

Abstract
Background
Selecting a parsimonious set of informative genes to build highly generalized performance classifier is the most important task for the analysis of tumor microarray expression data. Many existing gene pair evaluation methods cannot highlight diverse patterns of gene pairs only used one strategy of vertical comparison and horizontal comparison, while individual-gene-ranking method ignores redundancy and synergy among genes.
Results
Here we proposed a novel score measure named relative simplicity (RS). We evaluated gene pairs according to integrating vertical comparison with horizontal comparison, finally built RS-based direct classifier (RS-based DC) based on a set of informative genes capable of binary discrimination with a paired votes strategy. Nine multi-class gene expression datasets involving human cancers were used to validate the performance of new method. Compared with the nine reference models, RS-based DC received the highest average independent test accuracy (91.40 %), the best generalization performance and the smallest informative average gene number (20.56). Compared with the four reference feature selection methods, RS also received the highest average test accuracy in three classifiers (Naïve Bayes, k-Nearest Neighbor and Support Vector Machine), and only RS can improve the performance of SVM.
Conclusions
Diverse patterns of gene pairs could be highlighted more fully while integrating vertical comparison with horizontal comparison strategy. DC core classifier can effectively control over-fitting. RS-based feature selection method combined with DC classifier can lead to more robust selection of informative genes and classification accuracy.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-016-0893-0) contains supplementary material, which is available to authorized users.
Keywords: Microarray expression data, Gene selection, Direct classify, Relative simplicity, Binary-discriminative informative genes, Paired votes
Background
Microarray expression data of cancer tissue samples has the following properties: small sample size yet large number of features, high noise and redundancy, a remarkable level of background differences among samples and features, and nonlinearity [1, 2]. Selecting a parsimonious set of informative genes to build robust classifier with highly generalized performance is one of the most important tasks for the analysis of microarray expression data, as it can help to discover disease mechanisms, as well as improve the precision and reduce the cost of clinical diagnoses [3].
Gene selection depends on a given evaluation strategy and a defined score. The individual-gene-ranking methods rank genes by only comparing the expression values of the same individual gene between different classes (a vertical comparison evaluation strategy). This can be very far from the truth, as the deregulation of pathways, rather than individual genes, may be critical in triggering carcinogenesis [4]. If a gene has a remarkable joint effect on other genes, it should be selected as an informative gene, even though it may receive a lower rank in an individual-gene-ranking method. This joint effect of genes has been taken into account in most popular, existing algorithms, including top scoring pair (TSP) [5, 6], top scoring triplet(TST) [7], top-scoring ‘N’(TSN) [8], top scoring genes (TSG) [9] and doublet method [4]. However, the gene pairs score, that is the percentage of Δij in TSP [5, 6], cannot reflect size differences among samples. To fully utilize sample size information TSG introduces chi-square values as the score for gene pairs [9]. TSP and TSG are both pair-wise gene evaluations, which compare the expression values of the same sample between two different genes (a horizontal comparison evaluation strategy), and can help to eliminate the influence of sampling variability due to different subjects [5, 6, 9].
At the level of gene pairs, Merja et al. [10] defined two patterns based on rank data, rather than absolute expression, from data-driven perspective: the consistent reversal of relative expression and consistent relative expression. This premise allowed us to organize the cell types in to their ontogenetic lineage-relationships and may reflect regulatory relationships among the genes [10]. The first pattern can be subdivided into a consistent reversal of expression (Pattern I) and a consistent reversal of relative expression (Pattern II) based on absolute expression (see Table 1). Similarly, the second pattern can be subdivided in to a consistent expression (Pattern III) and a consistent relative expression (Pattern IV). Furthermore, a heterogeneous background expression of samples (Pattern V) and an interaction expression pattern (Pattern VI) can be defined, if the influence of sampling variability due to different subjects [9] and paired-gene interactions are considering [11]. Clearly, all twelve genes (G1 ~ G12) in Table 1 should be informative genes from data-driven perspective. However, individual-gene evaluations, which only detect different expression levels between positive samples and negative samples, cannot highlight Pattern V and Pattern VI. Pair-wise gene evaluation with vertical comparison can highlight most patterns except Pattern V. Only pair-wise gene evaluation with horizontal comparison can highlight Pattern V, even though it cannot detect most other patterns. Therefore, both vertical and horizontal comparisons need to be considered in pair-wise gene evaluation techniques.
Table 1.
Six patterns for joint effect of gene pairs in binary-class simulation data
| Class | Pattern I | Pattern II | Pattern III | Pattern IV | Pattern V | Pattern VI | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G1 | G2 | G3 | G4 | G5 | G6 | G7 | G8 | G9 | G10 | G11 | G12 | ||||||||||||||
| + | 50 | 100 | 5 | 100 | 50 | 50 | 5 | 50 | 50 | 100 | 50 | 50 | |||||||||||||
| + | 50 | 100 | 5 | 100 | 50 | 50 | 5 | 50 | 5 | 10 | 100 | 100 | |||||||||||||
| + | 50 | 100 | 5 | 100 | 50 | 50 | 5 | 50 | 50 | 100 | 50 | 50 | |||||||||||||
| + | 50 | 100 | 5 | 100 | 50 | 50 | 5 | 50 | 5 | 10 | 100 | 100 | |||||||||||||
| - | 100 | 50 | 10 | 50 | 100 | 100 | 10 | 100 | 100 | 50 | 50 | 100 | |||||||||||||
| - | 100 | 50 | 10 | 50 | 100 | 100 | 10 | 100 | 10 | 5 | 100 | 50 | |||||||||||||
| - | 100 | 50 | 10 | 50 | 100 | 100 | 10 | 100 | 100 | 50 | 50 | 100 | |||||||||||||
| - | 100 | 50 | 10 | 50 | 100 | 100 | 10 | 100 | 10 | 5 | 100 | 50 | |||||||||||||
| Background difference between gene pairs | Not exist | Exist | Not exist | Exist | Not exist | Not exist | |||||||||||||||||||
| Background difference among samples | Not exist | Not exist | Not exist | Not exist | Exist | Not exist | |||||||||||||||||||
| Vertical comparison of individual-gene |
G1 < 75 | G1 > 75 | G2 < 75 | G2 > 75 | G3 < 7 | G3 > 7 | G4 < 75 | G4 > 75 | G5 < 75 | G5 > 75 | G6 < 75 | G6 > 75 | G7 < 7 | G7 > 7 | G8 < 75 | G8 > 75 | G9 < 41 | G9 > 41 | G10 < 41 | G10 > 41 | G11 < 75 | G11 > 75 | G12 < 75 | G12 > 75 | |
| + | 4 | 0 | 0 | 4 | 4 | 0 | 0 | 04 | 4 | 0 | 4 | 0 | 4 | 0 | 4 | 0 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| - | 0 | 4 | 4 | 0 | 0 | 4 | 4 | 0 | 0 | 4 | 0 | 4 | 0 | 4 | 0 | 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| Highlight | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | No (χ 2 = 0.5) | No (χ 2 = 0.5) | No (χ 2 = 0.5) | No (χ 2 = 0.5) | |||||||||||||
| Horizontal comparison of pair-wise genes | G1 > G2 | G1 < G2 | G3 > G4 | G3 < G4 | G5 > G6 | G5 < G6 | G7 > G8 | G7 < G8 | G9 > G10 | G9 < G10 | G11 > G12 | G11 < G12 | |||||||||||||
| + | 0 | 4 | 0 | 4 | 2 | 2 | 0 | 4 | 0 | 4 | 2 | 2 | |||||||||||||
| - | 4 | 0 | 0 | 4 | 2 | 2 | 0 | 4 | 4 | 0 | 2 | 2 | |||||||||||||
| Highlight | Yes (χ 2 = 4.5*) | No (χ 2 = 0) | No (χ 2 = 0.5) | No (χ 2 = 0) | Yes (χ 2 = 4.5*) | No (χ 2 = 0.5) | |||||||||||||||||||
| Vertical comparison of pair-wise genes | G1 < 75 & G2 < 75 | G1 < 75 &G2 > 75 | G1 > 75 & G2 < 75 | G1 > 75 &G2 > 75 | G3 < 7 & G4 < 75 | G3 < 7 & G4 > 75 | G3 > 7 & G4 < 75 | G3 > 7 & G4 > 75 | G5 < 75 & G6 < 75 | G5 < 75 & G6 > 75 | G5 > 75 & G6 < 75 | G5 > 75 & G6 > 75 | G7 < 7 & G8 < 75 | G7 < 7 & G8 > 75 | G7 > 7 & G8 < 75 | G7 > 7 & G8 > 75 | G9 < 41 & G10 < 41 | G9 < 41 & G10 > 41 | G9 > 41 & G10 < 41 | G9 > 41 & G10 > 41 | G11 < 75 & G12 < 75 | G11 < 75 & G12 > 75 | G11 > 75 & G12 < 75 | G11 > 75 & G12 > 75 | |
| + | 0 | 4 | 0 | 0 | 0 | 4 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 2 | 0 | 0 | 2 | 2 | 0 | 0 | 2 | |
| - | 0 | 0 | 4 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 4 | 2 | 0 | 0 | 2 | 0 | 2 | 2 | 0 | |
| Highlight | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | Yes (χ 2 = 4.5*) | No (χ 2 = 0.5) | Yes ((χ 2 = 8*) | |||||||||||||||||||
Values in parenthesis are chi-square values, * denote p < 0.05
We first propose a novel score measure, in this paper, that of relative simplicity (RS), based on information theory. We adopt an integrated evaluation strategy to rank genes one by one, considering not only individual-gene effects, but also pair-wise joint effects between candidate gene and others. In particular, for pair-wise gene evaluations, vertical comparisons are integrated with horizontal comparisons to detect all six patterns of pair-wise joint effects. Ultimately, we construct a relative simplicity-based direct classifier (RS-based DC) to select binary-discriminative informative genes on training dataset and perform independent tests. The independent testing of nine multiclass tumor gene expression datasets showed that RS-based DC selects fewer informative genes and outperforms the referred models by a large margin, especially in larger m (total number of classes) datasets, such as Cancers (m = 11) [12]and GCM (m = 14) [13].
