

Abstract
Background
A key focus in cancer research is the discovery of biomarkers that accurately diagnose early lesions in non-invasive tissues. Several studies have identified malignancy-associated DNA methylation changes in blood, yet no general cancer biomarker has been identified to date. Here, we explore the potential of blood DNA methylation as a biomarker of pan-cancer (cancer of multiple different origins) in 41 female cancer discordant monozygotic (MZ) twin-pairs sampled before or after diagnosis using the Illumina HumanMethylation450 BeadChip.
Results
We analysed epigenome-wide DNA methylation profiles in 41 cancer discordant MZ twin-pairs with affected individuals diagnosed with tumours at different single primary sites: the breast, cervix, colon, endometrium, thyroid gland, skin (melanoma), ovary, and pancreas. No significant global differences in whole blood DNA methylation profiles were observed. Epigenome-wide analyses identified one novel pan-cancer differentially methylated position at false discovery rate (FDR) threshold of 10 % (cg02444695, P = 1.8 × 10−7) in an intergenic region 70 kb upstream of the SASH1 tumour suppressor gene, and three suggestive signals in COL11A2, AXL, and LINC00340. Replication of the four top-ranked signals in an independent sample of nine cancer-discordant MZ twin-pairs showed a similar direction of association at COL11A2, AXL, and LINC00340, and significantly greater methylation discordance at AXL compared to 480 healthy concordant MZ twin-pairs. The effects at cg02444695 (near SASH1), COL11A2, and LINC00340 were the most promising in biomarker potential because the DNA methylation differences were found to pre-exist in samples obtained prior to diagnosis and were limited to a 5-year period before diagnosis. Gene expression follow-up at the top-ranked signals in 283 healthy individuals showed correlation between blood methylation and gene expression in lymphoblastoid cell lines at PRL, and in the skin tissue at AXL. A significant enrichment of differential DNA methylation was observed in enhancer regions (P = 0.03).
Conclusions
We identified DNA methylation signatures in blood associated with pan-cancer, at or near SASH1, COL11A2, AXL, and LINC00340. Three of these signals were present up to 5 years prior to cancer diagnosis, highlighting the potential clinical utility of whole blood DNA methylation analysis in cancer surveillance.
Electronic supplementary material
The online version of this article (doi:10.1186/s13148-016-0172-y) contains supplementary material, which is available to authorized users.
Keywords: DNA methylation, Cancer, Discordant monozygotic twins, Epigenetics, Biomarker, Twin study
Background
Despite global research efforts, cancer remains one of the leading causes of death in economically developed countries, second only to cardiovascular disease [1, 2]. Early and accurate detection greatly increases the odds of successful treatment. Furthermore, considering the worldwide expected increase in cancer incidence [3], the need for new cost-effective detection or prediction methods to improve disease outcome, such as accurate and precise biomarkers is paramount. One key area of focus is the development of cancer biomarkers in non-invasive tissues, such as peripheral blood or serum, which can accurately diagnose early lesions, and hence, improve survival, and even identify individuals at risk [4–6].
DNA methylation is a molecular mark that has a great potential as biomarker for early cancer detection in non-invasive tissues. It is a relatively stable epigenetic mark that can be influenced by DNA sequence variation, but also environmental factors and stochastic changes that occur over a lifetime [7–11]. It provides a potential link between environmental conditions and exposures with changes in gene activity either directly or in combination with genetic susceptibility by influencing penetrance and expressivity [12]. Aberrant DNA methylation is associated with a broad range of diseases [13], age [14], environmental factors such as smoking [8, 15–18], and is especially prevalent in human cancer tissues [19, 20]. Epigenetic changes that occur in carcinogenesis can be detected in early neoplastic tissues, as well as tumour-derived DNA in plasma or serum of patients [21].
A complex interaction of environmental factors, stochastic events, and genetic susceptibility can lead to cancer development. Blood samples are known to reflect the health status of an individual and evaluating whole blood or blood cell types in particular, might reveal specific or systemic changes in the host that are associated with malignant disease. Indeed, DNA methylation signatures in blood have been associated with cancerous and pre-cancerous primary locations such as the breast [22, 23], colon [24], bladder [25], and ovary [26]. To the best of our knowledge, no study to date has attempted to identify pan-cancer epigenetic biomarkers, that is, epigenetic biomarkers indicative of cancer of multiple different origins, in whole blood samples. However, pan-cancer analyses have been conducted directly in tumour tissues, for example, the Cancer Genome Atlas Research Network launched a pan-cancer project in 2012 [27], and other recent studies that have identified pan-cancer DNA methylation patterns in different tumour tissues [28, 29]. The identification of a blood-based DNA methylation biomarker that can predict cancer or pan-cancer development would be a highly valuable asset to the current screening processes, as well as contributing to understanding potential common systemic changes associated with disease.
The aim of the present study was to explore whole blood DNA methylation patterns in cancer-discordant female monozygotic (MZ) twin-pairs to identify pan-cancer-associated epigenetic changes. MZ twins are matched for age, sex, cohort effects, many maternal influences, and early environment factors, and have nearly identical genomes. Discordant MZ studies are therefore a particularly powerful and less biased design for detecting disease-related epigenetic differences [30]. In the current study, we analysed blood samples that were taken from up to 11 years before or up to 5 years after cancer diagnosis, which allowed us to explore biomarker stability over time. This is the first study in MZ twin-pairs to explore pan-cancer-associated blood DNA methylation changes with a focus on the detection of early neoplastic development. Our results identify four CpG sites that are associated with cancer status, with follow-up replication and gene expression analyses, and highlight signals with promising biomarker potential.
Results
Global methylation profiles in cancer-discordant monozygotic twin-pairs
We analysed genome-wide DNA methylation profiles in whole blood samples of 41 female monozygotic twin-pairs discordant for cancers of the solid organs. Affected individuals in this sample had cancers at various primary sites: the breast, cervix, colon, endometrium, thyroid gland, skin (melanoma), ovary, and pancreas (Fig. 1a). To assess global DNA methylation variation, unsupervised hierarchical clustering of unadjusted normalised DNA methylation values was performed. Thirty-five of the 41 MZ twin-pairs (85.4 %) clustered with their co-twin (Fig. 1b). The remaining MZ twin-pairs were clustered by array, underlining the importance of correcting for technical covariates in downstream analyses. Subsequently, the top 1000 probes with the highest standard deviations were assessed with unsupervised hierarchical clustering, to determine if the most variable CpG sites combined were associated with cancer status. All MZ twin-pairs now clustered together and confirmed that MZ twin-pairs were globally more similar to each other, compared to unrelated individuals with the same affection status (cancer vs. non-cancer). Furthermore, MZ twin-pairs showed high within-pair correlations in normalised unadjusted DNA methylation values (mean Spearman’s rank correlation coefficient (rS) = 0.986), which was significantly greater than pairing at random, or pairing at random by affection status (Fig. 1c, P = 2.2 × 10−16). The high within-pair correlation is comparable to genome-wide average correlations estimated in newborn twins, ranging from 0.98 to 0.99 for the placenta, cord blood mononuclear cells, and human umbilical vascular endothelial cells [31]. A similar average correlation is seen in peripheral blood at 15 years (0.99) [32] and middle-aged individuals (0.98) [33] as well as in the adipose tissue of the middle-aged individuals (0.992) [34].
Fig. 1.
