturer’s instructions, and cDNA was prepared using a Superscript III Kit (Invitrogen). Primer sets were as follows: apbb1, 5′-GTGGAGGCGAGAACAGAG, 5′-CCAGCAGGAAGATCCGTGTC; dchs1a, 5′-GTTTCATGGAGGTTACAGC, 5′-CTTAATCCACCCCCATCCAC; dchs1b, 5′-GTTTCCTTGAGGTAAAGGCGG, 5′-GGCCACCCCCATCGGACG. qPCR was performed using SYBR Green (Applied Biosystems) in triplicate on an Applied Biosystems 7500 Fast Real-Time PCR instrument and normalized against β-actin. All zebrafish experiments were performed under protocols approved by the Institutional Animal Care and Use Committee at Mass General Hospital.
Danio rerio in situ hybridizations
In situ hybridizations were performed as previously described34 using a partial clone of dchs1b (Open Biosystems, Clone ID# 7136458) amplified with primers containing a T7 RNA polymerase site engineered onto the 3′ end of the reverse primer (Forward 5′-GGCAGTTCAAGTGGTGGT. Reverse: TAATACGACTCACTATAGGGTTAAATCCTCATCTCAGCCTCA, T7 site underlined.) The dchs1b probe was produced using a T7 RNA polymerase (Ambion,) and digoxygenin-labelled dNTPs (Roche). Other riboprobes utilized in the study have been previously described 35.
Generation of DCHS1 expression constructs
Human DCHS1 and the mutant containing the c.590C>T and c.7538G>A sequence changes were synthesized by Integrated DNA Technologies. A unique EcoRI site, a T7 polymerase site, and a Kozak sequence were added to the 5′ end of each gene, while a V5 tag and unique Xho1 site were added to the 3′ end. Each gene was then subcloned into the expression vector pcDNA3.1. Additional expression constructs were generated that contained only the c.590C>T (p.P197L) mutation or only the c.7538G>A (p.R2513H) mutation. These constructs were made using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) as per the manufacturers instructions. The P197L construct was generated from the double mutant construct by changing the R2513H (c.7538G>A) mutation back into the wild type sequence using the following primers: 5′-gctgatggaagccgcagccatgccgct, 3′-agcggcatggctgcggcttccatcagc. (The underlined bold base indicates the base pair changed.) The R2513H construct was generated by introducing the (c.7538G>A) mutation into the wild type DCHS1 construct using the following primers: 5′-gctgatggaagccacagccatgccgct, 3′-agcggcatggctgtggcttccatcagc.
Preparation and injection of DCHS1 mRNA
mRNA was prepared from the wild-type DCHS1 and DCHS1 mutant expression vectors using a T7 Message Machine Kit (Ambion) according to the manufacturer’s instructions. Injection mixtures containing 0.75 ng dchs1b MO alone, or 0.75 ng dchs1b MO plus 7 fg/μL of human DCHS1 mRNA (either wild type or mutant) were injected into one-cell embryos, and fish were scored for AV canal defects (failure to loop, and presence of regurgitation) 72 hours later. Data were collected from three independent experiments performed with 20–30 embryos each and comparisons made using Fisher’s Exact test.
Isolation of DCHS1 p.R2330C MVP and Control Patient Mitral Valve Tissue and Valvular Interstitial Cells
Resected posterior mitral valve tissue was utilized for culture and histology. For culture, valve pieces were minced in phosphate buffered saline (PBS) and washed in DMEM with antibiotics (Penicillin/Streptomycin (P/S) and fungizone) and incubated in DMEM with collagenase type II (Worthington) (1mg/ml) at 37°C for 12h. Following mechanical dissociation in DMEM, the cell suspension was filtered through a 40μm cell strainer and cells were cultured in DMEM with 15% fetal calf serum and antibiotics (P/S, fungizone). Although rare valve endothelial cells were present at P0, only cells with a fibroblastic phenotype (VICs) remained following P1–2. For all experiments, these valvular interstitial cells were utilized prior to passage 5. For Histology: valves were fixed in formalin, embedded in paraffin and sectioned at 5μm. Movat’s Pentachrome histological stain was performed using standard procedures.