Datasets and methods
Datasets
Ten multi-class datasets have been used in published previous TSP [5, 6] and TSG [9] papers. We did not include dataset Leukemia3 [14] in our study because 65 % of the expression values in it are zero. The remaining nine datasets references, sample sizes, numbers of genes, and numbers of classes are summarized in Table 2. Suppose that a training dataset has n samples and p genes, and that the data can be denoted as (Yi, Xi,j), i = 1,2,…, n; j = 1,2,…, p. Where Xi,j represents the expression value of the jth gene (Gj) in the ith sample; and Yi represents the class label of ith sample, where Yi∈{Class1, Class2, …, Classt, …, Classm}, t = 1,2,…,m.
Table 2.
Nine multi-class gene expression datasets
| Dataset | Platform | No. of classes | No. of genes | No. of samples | Source | |
|---|---|---|---|---|---|---|
| Training | Test | |||||
| Leukemia1 | Affy | 3 | 7129 | 38 | 34 | [15] |
| Lung1 | Affy | 3 | 7129 | 64 | 32 | [16] |
| Leukemia2 | Affy | 3 | 12582 | 57 | 15 | [17] |
| SRBCT | cDNA | 4 | 2308 | 63 | 20 | [18] |
| Breast | Affy | 5 | 9216 | 54 | 30 | [19] |
| Lung2 | Affy | 5 | 12600 | 136 | 67 | [20] |
| DLBCL | cDNA | 6 | 4026 | 58 | 30 | [21] |
| Cancers | Affy | 11 | 12533 | 100 | 74 | [12] |
| GCM | Affy | 14 | 16063 | 144 | 46 | [13] |
Data preprocessing
Adjustment for outliers
Outliers may exist in datasets. For example, in the Lung1 [16] training set, the expression value X54,4290 of the 54th sample in gene G4290 is 7396.1, while the average expression value of the other samples in gene G4290 is 80.15 (range from 16 to 197). The outliers overstate the differences among the classes, and need be adjusted before gene ranking. For gene Gj, we defined outliers as those values beyond the scope of [, ]. If or , then Xij is an outlier, where α is significance level, and σ. j represent the average value and standard deviation of X · j, respectively. Therefore, we adjust the outliers using the following formula:
| 1 |
Here and σ‐ i,j represent the average value and standard deviation of X · j without Xi,j, respectively. X"ij is the value of Xij after adjusting. represents the distribution interval of X-i,j. We generally set α to 0.05 (u0.05 = 1.96). Adjustment for outliers was only used with training set.
Transforming datasets from multi-class to binary-class with “one versus rest”
Suppose that Yi∈(Class1, Class2, …, Classt, …, Classm), and we adopt a “one versus rest” (OVR) approach to transform a multi-class training set to binary-class. This generates m binary-class datasets, denoted {Class1vs. non-Class1}, {Class2vs. non-Class2}, …, {Classtvs. non-Classt}, …, {Classmvs. non-Classm}. In each binary-class training dataset, Classt are positive samples {+}, and non-Classt are negative samples {−}.
Complexity and relative simplicity score
Entropy stands for disorder or uncertainty. For a discrete system with k events, its Shannon entropy is defined as:
| 2 |
Where ni denotes the frequency of event i, and N is the total frequency. Here we use base-2 logarithms. H only reflects the event ratios. Complexity (C) as proposed by Zhang [22] can reflect both event ratios and event frequencies:
| 3 |
For a given 2 × r Contingency table (Table 3), its complexity is the total of row complexities (Crow) and column complexities (Ccolumn). f+d (d = 1,…,r) and f−d in Table 3 represent the frequency of the event.
| 4 |
| 5 |
| 6 |
Table 3.
2×r Contingency table
| Class | Column1 | … | Columnd | … | Columnr | Total |
|---|---|---|---|---|---|---|
| + | f+1 | … | f+d | … | f+r | f+ |
| - | f-1 | … | f-d | … | f-r | f- |
| Total | f1 | … | fd | … | fr | n |
For contingency Table 1 (2 × r1) and contingency Table 2 (2 × r2), their complexities are incomparable if r1 is unequal to r2. Therefore we introduce a novel score, RS, according to their maximum complexity (Table 4). Table 4 cames directly from Table 3 directly, only the frequency of each column in the same class is set to be equal.
| 7 |
| 8 |
| 9 |
| 10 |
Table 4.
2×r Contingency table for maximum complexity
| Class | Column1 | … | Columnd | … | Columnr | Total |
|---|---|---|---|---|---|---|
| + | f+/r | … | f+/r | … | f+/r | f+ |
| - | f-/r | … | f-/r | … | f-/r | f- |
| Total | n/r | … | n/r | … | n/r | n |
Individual-gene evaluation
For a given gene Gj with continued expression values X. j in a binary-class training dataset, we partition X. j into two parts (X. j > EPj and X. j < EPj) with an endpoint (EP):
| 11 |
Where and are the average expression values of X. j for negative and positive samples, respectively. We then generate a 2 × 2 contingency table for gene Gj (Table 5).
Table 5.
2 × 2 contingency table for individual gene
| Class | X. j > EP j | X. j < EP j | Total |
|---|---|---|---|
| + | f+1 | f+2 | f+ |
| - | f−1 | f−2 | f− |
| Total | f1 | f2 | n |
f+1 is the number of positive samples with expression values larger than EP j, f+2 is the number of positive samples with expression values less than EP j, f−1 is the number of negative samples with expression values larger than EP j, and f−2 is the number of negative samples with expression values less than EP j. When X i,j equals EP j, and Y i belongs to positive sample {+}, f+1 and f+2 increase by 0.5 respectively; when X i,j equals EP j, and Y i belongs to negative sample {−}, f−1 and f−2 increase by 0.5 respectively
For the individual-gene evaluation of gene Gj, we then got its RS score, , according to Table 5 and formula (10).
Pair-wise gene evaluation
Horizontal comparison of gene pairs
For gene pairs Gj and Gq (j ≠ q) in a binary-class training dataset, we generate a 2 × 2 contingency table (Table 6) for the horizontal comparison with Xi,j > Xi,q and Xi,j < Xi,q, similar to TSP [2, 3] and TSG [9].
Table 6.
2 × 2 contingency table for gene pairs of horizontal comparison
| Class | X i,j > X i,q | X i,j < X i,q | Total |
|---|---|---|---|
| + | f+1 | f+2 | f+ |
| - | f−1 | f−2 | f− |
| Total | f1 | f2 | n |
X i,j represents the expression value of the j th gene (Gj) in the i th sample; f+1 is the number of positive samples with X i,j larger than X i,q, f+2 is the number of positive samples with X i,j less than X i,q, f−1 is the number of negative samples with X i,j larger than X i,q, and f−2 is the number of negative samples with X i,j less than X i,q
For horizontal comparison of gene pairs Gj and Gq, We generate the complexity Chor-Gj-Gq and the maximum complexity Chor-Gj-Gq-max, of gene pairs Gj and Gq, for the horizontal comparison, according to Table 6, formula (6), and formula (9).
Vertical comparison of gene pairs
For gene pairs Gj and Gq (j ≠ q) in a binary-class training dataset, we partition X. j and X. q into two parts with endpoint EPj and EPq, respectively. We then generate a 2 × 4 contingency table (Table 7) for the vertical comparison.
Table 7.
2 × 4 contingency table for gene pairs of vertical comparison
| Class | X. j > EP j & X. q > EP q | X. j > EP j & X. q < EP q | X. j < EP j & X. q > EP q | X. j < EP j & X. q < EP q | Total |
|---|---|---|---|---|---|
| + | f+1 | f+2 | f+3 | f+4 | f+ |
| - | f−1 | f−2 | f−3 | f−4 | f− |
| Total | f1 | f2 | f3 | f4 | n |
f+1 is the number of positive samples with X. j larger than EP j and X. qlarger than EP q, f+2 is the number of positive samples with X. j larger than EP j and X. q less than EP q, f+3 is the number of positive samples with X. j less than EP j and X. q larger than EP q, f+4 is the number of positive samples with X. j less than EP j and X. q less than EP q, f−1 is the number of positive samples with X. j larger than EP j and X. q larger than EP q, f−2 is the number of positive samples with X. j larger than EP j and X. q less than EP q, f−3 is the number of positive samples with X. j less than EP j and X. q larger than EP q, and f−4 is the number of positive samples with X. j less than EP j and X. q less than EP q
For vertical comparison of gene pairs Gj and Gq, We then generate the complexity Cver-Gj-Gq and the maximum complexity Cver -Gj-Gq-max of gene pairs Gj and Gq for the vertical comparison according to Table 7, formula (6), and formula (9).
RS score of gene pairs
For gene pairs Gj and Gq in a binary-class training dataset, we generate RS weight scores, RSGj_Gq, according to formula (12).
| 12 |
Integrated individual-gene ranking
For a given gene Gj in a binary-class training dataset, the integrated RS score, IRSGj, can be calculated with formula (13):
| 13 |
Here, RSGj represents vertical comparison of individual-gene; RSGj_Gq represents horizontal comparison and vertical comparison of pair-wise genes; represents the weight of Gj in the pair-wise comparison. According to IRSGj, the descending order of all p genes can be obtained and recorded as {GRank1, GRank2,…, GRankj,…, GRankp}. The integrated evaluation process of Gj is shown in Fig. 1.
Fig. 1.