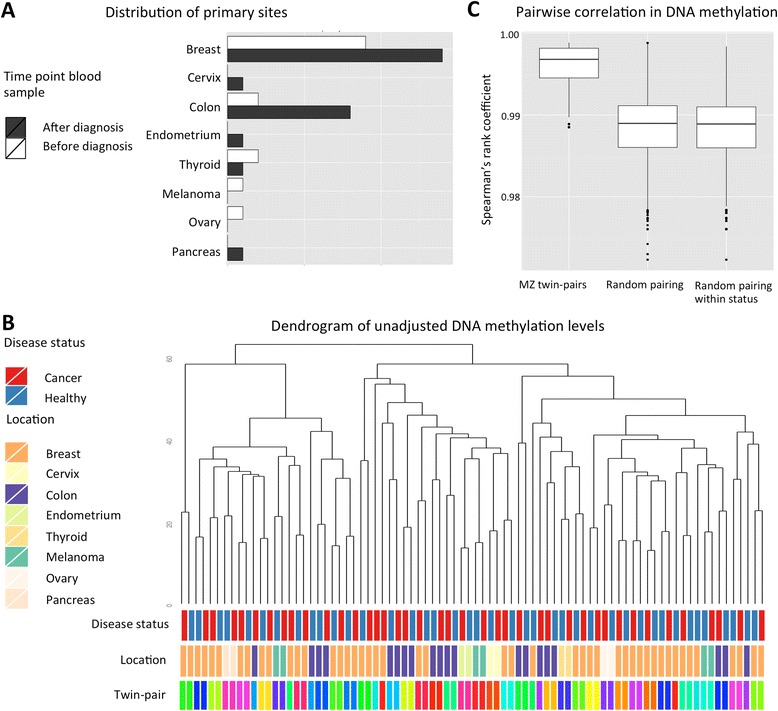
Diagnostic characteristics and global methylation profiles of 41 cancer-discordant MZ twin-pairs. a Number of cases for each primary location of cancer, where a blood sample was obtained before (white) and after (black) cancer diagnosis. b Dendrogram of the unadjusted global methylation profiles. Annotation bars denote each individual’s cancer status, type of cancer (identical for both twins in a pair), and family identifier (identical for both twins in a pair). c Pair-wise correlation in DNA methylation profiles shows greater similarity within MZ pairs, compared to pairs of unrelated individuals, either paired at random or at within affection status
Pan-cancer-associated differentially methylated positions
Differences in DNA methylation levels were next analysed at single CpG sites across the genome within 41 female MZ twin-pairs discordant for cancer development. DNA methylation values were adjusted for technical and biological covariates by using the first five principle components that explained 46 % of variance in the data. The first five principle components were significantly associated with variables that included technical covariates (batch and array), blood cell type composition, but not cancer status (see “Methods” section). The epigenome-wide association scan (EWAS) analysis identified one novel pan-cancer differentially methylated position (DMP) at a false discovery rate (FDR) threshold of 0.10 for probe cg02444695 (P = 1.8 × 10−7) located in an intergenic region. Additionally, three suggestive pan-cancer DMPs (P < 1.0 × 10−5) were identified for probes cg26079695 in COL11A2, cg27094856 in AXL, and cg21046959 in LINC00340 (Table 1, Fig. 2a).
Table 1.
Top-ranked results from EWAS of 41 cancer discordant MZ twin-pairs
| Discovery EWAS N = 41 MZ twin-pairs (prior to and after diagnosis) | Replication N = 9 MZ twin-pairs (NTR replication) | Discordant vs healthy MZ pairs N = 9 vs N = 480 (NTR variability) | EWAS prior to diagnosis N = 15 MZ twin-pairs (prior to diagnosis only) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CpG | Position (hg19) | Associated gene | Location | Rank EWAS | Mean difference* | P value | Mean difference* | P value | P value | Rank EWAS | Mean difference* | P value |
| cg02444695 | Chr6:148950185 | – | – | 1 | 0.70 | 1.80 × 10−7 | −0.64 | 0.26 | 0.09 | 10 | 0.88 | 2.40 × 10−5 |
| cg26079695 | Chr6:33143273 | COL11A2 | Intron | 2 | −0.67 | 3.32 × 10−6 | −050 | 0.23 | 0.34 | 1518 | −0.88 | 4.10 × 10-3 |
| cg27094856 | Chr19:41732589 | AXL | Intron | 3 | 0.56 | 3.41 × 10−6 | 0.02 | 0.96 | 0.05 | 3801 | 0.51 | 9.71 × 10−3 |
| cg21046959 | Chr6:22180833 | LINC00340 | Transcript | 4 | −0.53 | 8.89 × 10−6 | −0.43 | 0.37 | – | 407 | −0.73 | 1.21 × 10-3 |
*The mean differences are determined using adjusted DNA methylation values and calculated as cancer − unaffected twin
Fig. 2.
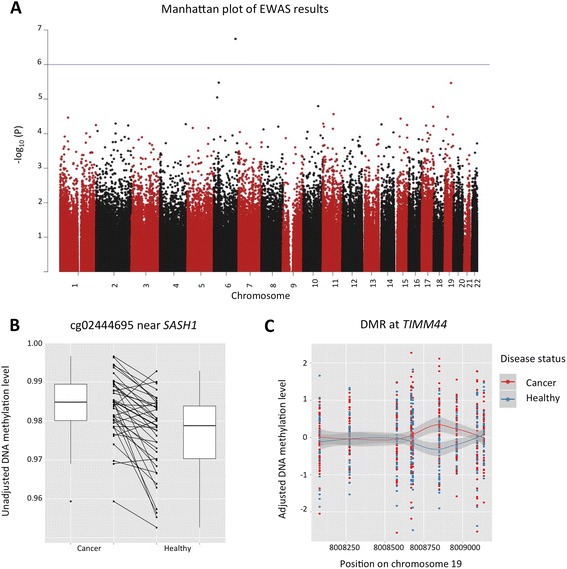
Pan-cancer epigenome-wide results in 41 discordant MZ twin-pairs. a Manhattan plot of the epigenome-wide association results in 41 pan-cancer-discordant MZ twin-pairs, where each point represents the observed −log10 P value at a CpG-site. b Direction of association at the top-ranked signal cg02444695, near SASH1. Results are plotted using normalised unadjusted beta values of cancer-affected individuals (left) and healthy individuals (right). The lines connect co-twins in twin-pairs and indicate a consistent direction of the effect with an average of 0.7 % within twin-pairs with a range of 0.9 to 3.0 %. The three suggestive probes are included in Additional file 1. c Pan-cancer DMR at TIMM44. Results are plotted using adjusted DNA methylation values at each CpG site in the DMR for individuals affected by cancer (red) and healthy co-twins (blue). Smooth (LOESS) lines with standard error are plotted for both groups. The CpG site driving the signal is at chr19:8,008,850 (hg19)
The most associated pan-cancer DMP, cg02444695, exhibited consistently higher DNA methylation levels in cancer-affected twins compared to the healthy co-twins (Fig. 2b). The CpG-site falls in an intergenic region, 70 kb upstream of the nearest gene, SASH1, a known tumour suppressor gene previously associated with aggressive tumour growth and metastasis formation in different types of cancer. Of the three genome-wide suggestive probes (Table 1, Additional file 1), cg27094856 was located in the fourth intron of the AXL gene, which is implicated in many cancers and is a therapeutic target for antibody-based therapies [35–37]. Another suggestive signal was obtained at cg21046959 directly in LINC00340, and the LINC00340 transcript has been linked to both neuroblastoma and ovarian tumours [38, 39].
We pursued replication at the four top-ranked results in an independent twin sample from the Netherlands Twin Registry (NTR). First, we analysed nine cancer-discordant MZ twin-pairs from NTR with the same analysis pipeline used for the discovery findings. We observed a similar direction of association at the probes, except for cg02444695 (Table 1, Additional file 2), but no results surpassed nominal significance in the replication sample, which may be due to the very small sample size. Second, we assessed how much variation in DNA methylation occurs at the top-ranked CpG sites in a healthy population. We expect if the observed changes are not stochastic effects and due to cancer status, to see less variation at these sites in a healthy population than cancer-discordant twin-pairs. To this end, data for the three of the four top-ranked probes were available for both the nine cancer-discordant MZ twin-pairs and 480 healthy MZ twin-pairs from NTR. We compared the absolute within-pair differences in DNA methylation at cg02444695, cg26079695, and cg27094856 (Table 1, Figures in Additional file 3). We observed greater within-pair difference in DNA methylation in cancer-discordant twins compared to healthy twins, with nominally significant effects at cg27094856 in AXL (healthy median 0.78 % vs. cancer median 1.44 %, P = 0.047), and near significant effects at cg02444695 near SASH1 (healthy median 1.48 % vs. cancer median 2.32 %, P = 0.091).
Pan-cancer-associated differentially methylated regions
We next aimed to identify potential pan-cancer DMRs, that is, larger genomic regions containing multiple CpG-sites that exhibit consistently different DNA methylation levels between the discordant MZ twin-pairs. We applied the ‘bump hunting’ method [40] to define DMRs with few modifications to account for twin structure present in our dataset. We kept the paired structure in the data and used the differences in PC-adjusted DNA methylation values, as described above, per twin-pair in the peak-calling algorithm. One DMR was identified on chromosome 19:8,008,080-8,009,137 (hg19) spanning ~1 kb (P = 0.01, Fig. 2c). The DMR is mainly driven by a single CpG (chromosome 19:8,008,850) ranked 24th in the single-CpG EWAS and its two adjacent CpGs that are hypermethylated in cancer-affected twins. This region overlaps a 5′ CpG island within an active promoter across multiple tissue types (according to ChromHMM), approximately ~1500 bp from the transcription start site (TSS) of TIMM44. TIMM44 has previously been associated with familial non-medullary thyroid carcinoma [41], aggressive serous ovarian cancers [42] and breast cancer recurrence [43].
