

Abstract
Residualizing labeling methods for internalizing peptides and proteins are designed to trap the radionuclide inside the cell after intracellular degradation of the biomolecule. The goal of this work was to develop a residualizing label for the 18F-labeling of internalizing biomolecules based on a template used successfully for radioiodination. N-succinimidyl 3-((4-(4-[18F]fluorobutyl)-1H-1,2,3-triazol-1-yl)methyl)-5-(bis-Boc-guanidinomethyl)benzoate (Boc2-[18F]SFBTMGMB) was synthesized by click reaction of an azide precursor and [18F]fluorohexyne in 8.5 ± 2.8% average decay-corrected radiochemical yield (n =15). An anti-HER2 nanobody 5F7 was labeled with 18F using [18F]SFBTMGMB ([18F]RL-I), obtained by the deprotection of Boc2-[18F]SFBTMGMB, in 31.2 ± 6.7% (n =5) conjugation efficiency. Thus labeled nanobody had a radiochemical purity of >95%, bound to the HER2-expressing BT474M1 breast cancer cells with an affinity of 4.7 ± 0.9 nM, and had an immunoreactive fraction of 62–80%. In summary, a novel residualizing prosthetic agent for labeling biomolecules with 18F has been developed. An anti-HER2 nanobody was labeled using this prosthetic group with retention of affinity and immunoreactivity to HER2.
Introduction
Radionuclide imaging using peptides and monoclonal antibodies (mAbs) is widely used for a number of applications. These include diagnosing cancers, quantifying expression of a particular target of interest, determination of radiation dosimetry prior to administering the same vector labeled with a therapeutic radionuclide, and to determine the pharmacokinetics of the labeled protein or peptide. The radiolabeling method has a significant influence on the biodistribution of the tracer.1 For molecules that undergo internalization after binding to receptors on the tumor cell, which subsequently undergo proteolysis within the lysosomal compartment, residualizing labels (RL) are preferred. Residualizing labels can potentially enable each of the radionuclide imaging applications noted above if the targeting vector undergoes extensive internalization after receptor binding. If higher tumor uptake and hence higher tumor-to-tissue ratios can be achieved using RLs compared with conventional prosthetic agents, then that will certainly be an advantage. While radiometal labeling is generally considered residualizing, radiohalogenation by the direct electrophilic approach is non-residualizing. This reflects the fact that monoiodotyrosine and free iodide, the primary radiolabeled catabolites from molecules radioiodinated by the direct method, wash out of the cell rather quickly resulting in lower cumulated radioactivity in tumor.
To overcome this problem, we and others have developed residualizing labels for the radiohalogenation of internalizing biomolecules.2–6 One of the agents we developed, N-succinimidyl 4-guanidinomethyl-3-iodobenzoate (SGMIB), contains a guanidine function that remains predominantly positively charged at lysosomal pH, and was designed on the basis that positively charged radiolabeled catabolites will remain trapped within the cells. Excellent results with respect to augmenting tumor retention of radioactivity from internalizing molecules radioiohalogenated using this template have been obtained.7, 8
Nanobodies (Nbs), a.k.a. VHH molecules and single domain antibody fragments (SdAbs), are the antigen-binding fragments of heavy-chain-only antibodies from Camelidae.9–11 Their molecular weight (12–15 kDa) is an order of magnitude less than that of intact mAbs, and considerably less than that of Fab (~50 kDa) or scFv (~25 kDa) fragments. SdAbs exhibit nanomolar affinities, high thermal and chemical stability, are more water-soluble compared with intact mAbs and their fragments, and have a lower tendency for aggregation. SdAbs specific to a number of molecular targets such as the growth factor receptor tyrosine kinase HER2 as well as epidermal growth factor receptor (EGFR) have been generated. There is an emerging interest in exploiting the excellent properties of SdAbs as a platform for the development of imaging agents for the quantification of HER2 and EGFR status by positron emission tomography (PET) in cancer patients. Based on the results we have obtained, maximum tumor uptake and contrast can be obtained within 2–4 hours of administration of labeled SdAbs,7, 12 making widely available 18F perhaps the ideal positron-emitter for labeling SdAbs.
Although a number of prosthetic agents have been reported for 18F-labeling of peptides and proteins,13–15 N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) — an agent originally developed by us16 — remains the most widely used agent for this purpose. There have been a few reports of [18F]SFB being utilized to label biomolecules reactive with internalizing molecular targets, and in these cases 18F activity in tumor in vivo reduced with time.17, 18 It seems likely that this behavior could in part reflect the lack of residualizing moieties in the structure of SFB, making this reagent less than ideal for labeling internalizing biomolecules such as anti-HER2 SdAbs. To investigate the potential advantages of a residualizing label for 18F-labeling, we have designed a new prosthetic agent combining the structural features of SGMIB and SFB.
Herein, we describe a method for the synthesis of N-succinimidyl 3-((4-(4-[18F]fluorobutyl)-1H-1,2,3-triazol-1-yl)methyl)-5-(guanidinomethyl)benzoate ([18F]SFBTMGMB), heretofore referred to as [18F]RL-I. An anti-HER2 SdAb 5F7 was labeled using [18F]RL-I and the integrity of the labeled SdAb as well as its affinity and immunoreactivity to HER2 were retained.
Results and Discussion
The goal of this work was to develop a residualizing agent for labeling internalizing biomolecules such as anti-HER2 nanobodies with 18F. Although we have developed a number of residualizing labels for radioiodination, unlike the case with intact mAbs, the best tumor targeting with SdAbs was seen when radioiodination was performed using the guanidine-containing prosthetic agent SGMIB.7 Radiosynthesis of an analogous agent — N-succinimidyl 3-[18F]fluoro-4-guanidinomethylbenzoate ([18F]SFGMB) — wherein the radioiodine in SGMIB is replaced with 18F, would be difficult. Introduction of 18F onto an aromatic ring by classical SNAr substitution is facilitated by the presence of electron-withdrawing groups at the ortho- and para-positions of the nucleofuge. In the SGMIB molecule, the iodine is present at the meta position of the activating ester group. Furthermore, because the active ester is susceptible under 18F-labeling conditions due to nucleophilic attack by fluoride or the constituent base,19 radiosynthesis of [18F]SFGMB by using SNAr substitution for 18F introduction will involve multiple steps. While higher radiochemical yields theoretically can be obtained for an isomeric compound wherein 18F is placed at the para-position of the ester group, its radiosynthesis will still involve multiple steps due to the lability of the active ester to 18F-labeling conditions. For these reasons, we resorted to a molecule containing a fluoroalkyl side chain because it is easier to introduce 18F on an sp3 carbon by SN2 substitution. The logical approach for this would be to have a precursor with an alkyl chain bearing a sulfonate leaving group, the displacement of which with nucleophilic [18F]fluoride would deliver the required reagent in a single step. However, because of the instability of the N-succinimidyl ester under 18F-labeling conditions as noted above, an alternative approach involving a click reaction was utilized (Scheme 1). An added advantage of this strategy is the presence of the polar triazole20 ring in its structure, which may contribute to its residualizing ability. Commercially available 3,5-dimethylbenzoic acid was converted to its TMSE ester 2 in 79% yield. Benzylic bromination and subsequent debromination of polybrominated side products with diethyl phosphite21, 22 rendered the bis-bromomethyl derivative 3 in 43% yield. Monoguanidinylation of 3 adapting a reported procedure23 delivered 4 in 33% yield. This guanidine derivative was treated with sodium azide to obtain 5 in almost quantitative yields. Compound 5 was converted to 7, the precursor for 18F-labeling, by fluoride-mediated deprotection of the TMSE ester, and in situ reesterification with N-hydroxysuccinimide in 35% yield for the two steps. Two approaches were taken to synthesize the protected standard 8. In the first approach, 5 was converted to 6 in 54% by its click reaction with 6-fluorohex-1-yne, which was synthesized by the deoxofluorination24 of hex-5-yn-1-ol or from the tosylate precursor as reported.25, 26 Removal of the TMSE group from 6 and in situ reesterification with N-hydroxysuccinimide resulted in 8 in 40% yield for the two steps. In the alternative approach, 7 was subjected to click reaction to obtain 8 in 44% yield. The final deprotected compound SFBTMGMB (9) was synthesized by TFA-mediated deprotection of 8 in 96% yield.
Scheme 1.

Synthesis of SFBTMGMB and its azide precursor.