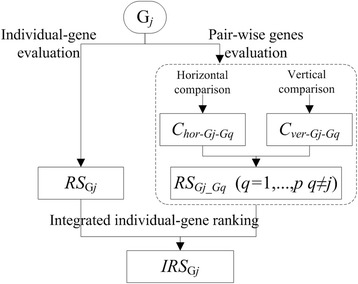
Integrated evaluation process of Gj
Informative gene selection
The IRS scores provide a list of top ranked genes. However, the combination of top ranked genes may not produce a top ranked combination of genes because of the redundancy and interaction among genes [23]. Therefore, we used a forward feature selection strategy to select informative gene subsets, along with our RS-based-DC classifier and leave-one-out cross-validation error estimates (LOOCV).
For a given binary-class training dataset with n samples and p ranked genes:
Step 1: Introduce gene GRank1, get dataset S∈(Yi, Xi), i = 1,2,…, n; Xi represents the expression value of gene GRank1 in the ith sample; Yi represents the class label of ith sample and Yi∈{+, −}. Leave out one sample as the validation data (S-validation) and the rest as the training data (S-train). First assign {+} to S-validation as a class label, merge S-validation and S-train, get RSGRank1(+); then assign {−} to S-validation as a class label, merge S-validation and S-train, get RSGRank1(−). If RSGRank1(+) is larger than RSGRank1(−), the S-validation sample belongs to the positive sample; otherwise, the S-validation sample belongs to the negative sample. Repeat prediction for all the samples in S to get the prediction class labels. Calculate the Matthew correlation coefficient (MCC) according to formula (14) and denote as MCC1.
| 14 |
Here TP, TN, FP, FN represent true positives, true negatives, false positives and false negatives, respectively.
Step 2: MCCbenchmark = MCC1.
Step 3: Introduce the next top ranked gene. In general, denote total number of the current genes as r. Get dataset S = (Yi, Xi,j), i = 1,2,…, n; j = 1,2,…, r. The network RS score of r gene can be calculated with formula (15).
| 15 |
Leave out one sample as the validation data (S-validation) and the rest as the training data (S-train). First assign {+} to S-validation as a class label, merge S-validation and S-train, get RSr-net(+); then assign {−} to S-validation as a class label, merge S-validation and S-train, get RSr-net (−). If RSr-net (+) is larger than RSr-net (−), the S-validation sample belongs to the positive sample; if RSr-net (+) is less than RSr-net (−), the S-validation sample belongs to the negative sample. Repeat prediction for all the samples in S to get the prediction class labels. Calculate MCC according to formula (14) and denote as MCCr.
Step 4: If MCCr ≤ MCCbenchmark delete X. r, else MCCbenchmark = MCCr.
Step 5: Repeat Step 3 and Step 4, until the top B rank genes are successively introduced (our experience suggests that it is sufficient to set the upper bound of B at 100).
We consequently generate the informative genes subset for the binary-class dataset (Pseudo-code see Table 8).
Table 8.
Pseudo-code of informative genes selection
| Algorithm 1 Informative gene selection (Dateset, GRank) |
| Require: Dateset is a binary-class training dataset with n samples |
| Require: GRank is the order of all p genes {GRank1, GRank2,…, GRankj,…, GRankp} |
| Ensure: Returns the binary-discriminative informative genes subset of Dateset |
| 1: ture_Y ← class lable of training samples |
| 2: j ← 1; MCCbenchmark ← 0; B ← 100 |
| 3: repeat |
| 4: S ← GRankj # introducing GRankj |
| 5: if |S| ≤ 1 then |
| 6: for i = 1 to n do # leave-one-out cross-validation |
| 7: Y i ← + |
| 8: get RS GRankj(+) |
| 9: Y i ← − |
| 10: get RS GRankj(−) |
| 11: if RS GRankj(+) > RS GRankj(−) then pred_Y i ← + |
| 12: else pred_Y i ← − |
| 13: end for |
| 14: MCCbenchmark ← get MCC (true_Y, pred_Y) from formula (14) |
| 15: else |
| 16: for i = 1 to n do # leave-one-out cross-validation |
| 17: Y i ← + |
| 18: get RS-net(+) from formula (15) |
| 19: Y i ← − |
| 20: get RS-net(−) from formula (15) |
| 21: if RS-net(+) > RS-net (−) then pred_Y i ← + |
| 22: else pred_Y i ← − |
| 23: end for |
| 24: MCC ← get MCC (true_Y, pred_Y) from formula (14) |
| 25: end if |
| 26: if MCC > MCCbenchmark then MCCbenchmark ← MCC |
| 27: else delete GRankj |
| 28: until j > B |
| 29: retrun S |
Paired votes prediction with RS-based DC
We generate an m binary-class training set, denoted as {Class1vs. non-Class1}, {Class2vs. non-Class2},…,{Classtvs. non-Classt},…,{Classmvs. non-Classm}, according our OVR approach; and the corresponding m binary-discriminative informative gene (BDIG) subsets, denoted as BDIGClass1, BDIGClass2, …, BDIGClasst, …, BDIGClassm, according to our individual-gene evaluation ~ informative gene selection sections.
For a test sample with m possible class labels, in general, for paired vote predictions between Classt and Classw, we merge the Classt and Classw samples into a new training set with r genes according to {BDIGClasst ∪ BDIGClassw}. We first assign {Classt} to the test sample as a class label, merge the test sample and the new training set, generating RSr ‐ net {Classt}; then we assign {Classw} to the test sample as class a label, merge the test sample and the new training set, generating RSr ‐ net {Classw}. If RSr-net {Classt} is larger than RSr-net {Classw}, the test sample belongs to Classt, else it belongs to Classw. The winner continues paired vote with the next class and the prediction class label of the test sample is the last winner.
After the predictions for all of the testing samples have been obtained, we calculate the test accuracy, expressed as the ratio of the number of correctly classified samples to the total number of samples, for multi-classification.
Results and analysis
Comparison of independent prediction accuracy and the number of informative genes among different models
We used nine reference models, HC-TSP [3], HC-K-TSP [3], DT [24], PAM [25], TSG [9], mRMR-SVM, SVM-RFE-SVM, Entropy-based DC and χ2-based DC, to evaluate the performance of RS-based DC. Results from the first five models are cited from the corresponding literature, and the results from the latter four models are presented in this paper.
As a feature selection method mRMR has two evaluation criterions: mutual information difference (MID) and mutual information quotient (MIQ). Here we used MIQ-mRMR, because MIQ is more robust than MID in general [26]. mRMR and SVM-RFE [27] only provide a list of ranked genes, therefore, we adopted the Library for Support Vector Machines (LIBSVM) as a classifier [28] to generate an informative gene subset. LIBSVM supports multiclass classification, and is available at http://www.csie.ntu.edu.tw/~cjlin/libsvm. We initially listed the top 2 % of informative genes according to mRMR or SVM-RFE. Second, we introduced these genes one by one and conducted 10-fold cross-validation for the training sets based on SVM. Third, we selected the genes with the highest cross-validation accuracy as our informative genes subset, and finally we performed independent predictions using SVM with informative genes, for the mRMR-SVM and SVM-RFE-SVM models. Four kernel functions, linear, radius basis function (RBF), sigmoid and polynomial in SVM, were evaluated, and the linear kernel produced optimal accuracy with the nine datasets. Therefore, we used linear kernel in this study, unless specifically stated. Different penalty parameters C (C∈[2−5, 215]) were optimized in different SVM models with the training set. Entropy-based DC and χ2-based DC uses the same modelling process as RS-based DC, except entropy [29] is used, rather than complexity, in Entropy-based DC, and χ2 is used, rather than RS, in χ2-based DC.
The test accuracy and informative gene number for nine different multi-class datasets are listed in Table 9. The best models based on average accuracy were RS-based DC (91.40 %), χ2-based DC (89.41 %), TSG (88.99 %), PAM (87.91 %), SVM-RFE-SVM (86.23 %) and HC-K-TSP (85.45 %). Of the six models, χ2-based DC, TSG and HC-K-TSP performed poorly in predictive power with GCM, Cancers and Breast datasets, respectively. PAM generated an unacceptable informative gene number (an average of 1450), and also demonstrated poor predictive performance with the Cancers dataset. RS-based DC and SVM-RFE-SVM performed robustly with all nine datasets. Compared with the nine reference models, RS-based DC received the least informative gene number (an average of 20.56), the highest average accuracy and the minimum standard deviation (9 %).
Table 9.
Independent test accuracy and the number of informative genes (in parenthesis) among different models
| Model | Leuk1 | Lung1 | Leuk2 | SRBCT | Breast | Lung2 | DLBCL | Cancers | GCM | Average |
|---|---|---|---|---|---|---|---|---|---|---|
| HC-TSPa | 97.06 | 71.88 | 80.00 | 95.00 | 66.67 | 83.58 | 83.33 | 74.32 | 52.17 | 78.22 ± 13.97 |
| (4) | (4) | (4) | (6) | (8) | (8) | (10) | (20) | (26) | (10.00) | |
| HC-K-TSPa | 97.06 | 78.13 | 100 | 100 | 66.67 | 94.03 | 83.33 | 82.43 | 67.39 | 85.45 ± 13.12 |
| (36) | (20) | (24) | (30) | (24) | (28) | (46) | (128) | (134) | (52.22) | |
| DTa | 85.29 | 78.13 | 80.00 | 75.00 | 73.33 | 88.06 | 86.67 | 68.92 | 52.17 | 76.40 ± 11.13 |
| (2) | (4) | (2) | (3) | (4) | (5) | (5) | (10) | (18) | (5.89) | |
| PAMa | 97.06 | 78.13 | 93.33 | 95.00 | 93.33 | 100 | 90.00 | 87.84 | 56.52 | 87.91 ± 13.34 |
| (44) | (13) | (62) | (285) | (4822) | (614) | (3949) | (2008) | (1253) | (1450) | |
| TSGb | 97.06 | 81.25 | 100 | 100 | 86.67 | 95.52 | 93.33 | 79.73 | 67.39 | 88.99 ± 11.11 |
| (6) | (20) | (44) | (13) | (63) | (60) | (16) | (81) | (112) | (46.11) | |
| mRMR-SVM | 76.47 | 78.13 | 100 | 75.00 | 96.67 | 95.52 | 96.67 | 71.62 | 45.65 | 81.75 ± 17.54 |
| (7) | (13) | (19) | (9) | (97) | (120) | (16) | (89) | (57) | (47.44) | |
| SVM-RFE-SVM | 85.29 | 78.13 | 93.33 | 95.00 | 90.00 | 88.06 | 90.00 | 93.24 | 63.04 | 86.23 ± 10.08 |
| (5) | (9) | (8) | (3) | (7) | (9) | (13) | (29) | (199) | (31.33) | |
| Entropy-based DC | 91.18 | 78.13 | 86.67 | 100 | 83.33 | 88.06 | 93.33 | 78.38 | 47.83 | 82.99 ± 14.93 |
| (7) | (14) | (13) | (9) | (13) | (39) | (15) | (73) | (93) | (30.67) | |
| χ 2-based DC | 94.12 | 81.00 | 100 | 100 | 90.00 | 97.02 | 93.33 | 90.54 | 58.70 | 89.41 ± 12.91 |
| (23) | (18) | (30) | (31) | (33) | (42) | (23) | (95) | (90) | (42.78) | |
| RS-based DC | 94.12 | 84.38 | 100 | 100 | 93.33 | 98.51 | 90.00 | 90.54 | 71.74 | 91.40 ± 9.00 |
| (7) | (12) | (13) | (11) | (15) | (21) | (16) | (36) | (54) | (20.56) |
The same modeling process was conducted for RS-based DC, Entropy-based DC and χ2-based DC to compare the merits of the defined score. As mentioned above, RS scores and χ2 scores utilize sample size information, whereas entropy scores only reflect the events ratio. Therefore, our RS-based DC and χ2-based DC have better predictive performance than Entropy-based DC method.
