

Abstract
We examine the hypothesis that the heritability of smoking has varied over the course of recent history as a function of associated changes in the composition of the smoking and non-smoking populations. Classical twin-based heritability analysis has suggested that that genetic basis of smoking has increased as the information about the harms of tobacco has become more prevalent—particularly after the issuance of the 1964 Surgeon General’s Report. In the present paper we deploy alternative methods to test this claim. We use data from the Health and Retirement Study to estimate cohort differences in the genetic influence on smoking using both genomic-relatedness-matrix restricted maximum likelihood and a modified DeFries-Fulker approach. We perform a similar exercise deploying a polygenic score for smoking using results generated by the Tobacco and Genetics consortium. The results support earlier claims that the genetic influence in smoking behavior has increased over time. Emphasizing historical periods and birth cohorts as environmental factors has benefits over existing GxE research. Our results provide additional support for the idea that anti-smoking policies of the 1980s may not be as effective because of the increasingly important role of genotype as a determinant of smoking status.
Keywords: Smoking, GCTA, GREML, Genome-wide, Polygenic score
1. Introduction
Nearly 400,000 Americans die every year from tobacco-related disease (CDC, 2008). Understanding why people initiate smoking is critical to prevention and cessation efforts and is thus a public health priority. One important factor in determining smoking behavior is that individuals differ in their genetic propensity to smoke. Twin studies have demonstrated that smoking is moderately to highly heritable (Li et al., 2003) and recent analyses with molecular genetic data have identified a number of single nucleotide polymorphisms (SNPs) of interest (Tobacco and Genetics Consortium, 2010). Better understanding this genetic propensity for smoking is important because of evidence suggesting that specific genotypes tied to the risk of smoking are also risk factors for subsequent smoking-related disease (Spitz et al., 2008). Such individuals are doubly at risk–more likely to smoke and more likely to develop cancer if they do smoke–so from a public health perspective it would clearly be useful to intervene with such individuals with early, effective intervention.
Research based on twins has shown that the genetic influences on smoking have changed in predictable ways across historical periods. For instance, Boardman et al. (2010) show that the heritability of smoking initiation bottomed out in roughly 1964 and climbed in successive periods. They argue that social factors were primarily responsible for smoking onset but then, following the surgeon general’s 1964 report “Smoking and Health: Report of the Advisory Committee of the Surgeon General of the Public Health Service”, the composition of smokers changed such that genetics became a more important determinant of smoking status. They followed this study up in an independent sample (Boardman et al. 2011) and showed that increasingly strict legislation regarding smoking (e.g., taxes, limits on public smoking, and so on) caused the composition of those who remain in the smoking group to be increasingly driven by genetic factors. Specifically, as the social and economic cost of smoking rose, those who were more able to leave the pool of smokers did so (e.g., those for whom smoking was not driven by biological pathways involving nicotine dependence), leaving behind a population of smokers with a potentially different biological relationship (partially based on genotype) with tobacco. Vink & Boomsma (2011) attempted to examine cohort effects in the heritability of smoking and found no evidence for a change. However, there are important differences in both the context (Europe versus the US) and the cohorts used in their research versus the earlier research by Boardman and colleagues. Vink & Boomsma focus on a younger cohort than the previous work and there are dramatically different rates of smoking in the US and Europe that could explain these differences (e.g., Cutler & Glaeser, 2009).
These studies are important because they suggest that policies that effectively limited smoking behaviors in the 1970s and 1980s—such as restricting public consumption of tobacco or increasing its cost—are likely to be less effective in the future, as the composition of smokers is fundamentally different now than when compared to smokers in the past; that is, smokers who continue to smoke in light of increased costs are the most inelastic consumers. The contributions of earlier research are limited in one crucial respect as they rely upon samples of identical and fraternal twins to estimate heritability. From the perspective of this paper, the key limitation is one of selection bias. Standard twin models require that both twins be alive at the time of data collection. Given the strong links between smoking and mortality, the selection of identical twins who are both still alive may be highly correlated with shared genotype. This form of gene-environment correlation has the potential to bias gene-by-period interaction analyses (Boardman & Fletcher, 2015).
Recent advances in statistical genetics extend techniques from the twin literature for estimation of trait heritability to the case of measured genetic relationships among unrelated persons (Yang et al. 2010). In particular, genomic-relatedness-matrix restricted maximum likelihood (GREML), as implemented in software (Genome-wide Complex Trait Analysis; GCTA) from Yang et al. (2011), have produced heritability estimates of smoking that are generally smaller than the estimates from twin studies, which is to be expected given that estimates derived in Yang et al. (2011) are shown to be attenuated due to regression dilution. That is, the lower GREML-based h2 estimates are largely the result of attenuation bias due to measurement error. In this case, the common genotyping platforms are picking up SNPs that are in incomplete linkage (i.e. not perfectly spatially correlated) with the true causal SNPs of genetic effects. Even given these limitations, GREML analyses generally support the notion that smoking onset is a moderately heritable phenotype (Lubke et al. 2012).
While the GREML approach largely translates the methodology underlying classical twin studies to molecular genetic data (genotypic similarity is associated with phenotypic similarity), an entirely new paradigm is possible with molecular genetic data. Genome-wide association studies (GWAS) are data mining approaches that tend to use large meta-analytic samples to estimate the effect of individual SNPs on an outcome. This approach has been applied to a large number of phenotypes in the past decade (Welter et al., 2014). The Tobacco and Genetics Consortium (2010) has conducted GWAS for having ever smoked, cigarettes per day, and other smoking traits. These findings can be used to create a polygenic score (PGS) that begins to quantify an individual’s genetic propensity towards smoking. PGSs have been used in studies of several traits including obesity (Belsky et al., 2012) and education (Rietveld et al. 2013; Conley et al., 2015) as well as smoking (Belsky et al., 2013) and are beginning to contribute to our knowledge of how these phenotypes progress over the lifecourse. We believe the replication of heritability-based conclusions about cohort effects in genetic influences on smoking using PGSs are important since they help ensure that the findings are robust and not due to, for example, subtle confounding of genetic relatedness with shared environmental influences (c.f. Conley et al. 2014).