No enrichment for cancer risk factors smoking and age
Enrichment for the two major risk factors for cancer development, age and smoking, was assessed in the 500 top-ranked pan-cancer DMP CpG sites. We obtained previously published age and smoking DMPs in whole blood, and assessed whether these CpG-sites tended to co-occur with the pan-cancer DMPs identified in the 41 discordant MZ pairs. The first five PCs used to adjust DNA methylation levels in the EWAS were not significantly associated with either smoking or age; however, they could account for some of the variation observed of these variables. Enrichment for age DMPs was assessed using the results of Steegenga et al. [44] that combined eight studies totalling 7318 age DMPs that were also available in our genome-wide dataset. There was no enrichment (eight probes, P = 1) within the top 500 ranked probes for known age DMPs, as compared to the remaining probes genome wide. To assess for enrichment of smoking signals, we used smoking DMP results from the largest whole blood DNA methylation study to date using the HumanMethylation450 BeadChip by Zeilinger et al. [8]. There were 948 smoking DMPs that were also available in our genome-wide dataset, and we did not observe an enrichment of smoking DMPs in the top 500 ranked EWAS associations (three probes, P = 0.089). None of the four top-ranked probes had been previously associated with age or smoking status. Finally, no significant enrichment was observed in the top 500 results from an EWAS performed correcting only for batch effects and estimated cell counts (see “Methods” section). Taken together, the pan-cancer DMPs seem indicative of a more complex representation of the risk factors or disease biology.
Biomarker potential: methylation analysis in samples obtained prior to diagnosis
To identify pan-cancer DMPs that could serve as biomarkers for early diagnosis, we performed analyses taking into account the time of cancer diagnosis. We selected a subset of 15 discordant MZ twin-pairs from the 41 female MZ twin-pairs where blood samples were obtained prior to date of official cancer diagnosis, in a 0–5-year period preceding diagnosis (Fig. 1a). EWAS in the 15 MZ pairs was performed using the same approach as the analyses in the 41 MZ twin pairs, and the top-ranked results included signals in the promoters (within 200 bp of the TSS) of genes COX7C and U2AF1 that have been previously linked to cancer (Table provided in Additional file 4). Specifically, the second ranked association in COX7C was located in a region previously identified as one of the nine loci that most significantly associated with bladder cancer in whole blood samples [45], and in both analyses, hyper-methylated effects were observed in cancer-affected individuals. In a pan-cancer tumour tissue analysis, recurrent somatic mutations identified in U2AF1 were shown to induce splicing inducing transcriptome changes [46]. Our previous most-associated pan-cancer DMP (cg02444695 near SASH1 in 41 MZ pairs) remained strongly significant (P = 2.40 × 10−5) and with the same direction of association in the new EWAS prior to cancer diagnosis, and was now ranked tenth overall (Table 1, Fig. 3a). The suggestive probes from the original 41 MZ pair EWAS also remained nominally significant in the new analysis and in the same direction of association (Table 1), and in the majority of cases (cg02444695, cg26079695, cg21046959), greater differences between MZ twin-pairs were identified in the samples collected before diagnosis.
Fig. 3.
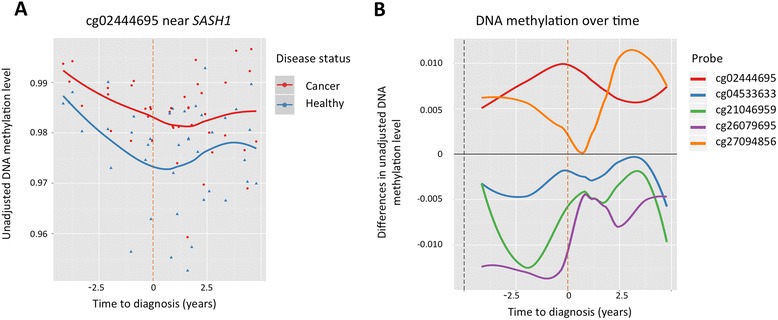
Differential methylation with respect to time of cancer diagnosis. a Unadjusted DNA methylation values at cg02444695 (near SASH1) in affected individuals (red) and healthy co-twins (blue), shown with respect to time of diagnosis (years) with smooth (LOESS) lines fitted for both groups. The orange vertical line represents the time of diagnosis. b Methylation differences within twin pairs at the four top-ranked DMPs and cg04533633 (at COX7C). Each smooth (LOESS) line represents the methylation difference (affected − unaffected twin) at an individual probe (see legend)
Pan-cancer-associated biomarker stability over time
We next explored the relationship between the time of blood sample extraction and the observed DNA methylation differences in cancer-discordant twins at the four top-ranked pan-cancer DMPs and the pan-cancer DMP located at COX7C identified above in more depth. The greatest DNA methylation difference at the top-ranked probe (cg02444695 near SASH1) between the MZ twin-pairs was observed when the DNA sample was obtained earlier in the same year as cancer diagnosis (Fig. 3b). The second- (cg26079695 in COL11A2) and fourth- (cg21046959 in LINC00340) ranked probes displayed greatest effects in the 5-year period prior to official diagnosis whereas the third probe (cg27094856 in AXL) showed the largest differences in the 5-year period after official diagnosis. There was no significant correlation between time to diagnosis and age at blood sample collection (P = 0.29, Additional file 5: Figure S1A); therefore, these results cannot be explained by the effect chronological age. The top-ranked probe shows differential methylation across all ages included in the large sample (Additional file 5: Figure S1B).
The observed early DNA methylation change between MZ twin-pairs was further explored for the top-ranked probes by including five additional cancer-discordant MZ twin-pairs where blood samples were obtained between 5 and 11 years prior to cancer diagnosis (Additional file 5: Figure S1C–F). At the top-ranked probe (cg02444695), the DNA methylation levels show a reverse pattern prior to 5 years to the diagnosis, the time window of the main study (Additional file 5: Figure S1C). Therefore, we conclude that DNA methylation differences at cg02444695 do not arise from treatment, as these changes can be observed in individuals specifically up to 5 years prior to cancer diagnosis.
Functional follow-up of pan-cancer differential methylation results
The four top-ranked pan-cancer DMPs and the pan-cancer DMR were first explored for association with gene expression levels of the closest available transcripts. Analyses were performed in 283 healthy individuals for whom both whole blood DNA methylation levels and gene expression profiles from the multiple tissues (the lymphoblastoid cell lines (LCL), skin, and adipose) were available. We identified two nominally significant correlations of DNA methylation at cg21046959 with LCL gene expression of the closest protein-coding transcript of PRL, located ~100 kb upstream of the CpG-site (r = 0.17, P = 4.5 × 10−3), and at cg27094856 with skin tissue expression levels of AXL (r = −0.15, P = 0.01) (Fig. 4a, b). We did not observe these correlations in the other available tissues (Table 2). ENCODE annotation data identified the CpG correlated to expression of PRL to be located in a heterochromatin block in the GM12878 B-lymphocyte cell line, although within an active promoter and weak enhancer in human embryonic stem cell line (H1-hESC) and leukaemia cell line (K562), respectively. The CpG site negatively associated with AXL expression levels in the skin tissue is identified by ENCODE to be located in a strong enhancer in epidermal keratinocytes and inactive or poised promoter in the GM12878 B-lymphocyte cell line. Higher expression levels of AXL, as described previously are implicated in proliferation, migration, and resistance to therapy of many cancers [35–37].
Fig. 4.