Comparison of feature selection methods
An excellent feature selection method should perform well with various classifiers. We used four reference feature selection methods, mRMR, SVM-RFE, TSG and HC-K-TSP, to evaluate the performance of RS.
As shown in Table 10, with the informative genes selected by the five feature selection methods, the average independent prediction precisions of Naïve Bayes (NB) [31] and K-nearest neighbor (KNN) [32] on the nine datasets were clearly improved. However, surprisingly, the four reference feature selection methods were ineffective in the SVM classifier. This seems to challenge the conventional wisdom that feature selection should be effective in improving the performance of the model. Fortunately, RS still performed well with the SVM classifier upholding the conventional wisdom. For the SVM classifier, in three (Lung1, SRBCT and GCM) out of nine datasets, there was basically no improvement in performing feature selection, regardless of the feature selection technique. However, the NB and KNN classifiers did not always show such a phenomenon, possibly because SVM is not sensitive to feature dimensions; therefore, SVM could obtain very precise prediction without feature selection. RS was the only strategy that was better than no feature selection, on average, when combined with SVM, because on the Leuk1, Breast and Cancers datasets it showed a sufficiently large improvement was large enough, while it slightly reduced the precision of the prediction on the other datasets. Thus, the results indicated that RS is superior to the other four feature selection methods.
Table 10.
Test accuracy of different classifiers with informative genes selected by different feature-selection methods
| Classifier | Feature-selection method | Leuk1 | Lung1 | Leuk2 | SRBCT | Breast | Lung2 | DLBCL | Cancers | GCM | Average |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NB | ALLa | 85.29 | 81.25 | 100 | 60.00 | 66.67 | 88.06 | 86.67 | 79.73 | 52.17 | 77.76 |
| RS | 94.12 | 84.38 | 100 | 85.00 | 93.33 | 88.06 | 90.00 | 85.14 | 71.74 | 87.97 | |
| mRMR | 79.41 | 68.75 | 100 | 90.00 | 93.33 | 97.01 | 96.67 | 70.27 | 45.65 | 82.34 | |
| SVM-RFE | 67.65 | 81.25 | 80.00 | 95.00 | 80.00 | 89.55 | 90.00 | 77.03 | 63.04 | 80.39 | |
| TSG | 91.18 | 84.38 | 93.33 | 100 | 86.67 | 94.03 | 100 | 71.62 | 65.22 | 87.38 | |
| HC-K-TSP | 91.18 | 81.25 | 100 | 80.00 | 80.00 | 95.52 | 86.67 | 77.03 | 65.22 | 84.10 | |
| KNN | ALLa | 67.65 | 75.00 | 86.67 | 70.00b | 63.33 | 88.06 | 93.33 | 64.86 | 34.78 | 71.71 |
| RS | 97.06 | 78.13 | 93.33 | 90.00 | 93.33 | 95.52 | 93.33 | 72.97 | 43.48 | 84.13 | |
| mRMR | 70.59 | 68.75 | 80.00 | 80.00 | 96.67 | 86.57 | 100 | 54.05 | 36.96 | 74.84 | |
| SVM-RFE | 76.47 | 68.75 | 86.67 | 100 | 90.00 | 86.57 | 90.00 | 58.11 | 45.65 | 78.02 | |
| TSG | 91.18 | 75.00 | 93.33 | 100 | 80.00 | 88.06 | 96.67 | 74.32 | 39.13 | 81.97 | |
| HC-K-TSP | 88.24 | 87.50 | 86.67 | 85.00 | 83.33 | 94.03 | 93.33 | 64.86 | 52.17 | 81.68 | |
| SVM | ALLa | 79.41 | 87.50 | 100 | 100 | 83.33 | 97.01 | 100 | 83.78 | 65.22 | 88.47 |
| RS | 94.12 | 84.38 | 100 | 95.00 | 93.33 | 95.52 | 96.67 | 89.19 | 65.22 | 90.38 | |
| mRMR | 76.47 | 78.13 | 100 | 75.00 | 96.67 | 95.52 | 96.67 | 71.62 | 45.65 | 81.75 | |
| SVM-RFE | 85.29 | 78.13 | 93.33 | 95.00 | 90.00 | 88.06 | 90.00 | 93.24 | 63.04 | 86.23 | |
| TSG | 91.18 | 81.25 | 93.33 | 80.00 | 80.00 | 94.03 | 100 | 68.92 | 54.35 | 82.56 | |
| HC-K-TSP | 85.29 | 84.38 | 100 | 90.00 | 86.67 | 98.51 | 96.67 | 82.43 | 60.87 | 87.20 |
Comparison of generalization performance among different models
Of the nine models in Table 9, PAM had an unacceptable informative gene number, DT had the lowest average accuracy (76.40 %), HC-TSP was similar to HC-K-TSP, and Entropy-based DC and χ2-based DC were similar to RS-based DC. Therefore, we selected five typical models, mRMR-SVM, SVM-RFE-SVM, HC-K-TSP, TSG and RS-based DC, for further evaluation of generalization performance by comparing the accuracy of fitting, LOOCV and independent testing. For LIBSVM[28], the LOOCV strategy was used to optimize penalty parameters C (C∈[2–5, 215]) and the gamma parameter γ(γ∈[2–15, 23]) in the kernel function. Suppose the training set has n samples, for a given combination of C and γ. We leave one as a validation sample and the other n-1 as sub-training samples, and acquire the LOOCV accuracy in this parameter combination after predicting n times. Traversing all parameter combinations, we acquire the highest LOOCV and the corresponding optimal C and γ. The optimal parameters and training set are used for constructing the predictive model. We apply this model to predict the training set and testing set, and obtain the fitting accuracy and independent testing accuracy, respectively. In sum, the fitting and LOOCV are the internal validation in this paper, and independent testing is the external validation. The results are shown in Fig. 2, 3, 45 and 6.
Fig. 2.
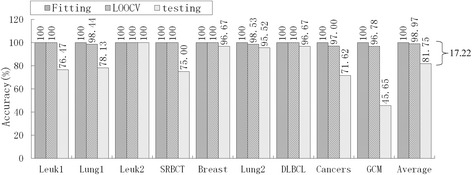
Accuracy of mRMR-SVM for fitting, LOOCV and independent test
Fig. 3.
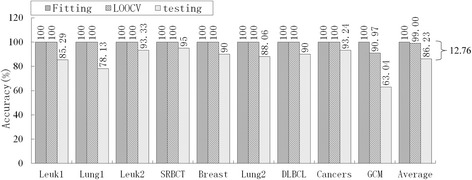
Accuracy of SVM-RFE-SVM for fitting, LOOCV and independent test
Fig. 4.
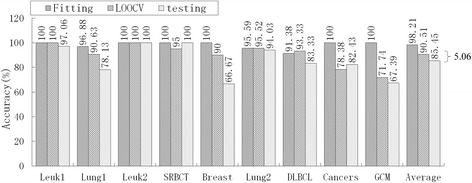
Accuracy of HC-K-TSP for fitting, LOOCV and independent test
Fig. 5.
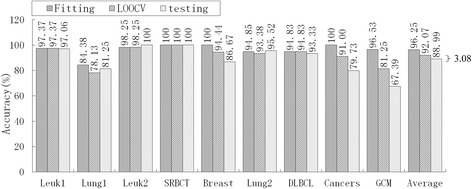
Accuracy of TSG for fitting, LOOCV and independent test
Fig. 6.
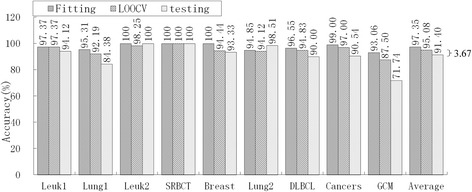
Accuracy of RS-based DC for fitting, LOOCV and independent test
Obviously, over-fitting occurred with all five models; average accuracy always decreased monotonically from fitting through LOOCV to the independent test. For the mRMR-SVM and SVM-RFE-SVM models, which require parameter optimizations, the gaps between LOOCV average accuracy and test average accuracy were 17.22 % and 12.76 %, respectively. However, HC-K-TSP, TSG and RS-based DC models, which adopted a DC core and were parameter-free, tended to generate smaller gaps (5.06 %, 3.08 % and 3.67 %, respectively). For those models that required parameter optimizations, the test accuracy was always systematically less than the LOOCV accuracy for each dataset. For the DC core model, the test accuracy was even higher than LOOCV accuracy for some datasets, for example, the HC-K-TSP model for the SRBCT and Cancers datasets, TSG model for Lung1, Leuk2 and Lung2 datasets, and RS-based DC model for Leuk2 and Lung2 datasets.