In this study, we utilize a nationally representative sample of older Americans from the Health and Retirement Study to evaluate the hypothesis that the relative contribution of genotype to smoking differs as a function of birth cohort using genetic information from unrelated persons. We focus on two research questions. First, do we observe a pattern in the genetic influence on smoking behavior over historical time and, if so, how does it compare to prior work? We anticipate that the heritability and association with the genetic risk score will have a loosely defined “U-shape” with a minimum for the 1940s cohort but overall, the genetic influences on smoking should generally increase over time. The second research question asks whether this result may be compromised due to selection bias in the sample. Building on the observation that genetic risks may not be randomly distributed across space (Rehkopf, 2014), we ask whether genetic risks are randomly distributed across time. In particular, given that smokers face increased risks of mortality, we hypothesize that the most genetically predisposed towards smoking may be underrepresented in the earlier birth cohorts in our study due to premature mortality. Concerns involving selection are explored throughout this paper.
2. Data
This study focuses on a set of 9,313 non-Hispanic white respondents from the Health and Retirement Study (HRS).1 HRS is a longitudinal survey of older Americans on issues related to health and the transition out of the workforce. These respondents were born between 1900 and 1970 with the IQR of birth years spanning from 1930 to 1950. Given the sample size limitations shown in Figure 1A, we focus on those born between 1919–1959 (inclusive) in our analyses (N=8,904). The respondents were majority female (58%) and 85% and 26% reported receiving more than 12 years or 16 years of education respectively. We focus on a binary indicator describing whether a respondent ever smoked. The majority, 57%, of our sample reported smoking at some point. Figure 1B shows the percentage of smokers as a function of birth cohort and demonstrates that the percentage of respondents who claim to have smoked has remained fairly constant over time with nearly half of the respondents from any given birth year saying they were smokers at one point. Figure 1C examines changes in the gender composition of the sample over time. The sample is majority female in our focal range of 1919–1959 but becomes predominantly women for those born after 1950. This has implications for the analyses we describe below.
Figure 1.
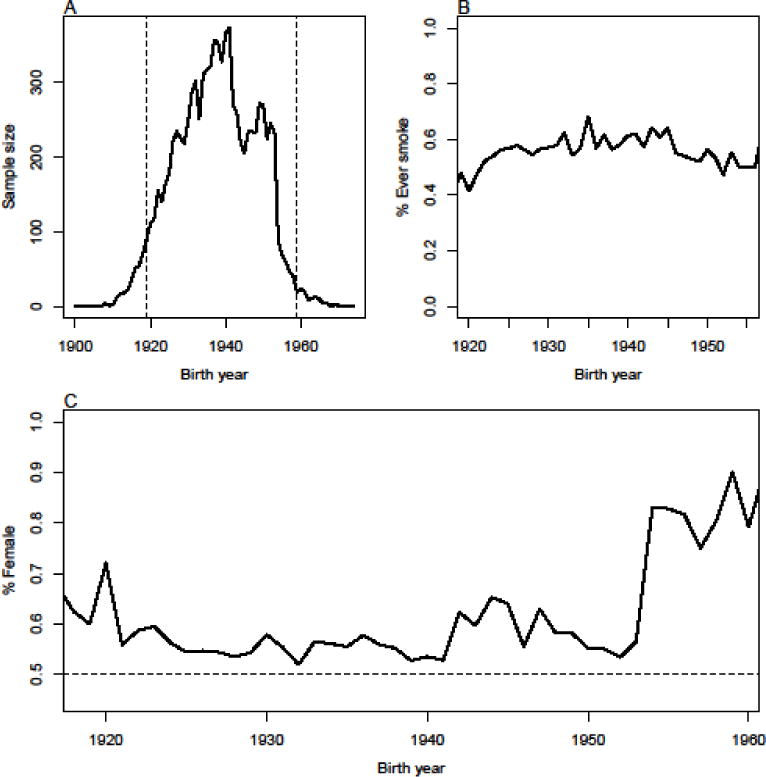
(A) Sample size, (B) % ever smokers, and (C) % female in our sample as a function of birth year cohort in HRS.
Genetic data for the HRS is based on DNA samples focus on single nucleotide polymorphisms (SNPs) collected in two phases. The first phase was collected via buccal swabs in 2006 using the QuiagenAutopure method. The second phase used saliva samples collected in 2008 and extracted with Oragene. Genotype calls were then made based on a clustering of both data sets using the Illumina HumanOmni2.5-4v1 array. SNPs are removed if they are missing in more than 5% of cases, have low MAF (0.01), and are not in HWE (p<0.001). We retained 1,698,845 SNPs after removing those which did not pass the QC filters.
3. Methods
Traditional behavioral genetics work leveraged the fact that pairs of individuals with known biological relationships have expected quantities of alleles shared by descent. For example, full siblings share an average of one-half of their alleles by descent and identical twins share all of their alleles by descent. In other words, although genetic similarity is unmeasured in such cases, the expected genetic covariance between family members of different genetic relatedness provides an entry point for understanding the relative contribution of genetic variance to overall phenotypic variance in the population. Here, we rely upon the fact that, given available molecular genetic data, the genetic similarity of two unrelated individuals can be computed. The proportion of alleles shared is assessed by state (IBS) rather than descent (IBD) and we use the genetic relationship measure implemented in GCTA (Yang et al., 2011). This is essentially a correlation between the count of minor alleles at each loci, weighted by the minor allele frequency. We consider only non-Hispanic whites due to the fact that these correlations can be problematic measures of genetic similarity when considered across racial groups. Consider Figure S5 of Domingue et al. (2014) which demonstrates a clear bias in the estimate of genetic similarity for black spouses (e.g., black spouses are estimated to be as genetically similar as close relatives). We also note that numerous other approaches are possible for computing genetic similarity (Speed & Balding, 2014 contains an overview of the available approaches).