The approach taken for the synthesis of [18F]SFBTMGMB-Boc2 is shown in Scheme 2. Various conditions were tried to optimize radiochemical yields for the two steps. In the first step, converting the tosylate precursor to 6-[18F]fluorohex-1-yne,26 initially, a large excess — up to 50 mg — of the tosylate precursor was used to offset its loss to side reactions like hydrolysis and β-elimination. Click reactions were initially performed without a ligand. Considerable improvement in radiochemical yields were obtained by including the water-soluble ligand bathophenanthroline disulphonate (BPDS).27 TLC (1:4 ethyl acetate:hexanes) of the HPLC injectate indicated [18F]SFBTMGMB-Boc2 (Rf = 0.5 – 0.6) was the predominant radioactive species (50–60%) with about 25% of radioactivity remaining at Rf = 0; however, up to 50% of the 18F activity was retained in the normal phase HPLC column. The same phenomenon was observed when HPLC-purified [18F]SFBTMGMB-Boc2 was reinjected onto the HPLC column. While the reason for this is not apparent, we think it might be due to the hydrolysis of the compound in the column. It is also likely that the active ester could be reacting with the amino groups on the stationary phase of the HPLC column, forming a covalent linkage. In future studies, further refinements in methods will be introduced to avoid HPLC purification without affecting the purity of the final product. Under optimized conditions (vide infra), [18F]SFBTMGMB-Boc2 was synthesized from aqueous fluoride in an overall decay-corrected radiochemical yield of 8.5 ± 2.8% (n =15); 207.2 ± 66.6 MBq (5.6 ± 1.8 mCi) could be obtained starting with 3.7 GBq (100 mCi) of aqueous [18F]fluoride in about 100 min, which includes HPLC purification. HPLC-purified [18F]SFBTMGMB-Boc2 was more than 95% radiochemically pure and generally no detectable UV peaks were seen in the quality control HPLC runs (Fig. 1). The specific activity of purified [18F]SFBTMGMB-Boc2 was greater than 9.3 TBq (250 Ci)/mmol. [18F]SFBTMGMB-Boc2 was deprotected by treatment with TFA to generate [18F]RL-I ([18F]9) and used as such for coupling with SdAb.
Scheme 2.
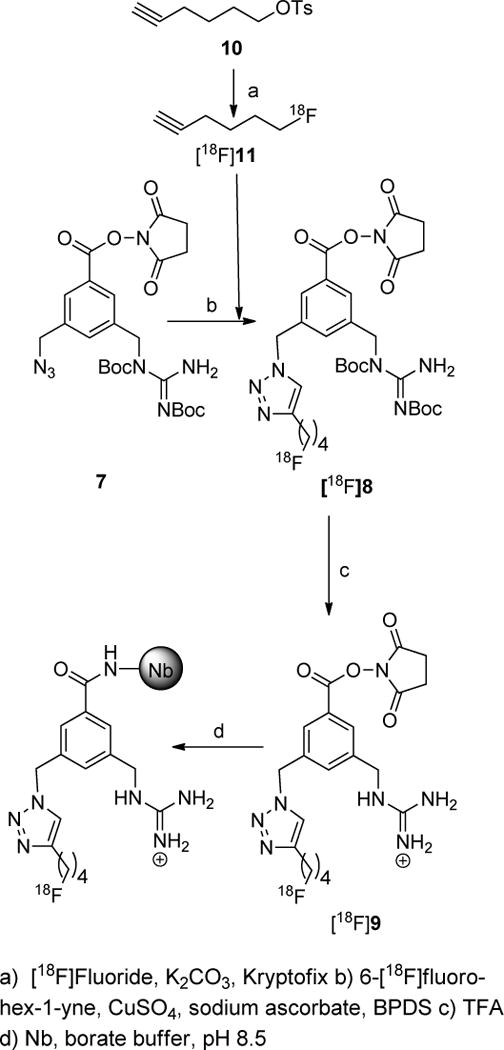
Synthesis of [18F]SFBTMGMB and its coupling to the Nanobody.
Figure 1.
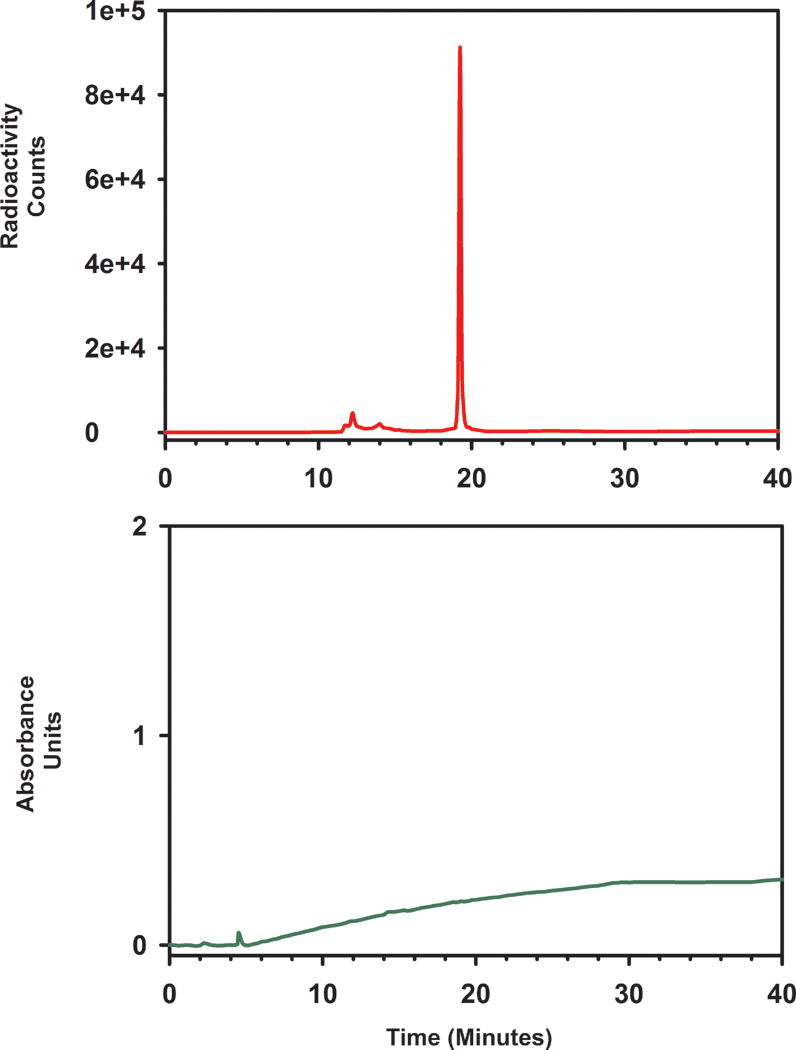
Quality Control HPLC of Boc2-[18F]SFBTMGMB.
Yields for coupling of [18F]SFBTMGMB to SdAb was erratic in early trials. It was thought that this might be due to the consumption of the SdAb by either the co-eluting unalabeled carrier potentially present in the mixture, or the azide precursor that could have bled into the radioactive peak, although, there was considerable difference in the retention time of 5 and 8 (~10 min and 20 min, respectively). In initial experiments, up to 7 mg of the azide precursor was used to facilitate the click reaction; subsequently, it was found that 3 mg was sufficient to get similar click reaction yields. This amount itself was probably too large a quantity to avoid HPLC co-elution. Attempts were made to scavenge the unreacted azide precursor by click reaction with a polymer-bound alkyne that we synthesized by coupling propiolic acid to 4-(bromomethyl)phenoxymethyl polystyrene following a reported procedure28. Parenthetically, while this work was in progress, a similar strategy for scavenging excess alkyne-modified peptide with a immobilized azide was reported.29 Scavenging unreacted azide precursor neither eliminated the presence of an unlabeled compound that closely eluted with [18F]SFBTMGMB-Boc2 in quality control HPLC nor gave the labeled SdAb in reasonable yields consistently.