Parameter optimizations may be responsible for SVM’s over-fitting? It could be argued that informative genes selected by mRMR and SVM-RFE are not the best feature subsets for mRMR-SVM and SVM-RFE-SVM models, respectively. RS resulted in better performance than the other four feature selection methods (Table 10). Therefore, we further compared the SVM performances with parameter optimizations or not, based on informative genes selected by RS. As shown in Table 11, parameter optimizations considerably improved the fitting and LOOCV accuracy of SVM. For the linear kernel and RBF kernel, the gaps between LOOCV average accuracy and test average accuracy with no parameter optimizations were 3.76 % and 1.90 %, respectively. However, the gaps with parameters optimization were 4.90 % and 9.43 %, respectively. That is, over-fitting is deepened by parameter optimizations in SVM.
Table 11.
SVM performances with parameters optimization or not based on informative genes selected by RS
| Parameters optimization | Kernel | Evaluation | Leuk1 | Lung1 | Leuk2 | SRBCT | Breast | Lung2 | DLBCL | Cancers | GCM | Average |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No (fixed C = 1) | linear | Fitting | 97.37 | 95.31 | 100 | 100 | 100 | 97.06 | 100 | 100 | 97.92 | 98.63 |
| LOOCV | 97.37 | 81.25 | 98.25 | 98.41 | 94.44 | 94.12 | 96.55 | 96 | 77.78 | 92.69 | ||
| Testing | 94.12 | 84.38 | 93.33 | 95 | 93.33 | 97.01 | 96.67 | 87.84 | 58.7 | 88.93 | ||
| Yes | linear | Fitting | 97.37 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | 100 | 99.6 |
| LOOCV | 97.37 | 90.63 | 100 | 100 | 98.15 | 94.12 | 100 | 96 | 81.25 | 95.28 | ||
| Testing | 94.12 | 84.38 | 100 | 95 | 93.33 | 95.52 | 96.67 | 89.19 | 65.22 | 90.38 | ||
| C | 0.25 | 32 | 0.03125 | 0.5 | 0.125 | 8 | 0.25 | 0.25 | 4 | |||
| No (fixed C = 1, γ = 1/m) | RBF | Fitting | 97.37 | 87.50 | 100 | 100 | 100 | 91.18 | 100 | 88.00 | 45.14 | 89.91 |
| LOOCV | 97.37 | 79.69 | 100 | 98.41 | 98.15 | 90.44 | 86.21 | 78.00 | 77.08 | 89.48 | ||
| Testing | 94.12 | 78.13 | 100 | 95.00 | 93.33 | 97.01 | 93.33 | 85.14 | 52.17 | 87.58 | ||
| Yes | RBF | Fitting | 97.37 | 100 | 100 | 100 | 100 | 95.59 | 100 | 100 | 100 | 99.22 |
| LOOCV | 97.37 | 90.63 | 100.00 | 98.00 | 98.15 | 94.12 | 100 | 98.00 | 82.64 | 95.43 | ||
| Testing | 94.12 | 84.38 | 86.67 | 90.00 | 93.33 | 95.52 | 90.00 | 87.84 | 52.17 | 86.00 | ||
| C | 8 | 2048 | 0.125 | 0.25 | 0.5 | 2 | 1 | 32768 | 32 | |||
| γ | 0.0125 | 0.0075125 | 0.25 | 0.125 | 0.25 | 0.0625 | 0.25 | 0.00390625 | 0.0625 |
C is penalty parameters and C∈[2−5, 215]; γ is gamma parameter in kernel function and γ∈[2−15, 23]; m is features number of each SVM models
Discussion
Outlier adjustment and endpoint selection
A small number of outliers may affect gene ranking by changing the endpoints. Although not all gene expression values fit the normal distribution, the standard deviation of a normal distribution has good robustness for outlier adjustment when the probability of that distribution is unknown [33]. We compared independent test accuracies of RS-based DC with different significance level α (i. no adjustment, ii. α = 0.01, iii. α = 0.05). As shown in Table 12, the significance level α had an evident effect on classification performance, and 0.05 is the most appropriate choice for α. Endpoint selection is the nature of the binarization procedure for the vertical comparison of gene evaluation. TSG uses the mean of gene expression values as its endpoint [9]. In this paper, the endpoint defined by formula (11) is based on Fisher’s discriminant principle. We also compared independent test accuracies of RS-based DC with different endpoint selection approaches. As shown in Table 12, the endpoint selection approach has very little influence on classification performance.
Table 12.
Independent test accuracy of RS-based DC with different outlier adjustment and endpoint selection approach
| α | EP selection | Leuk1 | Lung1 | Leuk2 | SRBCT | Breast | Lung2 | DLBCL | Cancers | GCM | Average |
|---|---|---|---|---|---|---|---|---|---|---|---|
| No adjustment | Formula (11) | 94.12 | 84.38 | 93.33 | 95.00 | 90.00 | 100.00 | 90.00 | 81.08 | 60.87 | 87.64 |
| 0.01 | Formula (11) | 94.12 | 84.38 | 93.33 | 100 | 90.00 | 97.02 | 90.00 | 90.54 | 63.04 | 89.16 |
| 0.05 | Formula (11) | 94.12 | 84.38 | 100 | 100 | 93.33 | 98.51 | 90.00 | 90.54 | 71.74 | 91.40 |
| 0.05 | Mean | 94.12 | 84.38 | 100 | 100 | 93.33 | 97.01 | 90.00 | 90.54 | 71.74 | 91.24 |
Entropy and complexity
In this study, a novel score measure, RS, is proposed based on complexity. Complexity and entropy are very similar. The former takes sample size information into account in addition to entropy. As scores are calculated based on percentages, sample size information is not fully utilized in the latter. For example, suppose three white balls and seven black balls are in a system, the entropy (H) is 0.88. In another case, suppose all the counts are multiplied by 10, i.e. 30 white balls and 70 black balls; H is identical to the previous case. The additional information related to the additional sample size is completely ignored in entropy measures. For Entropy-based DC, we used entropy in place of the complexity used in RS-based DC. The results are shown in Table 9. The same modeling process was conducted for the two models, but Entropy-based DC had poorer predictive performance than RS-based DC. This result shows that the additional information associated with sample size can improve a model’s predictive performance.
Horizontal and vertical evaluation of gene pairs
Background differences between pair-wise genes and among samples are fairly common in microarray expression data, and result in very diverse joint effect patterns. It is difficult to fairly evaluate all of the patterns with a single-strategy. As shown in Table 13, a vertical comparison cannot highlight gene G1141 and G4940 in the GCM dataset, and a horizontal comparison cannot highlight gene G6678 and G3330 in the Lung1 dataset. RS, however, highlighted the two pairs of genes by integrating vertical comparison with horizontal comparison.
Table 13.
Horizontal and vertical comparison of gene pairs in real data
| GCM dataset | Horizontal comparison | Vertical comparison | ||||
|---|---|---|---|---|---|---|
| X 1141 > X 4940 | X 1141 < X 4940 | X 1141 > 33 & X 4940 > 232 | X 1141 > 33 & X 4940 < 232 | X 1141 < 33 & X 4940 > 232 | X 1141 < 33 & X 4940 > 232 | |
| Class 11 | 2 | 9 | 3 | 1 | 1 | 6 |
| Class 12 | 10 | 1 | 0 | 0 | 0 | 11 |
| p-value | 0.0027 | 0.0908 | ||||
| Lung1 dataset | Horizontal comparison | Vertical comparison | ||||
| X 6678 > X 3330 | X 6678 < X 3330 | X 6678 > 7439 & X 3330 > 335 | X 6678 > 7439 & X 3330 < 335 | X 6678 < 7439 & X 3330 > 335 | X 6678 < 7439 & X 3330 < 335 | |
| Class 1 | 41 | 3 | 18 | 11 | 10 | 5 |
| Non-Class 1 | 19 | 1 | 0 | 2 | 1 | 17 |
| p-value | 0.7806 | 2.0716 × 10−7 | ||||
Direct classifier
Parameters need to be optimized and adjusted, e.g. the parameters of a kernel function in SVM, and the connection weights of neurons in an artificial neural network. This is the primary reason for classifier over-fitting. SVM integrates the minimum structure risk and the maximal margin and transduction inference, and thereby should be able to efficiently control over-fitting. SVM-RFE-SVM and mRMR-SVM have the highest LOOCV accuracies of those SVM classifiers we tested, 99 % and 98.97 %, respectively. Therefore, these two SVM variants should theoretically both receive high test accuracy. However, results were not as good as expected; obvious over-fitting still appeared (See Fig. 2 and Fig. 3) and deepened by parameter optimizations (See Table 11).
HC-K-TSP, TSG and RS-based DC models, on the other hand, simultaneously received high LOOCV accuracy, high independent test accuracy, and a small gap. Test accuracy higher than LOOCV accuracy appeared in different datasets for the three models, excluding the possibility that DC preferred a specific dataset. The three models have different defined scores and different feature selection methods, only having the same DC core; therefore, we believe that DC plays an important role in effectively controlling over-fitting.