Genetic similarity can then be compared to phenotypic similarity to compute heritability estimates (Yang et al., 2011). This technique has been used to study higher-level traits such as cognitive abilities (Plomin et al., 2013), alcohol consumption (Vrieze et al., 2013), and self-reported health (Boardman et al., 2014). For the non-Hispanic whites in HRS, we find a significant estimate for the heritability of having ever smoked (h2=0.22, SE=0.05). This result is inline with GREML estimates of smoking initiation among respondents of the Netherlands Twin Register Biobank Study who estimate a heritability of .19 for smoking initiation and .24 for current smoking (Lubke et al. 2012). Such estimates are narrow-sense, additive heritability estimates in that they do not account for dominant or epistatic effects, but they are also presumably biased downwards of the true narrow-sense heritability since they are focused on only the common variants included in the assay. That is, they suffer from attenuation bias to the extent that there is incomplete linkage between the true causal SNPs and those that are genotyped.
We also utilize a modified form of the DeFries-Fulker (1985) method to further explore whether hypothesized changes in the genetic architecture of smoking are present given the selection issues inherent in this sample. For twins, the basic equation underlying this method is
| (Eqn 1) |
where each pair i and j is double-entered (note that we are forcing the intercept to be zero). Traditionally, Aij is either 0.5 or 1 for DZ or MZ twins respectively. We now predict the phenotype for individual i using the phenotype for all other individuals i′ (where i ≠ i′) via:
| (Eqn 2) |
We again double-enter all pairs and also mean center yi within the sample. In Eqn 2, Aii′ is the estimated genetic similarity from GCTA (standardized across pairs) rather than the IBD between twins. Since this approach, which we describe as genome-wide DeFries-Fulker (GWDF) has not been previously used with unrelated individuals (to the best of our knowledge), we have included a small simulation study in the Appendix demonstrating that estimates of b are strongly correlated with GREML heritability estimates.
While the GWDF approach also suffers from attenuation bias due to incomplete linkage between causal and genotyped SNPs, there are several advantages to the GWDF approach. First, the estimation is essentially a generalized linear model that can be extended to include complex survey designs, sampling weights, and other complicated statistical techniques that are not available elsewhere. Others have extended the DeFries-Fulker model to the multilevel perspective (Boardman et al. 2008) and the same could be done here. Second, because of the flexibility of this model, known factors that may confound or mediate the link between genotype and phenotype can be adjusted and complex models of gene-environment correlation can be assessed empirically without relying on twins. The goal of this model is not to estimate heritability per se but to track changes in b3 when Eqn 2 is estimated in different birth cohorts. In particular, we hypothesize that b3 will be the smallest for birth cohorts in which smoking was largely driven by social factors but will increase across time for cohorts who were socialized about smoking following the 1964 Surgeon General’s Report. Note that individual i is included multiple times on the left-hand side of Equation 2 and thus there may be a violation of standard regression assumptions. In particular, eii′ may not be independent of eii″. This may cause underestimation of the relevant standard errors, so we consider models which account for the effect of this clustering within individual (Lumley, 2004). For reasons we now discuss, we also consider extensions of Equation 2 in which additional controls for both individual i and i′ are incorporated on the right-hand side of Equation 2.
An important concern is that the HRS is unlikely to have a consistent sample of respondents from the various birth cohorts given mortality effects (e.g., Zajacova & Burgard, 2013). Figure 2 shows the change in mean as a function of birth year for several key variables: having ever smoked, years of education, height (at wave 8), and mean BMI across all waves. The means of all non-smoking variables increase for both males and females as a function of birth year. While there are potentially cohort effects that underlie some of these changes, there are also known mortality effects within this sample (Zajacova & Burgard, 2013). The changes in smoking status are more complicated since they vary by gender. Younger males in the sample report smoking at lower rates than older males while the opposite is true for women. This gender-specific pattern has been observed in other research (Escobedo & Peddicord, 1996). Accordingly, we adjust our models for age and gender.
Figure 2.
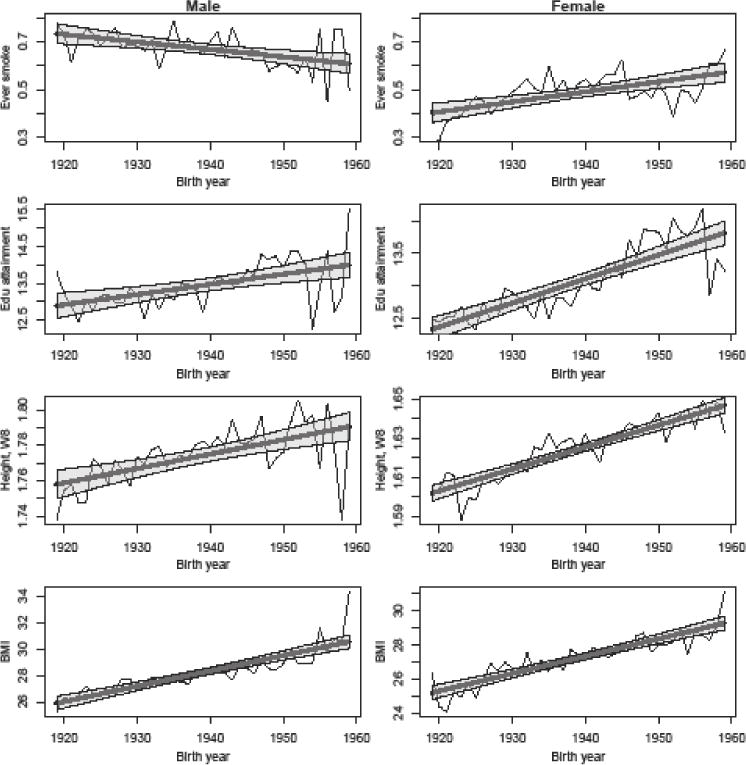
Changes in means by gender (along with fitted trends) for various variables as a function of birth year.
