

Abstract
Many proteins have modular design with multiple globular domains connected via flexible linkers. As a simple model of such system, we study a tandem construct consisting of two identical SH3 domains and a variable-length Gly/Ser linker. When the linker is short, this construct represents a dumbbell-shaped molecule with limited amount of domain-domain mobility. Due to its elongated shape, tandem SH3 efficiently aligns in steric alignment media. As the length of the linker increases, the two domains become effectively uncoupled and begin to behave as independent entities. Consequently, their degree of alignment drops, approaching that found in the (near-spherical) isolated SH3 domains.
To model the dependence of alignment parameters on the length of the interdomain linker, we have generated in silico a series of conformational ensembles representing SH3 tandems with different linker length. These ensembles were subsequently used as input for alignment prediction software PALES. To test the accuracy of theoretical predictions, we prepared a number of tandem-SH3 samples in PEG/hexanol alignment media. These samples were used to measure backbone 1HN-15N residual dipolar couplings associated with the two globular domains within the tandem. The experimental alignment tensors determined in this fashion were compared with the results of the PALES-based simulations, broadly confirming the anticipated trends.
The results of the comparison, however, fall short of quantitative agreement. In particular, it has been found that the isolated SH3 domain aligns much stronger than expected. This finding can be attributed to complex morphology of the PEG/hexanol media and/or to weak site-specific interactions between the protein and the media. In the latter case, there are strong indications that electrostatic interactions may play a role. The fact that PEG/hexanol does not behave as a simple steric media should serve as a caution for studies that use PALES as a quantitative prediction tool (especially for disordered proteins). Further progress in this area depends on our ability to accurately model the anisotropic media and its site-specific interactions with protein molecules. Once this ability is improved, it should be possible to use the alignment parameters as a measure of domain-domain cooperativity, thus identifying the situations where two domains transiently interact with each other or become coupled through a partially structured linker.
Keywords: Multidomain proteins, residual dipolar couplings, alignment tensor, generalized degree of order, domain-domain motion, flexible linker, conformational ensemble, conformational disorder, tandem SH3 construct, PALES software, steric alignment, electrostatic alignment, PEG/hexanol alignment media
Introduction
Modular design is a hallmark of eukaryotic proteins. A great number of them contain adaptor domains, such as SH2, SH3, WW, PDZ, FHA, etc., connected through stretches of predominantly flexible linker. These domains present a number of `generic' binding sites, facilitating assembly of protein complexes that prove to be of immense importance in signal transduction. Binding of adaptor domains to their molecular targets is typically characterized by relatively low affinity (low μM) and limited specificity. As a consequence, the complexes often have the ability to rapidly dissociate, offering fast dynamic response to external stimuli. Because many domains can bind multiple targets, this mechanism gives rise to sophisticated, non-linear signaling networks, efficiently serving the needs of higher organisms.1
As an illustration, let us briefly discuss the adaptor protein c-Crk. In mammalian cells, c-Crk is found in two different isoforms derived from alternative splicing. The smaller species, c-Crk I, are comprised of an SH2 domain followed by a ~15-residue linker and an SH3 domain (N-SH3). The larger form, c-Crk II, additionally contains a ~50-residue linker and an extra SH3 domain (C-SH3).2
Through its SH2 domain, Crk binds a variety of phosphotyrosine-containing proteins, most prominently p130Cas. In turn, p130Cas contains an N-terminal domain that interacts with focal adhesion kinase, the Crk binding region that contains 15 copies of YxxP motif, and the Src kinase binding domain.3 Under the effect of various external stimuli, such as growth factors, cytokines, and integrin-mediated mechanical forces, the YxxP tyrosines become phosphorylated by the p130Cas-associated Src kinase.4 The resulting phosphotyrosine-containing sequences are recognized by the SH2 domains from the Crk, paving the way to formation of the complex between p130Cas and multiple copies of Crk.