Paired votes based on binary-discriminative informative genes
In most cases, an informative gene can distinguish between just a few classes much more robustly than all of the classes in a multi-class dataset. Therefore, it is necessary to transform datasets from multi-class to binary-class with a “one versus one” (OVO) or an OVR approach. For an m-class dataset, OVO gets incredibly complicated, especially with a big m, as the OVO has to build m(m-1)/2 binary-classifiers. OVR only needs to build m binary-classifiers; however, a serious unbalance between the number of positive samples and negative samples may distort prediction resulting in non-unique calls. Therefore, we employ paired votes based on binary-discriminative informative genes that integrate OVO with OVR. We first build m binary-classifiers with OVR to select m BDIG subsets, then build m-1 binary-classifiers with OVO to perform paired votes. For each paired votes between Classt and Classw, feature subset {BDIGClasst ∪ BDIGClassw} was binary-discriminative and the sample sizes were balanced. Paired votes based on binary-discriminative informative genes only built 2 m-1 binary-classifiers and received robust prediction precision.
Biological relevance of informative genes selected by RS
Do informative genes selected by RS have any biological relevance for a particular tissue/cancer type? This is particularly relevant considering that even a random set of genes may be a good predictor for defining cancer samples [34]. In our study we scanned these potentially informative genes against PubMed. Two examples illustrate: for the Leuk2 dataset, 13 genes out of 12,582 were selected as informative genes by our method, of which ten genes are reported in PubMed as being related to tumors, and seven genes are reported as being related to leukemia (see Table 14). For the Cancers dataset (prostate, breast, lung, ovary, colorectum, kidney, liver, pancreas, bladder/ureter, and gastroesophagus), 36 genes out of 12,533 were selected as informative genes, of which 34 genes are reported related to be tumor related in PubMed (see Table 15). Clearly, most of informative genes selected by RS are supported by PubMed references (Informative genes selected by RS method of nine datasets see Additional file 1).
Table 14.
The 10 tumor related genes selected by RS on original training group of Leuk2 dataset
| Symbol | Synonym(s) | Entrez Gene Name | Related carcinoma and Ref. |
|---|---|---|---|
| FTL | LFTD, NBIA3 | ferritin, light polypeptide | breast cancer [35] |
| PDK1 | pyruvate dehydrogenase kinase, isozyme 1 | leukemia [36] | |
| POU2AF1 | BOB1, OBF-1, OBF1, OCAB | POU class 2 associating factor 1 | leukemia [37] |
| KLRK1 | CD314, D12S2489E, KLR, NKG2-D, NKG2D | killer cell lectin-like receptor subfamily K, member 1 | leukemia [38] |
| KCNH2 | ERG-1, ERG1, H-ERG, HERG, HERG1, Kv11.1, LQT2, SQT1 | potassium channel, voltage gated eag related subfamily H, member 2 | leukemia [39] |
| VLDLR | CAMRQ1, CARMQ1, CHRMQ1CH, VLDLR | very low density lipoprotein receptor | breast cancer [40] |
| MEIS1 | Meis homeobox 1 | leukemia [41] | |
| MLXIP | MIR, MONDOA, bHLHe36 | MLX interacting protein | leukemia [42] |
| NF2 | ACN, BANF, SCH | neurofibromin 2 (merlin) | tumor suppressor [43] |
| MAP3K5 | ASK1, MAPKKK5, MEKK5 | mitogen-activated protein kinase kinase kinase 5 | leukemia [44] |
Table 15.
The 34 tumor related genes selected by RS on original training group of Cancers dataset
| Symbol | Synonym(s) | Entrez Gene Name | Related carcinoma and Ref. |
|---|---|---|---|
| CYP1A1 | AHH, AHRR, CP11, CYP1, P1-450, P450-C, P450DX | cytochrome P450, family 1, subfamily A, polypeptide 1 | lung cancer [45] |
| PTPRZ1 | HPTPZ, HPTPzeta, PTP-ZETA, PTP18, PTPRZ, PTPZ, R-PTP-zeta-2, RPTPB, RPTPbeta, phosphacan | protein tyrosine phosphatase, receptor-type, Z polypeptide 1 | lung cancer [46] |
| WT1 | AWT1, EWS-WT1, GUD, NPHS4,WAGR, WIT-2, WT33 | Wilms tumor 1 | leukemic [47] |
| ANGPT2 | AGPT2, ANG2 | angiopoietin 2 | lung cancer [48] |
| LGALS1 | GAL1, GBP | lectin, galactoside-binding, soluble, 1 | hepatocellular carcinoma [49] |
| ACPP | 5'-NT, ACP-3, ACP3 | acid phosphatase, prostate | prostate cancer [50] |
| GC | DBP, DBP/GC, GRD3, HEL-S-51, VDBG, VDBP | group-specific component (vitamin D binding protein) | bladder cancer [51] |
| PRMT1 | ANM1, HCP1,HRMT1L2, IR1B4 | protein arginine methyltransferase 1 | breast cancer [52] |
| NOX1 | GP91-2, MOX1, NOH-1, NOH1 | NADPH oxidase 1 | colon cancer [53] |
| ADH7 | ADH4 | alcohol dehydrogenase 7 (class IV), mu or sigma polypeptide | gastric cancer [54] |
| DSG3 | CDHF6, PVA | desmoglein 3 | bladder carcinoma [55] |
| NKX2-1 | BCH, BHC, NK-2, NKX2.1, NKX2A, T/EBP, TEBP, TITF1, TTF-1, TTF1 | NK2 homeobox 1 | lung cancer [56] |
| EFHD1 | MST133, MSTP133, PP3051, SWS2 | EF-hand domain family, member D1 | colorectal cancer [57] |
| EREG | EPR, ER, Ep | epiregulin | colorectal cancer [58] |
| DHRS2 | HEP27, SDR25C1 | dehydrogenase/reductase (SDR family) member 2 | breast cancer [59] |
| ENPEP | APA, CD249, gp160 | glutamyl aminopeptidase (aminopeptidase A) | prostate cancer [60] |
| SCGB2A2 | MGB1, UGB2 | secretoglobin, family 2A, member 2 | breast cancer [61] |
| KRT13 | CK13, K13, WSN2 | keratin 13, type I | breast cancer [62] |
| SERPINC1 | AT3, AT3D, ATIII, THPH7 | serpin peptidase inhibitor, clade C (antithrombin), member 1 | bladder cancer [63] |
| SLC12A2 | BSC, BSC2, NKCC1, PPP1R141 | solute carrier family 12 (sodium/ potassium/chloride transporter),member 2 | esophageal squamous cell carcinoma [64] |
| IRF4 | LSIRF, MUM1, NF-EM5, SHEP8 | interferon regulatory factor 4 | hematological malignancies [65] |
| GPA33 | A33 | glycoprotein A33 (transmembrane) | colorectal cancer [66] |
| BCAT1 | BCATC, BCT1, ECA39, MECA39, PNAS121, PP18 | branched chain amino-acid transaminase 1, cytosolic | colorectal cancer [67] |
| COL10A1 | collagen, type X, alpha 1 | breast cancer [68] | |
| CEL | BAL, BSDL, BSSLL, CEase, FAP, FAPP, LIPA, MODY8, CEL | carboxyl ester lipase | pancreatic cysts [69] |
| NPC2 | EDDM1, HE1 | Niemann-Pick disease, type C2 | liver cancer [70] |
| CDH17 | CDH16, HPT-1, HPT1 | cadherin 17, LI cadherin (liver-intestine) | gastric cancer [71] |
| MEIS1 | Meis homeobox 1 | pancreatic cancer [72] | |
| KLK3 | APS, KLK2A1, PSA, hK3 | kallikrein-related peptidase 3 | prostrate [73] |
| CXCL13 | ANGIE, ANGIE2, BCA-1, BCA1, BLC, BLR1L, SCYB13 | chemokine (C-X-C motif) ligand 13 | breast cancer [74] |
| ELA3A | ELA3,ELA3A | chymotrypsin-like elastase family, member 3A | pancreatic carcinoma [75] |
| IRX5 | HMMS, IRX-2a, IRXB2 | iroquois homeobox 5 | prostate cancer [76] |
| VCAM1 | CD106, INCAM-100 | vascular cell adhesion molecule 1 | ovarian cancer [77] |
| P4HB | CLCRP1, DSI, ERBA2L, GIT, P4Hbeta, PDI, PDIA1, PHDB, PO4DB, PO4HB, PROHB | prolyl 4-hydroxylase, beta polypeptide | Glioblastoma multiforme [78] |
Conclusion
Gene selection and classifier choice are two key issues in the analysis of tumor microarray expression data. Gene selection depends on an evaluation strategy and on a defined score. Diverse patterns of gene pairs can be highlighted more fully by integrating a vertical comparison with a horizontal comparison strategy. The RS score and the χ2 score, which both consider events ratios as well as events frequencies, were superior to Δij scores and entropy scores. Parameter optimizations are the main reason for over-fitting classifiers, a DC core classifier can effectively control over-fitting. RS-based DC (Source code of RS-based DC see Additional file 2), which takes into account all of the above factors, receives the highest average independent test accuracy, the smallest informative average gene number, and the best generalization performance. This was confirmed by testing our method on nine bench-mark multi-class gene expression datasets, compared with the nine reference models and the four reference feature selection methods.
Acknowledgments
This work was supported by the Doctoral Foundation of Ministry of Education of China (No. 20124320110002), the Youth Project of Natural Science Foundation of China (No. 61300130), the Science and Technology Planning Projects of Changsha, China (No. K1406018-21), Scientific-Innovative team of Hunan Academy of Agricultural Sciences (2014TD01).
Abbreviations
- TSP
Top scoring pair
- TSG
Top score genes
- RS
Relative simplicity
- RS-based DC
relative simplicity-based direct classifier
- LOOCV
Leave-one-out cross validation
- OVR
One versus rest
- C
Complexity
- IRS
Integrated RS score
- MCC
Matthew correlation coefficient
- BDIG
Binary-discriminative informative genes. K-TSP, k top scoring pairs
- HC-TSP
Multi-class extension of TSP with hierarchical classification scheme
- HC-k-TSP
Multi-class extension of k-TSP with hierarchical classification scheme
- PAM
Prediction Analysis of Microarray
- SVM
Support Vector Machine classification
- NB
Naive bayes
- KNN
K-nearest neighbor
- OVO
One versus one
Additional files
The binary-discriminative informative genes selected by RS method of nine datasets. (XLS 26 kb)
Source code of RS-based DC. (RAR 768 kb)
Footnotes
Competing interest
The authors have declared that no competing interests exist.