PGSs were first introduced in 2009 (Purcell et al., 2009) as flexible tools for quantifying the genetic contribution to a phenotype. Their prime limitation is that they require much larger samples than currently exist to explain anywhere near the full narrow-sense heritability (see Dudbridge, 2013). PGSs for lifetime smoking status were calculated for each respondent using results from a recent GWAS on smoking (Tobacco and Genetics Consortium, 2010). Briefly, SNPs in the HRS genetic database were matched to SNPs with reported results in the GWAS. The matched set of SNPs was then “pruned” to account for linkage disequilibrium using the clumping procedure (which considers the level of association between the SNP and the phenotype, not simply LD) in the second-generation PLINK software (Chang et al., 2014).2 For each of these SNPs, a loading was calculated as the number of smoking associated alleles multiplied by the effect-size estimated in the original GWAS. SNPs with relatively large p-values will have small effects (and thus be down weighted in creating the composite), so we do not impose a p-value threshold. Loadings were summed across the SNP set to calculate the polygenic score. The score was then standardized to have a mean of 0 and SD of 1. Having ever smoked was correlated with its PGS at 0.088. We consider the estimated coefficient of the PGS in regression models where having ever been a smoker is predicted as a function of the PGS and other control variables.
4. Results
Figure 3A summarizes GREML heritability estimates for smoking from overlapping 12-year birth cohorts, the first such cohort centered in 1925 and the last such cohort centered in 1953. The pattern observed is comparable to what was shown in the Boardman et al. (2010) paper in which the heritability for smoking bottomed out for cohorts born in the early 1940s (those who would later be at the peak age of smoking when the Surgeon General’s report was released). Specifically, we estimate a heritability of smoking of roughly 0.4 for the earliest cohort (1925), a value of 0.13 for the 1939 cohort, and a return to 0.32 for the latest (1953) cohort. Despite the convergence of our findings with previously published work, it is important to note the very large confidence intervals for each birth cohort estimate. Recall that for our total sample of 9,313 we estimate a heritability of 0.22. Using the online GCTA-GREML power calculator3, the power to detect such a heritability is 1 in the full sample of 9,313 respondents. But our smallest cohort only contains 2,042 respondents and the estimate ranges from nearly zero to 0.6. For this group, we have a power of only 0.34. Accordingly, we caution readers to evaluate the pattern of the results from the GREML models in light of the fact that we have clearly limited power to detect significant heritability in some situations.
Figure 3.
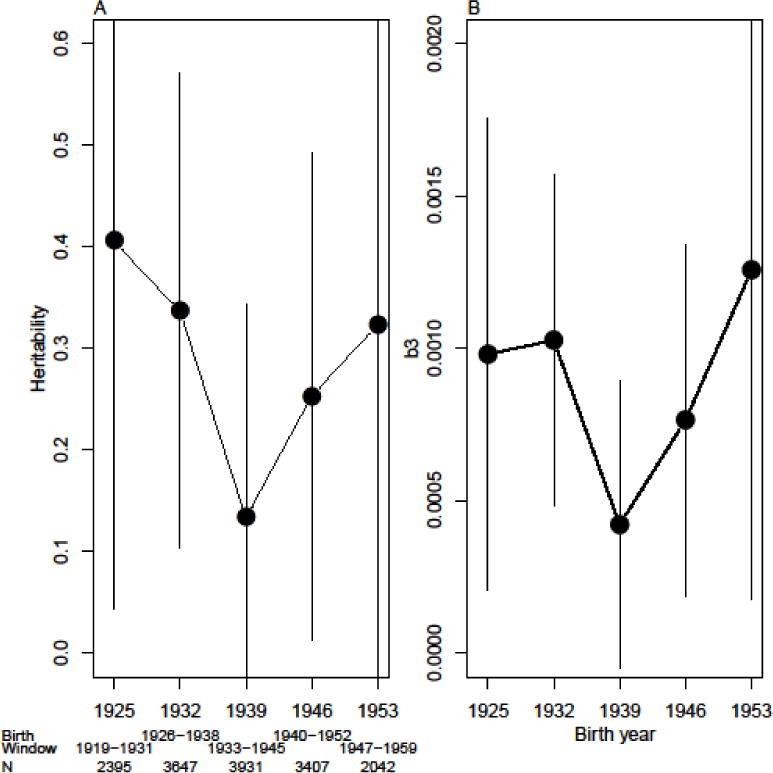
(A) Estimated heritability, adjusted for gender and birth year, of having ever been a smoker in HRS in overlapping birth windows centered at years show on x-axis. (B) Genome-wide DeFries-Fulker coefficients (b3 from Eqn 2) includes adjustments for multiple entry of individual outcomes and with controls for the birth year and gender of each individual as well as within-individual interactions between birth year and gender.
To further evaluate the pattern of the results using a different method, Figure 3B presents the estimates from the GWDF models. Results in Figure 3B are adjusted for the multiple entry of individual outcomes and with controls for the birth year and gender of each individual as well as within-individual interactions between birth year and gender. Given that the cohort effects might not be linear, we also replaced the birth year terms (both main effect and interactions) with set of 3 B-splines (results not shown). Results were virtually identical. Although the scale of the dependent variable is very different in the GWDF models, it is important to note that we continue to see a similar pattern in which the minimum value of genetic influence on smoking behaviors seems to be among those born between 1933 and 1945.
Figure 4A uses the genetic risk score for smoking to evaluate the correlation between specific polymorphisms and smoking behavior as a function of birth cohort. These results demonstrate an increase in the bivariate correlation between our PGS and smoking from roughly 0.05 in the earliest birth cohorts to above 0.1 in the latest cohorts. We do not observe the U-shaped association described in Figures 3A and 3A as well in the Boardman et al. (2010) paper but the results continue to support the notion that genetic influences on smoking have increased in successive birth cohorts. However, this increase could be partially due to the selection issues previously mentioned that are of concern. In particular, the most genetically predisposed to smoke from earlier cohorts may have died at higher rates due to their having smoked for longer periods. Thus, earlier cohorts would appear (in terms of who remains in the sample) less genetically susceptible to smoking. Figure 4B considers the average genetic risk score as a function of birth year. While it suggests that there might be selection involving the score, it is difficult to say authoritatively (i.e., the increase over time is consistent with random fluctuations as indicated by the confidence intervals). Allele frequencies for loci linked to smoking should be constant in the population over time because the risk for death occurs well after prime fertility years. Thus, we argue that this very slight rise may point to systematic bias in our sample such that the most genetically at risk from the earlier birth cohorts are not in the HRS data due to premature mortality or sample attrition.
