

Abstract
Tumor necrosis factor receptor–associated factor 3 (TRAF3) is an adaptor protein that inhibits signaling by CD40 and by the receptor for B cell–activating factor (BAFF) and negatively regulates homeostatic B cell survival. Loss-of-function mutations in TRAF3 are associated with human B cell malignancies, in particular multiple myeloma. The cytokine interleukin-6 (IL-6) supports the differentiation and survival of normal and neoplastic plasma cells. We found that mice with a deficiency in TRAF3 specifically in B cells (B-Traf3−/− mice) had about twice as many plasma cells as did their littermate controls. TRAF3-deficient B cells had enhanced responsiveness to IL-6, and genetic loss of IL-6 in B-Traf3−/− mice restored their plasma cell numbers to normal. TRAF3 inhibited IL-6 receptor (IL-6R)–mediated signaling by facilitating the association of PTPN22 (a nonreceptor protein tyrosine phosphatase) with the kinase Janus-activated kinase 1 (Jak1), which in turn blocked phosphorylation of the transcription factor STAT3 (signal transducer and activator of transcription 3). Consistent with these results, the number of plasma cells in the PTPN22-deficient mice was increased compared to that in the wild-type mice. Our findings identify TRAF3 and PTPN22 as inhibitors of IL-6R signaling in B cells and reveal a previously uncharacterized role for TRAF3 in the regulation of plasma cell differentiation.
INTRODUCTION
Tumor necrosis factor (TNF) receptor–associated factor 3 (TRAF3) is an adaptor protein that mediates signaling downstream of TNF receptor su-perfamily members (1). TRAF3 also participates in signaling through Toll-like receptors (TLRs), as well as through the T cell antigen receptor (TCR) complex (1, 2). In addition, TRAF3 plays a role in mediating interleukin-17 (IL-17) receptor (IL-17R) and IL-2R signaling (3, 4). Evidence is accumulating that the function of TRAF3 is highly receptor- and cell type–specific (1). Mice with a B cell–specific deletion of TRAF3 (B-Traf3−/− mice) show a marked B cell survival advantage in the absence of increased proliferation (5, 6). This enhanced survival causes accumulation of B cells in peripheral lymphoid organs, and ~30% of B-Traf3−/− mice develop lymphoma at >9 months of age (7). In humans, loss-of-function mutations in the TRAF3 gene have been reported in B cell lymphoma patients (8–11). Additionally, ~17% of multiple myeloma cell lines and ~12% of primary tumor samples from multiple myeloma patients show loss-of-function mutations in TRAF3 (12, 13). Together, these data from studies of mice and humans implicate TRAF3 as a tumor suppressor in B cells by restraining homeostatic B cell survival. However, how the loss of TRAF3 contributes to the differentiation of plasma cells (PCs) or the occurrence of multiple myeloma remains unexplored.
Naïve B cells encounter pathogens or cognate antigens in peripheral lymphoid organs, where they interact with follicular CD4+ helper T cells in the germinal center. These interactions result in the development of long-lived, antibody-secreting PCs and memory B cells (14, 15). After leaving the germinal center, PCs migrate into the bone marrow where they receive survival signals provided by bone marrow stroma and innate immune cells (16). These long-lived PCs continuously produce high-affinity antibodies for the lifetime of the host. IL-6 is a known B cell survival and PC differentiation factor (17–19), so it is not surprising that it also supports the growth of multiple myeloma cells and induces the development of plasmacytomas in mice in which the IL6 gene is overexpressed (20,21). Increased serum concentrations of IL-6 are frequently found in multiple myeloma patients and correlate with a poor prognosis (22). Dysregulated IL-6R signaling is observed in B cell malignancies and solid tumors (23, 24). Thus, the IL-6 signaling pathway is an attractive potential target for cancer therapies.
IL-6 binds to an IL-6R complex to initiate signaling in two alternative ways. In “classical activation,” IL-6 binds to the IL-6Rα chain that is in a complex with the cell surface signaling receptor glycoprotein 130 (gp130), which results in the activation of Janus-activated kinase 1 (Jak1) and the subsequent phosphorylation of gp130 (25, 26). Phosphorylated gp130 recruits signal transducer and activator of transcription 3 (STAT3), which is phosphorylated (and activated) by Jak1 (27). Activated STAT3 translocates into the nucleus to promote target gene expression. In “trans” signaling, IL-6 associates with soluble IL-6Rα (sIL-6Rα). The IL-6–sIL-6Rα complex then activates cells that have cell surface gp130 (25). In B cells, the IL-6–dependent activation of STAT3 is important for the initiation of PC differentiation programs, such as the generation of increased amounts of the transcription factors B lymphocyte–induced maturation protein 1 (BLIMP-1) and X box–binding protein 1 (Xbp-1) (28, 29).
The PTPN22 gene encodes protein tyrosine phosphatase nonreceptor type 22 (PTPN22), a phosphatase primarily found in lymphocytes and some myeloid cells (30). A variant of the PTPN22 gene (R620W) is highly associated with type 1 diabetes, rheumatoid arthritis, systemic lupus erythematosus, and other autoimmune diseases (30–32). PTPN22 regulates B cell receptor and TCR signaling by dephosphorylating downstream Src family kinases (33, 34); however, PTPN22 has not been previously implicated in cytokine-mediated Jak-STAT signaling. Here, we report that TRAF3 associates with PTPN22 in B cells to inhibit the IL-6–dependent activation of STAT3 by Jak1. This regulation restrains PC development in the spleen and bone marrow. These results have implications for the regulation of normal PC development, as well as for our understanding of the dysregulated signaling pathways that contribute to B cell malignancies, particularly multiple myeloma.
RESULTS
TRAF3 restricts the development of PCs
We previously showed that basal serum immunoglobulin (Ig) amounts in B-Traf3−/− mice are increased relative to those in TRAF3-sufficient littermate control mice, and these mice display spontaneous germinal centers in the spleen and lymph nodes (6). We thus investigated whether B-Traf3−/− mice had more PCs than did littermate control mice. We found that the spleen and bone marrow of B-Traf3−/− mice had ~1.5- to 2-fold more CD138+B220low PCs than did those of their littermate control mice (Fig. 1, A and B). Antibody-secreting cells (ASCs) in the spleen and bone marrow were also markedly increased in B-Traf3−/− mice compared to those in control mice (Fig. 1C). These data suggest that TRAF3 plays a role in restraining the size of the PC population in the spleen and bone marrow.
Fig. 1. TRAF3 inhibits the development of PCs.