It was reasoned that the closely-eluting peak was not the azide precursor as suspected but might be the product of click reaction between excess azide and the di-hexynyl ether (12; Scheme 3). The occurrence of side reactions — β-elimination and hydrolysis — during fluorination via SN2 reaction of aliphatic substrates, which result in the production of the corresponding alkene and alcohol, respectively, is often reported in the literature; however, although logical, formation of dialkyl ether has rarely been mentioned (Scheme 4). It can be generated by the reaction of alcohol/alkoxide, formed by the hydrolysis of substrates such as tosylates, with excess of the substrate. A careful search of the literature did lead to a report30 wherein such ether formation has been mentioned. During the synthesis of unlabeled 6-fluorohex-1-yne by the reaction of tosylate precursor 10 with TBAF, di-hexynyl ether 12 was isolated (see Electronic Supplementary Information) but no formation of the corresponding alkene, hex-1-en-5-yne was seen. Formation of the alkene resulting from β-elimination has been reported from 4-tosyloxy-1-butyne but not from 5-tosyloxy-1-pentyne upon treatment with potassium [18F]fluoride,31 suggesting that it is even less likely that the 6-fluorohex-1-yne will be formed from 6-tosyloxy-1-hexyne (hex-5-yn-1-yl 4-methylbenzenesulfonate). To explore whether 12 was formed during the radiochemical synthesis of 6-[18F]fluorohex-1-yne and whether it underwent click reaction with 7 during the synthesis of [18F]SFBTMGMB-Boc2, compound 13 was synthesized by the click reaction of azide 7 and 12, which was obtained by the reaction of the potassium salt of hex-5-yn-1-ol with the tosylate precursor 10. Indeed, the retention time of 13 on HPLC was the same as that of the byproduct that eluted very close to [18F]SFBTMGMB-Boc2. Although circumstantial, this suggests that the byproduct may very well be 13. Having an NHS moiety present in its structure, the deprotected derivative of 13 can compete with [18F]RL-I for reaction with the nanobody, which might explain the less than expected yields for the coupling of [18F]RL-I to SdAb. While there is a possibility of formation of the bis-adduct by the click reaction of 7 on both ends of 12, it was assumed that such a product would be considerably more polar than [18F]SFBTMGMB-Boc2. In considering the options to eliminate the nuisance byproduct, we first thought of clicking it with 4-azidomethyl- benzoic acid or benzene sulfonic acid to increase its polarity. Similar approaches have been reported recently for a peptide radiopharmaceutical32 and amino acids.33 We opted to evaluate a better scavenging agent, 4-(azidomethyl)-N,N,N-trimethylbenzenaminium triflate (14) assuming that the clicked product 15 can be easily washed away with water from the ethereal extract of reaction mixture. Compound 14 was synthesized by quaternization of the known compound 4-dimethylamino benzyl azide,34 which was synthesized starting from 4-dimethylamino benzaldehdye as detailed in the experimental section. An additional in situ click reaction using 14 as the azide partner, after the click reaction involving the 6-[18F]flurohex-1-yne and the azide precursor 7 considerably reduced the formation of the closely eluting byproduct in the synthesis of Boc2-[18F]SFBTMGMB, and improved the efficiency of coupling of [18F]RL-I to SdAb. This clearly indicates that the larger amounts of compound 13, which has an active ester in its structure, was consuming most of the SdAb, making it less available for conjugation with [18F]RL-I.
Scheme 3.

Path to the formation of side product (13) and the approach to scavenging it.
Scheme 4.
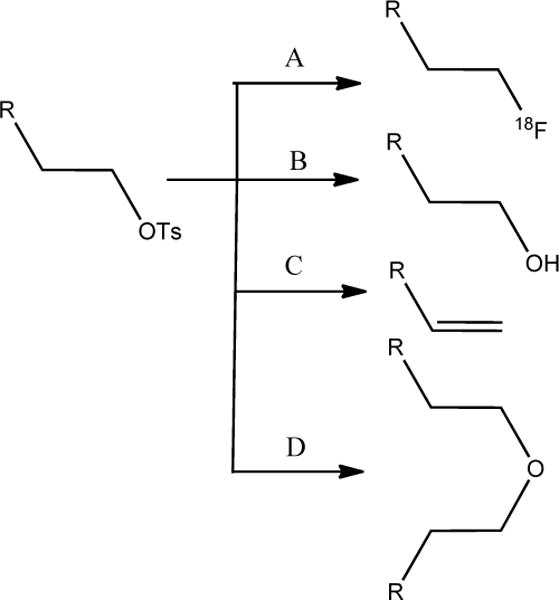
Four different possible products during the fluorination of a sulfonate precursor. A) Substitution B) Hydrolysis C) Elimination D) Ether formation
Even with the use of 14 for scavenging the putative 13, there were occasional failures in the coupling reaction. As noted before, like others,26 we used a fairly large amount of the tosylate precursor, which presumably led to the formation of 12 and in turn, 13. It was possible to reduce the amounts of precursor 10 considerably without concomitant loss of radiochemical yields for the initial labeling step. When only about 9 mg versus 50 mg of the tosylate precursor was used, radiochemical yields for the first step were not affected considerably. Although reduction in the amount of 10 itself may reduce the formation 13, we continued to do the second click reaction with 14 to ensure the absence of 13 during the coupling reaction with the SdAb.
An anti-HER2 SdAb (5F7) having no C-terminal cysteine and that exists exclusively in monomeric form was labeled with [18F]RL-I (Scheme 2). The efficiency for conjugation of 5F7 with [18F]RL-I synthesized using the optimized method (including the use of reduced amount of tosylate precursor) was 31.2 ± 6.7% based on the initial activity of [18F]RL-I (n = 5). The specific activity of the labeled protein was 74–444 MBq (2–12 mCi)/mg. TCA precipitation, SDS-PAGE/phosphor imaging (Fig. 2), and ITLC indicated that more than 95% of radioactivity was associated with the SdAb. The 5F7 SdAb labeled using [18F]RL-I ([18F]RL-I-5F7) bound specifically to the HER2-positive magnetic beads with an immunoreactive fraction (IRF) in the range of 62 – 84%. The [18F]RL-I-5F7 SdAb conjugate bound to BT474M1 cells with a Kd value of 4.7 ± 0.9 nM (Fig. 3). Similar low nanomolar affinity values have been obtained for another anti-HER2 SdAb radioiodinated by various methods.7 A Kd value of 0.29 nM, determined by surface plasmon resonance assay, has been reported for the unlabeled 5F7 SdAb.35 Our results indicate that SdAb 5F7 can be labeled using [18F]RL-I in reasonable yields, high specific activity and radiochemical purity, and with the preservation of affinity and immunoreactivity.
Figure 2.

SDS-PAGE (non-reducing)/Phophor image profile of [18F]RL-I-5F7 (lane 1) and of [18F]SFB-5F7 (lane 2).
Figure 3.
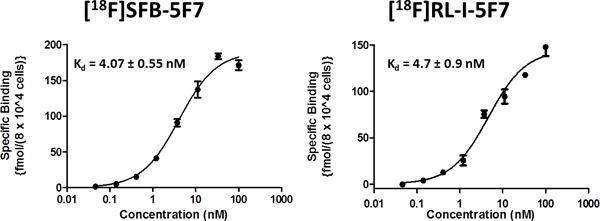
Saturation binding curves for [18F]SFB-5F7 and [18F]RL-I-5F7 obtained using BT474M1 cells.
For comparative evaluation, SdAb 5F7 was also conjugated with [18F]SFB ([18F]SFB-5F7) and [125I]SGMIB ([125I]SGMIB-5F7). SdAb 5F7 was conjugated with [18F]SFB in an average conjugation efficiency of 37.8 ± 11.5% (n = 2) with a specific activity of 370–740 MBq (10–20 mCi)/mg. TCA precipitation (96.8 ± 1.5%), SDS-PAGE/phosphor imaging (100%; Fig. 2), and ITLC (99.0 ± 0.5%) indicated that more than 95% of radioactivity was associated with the SdAb and it gave an IRF of 94.3%. Affinity determination by saturation binding assay gave a Kd value of 4.07 ± 0.55 nM (Fig. 3), not statistically different to that determined for [18F]RL-I-5F7. The prosthetic agent [125I]SGMIB was conjugated to 5F7 in 35.0 ± 11.2% (n = 4) yields with specific activity of 148 – 185 MBq (4–5 mCi)/mg. ITLC (97.6 ± 2.8%; n = 3) and TCA precipitation (96.7 ± 2.6%; n = 3) of [125I]SGMIB-5F7 indicated that more than 95% of radioactivity was associated with intact SdAb and its IRF was 79.3 ± 16.5% (n =2). Saturation binding assay of [125I]SGMIB-5F7 gave a Kd value of 5.02 ± 1.32 nM (Fig. S1).
Conclusion
A novel residualizing label for 18F-labeling was synthesized using a click reaction between [18F]fluorohexyne and an azide precursor containing an active ester and a guanidine moiety. An anti-HER2 SdAb was labeled with 18F using this prosthetic agent in reasonable yields with the preservation of affinity and immunoreactivity for HER2. The overall decay-corrected radiochemical yield of the final labeled nanobody from aqueous fluoride is only about 2–3%. A considerable improvement in radiochemical yield will be required before use of this agent on a routine basis can be considered. As an alternative, it may be possible to modify the protein with the deprotected derivative of azide precursor 7 first, and then perform the click reaction on the derivatized protein using the [18F]fluorohexyne. The copper-free click reaction using 18F-labeled cyclooctyne-containing derivatives that are reported in the literature36, 37 may fare better with proteins and we are currently pursuing such this strategy. As will be published elsewhere, higher retention of 18F activity in HER2 expressing tumor cells was observed both in cell culture and in a murine model for [18F]RL-I-5F7 compared with [18F]SFB-5F7.