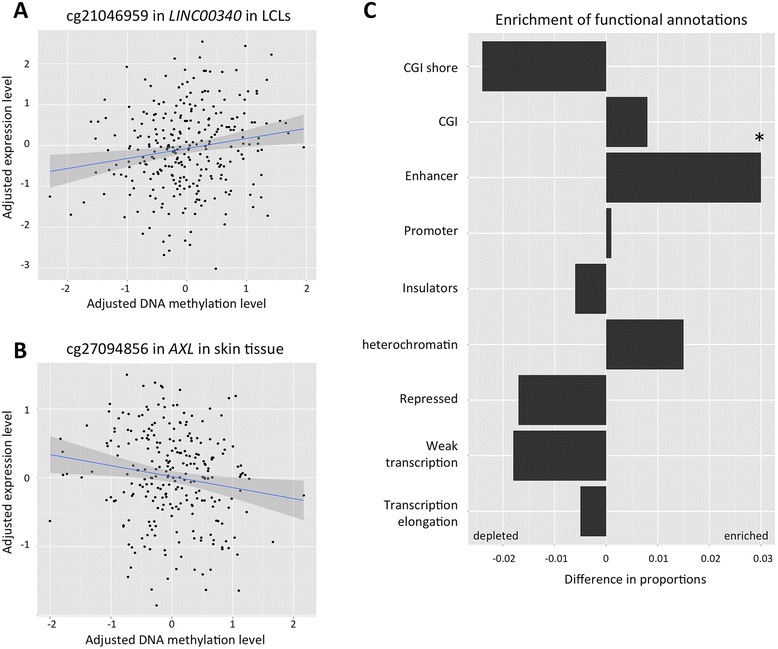
Functional follow-up of top-ranked pan-cancer DMPs. Adjusted whole blood DNA methylation profiles compared to adjusted gene expression levels for (a) cg21046959 in blood and ILMN_1809352 (PRL) in LCLs, and (b) cg27094856 in blood and ILMN_1701877 (AXL) in skin. Data are obtained from 283 healthy middle-aged females and lines represent the least squares regression fit. c Enrichment analysis of genomic annotation categories within the 500 top-ranked cancer DMPs. The bars indicate the difference in proportion of DMPs compared to the remainder of probes used in the study in the corresponding genomic annotation class. Nominally significant results were obtained for the ‘enhancer’ category (P = 0.03)
Table 2.
Gene expression analysis in of top ranked probes from EWAS in 283 healthy individuals
| LCL cell lines | Adipose tissue | Skin tissue | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CpG | Associated gene | Location | Nearest gene expression transcript probe | Name | CpG distance | r | P value | r | P value | r | P value |
| cg02444695 | – | – | ILMN_2185984 | SASH1 | 77 kb upstream | −0.02 | 0.79 | 0.01 | 0.91 | 0.03 | 0.59 |
| cg26079695 | COL11A2 | Intron | ILMN_2311456 | COL11A2 | – | 0.04 | 0.46 | 0.07 | 0.21 | 0.09 | 0.17 |
| cg27094856 | AXL | Intron | ILMN_1701877 | AXL | – | 0.01 | 0.91 | 0.07 | 0.20 | −0.15 | 0.01 |
| cg21046959 | LINC00340 | Transcript | ILMN_1809352 | PRL | 106 kb downstream | 0.17 | 4.5 × 10−3 | 0.03 | 0.59 | 0.02 | 0.74 |
| cg14044916 | TIMM44 | TSS1500 | ILMN_1784031 | TIMM44 | – | 0.02 | 0.75 | 0.05 | 0.44 | 0.04 | 0.55 |
We then tested for enrichment of functional annotations within the 500 top-ranked pan-cancer DMPs compared to the remaining CpG sites used within the study, hypothesising that this analysis could reveal systemic changes in the body associated with malignant tumour development (Fig. 4c). A nominally significant enrichment (P = 0.03) was observed for enhancers (pooled data from ENCODE) and driven by state 7 “weak enhancer” ChromHMM category (P = 0.03). In addition, we observed weak non-significant depletion of differential methylation in CGI shores, repressed regions, and weakly transcribed regions.
Discussion
This study examined MZ twin pairs discordant for multiple types of cancer to identify a pan-cancer DNA methylation signature in peripheral blood independent of host genetic variation. We identified one novel epigenome-wide significant pan-cancer DMP at a FDR threshold of 10 %, located in an intergenic region upstream of a known tumour suppressor gene SASH1, and three suggestive pan-cancer-associated signals in the genes COL11A2 and AXL, and in LINC00340, two of which have previously been linked to cancer (AXL, LINC00340). In a subset analysis of whole blood samples obtained before cancer diagnosis, we identified a signal in the promoter of COX7C, at the same site where whole blood DNA methylation was previously associated with bladder cancer [45]. We also considered regional-based DNA methylation changes, and observed one potential pan-cancer DMR in the TIMM44 gene, which was also previously linked to cancer. Despite cancer discordance, we did not observe global differences in DNA methylation profiles and found that MZ twin-pairs exhibited greater within-pair correlation than random pairing of individuals, as previously reported in healthy twins [31–34, 47].
The peak genome-wide significant pan-cancer DMP at cg02444695 upstream of the SASH1 tumour suppressor gene, showed consistently higher DNA methylation in the cancer-affected compared to their healthy co-twins, on average 0.7 % (range −0.9 to 3.0 % in normalised unadjusted DNA methylation levels). SASH1 expression has been negatively associated with aggressive tumour growth and metastasis formation in different types of cancer, including the breast, colon, and bone [48–50]. Annotation data from the ENCODE project identified the region harbouring cg02444695 as a weak transcribed region proximal (~500 bp) to a weak/poised enhancer, based on ChromHMM in the GM12878 B-lymphocyte cell line (LCL) which is biologically closest to our whole blood sample. Consistent with this, we observed a weak negative correlation between whole blood DNA methylation and LCL SASH1 expression (see section Gene expression analysis of top ranked probes, Table 2). However, Epstein-Barr virus (EBV) transformation for LCLs impacts DNA methylation profiles [51] and results in less variability in gene expression in mature LCLs [52]. Direct comparison between whole blood and LCL gene expression has revealed these sources of variation to be distinct, although LCL expression changes have been associated with phenotypes such as smoking [53]. Although we could not directly replicate the differential effect at cg02444695, this may be due to the small replication sample size of nine cancer-discordant MZ twin-pairs. However, we observed near significant greater blood methylation variability at this site in cancer-discordant twins compared to healthy concordant twins. The DNA methylation differences observed at this most associated pan-cancer DMP persisted in samples that were obtained prior to cancer diagnosis, indicating that this signal is not driven by cancer treatment and can be detected prior to or during early tumour development and could represent accrued environmental risk factor exposures, common systemic effects due to the presence of the tumour, or surrogate changes of the tumour. The effect is particularly strong in a critical time window of a maximum of 5 years prior to diagnosis, which makes it a very promising blood-based biomarker candidate.
Two of the genome-wide suggestive pan-cancer DMPs were also located in cancer-related gene AXL and lncRNA LINC00340. Cg27094856 was located in the fourth intron of the AXL gene, which is implicated in proliferation, migration, and resistance to therapy of many cancers and is a therapeutic target for antibody-based therapies [35–37]. Hypermethylation was observed in twins affected with cancer, and this effect was also observed in the replication sample of nine MZ twin-pairs, although it did not surpass nominal significance in this small sample. Furthermore, similar to the results for cg02444695, there was significantly greater blood methylation variability at cg27094856 in cancer-discordant twins compared to healthy concordant twins. A negative correlation was observed between DNA methylation at cg27094856 and the protein-coding transcript of AXL in the skin tissue only. Higher expression levels of AXL are found in many cancers tissues and implicated in proliferation, migration, and therapy resistance [35–37]. However, we did not observe a correlation in LCLs suggesting that this could be a biomarker in whole blood for common systemic effects. Exploring differential methylation results at this site with respect to time of diagnosis suggests that the majority of differential methylation arises in the samples after cancer diagnosis; therefore the methylation effect may be as a result of cancer treatment, systemic immune response to the presence of the tumour, or the tumour itself.
The second suggestive signal in a cancer-related gene was obtained at cg21046959 in LINC00340, and showed hypomethylation in cancer-affected twins. This LINC00340 transcript has been identified as a neuroblastoma susceptibility gene and was shown to be hypermethylated within its promoter in clear cell ovarian tumours [38, 39]. This is consistent with the observed direction of association in our results, as gene body hypomethylation and promoter hypermethylation are both associated with decreased expression [54]. We explored DNA methylation with the available gene expression data, but unfortunately, transcriptomic data was not available for LINC00340. However, we identified a positive correlation between DNA methylation at cg21046959 in whole blood and gene expression of PRL in LCL cell lines, but not in the skin or adipose tissue in 283 healthy female individuals. Previously, greater expression of PRL has been associated with progression of tumour development in different cancers [55–57]. Differential methylation effects at this locus were greatest in twin pairs sampled 2 years (range 0–5) prior to cancer diagnosis, and minimal after cancer diagnosis, suggesting that this locus also is of biomarker potential. The third genome-wide suggestive pan-cancer DMP was located in the COL11A2 gene, which to our knowledge has not been linked to cancer. This site also exhibited the greatest differential methylation effects within 0–5 years before diagnosis, therefore suggestive of biomarker potential.