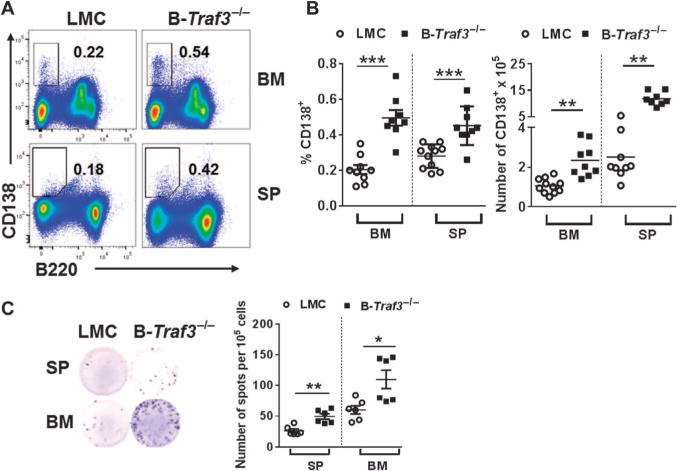
(A) Representative plots from the flow cytometric analysis of cells from the spleen (SP) and bone marrow (BM) of B-Traf3−/− mice and littermate control (LMC) mice. Outlined areas and numbers indicate the percentages of CD138+B220low PCs. Data are representative of four experiments. (B) Percentages (left) and numbers (right) of CD138+B220lowPCs in the spleens and bone marrow of littermate control mice and B-Traf3−/− mice based on data as identified in (A). Each symbol represents a single mouse, and the horizontal line indicates the mean value of each group. (C) Left: Representative wells from the enzyme-linked immunospot (ELISPOT) analysis of ASCs in the spleen and bone marrow of littermate control mice and B-Traf3−/− mice. Right: The numbers of ASCs from the spleen and bone marrow of mice of each strain. Each symbol represents the mean of technical triplicate samples from a single mouse, and the horizontal lines indicate mean values of six mice per group. ***P < 0.001, **P < 0.01, *P < 0.05 by Student’s t test.
TRAF3 inhibits the IL-6–mediated differentiation of PCs
IL-6 plays an important role in the development of PCs under normal conditions and in multiple myeloma (17, 19, 23, 29). To determine whether the increased number of PCs in B-Traf3−/− mice was IL-6–dependent, we bred B-Traf3−/− mice to IL-6–deficient (IL6−/−) mice. In the absence of IL-6, the number of PCs in B-Traf3−/− mice reverted to those of littermate control mice (Fig. 2, A to C); however, the overall increase in the number of B cells in B-Traf3−/− mice was not altered in the absence of IL-6 (fig. S1). This result suggests that IL-6 is required for the accumulation of PCs in B-Traf3−/− mice, but not for the increase in the total number of B cells. To explore whether TRAF3-deficient PCs had a survival advantage in response to IL-6, we purified CD138+ PCs by cell sorting and cultured them in the presence or absence of IL-6. We found that IL-6 equally increased the survival of TRAF3-deficient PCs and PCs from littermate controls (Fig. 2D), suggesting that the IL-6–dependent increase in PC numbers in B-Traf3−/− mice was not a result of the enhanced survival of PCs. Whereas the basal survival of TRAF3-deficient B cells is greater than that of B cells from littermate controls (5, 6), TRAF3-deficient PCs did not show a survival advantage, suggesting that although TRAF3 is important for restraining general B cell survival, it uses a different mechanism to limit increases in the numbers of PCs (Fig. 2D). To explore whether the increased number of PCs in B-Traf3−/− mice was a result of the increased basal amounts of IL-6 in these mice, we analyzed serum from B-Traf3−/− and littermate control mice but found that they had similar amounts of IL-6 (fig. S2).
Fig. 2. The enhanced development of PCs in B-Traf3−/− mice is IL-6–dependent.
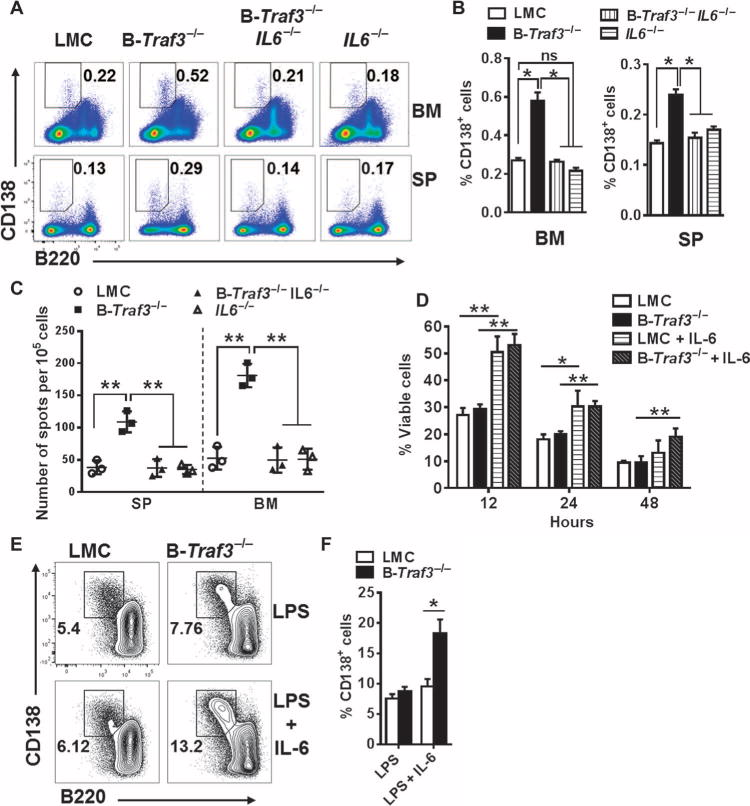
(A) Representative plots from the flow cytometric analysis of spleen and bone marrow cells from littermate control mice, B-Traf3−/−, IL6−/−, and B-Traf3−/− IL6−/− mice. Outlined areas and numbers indicate the percentages of CD138+B220low PCs. (B) Quantification of the percentages of PCs in the bone marrow (left) and spleens (right) of the indicated mice from the experiments depicted in (A). Data are means ± SEM of values from at least four mice from each group. *P < 0.05; ns, not statistically significant. (C) ELISPOT analysis of splenocytes and bone marrow cells from the indicated mouse strains. The graph shows the numbers of ASCs; each symbol represents a technical triplicate, whereas the lines indicate the mean values from three mice per group. **P < 0.01 by one-way analysis of variance (ANOVA). (D) Sorted PCs from the bone marrow of the indicated strains of mice were cultured in the presence or absence of IL-6 (50 ng/ml) for the indicated times. The percentage cell viability in each sample was determined by flow cytometric analysis of the percentage of propidium iodide (PI)–negative cells. Data are means ± SEM from three independent experiments. **P< 0.01, *P< 0.05 by one-way ANOVA. (E) Splenic B cells from the indicated mice were stimulated with LPS(10μg/ml) in the presence or absence of IL-6 (20 ng/ml) for 3 days before they were analyzed by flow cytometry to detect CD138+B220low PCs. Numbers in the plots represent the percentages of CD138+B220low PCs. (F) Quantification of the percentages of CD 138+B220low PCs in the experiments shown in (E). Data are means ± SEM values from three independent experiments. *P < 0.05 by Student’s t test.