Figure 4.
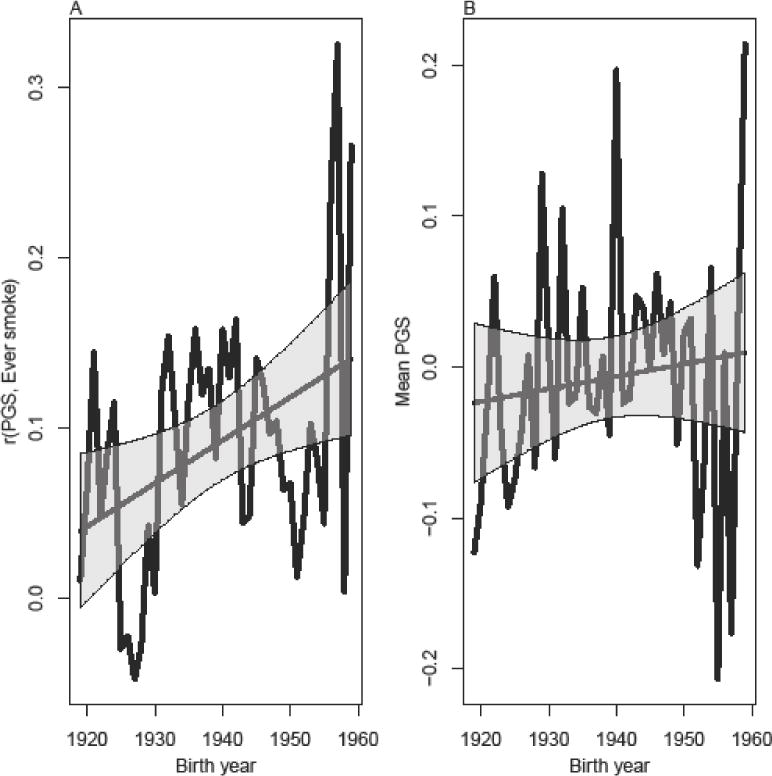
(A) Bivariate correlation between genetic risk of smoking and ever smoking. (B) Mean genetic risk for smoking as a function of birth year.
Figure 5 is a refinement of Figure 4A designed to adjust for this selection. Figure 5A shows the estimated “probability”4 of smoking based on a model that controls for the PGS, birth year, gender, interaction of PGS and birth year, and interaction of PGS and gender.5 Figure 5B allows for a more flexible main effect of birth year by modeling the three B-splines based on birth year while also including an interaction between birth year (modeled directly, not as the set of B-splines) and PGS.6 In both models, the interactions of birth year and PGS are marginally significant. The interaction between being female and the PGS is also significant for both models. Figure 5A shows that the estimated probabilities of smoking for males and females are higher for those born in later cohorts who have more genetic risk (PGS=1) compared to those with less genetic risk (PGS= −1). The effect is more pronounced for females than males. Fitted probabilities based on the more flexible model in Figure 5B are more difficult to interpret, but there is again an increasing probability of smoking at later birth cohorts, especially for females. Overall, these additional models support the notion that the genetic factors linked to smoking behaviors are stronger for more recent compared to older birth cohorts.
Figure 5.
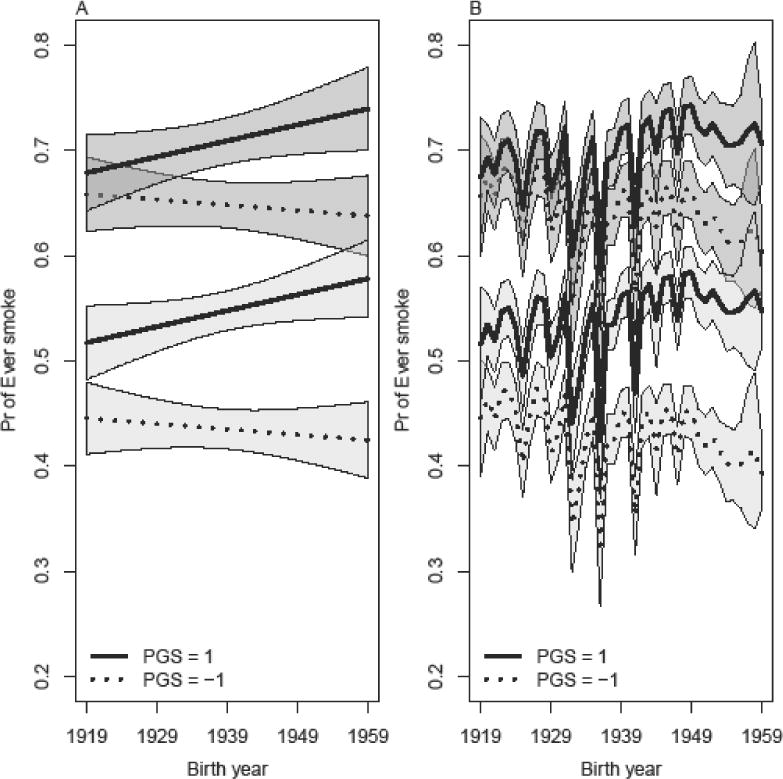
Estimated probability of ever smoking as a function of PGS, gender, and birth year. (A) includes controls for PGS, birth year, gender, interaction of PGS and gender, and the interaction of PGS and birth year. (B) includes controls PGS, main effects for three splines based on birth year, gender, interaction of PGS and gender, and interaction of PGS and birth year. Estimates for females are indicated via the darker gray confidence intervals.
5. Discussion
In this paper, we use three different statistical methods (GREML, GWDF, and PGS) and a nationally representative sample to evaluate the claim that the heritability of smoking has changed over recent birth cohorts. Our research relies on genetic inference using genome-wide similarity among unrelated persons rather than the use of twins as in previous work (Boardman et al. 2010; 2011). The most important result to emerge was a consistency with the direction and functional form of the work published earlier. Overall, we observe an increase in the relative contribution of genotype to explaining variation in smoking behavior. For the results based on genetic similarity (GREML and GWDF), the increase was from 1939 to 1953 whereas results from PGS approach were focused on an estimated increase over the entire timespan from 1919 to 1959. Confidence intervals for the genetic similarity results are quite large and limit our ability to make authoritative claims. Results based on the risk score analysis are stronger, although there are potential limitations for these findings as well. For example, it could be that the later birth cohorts are more comparable to the samples used in the TAG (2010) GWAS and that this is the reason for apparent rise in association between genotype and phenotype. Thus, we cannot say why the two sets of results differ, though we do want to emphasize that the upward trend is consistent across the middle of the twentieth century in both analyses.