Analysis of blood samples preceding diagnosis identified a signal in the promoter of the COX7C gene. The same site was previously associated with bladder cancer in whole blood samples [45]. Interestingly, none of the twins included in this study were diagnosed with cancer of the bladder as of yet. This suggests that the common observed effects prior to cancer diagnosis could include bladder cancer as well and requires further follow-up.
In addition to single-CpG-based analyses, we also considered differential methylation effects that spanned larger genomic regions. The genome-wide DMR analysis had attenuated findings, but highlighted a top-ranked pan-cancer DMR in the promoter of the TIMM44 gene. Germline genetic variants in this gene have been associated with familial non-medullary thyroid carcinoma [41]. Furthermore, an intragenic CpG island in TIMM44 has been found to be hypomethylated in aggressive serous ovarian cancers [42], and its expression is positively associated with recurrence after chemotherapy in breast cancer patients [43]. Given its implication in multiple cancers, this region requires further follow-up with higher resolution technologies.
To our knowledge, this is the first study to explore pan-cancer blood biomarkers in cancer-discordant MZ twin-pairs. One of the limitations of our study is that cancer is a heterogeneous disease with differing aetiology across many tissue types. Therefore, considering multiple types of cancer could potentially dilute cancer type-specific effects and may reduce power to detect true associations. However, pan-cancer signatures have previously been identified in tumour tissues spanning changes in DNA methylation as well as the proteome, somatic mutations, and somatic copy-number alterations [28, 58–61]. These findings show that shared signals across different tumour types can occur, and that these may also be associated with common systemic effects or surrogate changes.
The primary aim of our study was to identify blood-based pan-cancer biomarkers. These effects may capture either a general systemic (immune) response of the body to tumour development, accrued environmental risk exposures leading to cancer development, or changes that are present across cell types. A limitation is that although blood is an ideal sample for non-invasive biomarker screening, it is a surrogate tissue with a heterogeneous cell population. Here, we addressed cellular heterogeneity by correcting for biological covariates that capture the proportion of major white blood cell fractions. Our analyses of blood-based pan-cancer biomarkers detect signals in known tumour-associated genes, and extend previous findings of pan-cancer DNA methylation signatures to blood. Although we corrected for cellular heterogeneity, the identified signals may also reflect minor immune cell fractions or rare cell subtypes that are not covered by the applied cell-type correction, as shown for the GPR15 smoking findings in whole blood samples [62]. Further research is needed to investigate if these signals persist in sorted blood cell types, in the tissue of the primary tumour site, and tumour tissue itself, for example, to identify if these changes are present across cell types and could be surrogate changes of the tumour internal environment. Future studies are also needed to explore the longitudinal stability of these changes in different cell types over time.
By using a discordant MZ twin design, our aim was to identify systemic epigenetic changes that are independent of genotype or early environment. On the other hand, recent publications have identified genetic variants associated with DNA methylation (methylation quantitative trait loci), and these could potentially also interact with the environment. Future studies in larger population-based samples will be necessary to establish whether the DNA methylation signals identified here, also interact with genetic variants, or are subject to gene-environment interactions. We assessed whether our top-ranked results were associated with specific environmental disease risk factors, such as age and smoking. We therefore compared the most-associated pan-cancer signals to previously identified age and smoking methylation signals, but found no evidence that risk factors impact the identified cancer DMPs. Therefore the changes that we have detected are not simply biomarkers of these cancer risk factors. Whereas the BMI difference within a twin pair was not correlated with cancer incidence of the twins, three out of the 41 pairs had greater BMI in the twin diagnosed with cancer who was classified as obese within the BMI range of 30–40 kg/m2. However, the very small sample suggests that this concordance may have a negligent or very small effect on the results.
A MZ disease-discordant twin design of 41 twin-pairs in the discovery stage results in good power to detect moderate changes in DNA methylation. This study design is especially powerful in detecting differences in DNA methylation relatively independent of genetic variation with the need of fewer samples. On the other hand, the use of a surrogate tissue and the exploration of such a heterogeneous phenotype may reduce study power [63]. We estimate that we had 56 % power to detect the effect size of our top ranked probe at a conservative Bonferroni cut-off with the available sample [64], and would require 98 discordant twin pairs to reach 80 % power. The direct replication sample consisted of only 9 discordant MZ pairs of mixed sex, which provide low (10 %) mean power to detect the differential methylation effect observed at the top-ranked signal at nominal significance, and no power at a Bonferroni cut-off. However, access to these rare samples, can still give us an indication whether similar effects are observed in an independent dataset.
Conclusions
In conclusion, this is the first pan-cancer analysis of cancer-discordant twins using blood samples collected up to 5 years before the diagnosis. In this MZ cancer-discordant study, we identified one novel significant pan-cancer signal and three suggestive results in whole blood samples. The top-ranked pan-cancer signal was upstream of a known tumour suppressor gene, and the methylation effect was observed prior to diagnosis, making it a strong blood-based cancer biomarker candidate. Altogether, three of the four DNA methylation signals exhibited differential methylation effects prior to cancer diagnosis, and show potential, if can be robustly replicated by others, to have clinical utility as pan-cancer biomarkers.
Methods
Sample selection
Detailed information regarding cancer diagnosis was obtained from the National Cancer Registry at the Office for National Statistics (ONS) for twin-pairs registered with the TwinsUK Adult Twin Registry. Written informed consent from all subjects was obtained in accordance with Guy’s & St Thomas’ NHS Foundation Trust Ethics Committee (EC04/015—15-Mar-04). The twins in this registry were similar in means and ranges of quantitative phenotypes from an age-matched population in the UK [65]. DNA methylation data for 41 middle-aged (age range 42–79, median age 61 years old) female MZ twin-pairs of European descent were included in the study. Discordant MZ twin-pairs were selected based on the criteria that one twin was diagnosed at least once with malignant tumour development of a solid organ while the co-twin was never diagnosed with malignant tumour development to date, in the period ranging from 4 to 21 years after cancer diagnosis of the co-twin (median = 10.3 years). In total, cancers at eight different primary locations were included: the breast (23 pairs), cervix (1 pair), colon (10 pairs), endometrium (1 pair), thyroid gland (1 pair), melanoma (3 pairs), ovary (1 pair), and pancreas (1 pair). Whole blood samples for DNA methylation profiling were obtained within 5-years prior to diagnosis (15 pairs) and post diagnosis (26 pairs). Samples were excluded if individuals had blood and lymph system-related cancers, skin cancer except melanoma (i.e., basal cell carcinoma and squamous cell carcinoma), cervical cancers except adenocarcinoma, as well as blood samples obtained outside the 5-year window surrounding cancer diagnosis for the main study.
The 41 cancer-discordant MZ twin-pairs were assessed for discordance in body mass index (BMI), alcohol intake, and smoking, which are considered to be major risk factors for cancer development. The mean BMI across all subjects was 26.9, and 21 out of 41 pairs had a greater BMI in the cancer-affected twin than in the unaffected co-twin. The mean BMI within-pair difference was 1.6 kg/m2, with three pairs that had a difference greater than 6 kg/m2 concordant with cancer status. Self-reported alcohol intake did not differ significantly within twin-pairs. In terms of smoking habits, 29 MZ twin-pairs were concordant: 19 MZ twin-pairs were non-smokers, 1 MZ twin-pair was current smoker, and 9 MZ twin-pairs were ex-smokers (stopped smoking at least 3 years before blood sample collection). The smoking-discordant 12 MZ twin-pairs comprised of 7 MZ twin-pairs including an ex-smoker and non-smoker co-twin, and 5 MZ twin-pairs including an ex-smoker and current smoker co-twin.
To assess the biomarker potential of methylation signals, one follow-up analysis included five additional MZ twin-pairs where blood samples were obtained 5–11 years prior to the diagnosis only. The additional five cancer-discordant MZ twin-pairs (age range 38–62, median age 57) were discordant for cancers at two different primary locations, the breast (3 pairs) and colon (2 pairs). There was no discordance in smoking or alcohol intake habits. The BMI of these individuals ranged from 20.5 to 27.8 kg/m2, and the median BMI within-pair difference was 0.5 kg/m2.
Genome-wide DNA methylation data
Genome-wide DNA methylation profiles were obtained from 92 bisulfite-converted DNA whole blood samples, assayed by Illumina Infinium HumanMethylation450 BeadChip in two batches of 24 and 68 samples. The Infinium array targets 485,764 CpG sites across the genome and quantifies DNA methylation levels at a single CpG resolution as beta values, denoting the ratio of intensity signal from the methylated probes over the sum of intensity signals from both unmethylated and methylated probes, resulting in a beta value between 0 (unmethylated) and 1 (methylated). The probes cover 99 % of RefSeq genes and are distributed across the genome in the following manner: 20.75 % in promoter regions, 5 % in 5′ untranslated regions (UTR), 32.30 % in gene bodies, 3 % in 3′ UTR, and 24.60 % intergenic regions; and 14 % of CpG-sites not near genes in the genome [66].