Together, these results exclude increased numbers of total B cells, preferentially enhanced IL-6–mediated PC survival, and increased serum IL-6 concentrations as explanations for the increase in the number of PCs in B-Traf3−/− mice. We thus hypothesized that TRAF3-deficient B cells differentiate more readily than do their wild-type counterparts into PCs in response to IL-6. To test this hypothesis, we treated splenic B cells isolated from B-Traf3−/− or littermate control mice with LPS to induce their differentiation into PCs in vitro. LPS induced the generation of similar percentages of CD138+B220low PCs from Traf3−/− B cells and wild-type B cells (Fig. 2, E and F). However, Traf3−/− B cells treated with both LPS and IL-6 generated a greater percentage of CD138+ B220low PCs than did similarly treated B cells from littermate control mice, suggesting that Traf3−/− B cells more efficiently differentiated into PCs specifically in response to IL-6 (Fig. 2, E and F). Thus, TRAF3 inhibited PC development by restraining B cell responsiveness to IL-6.
TRAF3 inhibits the IL-6 signaling pathway
Most cell types, including B cells, constitutively display gp130 on their surface, but the surface presence of the IL-6Rα chain is regulated to prevent over-activation of the IL-6R pathway (35, 36). To explore why TRAF3-deficient B cells underwent enhanced IL-6–dependent differentiation into PCs, we first examined the relative abundances of the IL-6R components CD126 (IL-6Rα) and CD130 (gp130). Both gp130 and the IL-6Rα chain were present in similar amounts on the surface of B cells from B-Traf3−/− mice and their littermate controls (fig. S3). Thus, differential amounts of the IL-6R cannot account for the increased IL-6–dependent number of PCs in B-Traf3−/− mice.
TRAF3 inhibits cytokine-mediated signaling pathways in myeloid cells and T cells, including those stimulated by the receptors for IL-17 and IL-2, respectively (3, 4). Thus, we next investigated whether the absence of TRAF3 in B cells affected signaling by the IL-6R. In TRAF3-deficient subclones of the mouse B cell line A20, the IL-6–induced phosphorylation of STAT3 was enhanced compared to that in TRAF3-sufficient parent cells (Fig. 3A). To determine whether the increased IL-6–induced abundance of phosphorylated STAT3 (pSTAT3) occurred as a result of the deficiency in TRAF3, we stably transfected the TRAF3-deficient cell line to express Traf3 in response to isopropyl-β-D-thiogalactopyranoside (IPTG). The induction of TRAF3 protein production substantially reduced the IL-6–induced abundance of pSTAT3 in these cells (Fig. 3B). Furthermore, the abundance of pSTAT3 in response to IL-6 was greater in primary B cells from B-Traf3−/− mice than in primary B cells from littermate control mice (Fig. 3C). Additionally, we also observed that the IL-6–dependent activation of STAT3 was enhanced in normal human B cells in which TRAF3 abundance was reduced by small interfering RNA (siRNA) (Fig. 3D). These data suggest that TRAF3 inhibits the IL-6–stimulated activation of STAT3 in both mouse and human B cells. IL-21 is also important for the generation of PCs and memory B cells, and it activates the Jak-STAT signaling pathway (37, 38). However, we found that the absence of TRAF3 only minimally affected the IL-21–dependent phosphorylation of STAT3 (fig. S4). Thus, TRAF3 preferentially inhibits the IL-6–dependent activation of STAT3 in B cells.
Fig. 3. TRAF3 inhibits the IL-6–dependent activation of STAT3.
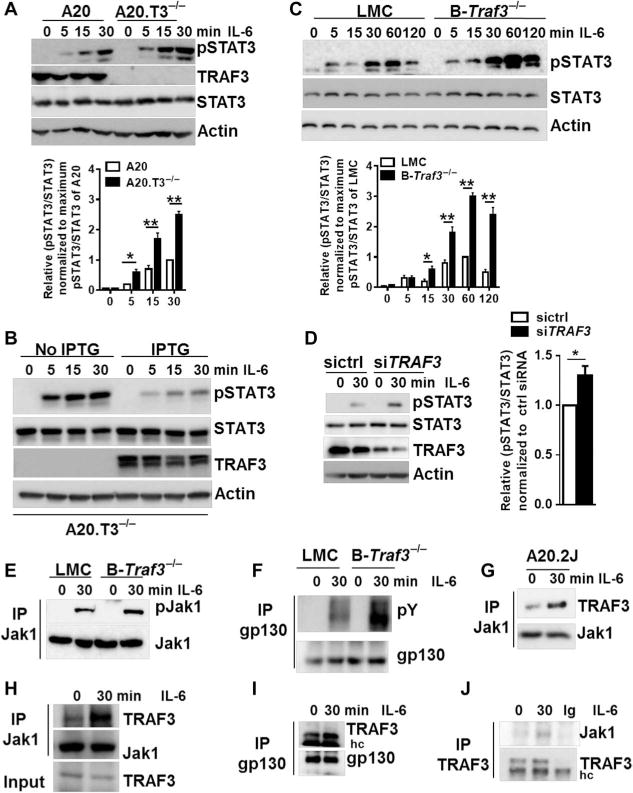
(A) Top: TRAF3-sufficient and TRAF3-deficient subclones of the mouse B cell line A20.2J (A20 and A20.T3−/− cells, respectively) were left untreated or were stimulated with IL-6 for the indicated times. Whole-cell lysates were then subjected to Western blotting analysis with antibodies against the indicated proteins. Western blots are representative of three independent experiments. Bottom: Densitometric analysis of the ratio of the abundance of pSTAT3 protein to that of total STAT3 protein, setting the maximum ratio in TRAF3-sufficient B cells at 1. Data are means ± SD of three independent experiments. (B) A20.T3−/− cells were stably transfected with a plasmid encoding IPTG-inducible Traf3. Cells were cultured in the presence or absence of IPTG for 12 hours before being stimulated with IL-6 for the indicated times and then analyzed by Western blotting with antibodies against the indicated proteins. Western blots are from one experiment and are representative of three independent experiments. (C) Top: Splenic B cells from the indicated mice were left unstimulated or were stimulated with IL-6 for the indicated times before being analyzed by Western blotting as described in (A). Western blots are representative of five individual experiments. Bottom: Densitometric analysis of the ratio of the abundance of pSTAT3 protein to that of total STAT3 protein, setting the maximum ratio in TRAF3-sufficient B cells at 1. Data are means ± SD of five independent experiments. (D) Left: Normal human peripheral B cells isolated as described in Materials and Methods were transfected with the indicated siRNAs before being left untreated or treated with recombinant human IL-6 for 30 min. Samples were then analyzed by Western blotting as described in (A). Western blots are from a single experiment and are representative of three experiments. Right: Data are means ± SEM of the ratio of the abundances of pSTAT3 protein to total STAT3 protein from three experiments, setting the maximum ratio in cells transfected with control siRNA (sictrl) at 1. *P < 0.05 by Student’s t test. (E and F) Splenic B cells from the indicated mice were left untreated or were stimulated with IL-6 for 30 min before being subjected to immunoprecipitation (IP) of (E) Jak1 or (F) gp130. Samples were then analyzed by Western blotting with antibodies specific for (E) pJak1 and total Jak1 or (F) phosphotyrosines (pY) and gp130. Western blots are from a single experiment and are representative of four experiments. (Gto I) A20.2J cell lines (G) or splenic B cells (H and I) were left untreated or were treated with IL-6 for the indicated times. Cell lysates were then subjected to immunoprecipitation with antibodies against the indicated proteins before being analyzed by Western blotting. Blots are from a single experiment and are representative of two independent experiments. (J) Human B cells were left untreated or were treated with recombinant human IL-6 for 30 min. Cell lysates were then subjected to immunoprecipitation with antibody against TRAF3, and the samples were then analyzed by Western blotting with antibodies against the indicated proteins. Western blots are from a single experiment and are representative of two independent experiments. hc, heavy chain.