Cell Culture Studies
Wild type, p.P197L, p.R2513H, and p.P197L/R2513H DCHS1 constructs were either synthesized by Integrated DNA Technologies or generated by site-directed mutagenesis (as described above), with an amino-terminal V5 epitope tag. Except where indicated, “mutant DCHS1” indicates the double mutant p.P197L/p.R2513H haplotype in Family 1. These constructs were expressed in mycoplasma-free HEK293 cells (ATCC) using cationic lipid-mediated transient transfection (Lipofectamine LTX, Invitrogen). Protein expression of transfected HEK cells was measured by quantifying Western blots using an antibody to the V5 epitope tag (Invitrogen). Patient Cells: For patient cells, control and p.R2330C valvular interstitial fibroblasts from posterior leaflets were plated at 2.5×104 cells were plated in a 24-well dish. 24 hours later, protein stability experiments were performed. Protein stability experiments involved addition of cycloheximide 24 hours after transfection (WT and p.R2513H transfectants) or plating (control or p.R2330C patient cells). For the cycloheximide experiments, media containing 100 ng/mL of cycloheximide was added at 24 hours post-transfection and each well was harvested as above at the indicated time points. Western blots were probed with either a mouse anti-V5 primary antibody (1:4000 dilution-Invitrogen) or a rabbit anti-Dchs1 antibody 36(1:1500 dilution), and an HRP-linked secondary at the same dilution (Thermo Scientific). Blots were also probed with a mouse anti-tubulin primary (Millipore) at a 1:4000 dilution, and the same secondary antibody as above. Blots were treated with Pierce ECL Substrate and visualized on film. For quantitation, blot pixel intensity was measured by ImageJ (NIH), and normalized to tubulin. Each sample was run in triplicate, and a regression curve was fit and half-lives calculated using SigmaPlot 12.
Mouse Studies
Dchs1 mice and genotyping were previously described22. All mice were blinded for genotype. Following phenotypic analyses, the genotypes of each sample were matched with the experimentally determined datasets. Histology, mitral valve analyses (echocardiography, MRI, and morphometric determination), and expression studies were performed on embryonic and adult (9-month male) wild-type (Dchs1+/+), heterozygote (Dchs1+/), and knockout (Dchs1−/−) hearts (C57/Bl6;Sv129 mixed background). For Histology: Fetal (E17.5) and adult (9-month) hearts were processed for hematoxylin and eosin stainings and immunohistochemistry (IHC) as previously described 37. For fetal analyses, Dchs1+/+, Dchs1+/−, and Dchs1−/− mice were analyzed.(N=5/genotype) Due to neonatal lethality of the Dchs1−/− mice and loss of function Dchs1 mutations in humans, adult analyses were restricted to Dchs1+/+ (N=7) and Dchs1+/− (N=5). For all analyses male mice were used. Antibodies used for IHC were: Hyaluronan Binding Protein (HABP) to stain proteoglycans (1:100) (Callbiochem), collagen I (1:100) (MDbio), and Hoescht to stain nuclei (1:10000) (Invitrogen). AMIRA 3D reconstructions were performed to generate volumetric measurements of fetal (E17.5) anterior and posterior mitral leaflets (N=5 for each genotype) as previously described 38. Length and width measurements of the mitral leaflets were obtained from histological sections. 25 consecutive 5 μM sections from anterior and posterior mitral leaflets of each genotype were used for measurements (Dchs1+/+, N=4, Dchs1+/−, N=7, Dchs1−/−, N=5). ImageJ software was used to measure the length of anterior and posterior leaflets from annulus to tip and the perpendicular width at the base, mid-region and tip. Measurements were compared to wild-type data to generate fold change and statistical significance (p<.01) was calculated using a Students t-test. For quantification of mitral valve interstitial cell alignment: hearts were initially dissected from E17.5 fetuses. The apex of the heart was dissected and discarded followed by removal of the left atrium. A cut was made cranial to caudal along the anterior aspect of the myocardium. The left ventricle was reflected to gain visualization of the leaflets. The left ventricle and interventricular septum were pinned such to stretch the papillary muscle and the chords. This resulted in obtainment of the leaflet as a planar sheet of tissue. The tissue was fixed in this position to ensure the leaflet was maintained in this orientation, as failure to do so results in curling of the leaflet making measurements and plane