Experimental
Chemistry
General
Chemicals and reagents were purchased from Sigma Aldrich (St. Louis, MO) unless noted otherwise. Sodium [125I]iodide in 0.1 N NaOH with specific activities of 81.4 TBq (2200 Ci)/mmol was purchased from Perkin-Elmer Life and Analytical Sciences (Boston, MA). The Boc-protected tin precursor of SGMIB and radioiodinated SGMIB were synthesized as reported before.8, 38 N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB)16, 39 was synthesized following a modified one-pot procedure.40 Aluminum-backed sheets (Silica gel 60 F254) used for analytical TLC and silica gel 60 for normal-phase column chromatography were obtained from EM Science (Gibbstown, NJ). In some cases, chromatography was also performed with the Biotage Isolera chromatography system (Charlotte, NC) using their pre-packed columns. Preparative thick layer chromatography was used for small-scale purification with plates obtained from Whatman (Clifton, NJ) or EM Science. High pressure liquid chromatography (HPLC) was performed using the following two systems: 1) for radiolabeled compounds: a Beckman Gold HPLC system equipped with a Model 126 programmable solvent module, a Model 166 NM variable wavelength detector, a Model 170 radioisotope detector and a Beckman System Gold remote interface module SS420X; data were acquired using the 32 Karat® software (Beckman Coulter, Inc., Brea, CA). Recently, the gamma detector in this system was replaced with a ScanRam RadioTLC scanner/HPLC detector combination (LabLogic; Brandon, Fl) and later radio HPLC analyses were performed with that detector. 2) for analytical and semi-preparative HPLC of unlabeled compounds, a Waters Model Delta 600 semi-preparative system with a Model 600 controller and a Model 2487 dual wavelength absorbance detector; data were acquired using Millenium software. Normal phase HPLC, used for purification of [18F]SFBTMGMB-Boc2, was performed using a 9.4 × 250 mm 5 μm Zorbax NH2 semi-preparative column obtained from Agilent Technologies (Santa Clara, CA). Reversed-phase HPLC was performed using a Waters 4.6 × 250-mm XTerra RP18 (5 μm) column and a 19 × 150-mm XTerra RP18 (7 μm) column for analytical and semi-preparative runs, respectively. PD-10 desalting columns for gel filtration were purchased from GE Healthcare (Piscataway, NJ). Instant thin layer chromatography (ITLC) was performed using silica gel impregnated glass fiber sheets (Pall Corporation, East Hills, NY) eluted with PBS, pH 7.4. The intact SdAb stays at the origin under these conditions. Developed sheets were analyzed for radioactivity either using the TLC scanner described above, or cutting the sheet into small strips and counting them in an automated gamma counter (LKB 1282, Wallac, Finland or Perkin Elmer Wizard II, Shelton, CT). Proton NMR spectra were obtained on a Varian 400 and 13C spectra on a Varian 500 (125.8 MHz) NMR spectrometer (Palo Alto, CA); chemical shifts are reported in δ units using the residual solvent peak as reference. Mass spectra were recorded using an Agilent LCMS-TOF with DART, a high resolution mass spectrometer used for ESI, DART and LC-MS. Elemental analyses were performed by Atlantic Microlabs, Inc. (Norcross, GA).
SdAb, cells, and culture conditions
Anti-HER2 SdAb 5F7, was a gift from Hilde Revets of Ablynx (Ghent, Belgium). All reagents used for cell studies were obtained from Invitrogen (Grand Island, NY). HER2-expressing BT474M1 human breast carcinoma cells41 were cultured in DMEM/F12 medium containing 10% fetal calf serum (FCS), streptomycin (100 μg/mL), and penicillin (100 IU/mL) (Sigma Aldrich, MO). Cells were cultured at 37°C in a humidified incubator under 5% CO2 with media changed every two days. When about 80% confluent, cells were sub-cultured by trypsinization (0.05 % Trypsin- EDTA).
2-(Trimethylsilyl)ethyl 3,5-dimethylbenzoate (2)
A mixture of 3,5-dimethylbenzoic acid (5.0 g, 33.3 mmol), 2-(trimethylsilyl)ethanol (3.9 g, 33.3 mmol), DMAP (1.5 mg, 0.01 mmol), and EDC (7.7 g, 40.0 mmol) in ethyl acetate (75 mL) was stirred at 20 °C overnight. The reaction mixture was washed with water and then with saturated NaHCO3. The ethyl acetate solution was separated, dried over MgSO4, and ethyl acetate removed by rotary evaporation. The residue was purified by chromatography using 10:1 hexanes:ethyl acetate to obtain 2 (6.6 g, 26.4 mmol, 79 % yield) as a an oil: 1H-NMR (CDCl3) δ 0.00 (s, 9H), 1.03 – 1.07 (t, 2H), 2.27 (s, 6H), 4.30 – 4.33 (t, 2H), 7.08 (s, 1H), 7.57 (s, 2H). 13C-NMR (CDCl3) δ 0.00, 19.50, 22.33, 64.10, 128.85, 132.05, 136.10, 139.48, 168.20. No tangible mass spectral data could be obtained for this compound. Anal. Calcd for C14H22O2Si: C, 67.15;. H, 8.86. Found: C, 67.28; H, 8.77.
2-(Trimethylsilyl)ethyl 3,5-bis(bromomethyl)benzoate (3)
A mixture of 2 (4.0 g, 16.0 mmol), NBS (11.4 g, 63.9 mmol), and benzoyl peroxide (0.4 g, 1.6 mmol) in dichloroethane (75 mL) was heated at reflux for 6 h. The reaction mixture was concentrated to dryness and the residue triturated with warm hexanes. The resultant free-flowing solid was filtered, and the filtrate was concentrated to dryness to give a semi-solid. It was dissolved in THF (100 mL) and the solution cooled to 0–5 °C. Diethyl phosphite (11.1 g, 80 mmol) and N,N-diisopropylethylamine (10.4 g, 80 mmol) were added to the above, and the reaction mixture stirred at 20 °C for 72 h. THF was evaporated, and the residue partitioned between water and ethyl acetate. The organic layer was separated, dried over MgSO4 and concentrated to dryness. The residue was purified by chromatography using 10:1 hexanes:ethyl acetate to afford 3 (2.8 g, 6.9 mmol, 42.9 % yield) as an oil: 1H-NMR (CDCl3) δ 0.07 (s, 9H), 1.11 – 1.23 (t, 2H), 4.38 – 4.43 (t, 2H), 4.48 (s, 4H), 7.58 (s, 1H), 7.96 (s, 2H). 13C-NMR (CDCl3) δ 0.00, 18.92, 33.37, 65.19, 131.40, 133.38, 135.12, 140.31, 167.02. No tangible mass spectral data could be obtained for this compound. Anal. Calcd for C14H20Br2O2Si: C, 41.19;. H, 4.94. Found: C, 41.25; H, 4.76.
2-(Trimethylsilyl)ethyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-(bromomethyl)benzoate (4)
A solution of potassium tert-butoxide in THF (1 M; 11 mL, 11 mmol) was added to a solution of 1,3-bis(tert-butoxycarbonyl)guanidine (1.4 g, 5.5 mmol) in DMF (25 mL), and the mixture stirred at 20 °C for 30 min. The above was added slowly over a period of 30 min to a stirred solution of 3 (4.5 g, 11.0 mmol) in DMF (25 mL), and the mixture stirred at 20 °C for 17 h. The mixture was partitioned between water and ethyl acetate, and the organics were dried over sodium sulfate, and concentrated. The residue was purified by chromatography using 5:1 hexanes:ethyl acetate to yield 4 (2.1 g, 3.6 mmol, 32.5 % yield) as solid: 1H-NMR (CDCl3) δ 0.08 (s, 9H), 1.11 – 1.15 (t, 2H), 1.37 (s, 9H), 1.49 (s, 9H), 4.38 – 4.46 (t, 2H), 4.41 (s, 2H), 5.22 (s, 2H), 7.51 (s, 1H), 7.88 (s, 1H), 7.95 (s, 1H), 9.25 (bs, 1H), 9.42 (bs, 1H). 13C-NMR (CDCl3) δ 0.01, 18.91, 29.34, 29.76, 33.97, 48.51, 64.96, 80.44, 86.05, 129.97, 130.18, 132.63, 133.98, 139.58, 141.41, 155.14, 162.02, 166.06, 167.53. LRMS (DART) m/z: 588.2 and 586.2 (M+H)+, 532.1 and 530.1 (M-tBu); HRMS (DART) calcd for C25H4179BrN3O6Si (M+H)+ 586.11948, found 586.1942 ± 0.0011 (n = 4).