The IL-6–dependent activation of gp130 and Jak1 is required for STAT3 phosphorylation. We observed the enhanced phosphorylation of both Jak1 and gp130 in Traf3−/− B cells upon stimulation with IL-6 (Fig. 3, E and F), which suggests that TRAF3 may regulate IL-6 signaling in a receptor-proximal manner. Consistent with this possibility, we found that stimulation of A20.2J or primary mouse splenic B cells with IL-6 increased the extent of the interactions between TRAF3 and either Jak1 or gp130 (Fig. 3, G to I). The associations between TRAF3 and either Jak1 or gp130 in unstimulated B cells suggest that TRAF3 also regulates ligand-independent signaling by the IL-6R. Immunoprecipitation of TRAF3 from primary human B cells showed that it interacted with Jak1 (Fig. 3J). Together, these data suggest that TRAF3 associates with components of the IL-6R signaling complex to inhibit the Jak1-STAT3 pathway.
TRAF3 interacts with PTPN22 to inhibit IL-6R signaling and PC development
We and others previously found that TRAF3 interacts with several phosphatases (3, 37). The association between TRAF3 and the phosphatase PTPN22 in myeloid cells potentiates the TLR-mediated production of type I interferon (IFN) (39, 40). We previously found that T cell–specific protein tyrosine phosphatase (TCPTP) is recruited by TRAF3 to inhibit IL-2R signaling in T cells (3). This finding prompted us to investigate whether TRAF3 limited IL-6 signaling in B cells by interacting with a phosphatase. Immunoprecipitation of Jak1 from mouse splenic B cells showed that it interacted with both TRAF3 and PTPN22 and that these interactions were enhanced in response to IL-6 (Fig. 4A). The IL-6–stimulated enhancement of the association between PTPN22 and Jak1 in cell lines and primary B cells was abrogated in the absence of TRAF3 (Fig. 4, B and C). However, we found no evidence of the binding of TCPTP to Jak1 in IL-6–stimulated B cells (fig. S5), in contrast to the association between TCPTP and Jak1 after the stimulation of T cells with IL-2 (3), which suggested that TRAF3 recruits distinct phosphatases to different cytokine receptors in a cell type–specific manner. To explore whether the recruitment of PTPN22 to the IL-6R signaling complex played a role in regulating IL-6 signaling, we stimulated splenic B cells isolated from wild-type or Ptpn22−/− mice with IL-6, which resulted in the enhanced phosphorylation of STAT3 in the PTPN22-deficient B cells (Fig. 4D), suggesting that PTPN22 was an inhibitor of IL-6R signaling. Together, our data suggest that TRAF3 restrains IL-6 signaling by facilitating the recruitment of PTPN22 to the IL-6R complex.
Fig. 4. TRAF3 interacts with PTPN22 to inhibit IL-6R signaling and PC development.
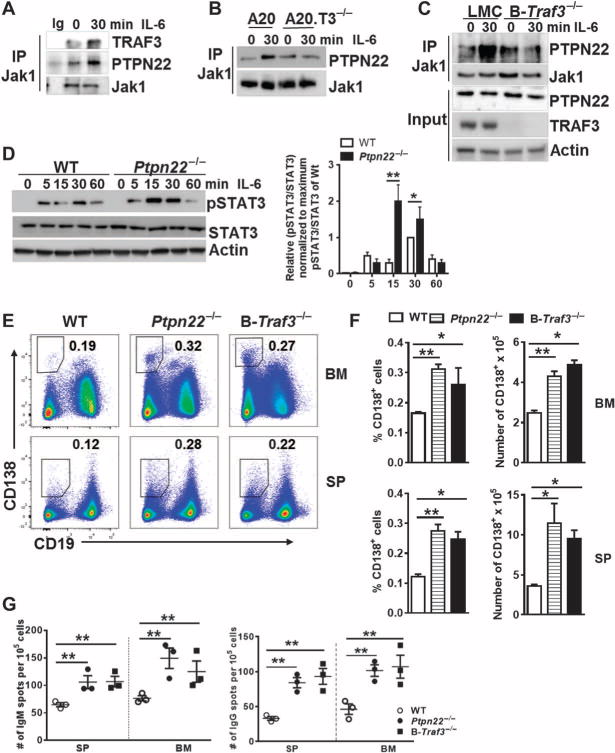
(A and B) Splenic B cells (A) and the indicated A20.2J cell lines (B) were left untreated or were treated with IL-6 for 30 min before being subjected to immunoprecipitation with an antibody specific for Jak1. An IgG control was included to control for the specificity of the anti-Jak1 antibody. Immunoprecipitates were then analyzed by Western blotting with antibodies specific for the indicated proteins. Blots are from a single experiment and are representative of three independent experiments. (C) Splenic B cells from the indicated mice were left untreated or were treated with IL-6 for 30 min. Whole-cell lysates were subjected to the immunoprecipitation of Jak1, and samples were then analyzed by Western blotting to detect PTPN22. As a control, whole-cell lysates (input) were also analyzed. Blots are from a single experiment and are representative of three independent experiments. (D) Left: Splenic B cells from the indicated mice were left untreated or were treated with IL-6 for the indicated times. Whole-cell lysates were then analyzed by Western blotting with antibodies against the indicated targets. Western blots are representative of three individual experiments. Right: Densitometric analysis of the ratio of the abundance of pSTAT3 protein to that of total STAT3 protein, setting the maximum ratio in PTPN22-sufficient B cells at 1. Data are means ± SD of five independent experiments. (E) Representative plots from the flow cytometric analysis of CD138+B220low PCs in the spleen and bone marrow of the indicated mice. The outlined areas and numbers indicate the percentages of the CD138+B220low PCs. (F) Quantification of the percentages of CD138+B220low PCs in the bone marrow (top) and spleens (bottom) of the indicated mouse strains from the experiments depicted in (E). Data are means ± SEM of six (Ptpn22−/−), four [wild type (WT)], and three (B-Traf3−/−) mice. (G) ELISPOT analysis of IgM-secreting (left) and IgG-secreting (right) ASCs in the spleen and bone marrow of the indicated mice. The graph represents the numbers of ASCs from the bone marrow and spleen. Each symbol represents the mean of technical triplicates from a single mouse, whereas the horizontal lines indicate means from three mice per group. *P < 0.05, **P < 0.01 by one-way ANOVA.