Pre-processing consisted of five initial stages of quality control. First, three sets of probes were removed: probes that failed detection in at least one sample and with a bead count less than three in more than 5 % of the samples (n = 3325), probes for which the 50 bp sequence aligned to multiple locations in the genome (n = 17,764), and probes located on the sex chromosomes (n = 11,650) [67]. The remaining number of probes for analysis was 453,627. Second, the data was inspected visually for outliers using beta density plots, boxplots, and a heatmap of pair-wise correlation coefficients of genome-wide DNA methylation patterns. Third, MZ twin-pairs were verified using their known genotype and the 57 autosomal control SNP probes on the array. Fourth, 203 genomic imprinted regions were assessed using the R package wateRmelon [68], and no extreme outliers were detected. Lastly, probes were removed if they contained SNPs occurring in European populations with a frequency of >1 % and where the SNPs were located within 10 bp of the probe CpG (n = 17,544) [69]. The beta values were normalised using the BMIQ method to correct for probe type bias [70].
Principal component analysis (PCA) was performed on normalised beta values (N(0,1) at each probe. The first five principle components (PCs) combined explained 46.8 % of the total variance and were tested for associations with cancer status and likely covariates for DNA methylation data including array, position on the array, age, batch, and blood cell counts (CD4+ T cells, CD8+ T cells, granulocytes, monocytes, natural killer (NK) cells, and B cells) estimated using a published algorithm [71]. No association between cancer status and the first five PCs could be detected; however, significant associations (P < 4.1 × 10−3) with CD8+ T cells, CD4+ T cells, natural killer cells, granulocytes, monocytes, batch, and array were identified.
Gene expression profiles
Gene expression profiles of the lymphoblastoid cell lines (LCLs), skin tissue, and adipose tissue from 283 healthy female individuals of European descent of the TwinsUK Adult Registry were obtained from the Multiple Tissue Human Expression Resource (MuTHER) project as previously described [72]. In short, punch biopsies (8 mm) were taken from a photo-protected area adjacent and inferior to the umbilicus of which the subcutaneous adipose tissue and skin tissue were dissected. LCLs were generated through EBV-mediated transformation of B-lymphocytes from peripheral blood samples collected at the same time point. RNA was extracted and expression profiling was performed using Illumina Human HT-12 v3 BeadChips. Probes with less than three beads present were excluded and log2-transformed expression signals were normalised separately per tissue, with quantile normalisation of the replicates of each individual followed by quantile normalisation across all individuals. Gene expression follow-up at each individual CpG site from the methylation analysis was performed using the nearest Illumina expression probe.
Whole blood DNA methylation in the same 283 healthy female individuals of European descent from TwinsUK was profiled using Illumina Infinium HumanMethylation450 BeadChip. Blood methylation data processing and quality control was performed as previously described.
Statistical analysis
Analysis of global DNA methylation variation was performed using unsupervised hierarchical clustering analysis with Euclidean distances and complete linkage method. Within twin-pair correlations where assessed for all CpG sites between discordant MZ twin-pairs and as individuals randomly assigned to one of 41 pairs (permuted 100 times) either within disease status or independent of disease status. Correlation was assessed using a Spearman’s rank test. A two sample t test was performed to test for significant differences in correlation between the groups.
To identify differentially methylated positions (DMPs) associated with cancer discordance, a linear model was fitted on the normalised beta values per probe and the first five PCs. The PCs capture not only known variance due to technical covariates, such as array and batch, but also variability introduced by the mixture of different blood cell types present in whole blood samples. This method is similar to reference-free computational approaches applied in recent published methods to control for cell heterogeneity and noise in large-scale datasets [73, 74]. The difference of DNA methylation residuals was calculated within twin-pairs in consistent direction (cancer-affected twin − healthy twin) and was followed by a one-sample t test on these data. Results were considered significant if they surpassed a false discovery rate (FDR) threshold of 10 %, estimated using qvalue in R. Suggestive results were considered if they surpassed nominal P value of 1 × 10−5. The top results were compared to DMP analyses using different methods for adjusting for blood cell types and technical covariates, including the computational approach ‘RefFreeEWAS’ (using PairsBootRefFreeEwasModel) [73] and an approach using the covariates identified by PCA earlier described (batch, granulocytes, CD8 T cells, and NK cells). The top-ranked probe, ranked first in all three methods, and the suggestive probes had P values <1 × 10−03 across the three approaches.
Differentially methylated regions (DMRs) associated with cancer discordance were identified using R package ‘bumphunter’ [40]. Regions were identified of at least three consecutive probes with a maximum gap of 500 bp between the locations of each probe. The difference in adjusted DNA methylation values (described above for DMP analyses) was calculated within twin-pairs in consistent direction (cancer-affected twin − healthy twin) and was compared to a group without DNA methylation differences using bumphunter. The cut-off used was set at 0.6 with 1000 permutations with potential DMRs identified with a P value <0.05 and family-wise error rate (FWER) P value <0.5, as estimated within bumphunter [40].
To assess whether pan-cancer DMPs were also likely to be associated with environmental cancer risk factors such as age and smoking, we selected previously published DMPs for age [44] and smoking [8] in whole blood. We then counted the occurrence of age or smoking DMPs within the top 500 ranked cancer DMP probes, and in the remainder of the probes in this study (453,127). Enrichment was assessed using Fisher’s exact test.
Gene expression analysis at genes in or near the top-ranked DNA methylation probes was performed in a sample of 283 healthy individuals in multiple tissues. A linear mixed-effects model was fitted on the expression data with age, BMI, batch, concentration (skin tissue only) as fixed effects, and family and zygosity as random effects. A similar linear mixed-effects model was fitted on the blood DNA methylation data in these individuals with age, BMI, array, position on the array, and granulocytes, monocytes, CD8 T cells (estimated) as fixed effects, and family and zygosity as random effects. Residuals from both models were compared with Pearson correlation.
Replication
Replication was pursued in MZ twin pairs registered with the Netherlands Twin Register (NTR). Detailed information regarding cancer diagnosis was obtained through record linkage with the Netherlands Cancer Registry (NKR). DNA methylation data and white blood cell counts were available for 703 MZ pairs who took part in the NTR biobank project [75] of which 15 were identified as discordant for any type of cancer. Discordant MZ twin-pairs for replication were selected based on the criteria that one twin was diagnosed at least once with malignant tumour development while the co-twin was never diagnosed with malignant tumour development to date. After excluding pairs with blood samples collected >5 years after the cancer diagnosis and pair discordant for Hodgkin’s disease, mature B cell or cervical cancer, nine middle-aged (age range 35–72, median age 52 years old) MZ twin-pairs were selected for the replication analysis, and these were four male and five female MZ twin-pairs. In total, cancers at six different primary locations were included: the breast (3 pairs), meninges (1 pair), pituitary gland (1 pair), prostate (2 pairs), rectum (1 pair), and soft tissue (1 pair).
Direct replication of the findings at the four top-ranked signals was pursued in the nine NTR cancer-discordant MZ pairs using the same quality control, normalisation, and analysis pipeline as the original discovery analysis in 41 MZ pairs. In addition, we also assessed whether cancer-discordant twins showed greater methylation variability compared to healthy concordant MZ twins at the top-ranked cancer probes. The variability analysis was performed on normalised (functional normalisation [76]) betas at three of the four top-ranked probes available in the overall NTR dataset, comparing 9 cancer-discordant to 480 healthy concordant MZ twin-pairs for which blood samples were obtained on the same time point. In the variability analysis, absolute within-pair methylation differences were determined of the 480 healthy MZ twin-pairs as well as the within-pair differences of the 9 cancer-discordant MZ twin-pairs and assessed significance by a Mann-Whitney U test.
Genomic annotation analysis
Significant and suggestive results were annotated with respect to defined CpG islands (CpG Islands [CGI], shores, shelves) and relative to RefSeq genes (promoter, 5′UTR end, gene body, 3′UTR, intergenic). The top-ranked results were further explored using data generated from the ENCODE project for histone modification signatures used in ChromHMM state analysis, DNase I hypersensitivity sites, and transcription factor binding sites.