Our work has implications for the gene-environment interaction literature. Our results suggest that the environment—birth cohort in our case—is critical for understanding genetic contributions to a particular health behavior. We hypothesize that those with the relevant genetic risk factors for smoking are smoking as a function of the genetic risk in a fairly consistent manner in all cohorts. However, those without genetic risks are responding to social cues that are changing over time. The environment is not causing genes to operate differently, rather the environment simply clarifies or masks when genotype is associated with smoking phenotype at a population level. Boardman et al. (2012) make a similar claim when they show that the link between the risky e4 allele in the ApoE gene more strongly predicts cognitive decline in the most organized and safe neighborhoods and has very little to do with cognitive decline in the most disorganized neighborhoods. Some have called this the social push perspective (Raine 2002). As the name suggests, when the environment is pushing the phenotype, such as for smoking amongst those born in the 1920s and cognitive decline for those in disorganized neighborhoods, genotype-phenotype associations are harder to observe.
Our guiding hypothesis has been that as more information and evidence emerged about the dangers of smoking during the second half of the Twentieth century, the biology of tobacco use become more salient since those who were able to heed the novel information (i.e. were less biochemically or behaviorally drawn to nicotine) dropped the habit or did not start, leaving those most biologically prone to smoke in the dwindling group of tobacco users. In this dynamic, the release of information like the 1964 Surgeon General’s Report on the dangers of smoking would have leveled the environmental playing field. That is, post-1964, we begin to see a general reduction in the environmental variation responsible for smoking, leaving a greater proportion of the overall variation to be explained by genetics. An alternative dynamic is that as the dangers of smoking became increasingly well known, this increased variance in information available to potential smokers (c.f., the fundamental cause hypothesis of Link and Phelan [1995]). Under this scenario heritability should fall over time as the variance in environment (i.e. information about smoking) increases. As shown here, we did not find evidence for this alternative scenario.
That does not mean, however, that multiple dynamics are not driving changes in heritability over time in our data. In addition to the changing informational landscape, other relevant aspects of the environment may be increasing or decreasing in salience (such as relative income shares and the cost of tobacco products). Furthermore, it could be the case that those who were the most biologically prone to smoking may also experience higher mortality incidence in our sample due to cigarette consumption. This would most likely lead to an underestimation of the increase in heritability that we document since those with the strongest genotypic influences are not observed due to premature death. However, if the same genotype that draws individuals to smoke more also increases their robustness to the ill-effects of tobacco products, the effect of selective mortality could work in the opposite direction: reducing the number of “environmentally induced smokers” earlier so that we do not observe them, leaving those who have the pro-smoking, pro-survival genotype in our sample. Thus, disentangling these forces across birth cohorts is a difficult task. We hope that the present paper has started on that worthy endeavor, and the issues we raise deserve further attention in future research.
Finally, the findings specific to females are worth further attention. Specifically, we show that there has been a larger divergence in the predicted smoking behavior of a genetically predisposed female to smoke versus a female without the same genetic predisposition than when compared to men (Figure 5A). That is, the role of genotype on smoking behavior is stronger for women compared to men and, more importantly, this association seems to have increased over time. These findings are somewhat contradictory to other research showing that genetic risk factors for substance use phenotypes are stronger for men compared to women (Hamilton et al. 2006; Perry et al. 2013). The GxE literature has found evidence for two very different mechanisms behind these somewhat contradictory results. The social trigger mechanism would anticipate that differences in norms and social roles played by men and women, trigger otherwise latent genetic tendencies to respond to stress with externalizing behaviors like cigarette smoking (Jackson et al. 2010). In this manner, genetic associations should be stronger for men. On the other hand, the social distinction perspective (Boardman, Freese, and Daw 2013) anticipates that cigarette smoking is driven by social factors for men more than women and thus genetic sensitivity to nicotine dependence may have a stronger signal among women compared to men. This is in line with the results that we present here. While this does merit further attention, the increase in smoking amongst later-born females in Figure 2 may also suggest that it may be due to the selection in our sample. That is, the increasing rates of smoking among women in our sample, compared to the decreasing rates of smoking among men, points to increasingly select groups of smokers who may differ by gender. We examined the sensitivity of our results to this type of attrition based selection (based on our models controlling for birth year, especially those which used the B-splines), but it is also possible that this pattern reflects a far more complicated story about the selection into the smoker status, the role of genes, the role of gender, and the role of broad and complex social structures. This should be the grist of future scholarship.
Acknowledgments
This research was supported, in part, by the following grants from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD): R21 HD078031, and R24 HD066613 which supports the CU Population Center. The Health and Retirement Study is sponsored by the National Institute on Aging (grant number NIA U01AG009740) and is conducted by the University of Michigan.
Appendix: GWDF Validation
We conducted a simulation to demonstrate a correspondence in relatively simple settings between GREML heritability estimates and the b estimate from Eqn 2. This simulation was based on a random sample of 5,000 respondents from the full set of respondents and random sample of 200,000 SNPs. Based on this set of respondents and SNPs, we simulated three sets of phenotypes using GCTA. The sets differed only in the true heritability of the phenotypes. The true heritabilites for the three sets were 0.25, 0.5, and 0.75. For each level of heritability, we simulated ten phenotypes. Thus, we have 30 simulated phenotypes in total.
Genetic similarities from the full set of markers (not the reduced set of 200,000 used to simulate the phenotypes) were then used to compute GREML heritability estimates as well as the b GWDF coefficient. Results are shown in Figure A1. Figures A1A and A1B show that the GREML heritability and GWDF coefficient estimates both increase along with the true heritability. The GREML heritability estimates and GWDF b3 estimates are correlated with the true heritabilities at 0.91 and 0.86 respectively. More importantly, the heritability estimates were strongly correlated (0.92) with the GWDF b3 estimates. The convergence of these results using two very different statistical techniques enhances our confidence in the validity of the GWDF approach and the empirical results that we present in the paper.