To investigate whether a deficiency in PTPN22 also affected PC development, we assessed the numbers of PCs in the spleens and bone marrow of Ptpn22−/− mice. Similar to B-Traf3−/− mice, Ptpn22−/− mice showed increased percentages and numbers of PCs in both the spleen and bone marrow (Fig. 4, E and F). Consistent with the increased numbers of CD138+ PCs in Ptpn22−/−and B-Traf3−/− mice, both strains of mice contained more IgM- and IgG-producing ASCs in the spleen and bone marrow than did the wild-type mice (Fig. 4G). Although there were no differences between the numbers of CD138+ PCs generated in vitro from PTPN22-deficient B cells and wildtype B cells (fig. S6), this might have been a result of impaired TLR signaling in the PTPN22-deficient B cells. Published studies showed that PTPN22 is essential for the TLR-dependent production of type I IFN in myeloid cells (39, 40). Collectively, these data suggest that the TRAF3-PTPN22 interaction in IL-6R signaling is a critical event in inhibiting the development of PCs.
DISCUSSION
Decreased amounts or complete loss of cellular TRAF3 protein is highly associated with B cell malignancies, particularly multiple myeloma (12, 13), yet very little is known about how TRAF3 contributes to multiple myeloma or to the normal development of PCs. Here, we report that TRAF3 is a critical inhibitor of PC development through its ability to inhibit IL-6R signaling. Mechanistically, TRAF3 limits IL-6R signaling by facilitating the association of PTPN22 with the IL-6R signaling complex, which in turn mediates the dephosphorylation of Jak1 and gp130, resulting in reduced STAT3 activation.
TRAF3 interacts with and inhibits the function of the IL-17R in myeloid cells (4). Our group demonstrated that in T cells, TRAF3 associates with the IL-2R signaling complex and inhibits IL-2R signaling and the development of T regulatory cells (3). Together with our current findings, these studies indicate that TRAF3 acts as an inhibitor of cytokine receptor–mediated signaling pathways; however, the mechanisms by which TRAF3 inhibits each of these signaling pathways are distinct. The association of TRAF3 with the IL-17R disrupts the formation of the signaling complex containing TRAF6, IL-17R, and the receptor-associated adaptor protein Act1 (4). In IL-2R–mediated signaling in T cells, TRAF3 is required for the binding of the phosphatase TCPTP to Jak1 and Jak3 (3, 4). Here, we showed that TRAF3 facilitated the association of PTPN22 with Jak1 to restrain IL-6R signaling in B cells. Thus, the physical binding of TRAF3 to the IL-17R impairs receptor signaling, whereas the major mechanism of action by which TRAF3 inhibits IL-2R and IL-6R signaling in lymphocytes is by influencing the associations of phosphatases with Jaks. These distinct molecular mechanisms of action further emphasize the multifaceted roles of TRAF3 in regulating different signaling pathways.
TRAF3 contains RING (really interesting new gene) and zinc finger domains, as well as a coiled-coil domain and a C-terminal TRAF domain. The TRAF domain of TRAF3 is required for its interaction with PTPN22 to potentiate the TLR-dependent production of type I IFN in myeloid cells (39, 41). However, the RING and zinc finger domains are essential for the ability of TRAF3 to interact with TCPTP during IL-2R signaling in T cells (3). Structural differences between these two phosphatases (42, 43) may direct their association with different domains of TRAF3. Additionally, PTPN22 resides primarily in the cytoplasm (44), which enables PTPN22 to readily interact with Jak1 in response to IL-6. In contrast, TCPTP localizes to the endoplasmic reticulum or the nucleus, and stimulation of the IL-2R causes the translocation of TCPTP out of these subcellular compartments to interact with its target substrates (45, 46). These differences may contribute to the involvement of PTPN22, but not TCPTP, in IL-6R signaling in B cells.
Activation of STAT3 is necessary to stimulate the differentiation of B cells into PCs in both humans and mice (47, 48). The importance of IL-6 for PC differentiation has at times been unclear, because the PC population seems relatively normal in IL6−/− mice (49). However, several cytokines of the IL-6 family can signal through gp130 to compensate for the loss of IL-6 (50) It should also be noted that although the number of PCs in IL6−/− mice appears to be normal, these mice display a blunted antibody response to infections with several types of viruses (51, 52). Overexpression of IL-6 leads to increased concentrations of serum Igs and excessive numbers of PCs in mice (20), also supporting the important role played by IL-6 in the generation of PCs and the production of antibodies. Our findings suggest that TRAF3 is a key regulator of the IL-6–dependent generation of PCs in addition to its role in restraining B cell homeostasis and survival (5, 6).
Consistent with the finding that TRAF3 facilitated PTPN22-mediated decreases in IL-6 signaling, the size of the PC population in Ptpn22−/− mice was increased compared to that in wild-type control mice. In the original report describing Ptpn22−/− mice, the authors attributed the increased serum antibody titers and the spontaneous formation of germinal centers in these mice to increased TCR signaling strength (33). A later study by Maine et al. showed that when challenged with T cell–dependent antigens, the Ptpn22−/− mice had more follicular CD4+ T cells than did wild-type mice, as well as having increased numbers of ASCs and increased concentrations of serum IgG (53). Given the previously uncharacterized function of PTPN22 that we demonstrated here, we propose that PTPN22-mediated restraint of IL-6R signaling may also contribute to these phenotypic features of the Ptpn22−/− mouse. Whereas our results showed that PTPN22-deficient B cells did not differentiate more efficiently into PCs in vitro in response to LPS and IL-6 than did wild-type cells, PTPN22 plays an important positive regulatory role in TLR signaling pathways in myeloid cells (39). It is thus possible that the absence of PTPN22 in B cells also affects TLR4 signaling to limit B cell differentiation into CD138+ cells in response to LPS and IL-6; additional receptors could compensate for this limitation in vivo.
That TRAF3 plays a role in restraining IL-6R signaling in B cells has implications for the contributions of both excessive IL-6 and TRAF3 deficiency to B cell cancers. Although IL-6 production by the tumor microenvironment is sufficient to induce the development of premalignant B cells into plasmacytomas in a mouse model (54), we suggest that premalignant TRAF3-deficient B cells, because they have enhanced responsiveness to IL-6, may require very low amounts of IL-6 for malignant transformation. Thus, our results have implications for treatment strategies for TRAF3-deficient B cell malignancies.
MATERIALS AND METHODS
Mice
Traf3fl/fl mice have been described previously (6) and were backcrossed with C57BL/6 mice for more than 10 generations. Traf3fl/fl mice were bred with CD19-Cre mice on a C57BL/6J background (Jackson Laboratory) to generate mice with B cell–specific loss of TRAF3. IL6−/− mice (on a C57BL/6 background) were obtained from Jackson Laboratory. Ptpn22−/− mice were previously described (33), and their spleens and bone marrow were used for the experiments described here. Mice of 12 to 16 weeks of age were used for all experiments. All mice were maintained under specific pathogen–free conditions at the University of Iowa or The Scripps Clinic and were used in accordance with guidelines of the U.S. National Institutes of Health under an animal protocol approved by the Animal Care and Use Committee of the University of Iowa.