After Crk is engaged by p130Cas, it can transmit signals downstream by binding various proteins through its N-SH3 domain.5, 6 In this manner Crk can recruit the guanine nucleotide exchange factor Sos, which carries a number of PxxP binding motifs in its C-terminal segment. Further downstream in the signaling cascade, Sos interacts with the small GTPase Ras, inducing the exchange of GDP for GTP.7 Alternatively, Crk can bind exchange factors C3G and DOCK1, which interact with GTPases Rap1 (Rap2) and Rac1, respectively.8, 9 In addition, Crk N-SH3 domain binds to JNK and Abl kinases, with Abl phosphorylating Crk and, at the same time, undergoing autophosphorylation.10–12
All of these interactions are associated with important signaling pathways. These pathways converge at the p130Cas / Crk assembly, which contains multiple copies of Crk and thus has an ability to bring together a number of different Crk binding partners. The major role of these (interconnected) pathways in cell growth, motility, proliferation, adhesion, and apoptosis has been well documented.5 There is also a strong link between Crk-mediated signaling processes and various human cancers, including lung and breast cancers, soft-tissue sarcomas and brain cancers.13–17
From a structural perspective, one of the key questions that can be asked about Crk and other modular proteins concerns the interactions between domains. On one hand, there is a scenario where two domains, connected through long and flexible linker, behave as essentially independent entities. The domain-domain coupling in this case is minimal. On the other hand, two domains can be significantly coupled through direct interaction and/or a conformationally restricted linker. In the case of Crk adaptor, recent structural study by Inagaki and co-workers showed that c-Crk I conforms more closely to the first scenario, while c-Crk II represents the second type of situation.2 More specifically, in the c-Crk I the two constitutive domains, SH2 and N-SH3, display significant motional freedom, whereas in the c-Crk II the three domains, SH2, N-SH3, and C-SH3, form a compact structure centered around the piece of the inter-SH3 linker. This result highlights the role of the extra domain: the binding surface in Crk C-SH3 has an unusual amino-acid composition, which degrades its ability to bind PxxP ligands; at the same time, C-SH3 occludes the binding site in N-SH3, thus downregulating a number of signaling events.2, 18–21 Another domain rearrangement, which has the effect of shutting down the Crk-mediated signals, occurs upon phosphorylation of the tyrosine residue in the inter-SH3 linker. The subsequent (intramolecular) binding of the pYxxP motif to the SH2 domain abrogates most of the Crk interactions.2, 22–24
The above example underscores the importance of domain-domain coupling. Note, however, that it is not always possible to investigate domain interactions by straightforward structural methods. Indeed, as already mentioned, these interactions are often dynamic in nature, corresponding to diverse and short-lived conformational species. Two different approaches have been developed to address this situation. The first method relies on backbone 15N relaxation. Indeed, domain cooperativity leads to increases in rotational correlation time τR, which can be sensitively detected by relaxation experiments.25, 26 This effect has been recently explored in-depth by Walsh et al.27 using protein constructs that are similar to those employed in our work. An accurate interpretation, however, can be challenging because spin relaxation is sensitive to many motional modes, including domain-domain motions.28, 29 It can also be difficult to `calibrate' this method since the existing algorithms for prediction of τR have limited accuracy.30 The second method makes use of residual dipolar couplings. In this approach, one of the domains is loaded with a paramagnetic ion, thus achieving a weak alignment in the external magnetic field. If the two domains are fully coupled, then the second domain shows the same degree of alignment as the first; conversely, if the domains are uncoupled, then the second domain fails to show any alignment. 31, 32 While elegant and potentially useful, this method faces certain technical difficulties as it depends on suitably engineered metal-binding sites or carefully designed chelate tags.33–35
In this study we turn to more conventional alignment methods involving steric mechanism. We further focus on the important limiting case where a pair of non-interacting domains is connected through a variable-length flexible linker. To model such a system, we manufactured a series of chimera proteins consisting of two copies of chicken α-spectrin SH3 domain (α-spc SH3)36 connected through a flexible (GGSGG)n-type linker.37 In the case of the short linker, this construct represents a dumbbell-shaped molecule which becomes efficiently oriented in a steric alignment media. Conversely, if the linker is long, then each SH3 domain behaves as an independent entity. Furthermore, since each individual domain has a nearly spherical shape, it displays only a small degree of alignment. Thus, we expect that the degree of alignment changes in a sigmoidal fashion – from high values (short linker) to low values (long linker). The broad goal of this paper is to quantitate this dependence, both experimentally and by means of simulations. In this manner we seek to establish an important reference case – that of a protein with minimal domain-domain coupling (due to a trivial linker connectivity). Anything beyond this minimal coupling would point toward domain cooperativity. Such cooperativity can arise from domain-domain interactions, an increasingly structured linker region, or a combination of these two factors.