2-(Trimethylsilyl)ethyl 3-(azidomethyl)-5-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)benzoate (5)
A mixture of 4 (1.2 g, 2.1 mmol) and sodium azide (0.13 g, 2.1 mmol) in DMF (25 mL) was stirred at 80 °C for 1.5 h. The reaction mixture was cooled to 20 °C, and partitioned between ethyl acetate and water. The pooled ethyl acetate solution was dried with MgSO4, and concentrated to dryness to afford 5 (1.1 g, 2.1 mmol, 98 % yield) as a white foamy solid. 1H-NMR (CDCl3) δ 0.08 (s, 9H), 1.11–1.15 (t, 2H), 1.37 (s, 9H), 1.49 (s, 9H), 4.37–4.45 (t, 2H), 4.27 (s, 2H), 5.20 (s, 2H), 7.50 (s, 1H), 7.87 (s, 1H), 7.93 (s, 1H), 9.25 (bs, 1H), 9.42 (bs, 1H). 13C NMR (CDCl3) δ 0.00, 18.92, 29.32, 29.76, 48.51, 55.80, 64.97, 80.43, 86.00, 129.35, 130.00, 132.61, 133.11, 137.29, 141.45, 156.16, 162.06, 165.08, 167.65. LRMS (DART) m/z: 549.3 (M+H)+, 493.2 (M-tBu)+; HRMS (DART) calcd for C25H41N6O6Si (M+H)+ 549.2857, found 549.2844 ± 0.0005 (n = 4).
2-(Trimethylsilyl)ethyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-((4-(4-fluorobutyl)-1H-1,2,3-triazol-1-yl)methyl)benzoate (6)
A solution of 5 (50 mg; 0.09 mmol) in DMF (3 mL) was mixed with a solution of 6-fluorohex-1-yne26 (excess) in dichloromethane. The click reaction reagent was prepared as follows: A solution of CuSO4·5H2O (10 mg, 0.04 mmol) in water (100 μL) was mixed with an aqueous solution of sodium ascorbate (30.0 mg, 0.15 mmol) (100μl). When the color of the mixture changed from black to yellow, a solution of the disodium salt of bathophenanthrolinedisulfonic acid (hydrate) (12.0 mg, 20.0 μmol) in 1:4 (v/v) DMF/H2O (200 μL) was added. A 100 μL portion of the click reaction reagent was added to the substrate solution, and the final mixture was heated at 80 °C for 30 min in a sealed reaction vessel. The reaction mixture was diluted with ethyl acetate and the solution washed with brine. The pooled ethyl acetate solution was dried using anhydrous MgSO4, ethyl acetate evaporated from the filtrate and the residue subjected to preparative TLC using 1:1 ethyl acetate:hexanes to obtain 32 mg (0.03 mmol, 54.1 % yield) of 6 as a solid: 1H-NMR (CDCl3) δ 0.06 (s, 9H), 1.08 – 1.16 (t, 2H), 1.32 (s, 9H), 1.43 (s, 9H), 1.56 – 1.93 (m, 2H), 1.97 – 2.15 (m, 2H), 2.97 – 3.05 (t, 2H), 4.38 – 4.60 (m, 6H), 5.72 (s, 2H), 7.62 (s, 1H), 7.98 (s, 1H), 8.05 (s, 1H), 8.13 (s, 1H), 9.18 – 9.48 (bs, 2H). 13C NMR (CDCl3) δ 0.08, 18.79, 26.72, 29.44, 29.71, 49.52, 49.67, 55.16, 65.15, 80.55, 84.16, 86.13, 121.72, 122.34, 129.33, 130.44, 133.04, 136.70, 141.91, 149.89, 156.10, 162.03, 165.06, 167.48. LRMS (DART) m/z: 649.35 (M+H)+, 549.30 (M-Boc)+, 449.25 (M-2Boc)+. HRMS (DART) calcd for C31H50FN6O6Si (M+H)+ 649.3545, found: 649.3537 ± 0.0002.
N-Succinimidyl 3-(azidomethyl)-5-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)benzoate (7)
A solution of tetrabutyl ammonium fluoride in THF (1 M; 2.2 mL, 2.2 mmol) was added to a solution of 5 (1.0 g, 1.8 mmol) in THF (25 mL) and the homogeneous mixture stirred at 20 °C for 16 h. THF was evaporated, and the residue was partitioned between water and ethyl acetate. The pooled ethyl acetate solution was dried over MgSO4, and the filtrate concentrated to dryness. The crude intermediate was taken in ethyl acetate (25 mL), and N-hydroxysuccinimide (0.2 g, 1.8 mmol), DMAP (0.02 g, 0.2 mmol), and EDC (0.35 g, 1.8 mmol) were added. The reaction mixture was stirred at 20 °C for 16 h. Water was added to the reaction mixture and layers were separated. The ethyl acetate layer was dried with Na2SO4 and the filtrate concentrated using a rotary evaporator. The crude product was purified by silica gel chromatography using 2:1 hexanes:ethyl acetate to obtain 7 (337 mg, 0.62 mmol, 33.9 % yield) as a white solid: 1H-NMR (CDCl3) δ 1.38 (s, 9H), 1.45 (s, 9H), 2.89 (s, 4H), 4.40 (s, 2H), 5.20 (s, 2H), 7.66 (s, 1H), 7.95 (s, 1H), 8.02 (s, 1H), 9.22 (bs, 1H), 9.42 (bs, 1H). 13C NMR (CDCl3) δ 25.69, 27.93, 28.28, 46.92, 54.04, 79.06, 84.93, 125.59, 128.70, 129.72, 133.97, 136.73, 140.63, 154.57, 160.40, 161.45, 163.52, 169.09. LRMS (LCMS-ESI) m/z: 584.2 (M+K)+, 568.2 (M+Na)+, 546.2 (M+H)+, 490.2 (M-Bu)+, 434.1, 390.2. HRMS (DART) calcd for C24H32N7O8 (M+H)+ 546.2312, found 546.2302 ± 0.0009 (n = 4).
N-succinimidyl 3-((1,2-bis(tert-butoxycarbonyl)guanidino)methyl)-5-((4-(4-fluorobutyl)-1H-1,2,3-triazol-1-yl)methyl)benzoate (8)
Method A. Tetrabutyl ammonium fluoride (1M in THF; 0.05 mL, 0.05 mmol) was added to a solution of 6 (30.0 mg, 0.05 mmol) in dry THF (25 mL) that was cooled to 0–5°C and the resultant solution was stirred under argon overnight. THF was evaporated and the residual material was partitioned between EtOAc and water. Pooled ethyl acetate fractions were dried over MgSO4, and the filtrate was concentrated to dryness. DMAP (0.85 mg, 6.94 μmol), EDC (13.30 mg, 0.07 mmol) and N-hydroxysuccinimide (7.98 mg, 0.07 mmol) were added a solution of the crude intermediate product dissolved in 25 mL of dichloromethane. The solution was stirred at 20°C for 4 h, washed sequentially with water and saturated NaHCO3. Dichloromethane solution was dried over MgSO4, filtered and the filtrate concentrated. The crude product was purified by preparative TLC using 1:1 hexanes:ethyl acetate as the mobile phase to yield 12 mg (0.02 mmol, 40.2% yield) of 8 as a foamy solid. Method B. A dichloromethane solution of 6-fluorohex-1-yne (excess) was added to a solution of 7 (35.0 mg, 0.06 mmol) in 3 mL of DMF. The click reagent (100 μL) prepared as described in a previous section was added to the above mixture and the sealed vial was heated at 80 °C for 30 min. The reaction mixture was partitioned between a large volume of ethyl acetate and water. The ethyl acetate solution was dried over MgSO4, filtered and the filtrate concentrated to dryness. The crude product was purified by preparative TLC using 2:1 hexanes:ethyl acetate to obtain 18 mg (0.03 mmol, 44 % yield) of 8. 1H-NMR (CDCl3) δ 1.36 (s, 9H), 1.46 (s, 9H), 1.57 – 1.62 (m, 2H), 1.68 – 1.76 (m, 2H), 2.70 – 2.73 (t, 2H), 2.88 (s, 4H), 3.60 – 3.64 (t, 2H) 5.16 (s, 2H), 5.51 (s, 2H), 7.22 (s, 1H), 7.63 (s, 1H), 7.86 (s, 1H), 8.05 (s, 1H), 9.30–9.45 (bs, 2H). 13C NMR (CDCl3) δ 24.00, 24.16, 24.63, 26.86, 27.24, 28.90, 45.80, 52.29, 78.06, 82.09, 83.04, 83.95, 119.59, 125.92, 127.55, 129.14, 133.01, 134.95, 140.01, 153.40, 159.25, 160.15, 162.39, 167.99. LRMS (LCMS-ESI) m/z: 645.29 (M+). HRMS (DART) calcd for C30H41FN7O8 (M+H)+ 646.3001, found 646.2990 ± 0.0012 (n = 4).