Cell lines
TRAF3-deficient A20.2J mouse B cell lines (A20.T3−/−) were previously described (55), as were subclones transfected with plasmid encoding Traf3 inducible by IPTG (Life Technologies) (56). All cell lines were maintained in B cell medium (BCM10) containing RPMI1640, penicillin (100 U/ml), streptomycin (100 U/ml), 2 mM L-glutamine (all from Life Technologies), 10 μM β-mercaptoethanol (Sigma), and 10% fetal calf serum (FCS). B cells expressing IPTG-inducible Traf3 were maintained in culture medium containing G418 disulfate (400 μg/ml; Research Products International) and hygromycin (200 μg/ml; Life Technologies).
B cell isolation and stimulation
B cells were isolated from single-cell suspensions of mouse splenocytes with anti-CD43 antibody–coated negative selection beads (Miltenyi) according to the manufacturer’s protocol. B cell purity was assessed by flow cytometric analysis with anti-B220 or anti-CD19 antibodies and was typically >97%. B cells were stimulated with recombinant murine IL-6 (20 ng/ml; PeproTech) or recombinant murine IL-21 (50 ng/ml; PeproTech) for the times indicated in the figure legends.
Flow cytometry and cell sorting
Single-cell suspensions were prepared from mouse spleens or from the bone marrow of femurs and tibias, and red blood cells were lysed with ACK buffer(0.15MNH4Cl, 10mMKHCO3,0.1 mMEDTA). For flow cytometric analysis, nonspecific staining was blocked by incubating the cells with monoclonal antibody against mouse CD16 and CD32 (clone 93), and cells were stained with fluorescently labeled monoclonal antibodies specific for B220, CD23, CD21/35, CD126 (IL-6Rα), and gp130 (all from eBioscience) or with antibody specific for CD138 (PharMingen). Flow cytometric analysis was performed on a FACSVerse flow cytometer (Becton Dickenson). To isolate PCs, CD138+ cells were pre-enriched with the Miltenyi CD138 Enrichment Kit according to the manufacturer’s protocol, and the pre-enriched CD138+ cells were sorted with a FACSAria flow cytometer (Becton Dickenson) at the University of Iowa Flow Cytometry Facility. The results were analyzed with Flow Jo software (Tree Star).
Human B cell isolation and depletion of TRAF3 with siRNA
Peripheral blood mononuclear cells were obtained from human whole-blood leukocyte reduction cones obtained from DeGowin Blood Center at the University of Iowa. Healthy donors of 18 to 55 years of age provided written consent for their blood to be used in research projects in compliance with the University of Iowa’s Institutional Review Board. Mononuclear cells were isolated with lymphocyte separation medium (Mediatech). CD19+ B cells were purified with Mltenyi positive selection beads according to the manufacturer’s protocol. To inhibit Traf3 expression, B cells were first stimulated for 48 hours with the F(ab′)2 fragments of anti-IgM antibody (5 μg/ml; Jackson ImmunoResearch), 5 μM CpG2006 (class B) purified oligonucleotides (Integrated DNA Technologies), and anti-human CD40 monoclonal antibody (2.5 μg/ml; G28.5), which was prepared in-house from a hybridoma provided by the American Type Culture Collection. Cells were transfected with the Traf3 Trilencer-27 siRNA (OriGene) with the siTran 1.0 reagent according to the manufacturer’s instructions. Recombinant human IL-4 protein (10 ng/ml; R&D Systems) was added to the culture medium during the transfection. Cells were harvested 48 hours after transfection and washed with BCM10, and then rested for 12 hours with additional washes at 6-hour intervals. After resting, cells were stimulated with recombinant human IL-6 (PeproTech) for the times indicated in the figure legends and then were analyzed by Western blotting to detect TRAF3 and pSTAT3.
Immunoprecipitation and Western blotting analysis
Antibodies used for immunoprecipitation included the following: anti-Jak1 (6G4, Cell Signaling Technology), anti-TRAF3 (M20, Santa Cruz Biotechnology), and anti-gp130 (MBL). Antibodies used for Western blotting analysis included the following: anti-Jak1, anti-pJak1, anti-pSTAT3 (Y705), anti-STAT3 (Cell Signaling Technology), anti-TRAF3 (H122), anti-gp130 (Santa Cruz Biotechnology), anti-PTPN22 (Proteintech), anti-phosphotyrosine (4G10, EMD Millipore), and anti–β-actin (Sigma). Cells were treated with IL-6 for the times indicated in the figure legends. Whole-cell lysates were prepared with radioimmunoprecipitation assay lysis buffer, resolved by SDS–polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride (PVDF) membranes for Western blotting analysis. For immunoprecipitation experiments, equal numbers of cells were lysed in lysis buffer [20 mM Tris-HCl (pH 7.5), 100 mM NaCl, 100 mM MgCl2, 100 mM CaCl2, supplemented with EDTA-free protease inhibitors (Roche), DNAse I (5 μg/ml; Roche), 2 mM Na3VO4 (Sigma)]. Antibodies used for immunoprecipitation (listed earlier) were added to the cell lysates and incubated overnight at 4°C with agitation, which was followed by a 2-hour incubation with Protein G magnetic beads (Life Technologies). Immunoprecipitates were used for Western blotting analysis to analyze interactions among various proteins. Multigauge software (FujiFilm) was used for the quantitative analysis of Western blots.
In vitro differentiation of PCs
Splenic B cells (1 × 106) were stimulated with LPS (10 μg/ml; Sigma) in the presence or absence of IL-6 (20 ng/ml; PeproTech) for 3 days. At the end of the culture period, the cells were collected and incubated with antibodies against B220 and CD138, as well as with the fixable viability dye eFluor-450 (eBioscience). Dead cells were excluded from analyses.
PC survival
Sorted PCs were cultured at a density of 1 × 104 cells per well in 96-well plates in the presence or absence of IL-6 (50 ng/ml; PeproTech) for the times indicated in the figure legends. The percentage cell survival was determined by calculating the percentage of the cell population that was negative for PI as determined by flow cytometric analysis.
ELISA and ELISpot analyses
Serum IL-6 was detected with the Quantikine Mouse IL-6 ELISA (enzyme-linked immunosorbent assay) Kit (R&D Systems) according to the manufacturer’s protocol. For ELISpot assays, 96-well plates with a hydrophobic PVDF membrane (Millipore) were coated with 100 μl of goat anti-mouse Ig (H+L) antibodies (15 μg/ml; Southern Biotech) overnight at 4°C. Coated plates were washed with phosphate-buffered saline (PBS) and blocked in BCM10 for at least 1 hour at room temperature. One hundred microliters of threefold serial dilutions of 2 × 105 splenocytes or 1 × 105 bone marrow cells was added to the antibody-coated wells, and the plates were then incubated at 37°C for 4 to 6 hours. The wells were washed three times with PBS and three times with PBS, 0.1% Tween 20 (PBS-T), and then were incubated with biotin-conjugated goat anti-mouse IgG or goat anti-mouse IgM antibodies (Southern Biotech) in PBS supplemented with 1% FCS overnight at 4°C. Plates were washed six times with PBS-T and incubated with alkaline phosphatase–conjugated streptavidin (at a 1:1000 dilution) in PBS for 1 hour in the dark at room temperature. Spots were visualized with bromochloroindolyl phosphate–nitro blue tetrazolium/nitro blue tetrazolium (BCIP/NBT) substrate (Sigma). There action was stopped by extensive washing with tap water, and the plates were air-dried. The number of spot-forming (antibody-secreting) cells was analyzed with the CTL-ImmunoSpot system.