of orientation inconsistent between animals. Once the planar leaflet tissue was fixed in 4%PFA for 5 minutes, the leaflet was released from the heart by cutting the chords and dissecting along the annulus fibrosae. The tissue was then processed through normal protocols and placed en-face on paraffin. This technique was performed blinded to genotype and performed in exactly the same manner for all valve isolates. Performing this type of dissection and tissue processing ensures that all leaflets are placed in nearly identically oriented planes. The vector maps and quantification of cell alignment were performed blinded by two independent researchers. Only after the data was generated did a third researcher perform the PCR genotyping. Vector maps were manually generated as previously described39. Cells that deviated >10 degrees from an average alignment plane were counted as misaligned. Interstitial cells within anterior leaflets from each genotype were measured. Total number of cells measured were: WT=1083, (N=4), Het=1118, (N=4), KO=1953, (N=4). Statistical significance was calculated using a Students t-test with a p value <.05 being significant. Very little variation existed between the independent valves of each genotype as is graphically depicted. For mouse echocardiography the Vevo2100 imaging system (VisualSonics, Toronto, Canada) was used with 22–55 MHz linear transducer probe (MS550D) and utilized for 2-D B-mode and M-mode analysis. Heart rate was maintained at 400–500 bpm via isoflurane anesthesia. The mitral valve leaflet was visualized and its function was assessed in parasternal long-axis B-mode view by placing the transducer on the left lateral chest wall. End-systolic and end-diastolic LV dimensions and wall thicknesses were measured according to the American Society of Echocardiography guidelines as applied to mice. LV wall thickness was measured at the level of interventricular septum and the posterior wall. LV volume was calculated from Simpson’s method of disks and ejection fraction determined from the formula (LV end-diastolic-end-systolic volume)/(LV end-diastolic volume). Offline image analyses were performed using dedicated VisualSonics Vevo2100 1.2.0 software. Mitral valve prolapse was determined based on superior systolic displacement of one or more leaflets above the line connecting the annular hinge points in the long-axis view (N=6/genotype). For MRI experiments: 9-month old male Dchs1+/+ and Dchs1+/− mice (n=4) were sacrificed and the hearts were perfusion fixed and immersed 1:40 (12.5 mmol) Gadolinium (ProHance) in 10% formalin overnight before imaging. MRI was undertaken at 7T using a Bruker Biospin console (Pavavision 5.1) with a volume transmitter coil and a phased array surface coil. Gradient echo FLASH 3D images were collected with TR/TE/=50ms/5.4ms, flip angle=30°, NEX=3, matrix=256×256×256 and pixel resolution=55×55×59μm. Images in DICOM format were imported into AMIRA 3D reconstruction software and volume quantification were performed as described previously 38. Pairwise comparison of littermates were performed and statistical significance was determined (Students t-test) with p<.01. All mouse experiments were performed under protocols approved by the Institutional Animal Care and Use Committee, Medical University of South Carolina. Prior to cardiac resection, mice were sacrificed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
RNA expression analyses of Dchs1 and Apbb1
Section in situ hybridization was performed as previously described 40 on 4 embryo’s at each timepoint to localize Dchs1 expressing cells throughout cardiac development. A Dchs1 digoxygenin-labeled riboprobe (Roche) was generated against region 9222–10180 of accession#: NM_00162943 and used for in situ hybridization at E11.5, E13.5, and E15.5. RNA in situ hybridization for Apbb1 at E14.5 was performed through GenePaint 14. Two separate riboprobes were used to analyze Apbb1 RNA expression at E14.5. These probes were generated against regions 1676–2506 and 370–1967 of accession#: NM_001253885.1. These probes span all known isoforms for Apbb1 and provide similar spatial RNA expression patterns.
Protein Expression
Dchs1 antibodies were generated by immunizing rabbits with a synthetic peptide corresponding to rat Dchs1 protein sequence: CSTYMVESPDLVEADSAA (region 1308–1324 of accession #: NP_001101014) 36. Immunohistochemistry was performed as described previously19 using a 1:100 dilution of primary antibody.