Enrichment of functional genomic elements was performed at the top 500 pan-cancer-ranked DMP probes, compared to the remainder of the probes in this study (453,127). Enrichment was performed with respect to the majority of annotation categories described above. We used ChromHMM classifications at 15 chromatin states of the GM12878 cell line from ENCODE data [77, 78]. Analyses were initially performed over 15 states, and subsequently, we also merged states active promoter (state 1), weak promoter (state 2), and poised promoter (state 3) were into a single ‘promoter’ category, and states strong enhancer (states 4 and 5), and weak enhancer (states 6 and 7) into a single ‘enhancer’ category. Enrichment tests were performed in the merged and individual categories, and nominally significant results were obtained for state weak enhancer (state 7) and merged enhancer category. Fisher’s exact test was used to assess significance.
Availability of supporting data
The dataset will be uploaded to GEO.
Acknowledgement
This work was funded by EU-FP7 project EpiTrain (316758). TwinsUK received funding from the Wellcome Trust; European Community’s Seventh Framework Programme (FP7/2007-2013), the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London. SNP Genotyping was performed by The Wellcome Trust Sanger Institute and National Eye Institute via NIH/CIDR. TDS is holder of an ERC Advanced Principal Investigator award. AB reports an Ambizione personal career fellowship from the Swiss National Science Foundation. The NTR methylation data were funded by the BBRMI-NL-financed BIOS Consortium (NWO 184.021.007).
Additional files
Pan-cancer top-ranked results in 41 discordant MZ twin-pairs. Direction of association at [A] cg26079695 in COL11A2, [B] cg27094856 in AXL, and [C] cg21046959 in LINC00340. Results are plotted using normalised unadjusted beta values of cancer-affected individuals (left) and healthy individuals (right). The lines connect co-twins in twin-pairs and indicate a consistent direction of effect. (PDF 78 kb)
Replication of four top-ranked pan-cancer DMPs in 9 cancer discordant MZ twin-pairs. Direction of association at [A] cg0244695 near SASH1, [B] cg26079695 in COL11A2, [C] cg27094856 in AXL, and [D] cg21046959 in LINC00340. Results are plotted using normalised unadjusted beta values of cancer-affected individuals (left) and healthy individuals (right). The lines connect co-twins in twin-pairs and indicate a consistent direction of effect. (PDF 88 kb)
Variability at three top-ranked pan-cancer DMPs in 9 cancer discordant and 480 healthy MZ twin-pairs. Histogram with density overlay of absolute differences of 480 healthy MZ twin-pairs with median (blue) and density and median of differences between 9 cancer discordant MZ twin-pairs (red). At [A] cg0244695 near SASH1, [B] cg26079695 in COL11A2, and [C] cg27094856 in AXL. (PDF 143 kb)
Top-ranked results from EWAS of 15 cancer discordant MZ twin-pairs prior to diagnosis. (XLSX 42 kb)
Differential methylation with respect to age and time of cancer diagnosis. [A] Time to diagnosis compared to age at blood sample collection, the line represent the least squares regression fit. [B] Unadjusted DNA methylation values at cg02444695 (near SASH1) in affected individuals (red) and healthy co-twins (blue), shown with respect to age at blood sample collection (years) with smooth (LOESS) lines fitted for both groups. [C–F] Unadjusted DNA methylation values in affected individuals (red) and healthy co-twins (blue), shown with respect to time of diagnosis (years) with smooth (LOESS) lines fitted for both groups with blood samples collected 5 to 11 years before cancer diagnosis. The orange and black vertical lines represent the time of diagnosis and time window of the main study respectively. At [C] cg0244695 near SASH1, [D] cg26079695 in COL11A2, [E] cg27094856 in AXL, and [F] cg21046959 in LINC00340. (PDF 185 kb)
Footnotes
Tim D. Spector and Jordana T. Bell joint contribution.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LR, CGB, TDS, and JTB conceived and designed the experiments. LR and JvD analysed the data. : PD, DIB, and TDS contributed data. LR, CGB, AB, TDS, and JTB interpreted the results and wrote the paper. All authors reviewed and approved the final manuscript.
Contributor Information
Leonie Roos, Email: leonie.roos@kcl.ac.uk.
Jenny van Dongen, Email: j.van.dongen@vu.nl.
Christopher G. Bell, Email: C.G.Bell@soton.ac.uk
Andrea Burri, Email: andrea.burri@kcl.ac.uk.
Panos Deloukas, Email: p.deloukas@qmul.ac.uk.
Dorret I. Boomsma, Email: di.boomsma@vu.nl
Tim D. Spector, Email: tim.spector@kcl.ac.uk
Jordana T. Bell, Email: jordana.bell@kcl.ac.uk
References
- 1.International Agency for Research on Cancer. World Cancer Report 2014. WHO Press; 2014
- 2.Gulland A. Global cancer prevalence is growing at “alarming pace”, says WHO. BMJ. 2014;348:g1338. doi: 10.1136/bmj.g1338. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 4.Hanash SM, Baik CS, Kallioniemi O. Emerging molecular biomarkers—blood-based strategies to detect and monitor cancer. Nat Rev Clin Oncol. 2011;8:142–50. doi: 10.1038/nrclinonc.2010.220. [DOI] [PubMed] [Google Scholar]
- 5.Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–92. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- 6.Ogino S, Lochhead P, Chan AT, Nishihara R, Cho E, Wolpin BM, et al. Molecular pathological epidemiology of epigenetics: emerging integrative science to analyze environment, host, and disease. Mod Pathol. 2013;26:465–84. doi: 10.1038/modpathol.2012.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12:R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeilinger S, Kühnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE. 2013;8. [DOI] [PMC free article] [PubMed]
- 9.Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5. [DOI] [PMC free article] [PubMed]
- 10.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah S, McRae AF, Marioni RE, Harris SE, Gibson J, Henders AK, et al. Genetic and environmental exposures constrain epigenetic drift over the human life course. Genome Res. 2014;24:gr.176933.114. doi: 10.1101/gr.176933.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31:142–7. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11:694–703. doi: 10.1111/j.1474-9726.2012.00835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27 K discovery and replication. Am J Hum Genet. 2011;88:450–7. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics. 2014;6:4. doi: 10.1186/1868-7083-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joubert BR, Håberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120:1425–31. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wan ES, Qiu W, Baccarelli A, Carey VJ, Bacherman H, Rennard SI, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012;21:3073–82. doi: 10.1093/hmg/dds135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 21.Yi JM, Guzzetta AA, Bailey VJ, Downing SR, Neste LV, Chiappinelli KB, et al. Novel methylation biomarker panel for the early detection of pancreatic cancer. Clin Cancer Res. 2013;19:6544–55. doi: 10.1158/1078-0432.CCR-12-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anjum S, Fourkala E-O, Zikan M, Wong A, Gentry-Maharaj A, Jones A, et al. A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med. 2014;6:47. doi: 10.1186/gm567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heyn H, Carmona FJ, Gomez A, Ferreira HJ, Bell JT, Sayols S, et al. DNA methylation profiling in breast cancer discordant identical twins identifies DOK7 as novel epigenetic biomarker. Carcinogenesis. 2013;34:102–8. doi: 10.1093/carcin/bgs321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim U, Flood A, Choi S, Albanes D, Cross AJ, Schatzkin A, et al. Genomic methylation of leukocyte DNA in relation to colorectal adenoma among asymptomatic women. Gastroenterology. 2008;134:47–55. doi: 10.1053/j.gastro.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langevin SM, Houseman EA, Accomando WP, Koestler DC, Christensen BC, Nelson HH, et al. Leukocyte-adjusted epigenome-wide association studies of blood from solid tumor patients. Epigenetics. 2014;9:884–95. doi: 10.4161/epi.28575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, Apostolidou S, et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS ONE. 2009;4:e8274. doi: 10.1371/journal.pone.0008274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, et al. The Cancer Genome Atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Witte T, Plass C, Gerhauser C. Pan-cancer patterns of DNA methylation. Genome Med. 2014;6. [DOI] [PMC free article] [PubMed]
- 29.Gevaert O, Tibshirani R, Plevritis SK. Pancancer analysis of DNA methylation-driven genes using MethylMix. Genome Biol. 2015;16. [DOI] [PMC free article] [PubMed]