Figure A1.
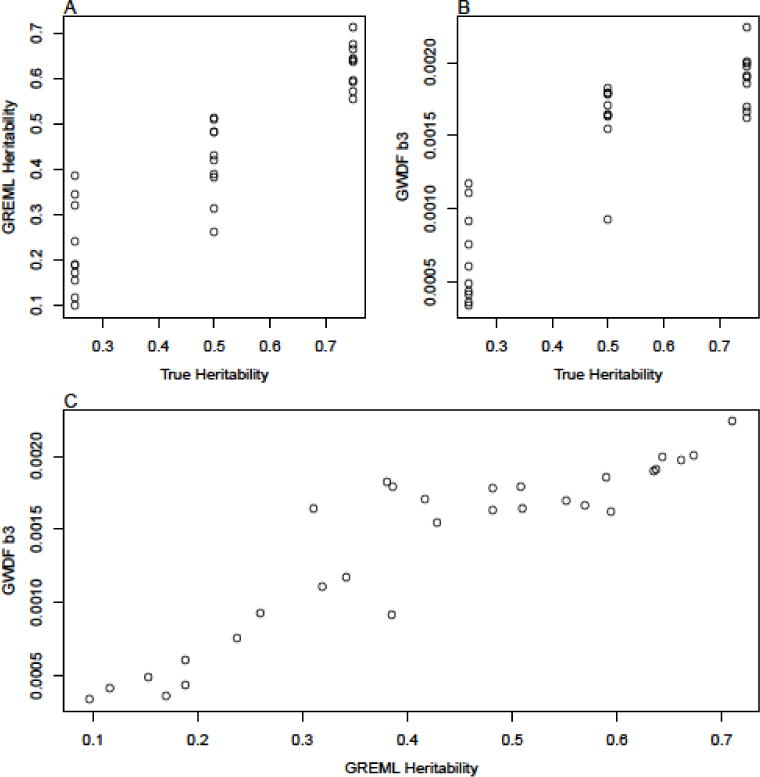
Comparison of true heritability (which is known since phenotypes are simulated), GREML estimates, and b3 estimates from GWDF models.
Footnotes
Specifically we use the RAND Fat Files (Clair et al., 2011).
Clumping takes place in two steps. The first pass is done in fairly narrow windows (250kb) for all SNPs (the p-value significance thresholds for both index and secondary SNPs is set to 1) with a liberal LD threshold (r2=0.5). In a second pass, SNPs remaining after the first prune are again pruned in broader windows (5000kb) but with a more conservative LD threshold (r2=0.2).
Available at http://spark.rstudio.com/ctgg/gctaPower/. The method is described in Visscher et al. (2014).
We used linear regression models instead of logistic regression models so as to ease interpretation of the relevant coefficients.
Estimates for PGS and its interaction with birth year and being female were 0.010 (p=0.43), 0.001 (p=0.06), and 0.026 (p=0.01) respectively.
Estimates for PGS and its interaction with birth year and being female were 0.010 (p=0.50), 0.001 (p=0.05), and 0.026 (p=0.01) respectively.
References
- Belsky DW, Moffitt TE, Houts R, Bennett GG, Biddle AK, Blumenthal JA, Caspi A. Polygenic risk, rapid childhood growth, and the development of obesity: evidence from a 4-decade longitudinal study. Archives of pediatrics & adolescent medicine. 2012;166(6):515–521. doi: 10.1001/archpediatrics.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Moffitt TE, Baker TB, Biddle AK, Evans JP, Harrington H, Caspi A. Polygenic risk and the developmental progression to heavy, persistent smoking and nicotine dependence: evidence from a 4-decade longitudinal study. JAMA psychiatry. 2013;70(5):534–542. doi: 10.1001/jamapsychiatry.2013.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Saint Onge JM, Haberstick BC, Timberlake DS, Hewitt JK. Do schools moderate the genetic determinants of smoking? Behavior genetics. 2008;38(3):234–246. doi: 10.1007/s10519-008-9197-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Blalock CL, Pampel FC. Trends in the genetic influences on smoking. Journal of health and social behavior. 2010;51(1):108–123. doi: 10.1177/0022146509361195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Blalock CL, Pampel FC, Hatemi PK, Heath AC, Eaves LJ. Population composition, public policy, and the genetics of smoking. Demography. 2011;48(4):1517–1533. doi: 10.1007/s13524-011-0057-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Barnes LL, Wilson RS, Evans DA, de Leon CFM. Social disorder, APOE-E4 genotype, and change in cognitive function among older adults living in Chicago. Social Science & Medicine. 2012;74(10):1584–1590. doi: 10.1016/j.socscimed.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Domingue BW, Daw J. What can genes tell us about the relationship between education and health? Social Science & Medicine. 2015;127:171–180. doi: 10.1016/j.socscimed.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman JD, Fletcher JM. To cause or not to cause? That is the question but identical twins might not have all of the answers. Social Science & Medicine. 2015;127:198–200. doi: 10.1016/j.socscimed.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) Smoking-attributable mortality, years of potential life lost, and productivity losses–United States, 2000–2004. MMWR. Morbidity and mortality weekly report. 2008;57(45):1226. [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. arXiv preprint. 2014;arXiv:1410.4803. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clair PS, Bugliari D, Campbell N, Chien S, Hayden O, Hurd M, Zissimopoulos J. RAND HRS Data Documentation, Version L. 2011. [Google Scholar]
- Conley D, Cesarini D, Dawes C, Domingue B, Boardman JD. Is the effect of parental education on offspring biased or moderated by genotype? Sociological Science. 2015;2:82–105. doi: 10.15195/v2.a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler DM, Glaeser EL. Developments in the Economics of Aging. University of Chicago Press; 2009. Why do Europeans smoke more than Americans? pp. 255–282. [Google Scholar]
- Daw J, Shanahan M, Harris KM, Smolen A, Haberstick B, Boardman JD. Genetic Sensitivity to Peer Behaviors 5HTTLPR, Smoking, and Alcohol Consumption. Journal of health and social behavior. 2013;54(1):92–108. doi: 10.1177/0022146512468591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFries JC, Fulker DW. Multiple regression analysis of twin data. Behavior genetics. 1985;15(5):467–473. doi: 10.1007/BF01066239. [DOI] [PubMed] [Google Scholar]
- Domingue BW, Fletcher J, Conley D, Boardman JD. Genetic and educational assortative mating among US adults. Proceedings of the National Academy of Sciences. 2014;111(22):7996–8000. doi: 10.1073/pnas.1321426111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS genetics. 2013;9(3):e1003348. doi: 10.1371/journal.pgen.1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobedo LG, Peddicord JP. Smoking prevalence in US birth cohorts: the influence of gender and education. American Journal of Public Health. 1996;86(2):231–236. doi: 10.2105/ajph.86.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AS, Lessov-Schlaggar CN, Cockburn MG, Unger JB, Cozen W, Mack TM. Gender differences in determinants of smoking initiation and persistence in California twins. Cancer Epidemiology Biomarkers & Prevention. 2006;15(6):1189–1197. doi: 10.1158/1055-9965.EPI-05-0675. [DOI] [PubMed] [Google Scholar]