In Vivo Lineage Trace
To trace the fate of epicardially derived cells in Dchs1+/+ and Dchs1+/− mitral leaflets, the mWt1/IRES/GFP-Cre mouse25 was bred with the Dchs1+/− and Dchs1+/+ mice (N=4/genotype). Mice were sacrificed at neonatal day 0 (P0) hearts were isolated and fixed overnight at 4°C in 4% paraformaldehyde dissolved in PBS. Hearts were processed through a series of graded ethanols, cleared in toluene, and embedded in Paraplast Plus (Fisherbrand, 23-021-400). Hearts were sectioned at 5 μm and slides were treated with 15mls of antigen unmasking solution (Vector Biolabs, H-3300) in 1600mls of distilled water for 10 minutes in a pressure cooker (Cuisinart) followed by incubation for 1 hour at room temperature with 1% BSA (Sigma, B4287) in PBS. Expression of EGFP after Cre recombination was detected by immunofluorescence using antibodies against GFP (Abcam, 13970) and Myosin heavy chain (MF20; DSHB). 5 μm sections throughout the entire valve were used for 3D reconstructions using Amira software. The volume of GFP positive cells and the volume of each mitral leaflet were measured using this software. Cell counting was done on GFP positive and GFP negative cells every 15 microns throughout the entire valve. Pairwise comparison of littermates were performed and statistical significance was determined (Students t-test) with p=.04 for posterior leaflet and p=.86 for anterior leaflet.
In Vitro Migration
Human mitral valve interstitial cells were isolated from a control and the patient with the DCHS1 mutation (p.R2230C) (proband Family 2) and seeded into the Radius 24-well Cell Migration Assay plate containing hydrogels (Cell Biolabs, CBA-125). Cells were allowed to adhere overnight and then gels were dissolved. Wells were imaged over a period of 24 hours and area of the cell free region was measured in photoshop v.10.0.01 and subtracted from the initial area of the hydrogel to generate area migrated over time. Migration in the Dchs1+/− and Dchs1+/+ mice was assessed by explanting P0 neonatal posterior mitral leaflets onto plastic. Images of the explants and migrating cells were captured at multiple timepoints. Distance migrated was measured as the distance from the explant to the farthest migrating cell. Measurements were taken at 5 points around the explant and averaged to calculate distance migrated. After 24 hours, cells were fixed in ice-cold 100% methanol for 10 minutes and immunofluorescence was performed using an antibody against N-cadherin (1:1000 dilution, BD Transduction Labs, 610920). Pairwise comparisons were performed and statistical significance was determined (Students t-test) with p<.05.
Statistical Consideration
In all experiments sample sizes were chosen to provide power of 0.8 to detect biological significant differences between test groups with two-sided α = 0.05. Specific statistical tests are listed in the methods for each individual experiment. Assumptions of normal distributions were made for quantitative biological measurements and comparison groups were assumed to have similar variances. For zebrafish experiments, fertilized oocytes were randomly selected within each clutch for injection with active compound vs. controls. Mouse experiments were interpreted blinded to genotype.
Extended Data
Extended Data Figure 1. Measurement Of Endogenous And Exogenous Gene Expression In Danio Rerio.

(A) Corresponding representative embryos of each morpholino knockdown on the left, with closeup of heart on the right. (B) To assess efficiency of morpholino knockdown, embryos were collected 72 hours after injection, mRNA was collected, and qPCR was performed. We demonstrate that morpholino (MO) knockdown of each indicated gene results in reduced mRNA expression, after normalization to beta-actin expression, compared to mock-injected controls (two-sided Student’s t-test). (C) Western blotting of embryos injected with DCHS1 mRNA demonstrates the production of protein. Mutant mRNA refers to the compound mutant P197L/R2513H. p-values are noted on graphs.
Extended Data Figure 2. Apbb1 Is Not Expressed During Cardiac Morphogenesis.
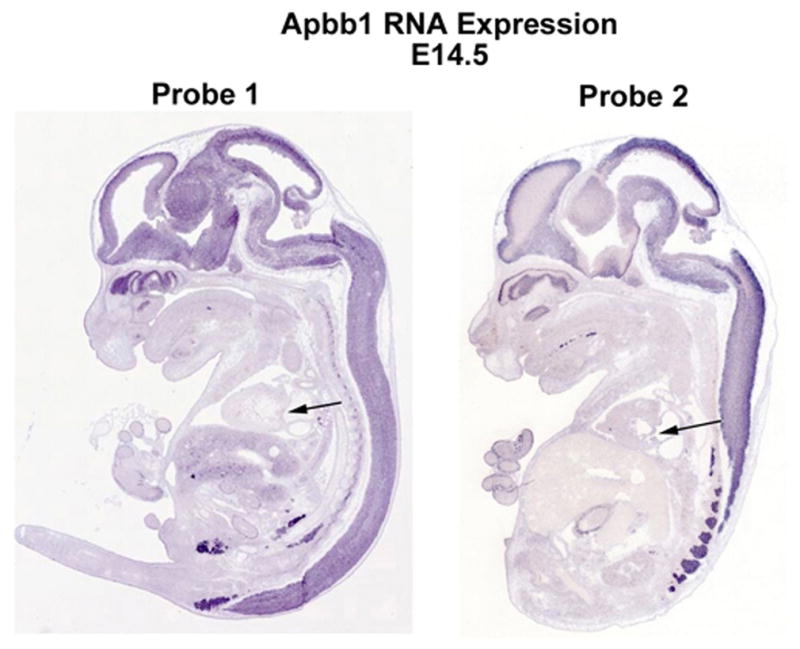
Apbb1 RNA expression was analyzed at E14.5 in sagittal sections using 2 separate antisense probes. Whereas strong cranial and neural expression is observed for Apbb1, no detectable cardiac expression or valve expression (arrow) is evident.