- 30.Roos L, Spector TD, Bell CG. Using epigenomic studies in monozygotic twins to improve our understanding of cancer. Epigenomics. 2014;6:299–309. doi: 10.2217/epi.14.13. [DOI] [PubMed] [Google Scholar]
- 31.Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, et al. Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res. 2012;22:1395–406. doi: 10.1101/gr.136598.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong CCY, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC, et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;19:495–503. doi: 10.1038/mp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell JT, Tsai P-C, Yang T-P, Pidsley R, Nisbet J, Glass D, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629. doi: 10.1371/journal.pgen.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, et al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet. 2013;93:876–90. doi: 10.1016/j.ajhg.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leconet W, Larbouret C, Chardès T, Thomas G, Neiveyans M, Busson M, et al. Preclinical validation of AXL receptor as a target for antibody-based pancreatic cancer immunotherapy. Oncogene. 2014;33:5405–14. doi: 10.1038/onc.2013.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bansal N, Mishra PJ, Stein M, DiPaola RS, Bertino JR. Axl receptor tyrosine kinase is up-regulated in metformin resistant prostate cancer cells. Oncotarget. 2015;6:15321. doi: 10.18632/oncotarget.4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinelli E, Martini G, Cardone C, Troiani T, Liguori G, Vitagliano D, et al. AXL is an oncotarget in human colorectal cancer. Oncotarget. 2015;5. [DOI] [PMC free article] [PubMed]
- 38.Cheung N-KV, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cicek MS, Koestler DC, Fridley BL, Kalli KR, Armasu SM, Larson MC, et al. Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum Mol Genet. 2013;22:3038–47. doi: 10.1093/hmg/ddt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41:200–9. doi: 10.1093/ije/dyr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonora E, Evangelisti C, Bonichon F, Tallini G, Romeo G. Novel germline variants identified in the inner mitochondrial membrane transporter TIMM44 and their role in predisposition to oncocytic thyroid carcinomas. Br J Cancer. 2006;95:1529–36. doi: 10.1038/sj.bjc.6603455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keita M, Wang Z-Q, Pelletier J-F, Bachvarova M, Plante M, Gregoire J, et al. Global methylation profiling in serous ovarian cancer is indicative for distinct aberrant DNA methylation signatures associated with tumor aggressiveness and disease progression. Gynecol Oncol. 2013;128:356–63. doi: 10.1016/j.ygyno.2012.11.036. [DOI] [PubMed] [Google Scholar]
- 43.Specht K, Harbeck N, Smida J, Annecke K, Reich U, Naehrig J, et al. Expression profiling identifies genes that predict recurrence of breast cancer after adjuvant CMF-based chemotherapy. Breast Cancer Res Treat. 2008;118:45–56. doi: 10.1007/s10549-008-0207-y. [DOI] [PubMed] [Google Scholar]
- 44.Steegenga WT, Boekschoten MV, Lute C, Hooiveld GJ, de Groot PJ, Morris TJ, et al. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age Dordr Neth. 2014;36:9648. doi: 10.1007/s11357-014-9648-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT. DNA methylation array analysis identifies profiles of blood-derived DNA methylation associated with bladder cancer. J Clin Oncol. 2011;29:1133–9. doi: 10.1200/JCO.2010.31.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS ONE. 2014;9:e87361. doi: 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaminsky ZA, Tang T, Wang S-C, Ptak C, Oh GHT, Wong AHC, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–5. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 48.Meng Q, Zheng M, Liu H, Song C, Zhang W, Yan J, et al. SASH1 regulates proliferation, apoptosis, and invasion of osteosarcoma cell. Mol Cell Biochem. 2012;373:201–10. doi: 10.1007/s11010-012-1491-8. [DOI] [PubMed] [Google Scholar]
- 49.Zeller C, Hinzmann B, Seitz S, Prokoph H, Burkhard-Goettges E, Fischer J, et al. SASH1: a candidate tumor suppressor gene on chromosome 6q24.3 is downregulated in breast cancer. Oncogene. 2003;22:2972–83. doi: 10.1038/sj.onc.1206474. [DOI] [PubMed] [Google Scholar]
- 50.Rimkus C, Martini M, Friederichs J, Rosenberg R, Doll D, Siewert JR, et al. Prognostic significance of downregulated expression of the candidate tumour suppressor gene SASH1 in colon cancer. Br J Cancer. 2006;95:1419–23. doi: 10.1038/sj.bjc.6603452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grafodatskaya D, Choufani S, Ferreira JC, Butcher DT, Lou Y, Zhao C, et al. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics. 2010;95:73–83. doi: 10.1016/j.ygeno.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 52.Çalışkan M, Pritchard JK, Ober C, Gilad Y. The effect of freeze-thaw cycles on gene expression levels in lymphoblastoid cell lines. PLoS ONE. 2014;9:e107166. doi: 10.1371/journal.pone.0107166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joehanes R, Johnson AD, Barb JJ, Raghavachari N, Liu P, Woodhouse KA, et al. Gene expression analysis of whole blood, peripheral blood mononuclear cells, and lymphoblastoid cell lines from the Framingham Heart Study. Physiol Genomics. 2012;44:59–75. doi: 10.1152/physiolgenomics.00130.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 55.Bessette DC, Qiu D, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev. 2008;27:231–52. doi: 10.1007/s10555-008-9121-3. [DOI] [PubMed] [Google Scholar]
- 56.Dumaual CM, Sandusky GE, Soo HW, Werner SR, Crowell PL, Randall SK. Tissue-specific alterations of PRL-1 and PRL-2 expression in cancer. Am J Transl Res. 2012;4:83–101. [PMC free article] [PubMed] [Google Scholar]
- 57.Hardy S, Wong NN, Muller WJ, Park M, Tremblay ML. Overexpression of the protein tyrosine phosphatase PRL-2 correlates with breast tumor formation and progression. Cancer Res. 2010;70:8959–67. doi: 10.1158/0008-5472.CAN-10-2041. [DOI] [PubMed] [Google Scholar]
- 58.Jandaghi P, Hoheisel JD, Riazalhosseini Y. GHSR hypermethylation: a promising pan-cancer marker. Cell Cycle. 2015, 0(ja):00–00. [DOI] [PMC free article] [PubMed]
- 59.Akbani R, Ng PKS, Werner HMJ, Shahmoradgoli M, Zhang F, Ju Z, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun. 2014;5. [DOI] [PMC free article] [PubMed]
- 60.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–40. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bauer M, Linsel G, Fink B, Offenberg K, Hahn AM, Sack U, et al. A varying T cell subtype explains apparent tobacco smoking induced single CpG hypomethylation in whole blood. Clin Epigenetics. 2015;7:1–11. doi: 10.1186/s13148-015-0113-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai P-C, Bell JT. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. Int J Epidemiol 2015:dyv041. [DOI] [PMC free article] [PubMed]
- 64.Cohen J: Power Analysis for the Behavioral Sciences. 2nd edition.
- 65.Andrew T, Hart DJ, Snieder H, de Lange M, Spector TD, MacGregor AJ. Are twins and singletons comparable? A study of disease-related and lifestyle characteristics in adult women. Twin Res Hum Genet. 2001;4:464–77. doi: 10.1375/1369052012803. [DOI] [PubMed] [Google Scholar]
- 66.Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics. 2011;3:771–84. doi: 10.2217/epi.11.105. [DOI] [PubMed] [Google Scholar]
- 67.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schalkwyk LC, Pidsley R, Wong CC, Touleimat N, Defrance M, Teschendorff A, Maksimovic J. wateRmelon: Illumina 450 Methylation Array Normalization and Metrics. 2013. [Google Scholar]
- 69.Butcher L. Illumina450ProbeVariants.db: annotation package combining variant data from 1000 genomes project for Illumina HumanMethylation450 Bead Chip probes. R package version 1.3.1. 2013. [Google Scholar]
- 70.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450k DNA methylation data. Bioinforma Oxf Engl. 2013;29:189–96. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grundberg E, Small KS, Hedman ÅK, Nica AC, Buil A, Keildson S, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–9. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinforma Oxf Engl. 2014;30:1431–9. doi: 10.1093/bioinformatics/btu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zou J, Lippert C, Heckerman D, Aryee M, Listgarten J. Epigenome-wide association studies without the need for cell-type composition. Nat Methods. 2014;11:309–11. doi: 10.1038/nmeth.2815. [DOI] [PubMed] [Google Scholar]
- 75.Willemsen G, de Geus EJC, Bartels M, van Beijsterveldt CEMT, Brooks AI, Estourgie-van Burk GF, et al. The Netherlands twin register Biobank: a resource for genetic epidemiological studies. Twin Res Hum Genet. 2010;13:231–45. doi: 10.1375/twin.13.3.231. [DOI] [PubMed] [Google Scholar]
- 76.Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Systematic analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]