Authors’ contributions
YC designed the RS-based DC algorithm and drafted the manuscript. LFW participated in the numerical experiments and helped to draft the manuscript. LZL participated in the numerical experiments. HYZ conducted the reference models. ZMY conceived and designed the experiments. All authors read and approved the final manuscript.
Contributor Information
Yuan Chen, Email: chenyuan0510@126.com.
Lifeng Wang, Email: wanglifeng112358@163.com.
Lanzhi Li, Email: lancy0829@163.com.
Hongyan Zhang, Email: hongyan_zhang6@aliyun.com.
Zheming Yuan, Email: zhmyuan@sina.com.
References
- 1.Tang Y, Zhang YQ, Huang Z. Development of two-stage SVM-RFE gene selection strategy for microarray expression data analysis. IEEE Acm T Comput Bi. 2007;4:365–381. doi: 10.1109/TCBB.2007.70224. [DOI] [PubMed] [Google Scholar]
- 2.Cox B, Kislinger T, Emili A. Integrating gene and protein expression data: pattern analysis and profile mining. Methods. 2005;35:303–314. doi: 10.1016/j.ymeth.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 3.Martínez E, Yoshihara K, Kim H, Mills GM, Treviño V, Verhaak RGW. Comparison of gene expression patterns across 12 tumor types identifies a cancer supercluster characterized by TP53 mutations and cell cycle defects. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chopra P, Lee J, Kang J, Lee S. Improving cancer classification accuracy using gene pairs. PLoS One. 2010;5:e14305. doi: 10.1371/journal.pone.0014305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geman D, d’Avignon C, Naiman DQ, Winslow RL. Classifying gene expression profiles from pairwise mRNA comparisons. Stat Appl Genet Mol. 2004;3: Article19. doi:10.2202/1544-6115.1071. [DOI] [PMC free article] [PubMed]
- 6.Tan AC, Naiman DQ, Xu L, Winslow RL, Geman D. Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics. 2005;21:3896–3904. doi: 10.1093/bioinformatics/bti631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin X, Afsari B, Marchionni L, Cope L, Parmigiani G, Naiman D, et al. The ordering of expression among a few genes can provide simple cancer biomarkers and signal BRCA1 mutations. BMC Bioinformatics. 2009;10:256. doi: 10.1186/1471-2105-10-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magis AT, Price ND. The top-scoring ‘N’algorithm: a generalized relative expression classification method from small numbers of biomolecules. BMC Bioinformatics. 2012;13:227. doi: 10.1186/1471-2105-13-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Zhang H, Dai Z, Chen MS, Yuan Z. TSG: a new algorithm for binary and multi-class cancer classification and informative genes selection. BMC Med Genomics. 2013;6:S3. doi: 10.1186/1755-8794-6-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinäniemi M, Nykter M, Kramer R, Wienecke-Baldacchino A, Sinkkonen L, Zhou JX, et al. Gene-pair expression signatures reveal lineage control. Nat Methods. 2013;10:577–583. doi: 10.1038/nmeth.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ignac TM, Skupin A, Sakhanenko NA, Galas DJ. Discovering Pair-Wise Genetic Interactions: An Information Theory-Based Approach. PLoS One. 2014;9:e92310. doi: 10.1371/journal.pone.0092310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su AI, Welsh JB, Sapinoso LM, Kern SG, Dimitrov P, Lapp H, et al. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 2001;61:7388–7393. [PubMed] [Google Scholar]
- 13.Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, et al. Multiclass cancer diagnosis using Tumor gene expression signatures. Proc Natl Acad Sci U S A. 2001;98:15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–143. doi: 10.1016/S1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 15.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 16.Beer DG, Kardia SLR, Huang CC, Giordano TJ, Levin AM, Misek DE, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 18.Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast Tumors. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 20.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 22.Zhang XW. Constitution Theory. Hefei: Press of University of Science and Technology of China; 2003. [Google Scholar]
- 23.Zhang H, Wang H, Dai Z, Chen MS, Yuan Z. Improving accuracy for cancer classification with a new algorithm for genes selection. BMC Bioinformatics. 2012;13:298. doi: 10.1186/1471-2105-13-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehenni T, Moussaoui A. Data mining from multiple heterogeneous relational databases using decision tree classification. Pattern Recogn Lett. 2012;33:1768–1775. doi: 10.1016/j.patrec.2012.05.014. [DOI] [Google Scholar]
- 25.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng H, Long F, Ding C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE T Pattern Anal. 2005;27:1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 27.Liu Q, Sung AH, Chen Z, Liu J, Chen L, Qiao M, et al. Gene selection and classification for cancer microarray data based on machine learning and similarity measures. BMC Genomics. 2011;12:S1. doi: 10.1186/1471-2164-12-S5-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang CC, Lin CJ. LIBSVM: a library for support vector machines. ACM T Intel Syst Tec. 2011;2:27. [Google Scholar]
- 29.Zhu S, Wang D, Yu K, Li T, Gong Y. Feature selection for gene expression using model-based entropy. IEEE ACM T Comput Bi. 2010;7:25–36. doi: 10.1109/TCBB.2008.35. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Lo SH, Zheng T, Hu I. Interaction-based feature selection and classification for high-dimensional biological data. Bioinformatics. 2012;28:2834–2842. doi: 10.1093/bioinformatics/bts531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei W, Visweswaran S, Cooper GF. The application of naive Bayes model averaging to predict Alzheimer’s disease from genome-wide data. J Am Med Inform Assn. 2011;18:370–375. doi: 10.1136/amiajnl-2011-000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parry RM, Jones W, Stokes TH, Phan JH, Moffitt RA, Fang H, et al. k-Nearest neighbor models for microarray gene expression analysis and clinical outcome prediction. Pharmacogenomics J. 2010;10:292–309. doi: 10.1038/tpj.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng YH. A novel ensemble machine learning for robust microarray data classification. Comput BiolMed. 2006;36:553–573. doi: 10.1016/j.compbiomed.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol. 2011;7:e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orlandi R, De Bortoli M, Ciniselli CM, Vaghi E, Caccia D, Garrisi V, et al. Hepcidin and ferritin blood level as noninvasive tools for predicting breast cancer. Ann Oncol. 2014;25:352–357. doi: 10.1093/annonc/mdt490. [DOI] [PubMed] [Google Scholar]
- 36.Zabkiewicz J, Pearn L, Hills RK, Morgan RG, Tonks A, Burnett AK, et al. The PDK1 master kinase is over-expressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica. 2014;99:858–864. doi: 10.3324/haematol.2013.096487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Auer RL, Starczynski J, McElwaine S, Bertoni F, Newland AC, Fegan CD, et al. Identification of a potential role for POU2AF1 and BTG4 in the deletion of 11q23 in chronic lymphocytic leukemia. Gene Chromosome Canc. 2005;43:1–10. doi: 10.1002/gcc.20159. [DOI] [PubMed] [Google Scholar]
- 38.Huergo-Zapico L, Acebes-Huerta A, Gonzalez-Rodriguez AP, Contesti J, Gonzalez-García E, Payer AR, et al. Expansion of NK cells and reduction of NKG2D expression in chronic lymphocytic leukemia. Correlation with progressive disease. PloS One. 2014;9:e108326. [DOI] [PMC free article] [PubMed]
- 39.Marcucci G, Baldus CD, Ruppert AS, Radmacher MD, Mrózek K, Whitman SP, et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype. a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:9234–9242. doi: 10.1200/JCO.2005.03.6137. [DOI] [PubMed] [Google Scholar]
- 40.He L, Lu Y, Wang P, Zhang J, Yin C, Qu S. Up-regulated expression of type II very low density lipoprotein receptor correlates with cancer metastasis and has a potential link to β-catenin in different cancers. BMC Cancer. 2010;10:601. doi: 10.1186/1471-2407-10-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q, Li Y, Dong J, Li B, Kaberlein JJ, Zhang L, et al. Regulation of MEIS1 by distal enhancer elements in acute leukemia. Leukemia. 2014;28:138–146. doi: 10.1038/leu.2013.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wernicke CM, Richter GH, Beinvogl BC, Plehm S, Schlitter AM, Bandapalli OR, et al. MondoA is highly overexpressed in acute lymphoblastic leukemia cells and modulates their metabolism, differentiation and survival. Leukemia Res. 2012;36:1185–1192. doi: 10.1016/j.leukres.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 43.Cooper J, Giancotti FG. Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett. 2014;588:2743–2752. doi: 10.1016/j.febslet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan W, Arai A, Aoki M, Ichijo H, Miura O. ASK1 is activated by arsenic trioxide in leukemic cells through accumulation of reactive oxygen species and may play a negative role in induction of apoptosis. Biochem Bioph Res Co. 2007;355:1038–1044. doi: 10.1016/j.bbrc.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 45.Lin J, He B, Cao L, Zhang Z, Liu H, Rao J, et al. CYP1A1 Ile462Val polymorphism and the risk of non-small cell lung cancer in a Chinese population. Tumori. 2013;100:547–552. doi: 10.1700/1660.18178. [DOI] [PubMed] [Google Scholar]