Extended Data Figure 3. Dachsous1b Expression At The AV Junction.

In situ hybridization reveals the presence of dchs1b in the AV canal (avc) at 54 hpf (A,B) and 72 hpf (C). The dchs1b expression is purple while a counterstain for cardiac tissue is brown (panel A). White arrows highlight the dchs1b signal in the AV canal.
Extended Data Figure 4. Dachsous1b knockdown alters AV ring markers.

In situ hybridization at 48hpf and 72hpf, as indicated, was performed for known AV ring markers. In contrast to wt (A) bmp4 expression is expanded into the ventricle at 48 hpf in dchs1 knockdown embryos at 48 hpf (B), spp1 and notch1b expression was largely unperturbed (C–F), and has2 expression was not detected at 48 hpf, and is faint at 72 hpf in dchs1 knockdown, compared to identically handled and stained controls (I–L).
Extended Data Figure 5. Histopathology Mitral Valves.

Human posterior leaflets of control, Barlow’s with MVP, and DCHS1 p.R2330C were isolated, fixed and stained with Movats Pentachrome. Leaflet thickening, elongation and myxomatous degeneration is observed in the Barlow’s and DCHS1 p.R2330C leaflets compared to controls. Expansion of the proteoglycan layer (blue) and disruption of the normal stratification of matrix boundaries is observed in the Barlow’s and DCHS1 p.R2330C leaflets. Blue=proteoglycan, Yellow=collagen, Black=elastin, Red=fibrin or cardiac muscle. Scale bars = 0.5cm
Extended Data Figure 6. Protein Expression Of Uncoupled Mutations.

In order to determine which Family 1 DCHS1 mutation is leading to the observed decrease in protein expression, constructs were generated that harbored only the p.P197L or the p.R2513H variant. Mutant refers to the double mutant P197L/R2513H construct. Western blot analyses from transfected HEK293 cells demonstrate that the p.R2513H mutation causes a significant decrease in DCHS1 protein expression, similar to that of the construct with both variants (mutant), suggesting pathogenicity. Percent difference in protein levels is depicted. Normalization of data was accomplished by qPCR specific to the transfected constructs. p-values are denoted in graphs.
Extended Data Figure 7. Cardiac Function Is Not Altered In Dchs1+/− Mice.

M-Mode analyses were performed to determine whether cardiac structure and/or function were perturbed in the Dchs1+/− mice. No statistically significant differences were observed in either cardiac structure or calculated cardiac function (N=6 for each genotype). Abbreviations: IVS-interventricular septum, d-diastole, s-systole, LVID-left ventricular internal dimension, LVPW-left ventricular posterior wall, EF-ejection fraction, FS-fractional shortening, LV-left ventricle
Extended Data Figure 8. Dchs1 Expression During Cardiac Development.

Upper Panel: RNA expression of Dchs1 was analyzed during embryonic gestation (E11.5, E13.5, and E15.5) by section in situ hybridization. At E11.5 Dchs1 RNA (blue staining) expression is observed in the endocardium and mesenchyme of the superior and inferior cushions (sAVC and iAVC, respectively). A gradient pattern of expression is observed at this time point with more intense expression near the endocardium. At E13.5 and E15.5, a similar pattern is observed in the forming anterior and posterior mitral leaflets (AL and PL, respectively). Lower Panel: Dchs1 protein expression (red) is observed throughout cardiac development in the endothelial cells and interstitial cells of the developing valves. Dchs1 shows asymmetric expression in the valvular interstitial cell bodies around E15.5 (arrowheads). Dchs1 protein is also observed in the epicardium and AV sulcus (arrows). (Red-Dchs1, Green-MF20, Blue-Hoescht).