N-Succinimidyl 3-((4-(4-fluorobutyl)-1H-1,2,3-triazol-1-yl)methyl)-5-(guanidinomethyl)benzoate (9)
TFA (1 mL) was added to 50 mg (0.08 mmol) of 8 and the resultant clear solution was left at 20°C. The reaction was followed by reversed-phase HPLC. For this an analytical column was eluted with a gradient of mobile phase consisting of 0.1% TFA in water (solvent A) and 0.1% TFA in acetonitrile (solvent B) at a flow rate of 1 mL/min; the proportion of B was linearly increased from 5% to 100% over a period of 30 min. Under these conditions, the retention times of 8 and 9 were 25.0 min and 22.2 min, respectively. HPLC of an aliquot of reaction mixture at 30 min indicated a single peak (>98%) corresponding to the product. TFA was evaporated from the reaction mixture. To insure complete removal of TFA, three 100 μL aliquots of dichloromethane were added, evaporating each time. The sample was taken in acetonitrile and subjected to semi-preparative HPLC using the same gradient but at a flow rate of 7 mL/min. HPLC fractions containing the product (tR = 14.3 min) were pooled, and solvents evaporated to obtain 41 mg (95%) of a glassy solid of 9: Analytical HPLC of this indicated a purity of 95.5%; a minor peak of lower tR, presumably the acid resulting from hydrolysis of the product during HPLC purification or concentration was seen. 1H-NMR (CD3CN) δ 1.72 (br m, 4 H), 2.72 (m, 2H), 2.86 (s, 4H), 4.35 – 4.45 (m, 4H), 5.64 (s, 2H), 6.60 (br s, 4H), 7.45–8.03 (m, 4H), 13.5 (br s, 1H). 13C NMR (CDCl3) δ 24.13, 24.98, 25.66, 29.5, 44.08, 53.08, 83.39, 84.80, 125.96, 128.88, 129.18, 133.31, 137.69, 138.82, 157.63, 161.74, 170.29. LRMS (LCMS-ESI) m/z: 446.3 (M+H)+. HRMS (DART) calcd for C20H25FN7O4 (M+H)+ 446.1952, found 446.1948 ± 0.0003 (n = 4).
6-(Hex-5-yn-1-yloxy)hex-1-yne (12)
The title compound42 was synthesized as follows: Potassium tert-butoxide (1M in THF; 41.6 mL, 41.6 mmol) was added to a solution of hex-5-yn-1-ol (3.27 g, 33.3 mmol) in 50 mL of DMF and the mixture stirred at 20°C for 30 min. Hex-5-yn-1-yl 4-methylbenzenesulfonate (7.00 g, 27.7 mmol), prepared by adapting literature protocols,26, 43 was added to the above, and the mixture stirred at 20°C overnight. The reaction mixture was partitioned between ether and water, the pooled ethereal layers were dried over Na2SO4 and the filtrate concentrated to dryness. The residue was purified by silica gel chromatography using 5:1 (v/v) hexanes:ethyl acetate as the mobile phase to afford 12 (3.3 g, 18.51 mmol, 66.7 % yield) as a clear oil. 1H-NMR (CDCl3) δ 1.53 – 1.69 (m, 8H), 1.91 (t, 2H), 2.17 – 2.21 (m, 4H), 3.39 (s, 4H).
N-Succinimidyl 3-((1,3-bis(tert-butoxycarbonyl)guanidino)methyl)-5-((4-(4-(hex-5-yn-1-yloxy)butyl)-1H-1,2,3-triazol-1-yl)methyl)benzoate (13)
Compounds 7 (50.0 mg, 0.09 mmol) and 12 (49.0 mg, 0.28 mmol) were dissolved in 3.0 mL of DMF. A 100 μL aliquot of click reaction reagent (vide infra) was added and the sealed vial was heated at 80 °C for 30 min. The reaction mixture was partitioned between ethyl acetate and water. The ethyl acetate layer was dried with MgSO4, filtered, and the filtrate was concentrated to dryness. The crude product was purified by preparative TLC using using 2:1 hexanes:ethyl acetate as the mobile phase to yield 21 mg (0.03 mmol, 31.7%) of a solid: 1H-NMR (CDCl3) δ 1.38 (s, 9H), 1.48 (s, 9H), 1.53 – 1.74 (m, 10H), 1.94 (t, 1H), 2.20 (m, 2H), 2.91 (s, 4H), 3.39 – 3.42 (tt, 4H) 5.18 (s, 2H), 5.62 (s, 2H), 7.22 (s, 1H), 7.65 (s, 1H), 7.93 (s, 1H), 8.09 (s, 1H), 9.22 (bs, 1H), 9.43 (bs, 1H). 13C NMR (CDCl3) δ 18.24, 25.24, 25.49, 25.68, 27.81, 28.00, 28.19, 28.38, 28.77, 53.31, 70.24, 70.48, 79.09, 84.98, 120.23, 120.88, 125.94, 128.71, 130.08, 130.24, 133.97, 136.06, 141.05, 148.99, 154.46, 160.31, 161.22, 163.44, 169.04. LRMS (DART) m/z: 746.3 (M+Na)+, 724.4 (M+H)+, 624.3 (M-Boc)+. HRMS (DART) calcd for C36H50N7O9 (M+H)+ 724.3670, found 724.3655 ± 0.0012 (n = 4).
4-(azidomethyl)-N,N,N-trimethylbenzenaminium trifluoromethane sulfonate (14)
The known compound 4-dimethylamino benzyl alcohol44, 45 was synthesized by sodium borohydride reduction of the corresponding aldehyde. Sodium borohydride (1.18 g, 31.2 mmol) was added to a cooled (0 – 5°C) solution of 4-(dimethylamino)benzaldehyde (3.10 g, 20.78 mmol) in THF (55 mL) containing 100 μL MeOH. The mixture was stirred at 20°C overnight, partitioned between ether and water and the ethereal layer was separated and dried over MgSO4. The drying agent was filtered, and the solvents from the filtrate were evaporated to afford 3.0 g (19.84 mmol, 95 % yield) of the alcohol as a pure solid: 1H-NMR (CDCl3) δ 2.2 (s, 6H), 3.81 (s, 2H), 6.61 (d, 2H), 7.05 (d, 2H). DBU (2.72 ml, 18.05 mmol) and 2-azido-1,3-dimethyl-4,5-dihydro-1H-imidazol-3-ium hexafluorophosphate(V) (4.75 g, 16.67 mmol) were added to a solution of the above alcohol (2.10 g, 13.89 mmol) in 65 mL THF, and the reaction mixture was stirred at 20°C for 10 min. The reaction was quenched with the addition of saturated aqueous NH4Cl, and organic materials were extracted from it with dichloromethane. The combined extracts were washed with brine and then dried over anhydrous Na2SO4. The solvent from the filtrate was removed in vacuo to afford the crude compound, which was purified by silica gel chromatography using 5:1 (v/v) hexanes/ethyl acetate to give 4-(azidomethyl)-N,N-dimethylaniline34, 46 (2.1 g, 11.92 mmol, 86% yield) as a white solid: 1H-NMR (CDCl3) δ 2.87 (s, 6H), 3.78 (s, 2H), 6.66 (d, 2H), 7.25 (d, 2H). Methyl trifluoromethanesulfonate (1.02 g, 6.24 mmol) was added to a rapidly stirred solution of 4-(azidomethyl)-N,N-dimethylaniline (1.0 g, 5.67 mmol) in 60 mL of anhydrous ether. There was an immediate formation of an off white precipitate. The heterogeneous mixture was stirred for 15 min and the precipitate was filtered, washed with a large volume of ether, and dried under high vacuum to obtain 1.50 g (4.41 mmol, 78 % yield) of 14 as a tan solid: 1H-NMR (CD3OD) δ 3.65 (s, 9H), 4.49 (s, 2H), 7.62 (d, 2H), 7.92 (d, 2H). 13C-NMR (CD3CN) δ 52.98, 57.10, 120.66, 130.04, 138.98. LRMS (LCMS-ESI) m/z: 191.1 (M+), 173.1, 163.1 (M-N2)+, 148.1. HRMS (DART) calcd for C10H15N4 (M+) 191.1291; found 191.1295 ± 0.0001 (n = 4).