Statistical analysis
Statistical differences between two means of various experimental groups were evaluated with the two-tailed unpaired Student’s t test. For comparisons of multiple groups, one-way ANOVA was used. Statistical significance was set at a P value of ≤0.05. All values were calculated with Prism software (GraphPad Software).
Supplementary Material
Fig. S1. The increase in the number of B cells caused by TRAF3 deficiency is independent of IL-6.
Fig. S2. Loss of TRAF3 in B cells does not alter the amounts of serum IL-6 in mice.
Fig. S3. Loss of TRAF3 does not affect IL-6R abundance on B cells.
Fig. S4. TRAF3 is not required for the IL-21–dependent activation of STAT3.
Fig. S5. TCPTP does not associate with Jak1 in B cells in response to IL-6.
Fig. S6. PTPN22-deficient B cells do not show enhanced PC differentiation in vitro in response to LPS and IL-6.
Acknowledgments
We are grateful to B. Hostager for helpful discussion and critical review of the manuscript.
Funding: This study was supported by NIH R01 awards CA099997 and AI28847 and a developmental pilot award from P50 CA97274 to G.A.B., NIH DK050824 to L.A.S., and core facilities supported by P30CA086862. Z.Y. received support from NIH T32 AI007260. This material is based on work supported in part by the Department of Veterans Affairs, Veterans Health Administration (VHA), Office of Research and Development. G.A.B. is a Senior Research Career Scientist of the VHA.
Footnotes
Author contributions: W.W.L. designed and performed experiments, analyzed the data, and wrote the manuscript; Z.Y. and L.L.S. performed experiments, provided conceptual input for data interpretation, and edited the manuscript; L.A.S. and C.J.M. produced and provided the Ptpn22−/− mice; and G.A.B. contributed to the conception and design of the experiments, interpreted the data, and edited the manuscript.
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Yi Z, Lin WW, Stunz LL, Bishop GA. Roles for TNF-receptor associated factor 3 (TRAF3) in lymphocyte functions. Cytokine Growth Factor Rev. 2014;25:147–156. doi: 10.1016/j.cytogfr.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie P, Kraus ZJ, Stunz LL, Liu Y, Bishop GA. TNF receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol. 2011;186:143–155. doi: 10.4049/jimmunol.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yi Z, Lin WW, Stunz LL, Bishop GA. The adaptor TRAF3 restrains the lineage determination of thymic regulatory T cells by modulating signaling via the receptor for IL-2. Nat Immunol. 2014;15:866–874. doi: 10.1038/ni.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu S, Pan W, Shi P, Gao H, Zhao F, Song X, Liu Y, Zhao L, Li X, Shi Y, Qian Y. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J Exp Med. 2010;207:2647–2662. doi: 10.1084/jem.20100703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391–401. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253–267. doi: 10.1016/j.immuni.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore CR, Liu Y, Shao C, Covey LR, Morse HC, III, Xie P. Specific deletion of TRAF3 in B lymphocytes leads to B-lymphoma development in mice. Leukemia. 2012;26:1122–1127. doi: 10.1038/leu.2011.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braggio E, Keats JJ, Leleu X, Van Wier S, Jimenez-Zepeda VH, Valdez R, Schop RF, Price-Troska T, Henderson K, Sacco A, Azab F, Greipp P, Gertz M, Hayman S, Rajkumar SV, Carpten J, Chesi M, Barrett M, Stewart AK, Dogan A, Bergsagel PL, Ghobrial IM, Fonseca R. Identification of copy number abnormalities and inactivating mutations in two negative regulators of NF-κB signaling pathways in Waldenstrom’s macroglobulinemia. Cancer Res. 2009;69:3579–3588. doi: 10.1158/0008-5472.CAN-08-3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braggio E, Keats JJ, Leleu X, Wier SV, Jimenez-Zepeda VH, Schop RF, Chesi M, Barrett M, Stewart AK, Dogan A, Bergsagel PL, Ghobrial IM, Fonseca R. High-resolution genomic analysis in Waldenström’s macroglobulinemia identifies disease-specific and common abnormalities with marginal zone lymphomas. Clin Lymphoma Myeloma. 2009;9:39–42. doi: 10.3816/CLM.2009.n.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Calado DP, Wang Z, Fröhler S, Köchert K, Qian Y, Koralov SB, Schmidt-Supprian M, Sasaki Y, Unitt C, Rodig S, Chen W, Dalla-Favera R, Alt FW, Pasqualucci L, Rajewsky K. An oncogenic role for alternative NF-κB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Rep. 2015;11:715–726. doi: 10.1016/j.celrep.2015.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, Gaidano G, Mullighan CG, Rabadan R, Dalla-Favera R. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt EM, Tan B, Zhao H, Stephens O, Santra M, Williams DR, Dang L, Barlogie B, Shaughnessy JD, Jr, Kuehl WM, Staudt LM. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, Kelsoe G. Cellular interaction in germinal centers. Roles of CD40 ligand and B7-2 in established germinal centers. J Immunol. 1995;155:556–567. [PubMed] [Google Scholar]
- 15.Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, Brink R. High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med. 2006;203:2419–2424. doi: 10.1084/jem.20061254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu VT, Berek C. The establishment of the plasma cell survival niche in the bone marrow. Immunol Rev. 2013;251:177–188. doi: 10.1111/imr.12011. [DOI] [PubMed] [Google Scholar]
- 17.Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for nonmalignant human plasmablasts. Blood. 2001;97:1817–1822. doi: 10.1182/blood.v97.6.1817. [DOI] [PubMed] [Google Scholar]
- 18.Rousset F, Garcia E, Banchereau J. Cytokine-induced proliferation and immunoglobulin production of human B lymphocytes triggered through their CD40 antigen. J Exp Med. 1991;173:705–710. doi: 10.1084/jem.173.3.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin-6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 20.Suematsu S, Matsuda T, Aozasa K, Akira S, Nakano N, Ohno S, Miyazaki JI, Yamamura KI, Hirano T, Kishimoto T. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc Natl Acad Sci USA. 1989;86:7547–7551. doi: 10.1073/pnas.86.19.7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawano M, Hirano T, Matsuda T, Taga T, Horii Y, Iwato K, Asaoku H, Tang B, Tanabe O, Tanaka H, Kuramoto A, Kishimoto T. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332:83–85. doi: 10.1038/332083a0. [DOI] [PubMed] [Google Scholar]
- 22.Ludwig H, Nachbaur DM, Fritz E, Krainer M, Huber H. Interleukin-6 is a prognostic factor in multiple myeloma. Blood. 1991;77:2794–2795. [PubMed] [Google Scholar]
- 23.Burger R. Impact of interleukin-6 in hematological malignancies. Transfus Med Hemother. 2013;40:336–343. doi: 10.1159/000354194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K, Taga T, Kishimoto T. IL-6–induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 1993;260:1808–1810. doi: 10.1126/science.8511589. [DOI] [PubMed] [Google Scholar]