- Jackson JS, Knight KM, Rafferty JA. Race and unhealthy behaviors: chronic stress, the HPA axis, and physical and mental health disparities over the life course. American journal of public health. 2010;100(5):933–939. doi: 10.2105/AJPH.2008.143446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MD, Cheng R, Ma JZ, Swan GE. A meta-analysis of estimated genetic and environmental effects on smoking behavior in male and female adult twins. Addiction. 2003;98(1):23–31. doi: 10.1046/j.1360-0443.2003.00295.x. [DOI] [PubMed] [Google Scholar]
- Link BG, Phelan J. Social conditions as fundamental causes of disease. Journal of health and social behavior. 1995:80–94. [PubMed] [Google Scholar]
- Lubke GH, Hottenga JJ, Walters R, Laurin C, De Geus EJ, Willemsen G, Boomsma DI. Estimating the genetic variance of major depressive disorder due to all single nucleotide polymorphisms. Biological psychiatry. 2012;72(8):707–709. doi: 10.1016/j.biopsych.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumley T. Analysis of complex survey samples. Journal of Statistical Software. 2004;9(1):1–19. [Google Scholar]
- Perry BL, Pescosolido BA, Bucholz K, Edenberg H, Kramer J, Kuperman S, Nurnberger JI., Jr Gender-specific gene–environment interaction in alcohol dependence: the impact of daily life events and GABRA2. Behavior genetics. 2013;43(5):402–414. doi: 10.1007/s10519-013-9607-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, Haworth CM, Meaburn EL, Price TS, Davis OS. Common DNA markers can account for more than half of the genetic influence on cognitive abilities. Psychological Science. 2013;0956797612457952 doi: 10.1177/0956797612457952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Fraser G. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehkopf DH. Understanding the role of social and economic factors in GCTA heritability estimates. Presented at the 5th Annual IGSS Conference; Boulder, CO. 2014. [Google Scholar]
- Raine A. Biosocial studies of antisocial and violent behavior in children and adults: A review. Journal of abnormal child psychology. 2002;30(4):311–326. doi: 10.1023/a:1015754122318. [DOI] [PubMed] [Google Scholar]
- Rietveld CA, Medland SE, Derringer J, Yang J, Esko T, Martin NW, McMahon G. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science. 2013;340(6139):1467–1471. doi: 10.1126/science.1235488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenquist JN, Lehrer SF, O’Malley AJ, Zaslavsky AM, Smoller JW, Christakis NA. Cohort of birth modifies the association between FTO genotype and BMI. Proceedings of the National Academy of Sciences. 2015;112(2):354–359. doi: 10.1073/pnas.1411893111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan MJ, Hofer SM. Social context in gene–environment interactions: Retrospect and prospect. The Journals of Gerontology Series B: Psychological Sciences and Social Sciences. 2005;60(Special Issue 1):65–76. doi: 10.1093/geronb/60.special_issue_1.65. [DOI] [PubMed] [Google Scholar]
- Speed D, Balding DJ. Relatedness in the post-genomic era: is it still useful? Nature Reviews Genetics. 2015;16(1):33–44. doi: 10.1038/nrg3821. [DOI] [PubMed] [Google Scholar]
- Spitz MR, Amos CI, Dong Q, Lin J, Wu X. The CHRNA5-A3 region on chromosome 15q24-25.1 is a risk factor both for nicotine dependence and for lung cancer. Journal of the National Cancer Institute. 2008;100(21):1552–1556. doi: 10.1093/jnci/djn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nature genetics. 2010;42(5):441–447. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink JM, Boomsma DI. Interplay between heritability of smoking and environmental conditions? A comparison of two birth cohorts. BMC public health. 2011;11(1):316. doi: 10.1186/1471-2458-11-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Hemani G, Vinkhuyzen AA, Chen GB, Lee SH, Wray NR, Yang J. Statistical power to detect genetic (co) variance of complex traits using SNP data in unrelated samples. PLoS genetics. 2014;10(4):e1004269. doi: 10.1371/journal.pgen.1004269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, McGue M, Miller MB, Hicks BM, Iacono WG. Three mutually informative ways to understand the genetic relationships among behavioral disinhibition, alcohol use, drug use, nicotine use/dependence, and their co-occurrence: Twin biometry, GCTA, and genome-wide scoring. Behavior genetics. 2013;43(2):97–107. doi: 10.1007/s10519-013-9584-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research. 2014;42(D1):D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Visscher PM. Common SNPs explain a large proportion of the heritability for human height. Nature genetics. 2010;42(7):565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajacova A, Burgard SA. Healthier, wealthier, and wiser: a demonstration of compositional changes in aging cohorts due to selective mortality. Population research and policy review. 2013;32(3):311–324. doi: 10.1007/s11113-013-9273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]