N-Succinimidyl 3-((4-(4-[18F]fluorobutyl)-1H-1,2,3-triazol-1-yl)methyl)-5-(guanidinomethyl)benzoate ([18F]9)-Optimized procedure
A) Synthesis of 6-[18F]fluorohex-1-yne
Fluorine-18 was obtained either by in house cyclotron irradiation of [18O]H2O as described before47 or from PET-NET solutions (Durham, NC). For labeling reactions, 18F activity trapped in a QMA cartridge was eluted with a mixture of Kryptofix (28 mg) and potassium carbonate (2.4 mg) in 0.75 mL of 95% acetonitrile, and dried by azeotroping with acetonitrile three times. A solution of hex-5-yn-1-yl 4-methylbenzenesulfonate (9 mg, 35 μmol) was added to the reaction vial containing dried 18F activity (1.85 – 7.4 GBq; 50 – 200 mCi), and the reaction vial heated at 110ºC for 20 min. The reaction vial was constantly purged with a gentle stream of argon to hasten the distillation of the product. Collection of 6-[18F]fluorohex-1-yne was facilitated using a 19-guage teflon tubing inserted into the headspace of the reaction vial through a septum. The other end of the tubing was attached to the first of two 3 mL Wheaton vials, each containing 200 μL of dimethylformamide. The two vials were connected with another piece of the tubing. In each case, the tubing was immersed into the dimethylformamide so as to trap 6-[18F]fluorohex-1-yne. After 20 min, the first Wheaton vial containing most of the radioactivity was used for the next step.
B) Click reaction
Compound 7 (3 mg, 11.00 μmol) was added to the above DMF solution of 6-[18F]fluorohex-1-yne. The click reaction reagent prepared as described above (25 μL) was added, and the mixture heated at 50°C for 15 min. A 7 mg aliquot of 4-(azidomethyl)-N,N,N-trimethylbenzenaminium trifluoromethane sulfonate (14) and 100 μL of click reaction reagent were added and the mixture heated for an additional 15 min. The reaction mixture was diluted with ether, and the ethereal layer washed with brine and dried by passage over an anhydrous sodium sulfate cartridge (Agilent Technologies, Santa Clara, CA; part no. 12132044). Ether was evaporated to less than 500 μL using a stream of argon and the concentrate was purified using normal phase HPLC. For this, the normal phase semi-preparative column was eluted at a flow rate of 3 mL/min with a gradient consisting of 0.2% (v/v) acetic acid in both hexane (solvent A) and ethyl acetate (solvent B); the composition of solvent B was linearly increased from 30% to 100% over 30 min. A couple of experiments was performed to investigate whether unreacted 7 could be scavenged with polymer-bound alkyne.28 For this, after the initial click reaction with 7 and 6-[18F]fluoro-hex-1-yne, 10 mg (>1 equiv.) of the polymer and 100 μL of the click reaction reagent were added and the mixture heated at 50°C for 15 min. The mixture was processed as above but using ethyl acetate instead of ether for extraction. Ethyl acetate extract was filtered to remove any residual polymer before concentration and injection onto HPLC.
Radiolabeling SdAb 5F7
SdAb 5F7 was radioiodinated using [125I]SGMIB as reported before7 and the SdAb was labeled with 18F using [18F]SFB adapting procedures used for conjugation labeling of proteins with N-succinimidyl ester-containing prosthetic groups.48, 49 In these two cases, 50 μL of 5F7 (2 mg/mL) solution in 0.1M borate buffer, pH 8.5 was used. For labeling 5F7 using [18F]RL-I, the HPLC fractions containing Boc2-[18F]SFBTMGMB were evaporated to dryness using a stream of argon and the residual radioactivity was treated with 100 μL of TFA. After 10 min at 20°C, TFA was evaporated from the mixture, and to insure complete removal of TFA, 50 μL of ethyl acetate was added to the reaction mixture and evaporated three times. SdAb 5F7 in borate as above (100 μL; 200 μg) was added to [18F]RL-I as obtained above, and the mixture incubated at 20°C for 20 min. The entire mixture was loaded onto a gel filtration column (PD10; GE Healthcare, Piscataway, NJ), and the column eluted with PBS, pH 7.4, collecting 250 μL fractions. The protein typically eluted in fractions 5–10.
Determination of radiochemical purity
Three tests were performed to determine protein-associated radioactivity in a single, or when appropriate, in a paired-label format. TCA precipitability was determined by incubating about 5 ng of each labeled SdAb with 800 μL of 2% bovine serum albumin (BSA) and 100 μL of 20% TCA at 20 °C for 15 min. The mixtures were pelletted, and both pellets and supernatants counted for radioactivity. Protein-associated radioactivity was the percentage of total radioactivity (pellet plus supernatants) that was present in the pellet. ITLC was performed by eluting silica gel-impregnated glass fiber sheets (Pall Corporation, East Hills, NY) with PBS, pH 7.4. Under these conditions, the labeled protein remained at the origin and small molecular weight components eluted with an Rf of 0.7 – 0.8. The sheets were cut into small strips and counted for radioactivity. The integrity of labeled proteins was further assessed by SDS-PAGE under nonreducing conditions and subsequent phosphor imaging using a Storage Phosphor System Cyclone Plus phosphor imager (Perkin-Elmer Life and Analytical Sciences, Downers Grove, IL, USA) as previously described.12
Immunoreactivity of 5F7 after labeling
The immunoreactivity of the labeled proteins was determined by the Lindmo assay using magnetic beads coated with the extracellular domain of HER2, or as control for nonspecific binding, with BSA.12, 48, 50
Determination of affinity (KD)
The binding affinity of [18F]RL-I-5F7 and [18F]SFB-5F7 to HER2 on BT747M1 cells was determined using a saturation binding assay. BT474M1 cells were plated in 24-well plates at a density of 8 × 104 cells/well and incubated at 37°C for 24 h. The plates were kept at 4°C for 30 min, supernatants were removed, and the cells were washed twice with 1 mL cold PBS. Cells were then incubated in triplicate with increasing concentrations (0.05 – 100 nM; final volume 600 μL) of [18F]RL-I-5F7 or [18F]SFB-5F7 for 1 h at 4°C. A parallel assay was performed in which cells were co-incubated with 10 μM of HER2-specific trastuzumab to determine nonspecific binding at each concentration. The medium containing unbound radioactivity was removed, and the cells were washed twice with cold PBS. Finally, the cells were solubilized by treatment with 1N NaOH (0.5 mL) at 37°C for 10 min. Cell-associated 18F activity was determined using the automated gamma counter. The data were fitted using GraphPad Prism software to determine KD values.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grants CA188177 and CA42324. The authors want to thank Hilde Revets of Ablynx (Brussels, Belgium) for providing 5F7 SdAb and Marc Hens, who helped synthesizing some intermediates.