Extended Data Figure 9. Dchs1 Deficiency Causes Altered Valvular Interstitial Cell Patterning In Vivo.

(A) IHC for eGFP of postnatal day 0 (P0) lineage traced Wt1-Cre/Rosa-eGFP/Dchs1+/+ neonatal mice show epicardial-derived cells (EPDCs) migrating into the posterior leaflet as a sheet of cells directly under the endothelium of the atrialis. This normal patterning is perturbed in the Wt1-Cre/Rose-eGFP/Dchs1+/− mice. 3D reconstructions were used to examine all EPDCs in the posterior leaflet of both genotypes to obtain a complete fate map of these cells. (B, C) Total volume of the leaflet is unchanged at this time point. However, the total volume of EPDCs as well as total EPDC cell number is significantly increased. There is a significant decrease in the number of non-EPDCs in the posterior leaflet with no overall change in total cell number. These data demonstrate that a minimum threshold of Dchs1 expression is required for normal migration of EPDCs into the posterior leaflet, normal patterning of this cell population, and cross-talk between EPDC and non-EPDC cell types in the valve. (**p<.01) (D) Isolated anterior mitral leaflet from fetal (E17.5) Dchs1+/+, Dchs1+/−, and Dchs1−/− mice were used to quantify cellular alignment of valvular interstitial cells. Vector maps were generated from histological (H&E) stains to show orientation and alignment of cells in relationship to each other. Boxes in each vector map panel are represented as zoomed images of regions within each of the valves to show cell orientation. (E) Cell alignment and polarity were quantified as the number of cells that deviate >10 degrees from the proximal-distal (P–D) axis of the leaflet. 90% of the cells in Dchs1+/+ show proper alignment with each other and along this P–D axis. Haploinsufficiency (Dchs1+/−) results in a 50% reduction in cell alignment, which is further reduced in Dchs1−/− (*=p-values<.01).
Extended Data Figure 10. Mice And MVP Patients With Dchs1 Deficiency Exhibit Migratory Defects In Vitro.

(A) Posterior Leaflets of P0 neonatal Dchs1+/+ and Dchs1+/− mice were explanted and interstitial cells were allowed to migrate out for 24 hours. Dchs1+/− mice exhibit increased migration (black lines drawn from explants) coincident with loss of cell-cell contacts and N-Cadherin expression at focal adhesions. Whereas N-Cadherin expression (red) is found at the membrane at points of cell-cell contract in Dchs1+/+ valvular interstitial cells (arrows), this membrane expression is lost in the Dchs1+/− cells and is prominently expressed in the cytoplasm (arrows). (Nuclei-blue). (B) Migration assays using control and MVP patient (p.R2330C) valvular interstitial cells exhibit a similar affect as observed in the mouse cells whereby the p.R2330C cells exhibit an increase in migration. p-values are denoted in graphs.
Supplementary Material
Acknowledgments
This work was supported by the Fondation Leducq (Paris, France) Mitral Transatlantic Network of Excellence grant 07CVD04. MVP patient studies were supported by an Innovation in Clinical Research award of the Doris Duke Charitable Foundation (SAS and RAL). Sequencing of the candidate region was performed at the Venter Institute through a grant from the National Heart Lung and Blood Institute Resequencing and Genotyping (RS&G) Service (SAS). The work at MUSC was performed in a facility constructed with support from the National Institutes of Health, Grant Number C06 RR018823 from the Extramural Research Facilities Program of the National Center for Research Resources. Other funding sources: National Heart Lung and Blood Institute: R01HL122906-01 (AW), R01-HL33756 (RRM), COBRE 1P30 GM103342 (RRM, RAN, AW), 8P20 GM103444-07 (RRM and RAN), R01-HL109004 (DJM), R01-HL127692 (DJM, SAS, RAN, RAL); RO1-HL095696 (DRM), VA Merit Review BX002327 (DRM); National Institute of Mental Health R00-MH095867 (MET) The Hassenfeld Scholar Program (DJM); The March of Dimes (MT); MGH Scholars Program (SAS); American Heart Association: 09GRNT2060075 (AW), 11SDG5270006 (RAN), 2261354 (DJM); National Science Foundation: EPS-0903795 (RRM); NHLBI K24 HL67434, R01HL72265 and R01HL109506 and the Ellison Foundation, Boston, MA (RAL), NIMH MH095867 (MET), Howard Hughes Medical Institute (KDI), and a gift from Michael Zak (DJM). Special thanks to Dr. Truman Brown (MUSC) for his guidance on MRI studies, Ms. Carrie Hanscom (MGH) for assistance with genomic libraries, and Elaine Lim (HMS) for contributions in interpreting the exome mutation data. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author Contributions: RD, ML, CS, CJ, MP, XJ, J-JS, DT, SO, XE, FC, and SAS participated in genetic analysis, sequencing and mutation cloning. RD, FND, LAF, TLT, HLM, LF-F, JS, CT and RAL participated in patient collection and phenotyping using echocardiography. DSP, SNL and DJM performed Zebrafish and cell culture experiments. MT, MRS, NBN, CD, HB and CC performed bioinformatics and statistical analysis. AC, PC, and PB established human patient cell cultures and histology on human tissue. AdV, KW, KDI, YM, KS, AW, TM, KT, RRM and RAN performed mouse embryo and knockout experiments and cell alignment and migration assays. XN, A-MB, DRM, HK performed mouse in vivo imaging. RD, DJM, RAL, RAN and SAS wrote the manuscript. RAL, AH, SAS and JJS coordinated the Leducq Mitral Network.
Competing Interest Statement: The authors declare no competing financial interests
References
- 1.Freed LA, et al. Mitral valve prolapse in the general population: the benign nature of echocardiographic features in the Framingham Heart Study. J Am Coll Cardiol. 2002;40:1298–1304. doi: 10.1016/s0735-1097(02)02161-7. S0735109702021617 [pii] [DOI] [PubMed] [Google Scholar]
- 2.Avierinos JF, et al. Natural history of asymptomatic mitral valve prolapse in the community. Circulation. 2002;106:1355–1361. doi: 10.1161/01.cir.0000028933.34260.09. [DOI] [PubMed] [Google Scholar]
- 3.Freed LA, et al. Prevalence and clinical outcome of mitral-valve prolapse. N Engl J Med. 1999;341:1–7. doi: 10.1056/NEJM199907013410101. [DOI] [PubMed] [Google Scholar]
- 4.Vaishnava P, Fuster V, Goldman M, Bonow RO. Surgery for asymptomatic degenerative aortic and mitral valve disease. Nature reviews. Cardiology. 2011;8:173–177. doi: 10.1038/nrcardio.2010.203. [DOI] [PubMed] [Google Scholar]
- 5.Waller BF, Maron BJ, Del Negro AA, Gottdiener JS, Roberts WC. Frequency and significance of M-mode echocardiographic evidence of mitral valve prolapse in clinically isolated pure mitral regurgitation: analysis of 65 patients having mitral valve replacement. The American journal of cardiology. 1984;53:139–147. doi: 10.1016/0002-9149(84)90698-2. [DOI] [PubMed] [Google Scholar]
- 6.Freed LA, et al. A locus for autosomal dominant mitral valve prolapse on chromosome 11p15.4. Am J Hum Genet. 2003;72:1551–1559. doi: 10.1086/375452. S0002-9297(07)60454-6 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine RA, et al. Three-dimensional echocardiographic reconstruction of the mitral valve, with implications for the diagnosis of mitral valve prolapse. Circulation. 1989;80:589–598. doi: 10.1161/01.cir.80.3.589. [DOI] [PubMed] [Google Scholar]
- 8.Levine RA, Stathogiannis E, Newell JB, Harrigan P, Weyman AE. Reconsideration of echocardiographic standards for mitral valve prolapse: lack of association between leaflet displacement isolated to the apical four chamber view and independent echocardiographic evidence of abnormality. J Am Coll Cardiol. 1988;11:1010–1019. doi: 10.1016/s0735-1097(98)90059-6. [DOI] [PubMed] [Google Scholar]
- 9.Perloff JK, Child JS. Clinical and epidemiologic issues in mitral valve prolapse: overview and perspective. American heart journal. 1987;113:1324–1332. doi: 10.1016/0002-8703(87)90961-6. 0002-8703(87)90961-6 [pii] [DOI] [PubMed] [Google Scholar]
- 10.Clark HF, et al. Dachsous encodes a member of the cadherin superfamily that controls imaginal disc morphogenesis in Drosophila. Genes Dev. 1995;9:1530–1542. doi: 10.1101/gad.9.12.1530. [DOI] [PubMed] [Google Scholar]