- 26.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 28.Rodríguez-Bayona B, Ramos-Amaya A, López-Blanco R, Campos-Caro A, Brieva JA. STAT-3 activation by differential cytokines is critical for human in vivo–generated plasma cell survival and Ig secretion. J Immunol. 2013;191:4996–5004. doi: 10.4049/jimmunol.1301559. [DOI] [PubMed] [Google Scholar]
- 29.Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: From B-cell subsets to long-term survival niches. Semin Immunol. 2008;20:49–58. doi: 10.1016/j.smim.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Bottini N, Peterson EJ. Tyrosine phosphatase PTPN22: Multifunctional regulator of immune signaling, development, and disease. Annu Rev Immunol. 2014;32:83–119. doi: 10.1146/annurev-immunol-032713-120249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai X, James RG, Habib T, Singh S, Jackson S, Khim S, Moon RT, Liggitt D, Wolf-Yadlin A, Buckner JH, Rawlings DJ. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest. 2013;123:2024–2036. doi: 10.1172/JCI66963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menard L, Saadoun D, Isnardi I, Ng YS, Meyers G, Massad C, Price C, Abraham C, Motaghedi R, Buckner JH, Gregersen PK, Meffre E. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121:3635–3644. doi: 10.1172/JCI45790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. 2004;303:685–689. doi: 10.1126/science.1092138. [DOI] [PubMed] [Google Scholar]
- 34.Negro R, Gobessi S, Longo PG, He Y, Zhang ZY, Laurenti L, Efremov DG. Overexpression of the autoimmunity-associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood. 2012;119:6278–6287. doi: 10.1182/blood-2012-01-403162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oberg HH, Wesch D, Grüssel S, Rose-John S, Kabelitz D. Differential expression of CD126 and CD130 mediates different STAT-3 phosphorylation in CD4+CD25− and CD25high regulatory T cells. Int Immunol. 2006;18:555–563. doi: 10.1093/intimm/dxh396. [DOI] [PubMed] [Google Scholar]
- 37.Kwon H, Thierry-Mieg D, Thierry-Mieg J, Kim HP, Oh J, Tunyaplin C, Carotta S, Donovan CE, Goldman ML, Tailor P, Ozato K, Levy DE, Nutt SL, Calame K, Leonard WJ. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. 2009;31:941–952. doi: 10.1016/j.immuni.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, Corcoran LM, Godfrey DI, Toellner KM, Smyth MJ, Nutt SL, Tarlinton DM. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell–intrinsic mechanism. J Exp Med. 2010;207:365–378. doi: 10.1084/jem.20091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, Shaheen ZR, Cheng G, Sawatzke K, Campbell AM, Auger JL, Bilgic H, Shoyama FM, Schmeling DO, Balfour H, Jr, Hasegawa K, Chan AC, Corbett JA, Binstadt BA, Mescher MF, Ley K, Bottini N, Peterson EJ. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity. 2013;39:111–122. doi: 10.1016/j.immuni.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maine CJ, Marquardt K, Scatizzi JC, Pollard KM, Kono DH, Sherman LA. The effect of the autoimmunity-associated gene, PTPN22, on a BXSB-derived model of lupus. Clin Immunol. 2015;156:65–73. doi: 10.1016/j.clim.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Müller S, Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136:352–363. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiganis T, Bennett AM. Protein tyrosine phosphatase function: The substrate perspective. Biochem J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gjörloff-Wingren A, Saxena M, Han S, Wang X, Alonso A, Renedo M, Oh P, Williams S, Schnitzer J, Mustelin T. Subcellular localization of intracellular protein tyrosine phosphatases in T cells. Eur J Immunol. 2000;30:2412–2421. doi: 10.1002/1521-4141(2000)30:8<2412::AID-IMMU2412>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 45.Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. The T cell protein tyrosine phosphatase is a negative regulator of Janus family kinases 1 and 3. Curr Biol. 2002;12:446–453. doi: 10.1016/s0960-9822(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 46.ten Hoeve J, de Jesus Ibarra Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K, ten Hoeve J, de Jesus Ibarra Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K, ten Hoeve J, de Jesus Ibarra Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 48.Diehl SA, Schmidlin H, Nagasawa M, van Haren SD, Kwakkenbos MJ, Yasuda E, Beaumont T, Scheeren FA, Spits H. STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol. 2008;180:4805–4815. doi: 10.4049/jimmunol.180.7.4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Köhler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 50.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- 51.Harker JA, Lewis GM, Mack L, Zuniga EI. Late IL-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science. 2011;334:825–829. doi: 10.1126/science.1208421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dienz O, Eaton SM, Bond JP, Neveu W, Moquin D, Noubade R, Briso EM, Charland C, Leonard WJ, Ciliberto G, Teuscher C, Haynes L, Rincon M. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J Exp Med. 2009;206:69–78. doi: 10.1084/jem.20081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maine CJ, Marquardt K, Cheung J, Sherman LA. PTPN22 controls the germinal center by influencing the numbers and activity of T follicular helper cells. J Immunol. 2014;192:1415–1424. doi: 10.4049/jimmunol.1302418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosean TR, Tompkins VS, Olivier AK, Sompallae R, Norian LA, Morse HC, III, Waldschmidt TJ, Janz S. The tumor microenvironment is the main source of IL-6 for plasma cell tumor development in mice. Leukemia. 2015;29:233–237. doi: 10.1038/leu.2014.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie P, Hostager BS, Bishop GA. Requirement for TRAF3 in signaling by LMP1 but not CD40 in B lymphocytes. J Exp Med. 2004;199:661–671. doi: 10.1084/jem.20031255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin WW, Hildebrand JM, Bishop GA. A complex relationship between TRAF3 and non-canonical NF-κB2 activation in B lymphocytes. Front Immunol. 2013;4:477. doi: 10.3389/fimmu.2013.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The increase in the number of B cells caused by TRAF3 deficiency is independent of IL-6.
Fig. S2. Loss of TRAF3 in B cells does not alter the amounts of serum IL-6 in mice.
Fig. S3. Loss of TRAF3 does not affect IL-6R abundance on B cells.
Fig. S4. TRAF3 is not required for the IL-21–dependent activation of STAT3.
Fig. S5. TCPTP does not associate with Jak1 in B cells in response to IL-6.
Fig. S6. PTPN22-deficient B cells do not show enhanced PC differentiation in vitro in response to LPS and IL-6.