- 46.Makinoshima H, Ishii G, Kojima M, Fujii S, Higuchi Y, Kuwata T, et al. PTPRZ1 regulates calmodulin phosphorylation and tumor progression in small-cell lung carcinoma. BMC Cancer. 2012;12:537. doi: 10.1186/1471-2407-12-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Wang J, Li X, Jia Y, Huai L, He K, et al. Role of the Wilms’ tumor 1 gene in the aberrant biological behavior of leukemic cells and the related mechanisms. Oncol Rep. 2014;32:2680–2686. doi: 10.3892/or.2014.3529. [DOI] [PubMed] [Google Scholar]
- 48.Coelho AL, Araújo A, Gomes M, Catarino R, Marques A, Medeiros R. Circulating Ang-2 Mrna Expression Levels: Looking ahead to a New Prognostic Factor for NSCLC. PloS One. 2014;9:e90009. [DOI] [PMC free article] [PubMed]
- 49.Bacigalupo ML, Manzi M, Espelt MV, Gentilini LD, Compagno D, Laderach DJ, et al. Galectin‐1 Triggers Epithelial‐Mesenchymal Transition in Human Hepatocellular Carcinoma Cells. J Cell Physiol. 2015;230:1298–1309. doi: 10.1002/jcp.24865. [DOI] [PubMed] [Google Scholar]
- 50.Kirschenbaum A, Liu XH, Yao S, Leiter A, Levine AC. Prostatic acid phosphatase is expressed in human prostate cancer bone metastases and promotes osteoblast differentiation. Ann Ny Acad Sci. 2011;1237:64–70. doi: 10.1111/j.1749-6632.2011.06198.x. [DOI] [PubMed] [Google Scholar]
- 51.Li F, Chen DN, He CW, Zhou Y, Olkkonen VM, He N, et al. Identification of urinary Gc-globulin as a novel biomarker for bladder cancer by two-dimensional fluorescent differential gel electrophoresis (2D-DIGE) J Proteomics. 2012;77:225–236. doi: 10.1016/j.jprot.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 52.Baldwin RM, Morettin A, Paris G, Goulet I, Côté J. Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle. 2012;11:4597–4612. doi: 10.4161/cc.22871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang R, Dashwood WM, Nian H, Löhr CV, Fischer KA, Tsuchiya N, et al. NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2‐amino‐1‐methyl‐6‐phenylimidazo [4, 5‐b] pyridine (PhIP) Int J Cancer. 2011;128:2581–2590. doi: 10.1002/ijc.25610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jelski W, Chrostek L, Zalewski B, Szmitkowski M. Alcohol dehydrogenase (ADH) isoenzymes and aldehyde dehydrogenase (ALDH) activity in the sera of patients with gastric cancer. Digest Dis Sci. 2008;53:2101–2105. doi: 10.1007/s10620-007-0135-4. [DOI] [PubMed] [Google Scholar]
- 55.Huang W, Williamson SR, Rao Q, Lopez-Beltran A, Montironi R, Eble JN, et al. Novel markers of squamous differentiation in the urinary bladder. Hum Pathol. 2013;44:1989–1997. doi: 10.1016/j.humpath.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 56.Yang L, Lin M, Ruan WJ, Dong LL, Chen EG, Wu XH, et al. Nkx2-1: a novel tumor biomarker of lung cancer. J Zhejiang Univ Sci B. 2012;13:855–866. doi: 10.1631/jzus.B1100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takane K, Midorikawa Y, Yagi K, Sakai A, Aburatani H, Takayama T, et al. Aberrant promoter methylation of PPP1R3C and EFHD1 in plasma of colorectal cancer patients. Cancer Med-Us. 2014;3:1235–1245. doi: 10.1002/cam4.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jonker DJ, Karapetis CS, Harbison C, O’Callaghan CJ, Tu D, Simes RJ, et al. Epiregulin gene expression as a biomarker of benefit from cetuximab in the treatment of advanced colorectal cancer. Brit J Cancer. 2014;110:648–655. doi: 10.1038/bjc.2013.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thorner AR, Parker JS, Hoadley KA, Perou CM. Potential tumor suppressor role for the c-Myb oncogene in luminal breast cancer. PLoS One. 2010;5:e13073. doi: 10.1371/journal.pone.0013073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teranishi JI, Ishiguro H, Hoshino K, Noguchi K, Kubota Y, Uemura H. Evaluation of role of angiotensin III and aminopeptidases in prostate cancer cells. Prostate. 2008;68:1666–1673. doi: 10.1002/pros.20835. [DOI] [PubMed] [Google Scholar]
- 61.Classen‐Linke I, Moss S, Gröting K, Beier HM, Alfer J, Krusche CA. Mammaglobin 1: not only a breast‐specific and tumour‐specific marker, but also a hormone‐responsive endometrial protein. Histopathology. 2012;61:955–965. doi: 10.1111/j.1365-2559.2012.04290.x. [DOI] [PubMed] [Google Scholar]
- 62.Sheng S, Barnett DH, Katzenellenbogen BS. Differential estradiol and selective estrogen receptor modulator (SERM) regulation of Keratin 13 gene expression and its underlying mechanism in breast cancer cells. Mol Cell Endocrinol. 2008;296:1–9. doi: 10.1016/j.mce.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meyer-Siegler KL, Cox J, Leng L, Bucala R, Vera PL. Macrophage migration inhibitory factor anti-thrombin III complexes are decreased in bladder cancer patient serum: Complex formation as a mechanism of inactivation. Cancer Lett. 2010;290:49–57. doi: 10.1016/j.canlet.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shiozaki A, Nako Y, Ichikawa D, Konishi H, Komatsu S, Kubota T, et al. Role of the Na+/K+/2Cl-cotransporter NKCC1 in cell cycle progression in human esophageal squamous cell carcinoma. World J Gastroentero. 2014;20:6844. doi: 10.3748/wjg.v20.i22.6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L, Yao ZQ, Moorman JP, Xu Y, Ning S. Gene Expression Profiling identifies IRF4-associated molecular Signatures in Hematological Malignancies. PloS One. 2014;9:e106788. [DOI] [PMC free article] [PubMed]
- 66.Infante JR, Bendell JC, Goff LW, Jones SF, Chan E, Sudo T, et al. Safety, pharmacokinetics and pharmacodynamics of the anti-A33 fully-human monoclonal antibody, KRN330, in patients with advanced colorectal cancer. Eur J Cancer. 2013;49:1169–1175. doi: 10.1016/j.ejca.2012.11.033. [DOI] [PubMed] [Google Scholar]
- 67.Yoshikawa R, Yanagi H, Shen CS, Fujiwara Y, Noda M, Yagyu T, et al. ECA39 is a novel distant metastasis-related biomarker in colorectal cancer. World J Gastroentero. 2006;12:5884–5889. doi: 10.3748/wjg.v12.i36.5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chang HJ, Yang MJ, Yang YH, Hou MF, Hsueh EJ, Lin SR. MMP13 is potentially a new tumor marker for breast cancer diagnosis. Oncol Rep. 2009;22:1119–1127. doi: 10.3892/or_00000544. [DOI] [PubMed] [Google Scholar]
- 69.Ræder H, McAllister FE, Tjora E, Bhatt S, Haldorsen I, Hu J, et al. Carboxyl-ester lipase maturity-onset diabetes of the young is associated with development of pancreatic cysts and upregulated MAPK signaling in secretin-stimulated duodenal fluid. Diabetes. 2013;DB_131012:2-61. [DOI] [PMC free article] [PubMed]
- 70.Liao YJ, Lin MW, Yen CH, Lin YT, Wang CK, Huang SF, et al. Characterization of Niemann-Pick Type C2 protein expression in multiple cancers using a novel NPC2 monoclonal antibody. PLoS One. 2013;8:e77586. doi: 10.1371/journal.pone.0077586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hb Q, Ly Z, Ren C, Zl Z, Wj W. Targeting CDH17 suppresses tumor progression in gastric cancer by downregulating Wnt/β-catenin signaling. PLoS One. 2013;8:e56959. doi: 10.1371/journal.pone.0056959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tomoeda M, Yuki M, Kubo C, Yoshizawa H, Kitamura M, Nagata S, et al. Role of Meis1 in mitochondrial gene transcription of pancreatic cancer cells. Biochem Bioph Res Co. 2011;410:798–802. doi: 10.1016/j.bbrc.2011.06.066. [DOI] [PubMed] [Google Scholar]
- 73.Zhang HM, Yan Y, Wang F, Gu WY, Hu GH, Zheng JH. Ratio of prostate specific antigen to the outer gland volume of prostrate as a predictor for prostate cancer. Int J Clin Exp Patho. 2014;7:6079. [PMC free article] [PubMed] [Google Scholar]
- 74.Panse J, Friedrichs K, Marx A, Hildebrandt Y, Luetkens T, Bartels K, et al. Chemokine CXCL13 is overexpressed in the tumour tissue and in the peripheral blood of breast cancer patients. Brit J Cancer. 2008;99:930–938. doi: 10.1038/sj.bjc.6604621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shimada SHINYA, Yamaguchi KENJI, Takahashi MASAYUKI, Ogawa MICHIO. Pancreatic elastase IIIA and its variants are expressed in pancreatic carcinoma cells. Int J Mol Med. 2002;10:599–603. [PubMed] [Google Scholar]
- 76.Myrthue A, Rademacher BL, Pittsenbarger J, Kutyba-Brooks B, Gantner M, Qian DZ, et al. The iroquois homeobox gene 5 is regulated by 1, 25-dihydroxyvitamin D3 in human prostate cancer and regulates apoptosis and the cell cycle in LNCaP prostate cancer cells. Clin Cancer Res. 2008;14:3562–3570. doi: 10.1158/1078-0432.CCR-07-4649. [DOI] [PubMed] [Google Scholar]
- 77.Huang J, Zhang J, Li H, Lu Z, Shan W, Mercado-Uribe I, et al. VCAM1 expression correlated with tumorigenesis and poor prognosis in high grade serous ovarian cancer. Am J Transl Res. 2013;5:336. [PMC free article] [PubMed] [Google Scholar]
- 78.Sun S, Lee D, Ho AS, Pu JK, Zhang XQ, Lee NP, et al. Inhibition of prolyl 4-hydroxylase, beta polypeptide (P4HB) attenuates temozolomide resistance in malignant glioma via the endoplasmic reticulum stress response (ERSR) pathways. Neuro-oncology. 2013;not005:1-16. [DOI] [PMC free article] [PubMed]