References
- 1.Tolmachev V, Stone-Elander S. Biochim Biophys acta. 2010;1800:487–510. doi: 10.1016/j.bbagen.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Tolmachev V, Orlova A, Lundqvist H. Curr Med Chem. 2003;10:2447–2460. doi: 10.2174/0929867033456666. [DOI] [PubMed] [Google Scholar]
- 3.Vaidyanathan G, White BJ, Affleck DJ, Zhao XG, Welsh PC, McDougald D, Choi J, Zalutsky MR. Bioorg Med Chem. 2012;20:6929–6939. doi: 10.1016/j.bmc.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Schaijk FG, Broekema M, Oosterwijk E, van Eerd JE, McBride BJ, Goldenberg DM, Corstens FH, Boerman OC. J Nucl Med. 2005;46:1016–1022. [PubMed] [Google Scholar]
- 5.Vaidyanathan G, Alston KL, Bigner DD, Zalutsky MR. Bioconjugate Chem. 2006;17:1085–1092. doi: 10.1021/bc0600766. [DOI] [PubMed] [Google Scholar]
- 6.Boswell CA, Marik J, Elowson MJ, Reyes NA, Ulufatu S, Bumbaca D, Yip V, Mundo EE, Majidy N, Van Hoy M, Goriparthi SN, Trias A, Gill HS, Williams SP, Junutula JR, Fielder PJ, Khawli LA. J Med Chem. 2013;56:9418–9426. doi: 10.1021/jm401365h. [DOI] [PubMed] [Google Scholar]
- 7.Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, Lyerly HK, Zalutsky MR. J Nucl Med. 2014;55:650–656. doi: 10.2967/jnumed.113.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaidyanathan G, Zalutsky MR. Nat Protoc. 2007;2:282–286. doi: 10.1038/nprot.2007.20. [DOI] [PubMed] [Google Scholar]
- 9.Chakravarty R, Goel S, Cai W. Theranostics. 2014;4:386–398. doi: 10.7150/thno.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Meyer T, Muyldermans S, Depicker A. Trends Biotechnol. 2014;32:263–270. doi: 10.1016/j.tibtech.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 11.D’Huyvetter M, Xavier C, Caveliers V, Lahoutte T, Muyldermans S, Devoogdt N. Expert Opin Drug Deliv. 2014;11:1939–1954. doi: 10.1517/17425247.2014.941803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, Zalutsky MR. Nucl Med Biol. 2013;40:52–59. doi: 10.1016/j.nucmedbio.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richter S, Wuest F. Molecules. 2014;19:20536–20556. doi: 10.3390/molecules191220536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobson O, Kiesewetter DO, Chen X. Bioconjugate Chem. 2015;26:1–18. doi: 10.1021/bc500475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Z, Kandeel F. Curr Pharm Biotechnol. 2010;11:572–580. doi: 10.2174/138920110792246564. [DOI] [PubMed] [Google Scholar]
- 16.Vaidyanathan G, Zalutsky MR. Nat Protoc. 2006;1:1655–1661. doi: 10.1038/nprot.2006.264. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Park R, Hou Y, Khankaldyyan V, Gonzales-Gomez I, Tohme M, Bading JR, Laug WE, Conti PS. Eur J Nucl Med Mol Imaging. 2004;31:1081–1089. doi: 10.1007/s00259-003-1452-2. [DOI] [PubMed] [Google Scholar]
- 18.Ren G, Liu Z, Miao Z, Liu H, Subbarayan M, Chin FT, Zhang L, Gambhir SS, Cheng Z. J Nucl Med. 2009;50:1865–1872. doi: 10.2967/jnumed.109.062877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaidyanathan G, Zalutsky MR. Nucl Med Biol. 1992;19:275–281. [Google Scholar]
- 20.Waldmann CM, Hermann S, Faust A, Riemann B, Schober O, Schafers M, Haufe G, Kopka K. Bioorg Med Chem. 2015;23:5734–5739. doi: 10.1016/j.bmc.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Liu PN, Chen YC, Deng JG, Tu YQ. Synthesis. 2001:2078–2080. [Google Scholar]
- 22.Maeda DY, Mahajan SS, Atkins WM, Zebala JA. Bioorg Med Chem Lett. 2006;16:3780–3783. doi: 10.1016/j.bmcl.2006.04.041. [DOI] [PubMed] [Google Scholar]
- 23.Vaidyanathan G, Zalutsky MR. J Org Chem. 1997;62:4867–4869. [Google Scholar]
- 24.L’Heureux A, Beaulieu F, Bennett C, Bill DR, Clayton S, Laflamme F, Mirmehrabi M, Tadayon S, Tovell D, Couturier M. J Org Chem. 2010;75:3401–3411. doi: 10.1021/jo100504x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mindt TL, Muller C, Stuker F, Salazar JF, Hohn A, Mueggler T, Rudin M, Schibli R. Bioconjugate Chem. 2009;20:1940–1949. doi: 10.1021/bc900276b. [DOI] [PubMed] [Google Scholar]
- 26.Ross TL, Honer M, Lam PY, Mindt TL, Groehn V, Schibli R, Schubiger PA, Ametamey SM. Bioconjugate Chem. 2008;19:2462–2470. doi: 10.1021/bc800356r. [DOI] [PubMed] [Google Scholar]
- 27.Lewis WG, Magallon FG, Fokin VV, Finn MG. J Am Chem Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 28.Harju K, Kylanlahti I, Paananen T, Polamo M, Nielsen J, Yli-Kauhaluoma J. J Comb Chem. 2006;8:344–349. doi: 10.1021/cc050138j. [DOI] [PubMed] [Google Scholar]
- 29.Sachin K, Jadhav VH, Kim EM, Kim HL, Lee SB, Jeong HJ, Lim ST, Sohn MH, Kim DW. Bioconjugate Chem. 2012;23:1680–1686. doi: 10.1021/bc3002425. [DOI] [PubMed] [Google Scholar]
- 30.Seo JW, Lee BS, Lee SJ, Oh SJ, Chi DY. B Korean Chem Soc. 2011;32:71–76. [Google Scholar]
- 31.Kim DH, Choe YS, Kim BT. Appl Radiat Isot. 2010;68:329–333. doi: 10.1016/j.apradiso.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 32.George GPC, Pisaneschi F, Stevens E, Nguyen QD, Aberg O, Spivey AC, Aboagye EO. J Labelled Compd Rad. 2013;56:679–685. doi: 10.1002/jlcr.3095. [DOI] [PubMed] [Google Scholar]
- 33.Yook CM, Lee SJ, Oh SJ, Ha HJ, Lee JJ. J Labelled Compd Radiopharm. 2015;58:317–326. doi: 10.1002/jlcr.3297. [DOI] [PubMed] [Google Scholar]
- 34.Barluenga J, Tomas-Gamasa M, Valdes C. Angew Chem Intl Ed. 2012;51:5950–5952. doi: 10.1002/anie.201200313. [DOI] [PubMed] [Google Scholar]
- 35.Revets MHAP, Boutton WC, Hoogenboom MHRJM. 0059090 A1. US Pat. 2011
- 36.Arumugam S, Chin J, Schirrmacher R, Popik VV, Kostikov AP. Bioorg Med Chem Lett. 2011;21:6987–6991. doi: 10.1016/j.bmcl.2011.09.126. [DOI] [PubMed] [Google Scholar]
- 37.Bouvet V, Wuest M, Wuest F. Org Biomol Chem. 2011;9:7393–7399. doi: 10.1039/c1ob06034a. [DOI] [PubMed] [Google Scholar]
- 38.Vaidyanathan G, Affleck DJ, Li J, Welsh P, Zalutsky MR. Bioconjugate Chem. 2001;12:428–438. doi: 10.1021/bc0001490. [DOI] [PubMed] [Google Scholar]
- 39.Vaidyanathan G, Zalutsky MR. Bioconjugate Chem. 1994;5:352–356. doi: 10.1021/bc00028a012. [DOI] [PubMed] [Google Scholar]
- 40.Tang G, Zeng WB, Yu MX, Kabalka G. J Labelled Compd Radiopharm. 2008;51:68–71. [Google Scholar]
- 41.Yu Z, Xia W, Wang HY, Wang SC, Pan Y, Kwong KY, Hortobagyi GN, Hung MC. Mol Carcinog. 2006;45:667–675. doi: 10.1002/mc.20212. [DOI] [PubMed] [Google Scholar]
- 42.Wotiz JH, Adams RF, Parsons CG. J Am Chem Soc. 1961;83:373–376. [Google Scholar]
- 43.Findlay B, Zhanel GG, Schweizer F. Bioorg Med Chem Lett. 2012;22:1499–1503. doi: 10.1016/j.bmcl.2012.01.025. [DOI] [PubMed] [Google Scholar]
- 44.Naimi-Jamal MR, Mokhtari J, Dekamin MG, Kaupp G. Eur J Org Chem. 2009:3567–3572. [Google Scholar]
- 45.Shi L, Liu YY, Liu QF, Wei B, Zhang GS. Green Chem. 2012;14:1372–1375. [Google Scholar]
- 46.Kitamura M, Koga T, Yano M, Okauchi T. Synlett. 2012;23:1335–1338. [Google Scholar]
- 47.Vaidyanathan G, White BJ, Zalutsky MR. Curr Radiopharm. 2009;2:63–74. doi: 10.2174/1874471010902010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi J, Vaidyanathan G, Koumarianou E, McDougald D, Pruszynski M, Osada T, Lahoutte T, Lyerly HK, Zalutsky MR. Nucl Med Biol. 2014;41:802–812. doi: 10.1016/j.nucmedbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaidyanathan G, Bigner DD, Zalutsky MR. J Nucl Med. 1992;33:1535–1541. [PubMed] [Google Scholar]
- 50.Foulon CF, Reist CJ, Bigner DD, Zalutsky MR. Cancer Res. 2000;60:4453–4460. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.