- 11.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome research. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 14.Visel A, Thaller C, Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32:D552–556. doi: 10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guenette S, et al. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. The EMBO journal. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margolin DH, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368:1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Golzio C, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaki M, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norris RA, et al. Expression of the familial cardiac valvular dystrophy gene, filamin-A, during heart morphogenesis. Dev Dyn. 2010;239:2118–2127. doi: 10.1002/dvdy.22346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rabkin E, et al. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 21.Nesta F, et al. Leaflet concavity: a rapid visual clue to the presence and mechanism of functional mitral regurgitation. J Am Soc Echocardiogr. 2003;16:1301–1308. doi: 10.1067/j.echo.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Mao Y, et al. Characterization of a Dchs1 mutant mouse reveals requirements for Dchs1-Fat4 signaling during mammalian development. Development. 2011;138:947–957. doi: 10.1242/dev.057166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nesta F, et al. New locus for autosomal dominant mitral valve prolapse on chromosome 13: clinical insights from genetic studies. Circulation. 2005;112:2022–2030. doi: 10.1161/CIRCULATIONAHA.104.516930. CIRCULATIONAHA.104.516930 [pii] [DOI] [PubMed] [Google Scholar]
- 24.Cho E, Irvine KD. Action of fat, four-jointed, dachsous and dachs in distal-to-proximal wing signaling. Development. 2004;131:4489–4500. doi: 10.1242/dev.01315. [DOI] [PubMed] [Google Scholar]
- 25.Wessels A, et al. Epicardially-derived Fibroblasts and their Contribution to the Parietal Leaflets of the Atrioventricular Valves in the Murine Heart. Developmental Biology. 2012;366:111–124. doi: 10.1016/j.ydbio.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuppiroli A, Roman MJ, O’Grady M, Devereux RB. A family study of anterior mitral leaflet thickness and mitral valve prolapse. The American journal of cardiology. 1998;82:823–826. A810. doi: 10.1016/s0002-9149(98)00454-8. [DOI] [PubMed] [Google Scholar]
- 27.Kolibash AJ, Jr, et al. Evidence for progression from mild to severe mitral regurgitation in mitral valve prolapse. The American journal of cardiology. 1986;58:762–767. doi: 10.1016/0002-9149(86)90352-8. 0002-9149(86)90352-8 [pii] [DOI] [PubMed] [Google Scholar]
- 28.Li K, Stockwell TB. VariantClassifier: A hierarchical variant classifier for annotated genomes. BMC research notes. 2010;3:191. doi: 10.1186/1756-0500-3-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome research. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome research. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome research. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genomes Project C, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison TB, Weis JJ, Wittwer CT. Quantification of low-copy transcripts by continuous SYBR Green I monitoring during amplification. BioTechniques. 1998;24:954–958. 960–962. [PubMed] [Google Scholar]
- 34.Thisse B, et al. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods in cell biology. 2004;77:505–519. doi: 10.1016/s0091-679x(04)77027-2. [DOI] [PubMed] [Google Scholar]
- 35.Kolpa HJ, et al. miR-21 represses Pdcd4 during cardiac valvulogenesis. Development. 2013;140:2172–2180. doi: 10.1242/dev.084475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukasaki Y, et al. Giant cadherins Fat and Dachsous self-bend to organize properly spaced intercellular junctions. Proc Natl Acad Sci U S A. 2014;111:16011–16016. doi: 10.1073/pnas.1418990111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Norris RA, et al. Periostin regulates atrioventricular valve maturation. Developmental Biology. 2008;316:200–213. doi: 10.1016/j.ydbio.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sauls K, et al. Developmental basis for filamin-A-associated myxomatous mitral valve disease. Cardiovasc Res. 2012;96:109–119. doi: 10.1093/cvr/cvs238. cvs238 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu F, Beyazoglu T, Hefner E, Gurkan UA, Demirci U. Automated and adaptable quantification of cellular alignment from microscopic images for tissue engineering applications. Tissue engineering. Part C, Methods. 2011;17:641–649. doi: 10.1089/ten.tec.2011.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norris RA, et al. Identification and detection of the periostin gene in cardiac development. Anat Rec A Discov Mol Cell Evol Biol. 2004;281:1227–1233. doi: 10.1002/ar.a.20135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.