Materials and methods
Plasmid construction
The original pET3d plasmid encoding chicken α-spectrin SH3 domain (full-length construct fSH3) was a gift from Prof. Bernd Reif. Since the N-terminus of α-spc SH3 is highly disordered,36, 38, 39 we truncated the construct by deleting residues 2–6 (deletion construct dSH3). We have also manufactured three chimera proteins, each of which contained two copies of dSH3 connected via a glycine-serine linker. To link the domains we have used the GSG, GG(GGSGG)GG, and GG(GGSGG)5GG sequences (the latter contains 29 residues). In what follows, these tandem constructs are referred to as dSH3-sl-dSH3, dSH3-ml-dSH3, and dSH3-ll-dSH3, alluding to short, medium-length, and long linker, respectively. To produce the cDNA for tandem constructs, two separate PCR reactions were carried out. One PCR product encoded N-terminal dSH3 plus half of the linker region; it was digested with restriction enzymes NcoI and BspEI. The other PCR product encoded the remaining half of the linker plus C-terminal SH3 domain; it was digested with BspEI and BamHI. In the next step, the two digested PCR products were cloned into pET3d vector and, at the same time, fused by means of the T4 DNA ligase. Note that the use of BspEI sites and T4 DNA ligase does not introduce any unwanted residues in the linker sequence. All plasmids were transformed into BL21(DE3) E. coli strain and sequenced to check for accuracy. Primer sequences used to produce the DNA vectors and amino-acid sequences of the SH3 constructs are listed in Tab. S1.
Protein expression and purification
The bacteria transfected with plasmids were grown in 1 L of M9 minimal media enriched with 15NH4Cl at the temperature 37 °C. Protein expression was induced at OD600~0.6 by adding 0.5 mM IPTG. After 4 hours of incubation the cells were harvested by centrifugation (15 min at 8000 rpm) and stored at −80 °C. The cell pellet was then resuspended in pH 8 lysis buffer and lysed using a French Press. fSH3 was contained in the supernatant; it has been purified using the previously described protocol.39 All other constructs formed inclusion bodies during the expression. To purify the precipitated protein material, we first washed the French press lysis pellet four times using 20 mM Tris (pH 8), 1 mM EDTA, 100 mM NaCl, 0.5% Triton X-100 buffer. Each wash was carried out in 30 mL of the buffer (30 min at 18000 rpm); the last wash was done without the Triton. The pellet was then dissolved in 30 mL of 8 M urea, 20 mM Na-citrate, pH 3.5 denaturing buffer and heated in a water bath at 70 °C for 30 mins. The solution was subsequently dialyzed over the course of 1 day against 20 mM Na-citrate, pH 3.5 refolding buffer using the dialysis bag with MW cutoff 3 kDa. The refolded protein was purified using the Superdex 75 gel filtration column and then concentrated using the Millipore Amicon devices with 3 kDa MW cutoff. All samples have been analyzed by SDS-PAGE to check the protein molecular weight. Protein concentration determined by UV absorbance at 280 nm was in the range from 0.5 to 1.0 mM. In addition to α-spectrin SH3 domain, we have also prepared two samples of human ubiquitin. The plasmid was supplied by Rachel Klevit laboratory through Addgene repository; the expression and purification procedure was adapted from the work of Lazar et al.40
NMR Spectroscopy
The experimental measurements involved five constructs of α-spectrin SH3 domain (fSH3, dSH3, dSH3-sl-dSH3, dSH3-ml-dSH3, and dSH3-lldSH3) in solution with 20 mM Na-citrate, pH 3.5, 0.02% (w/v) NaN3. One extra sample of fSH3 additionally included 100 mM NaCl (see Tab. 1). The oriented samples were prepared by using 5% penta(ethylene glycol) monododecyl ether (C12E5) and 1-hexanol.41 Two series of samples were prepared with the C12E5-to-hexanol molar ratio r = 0.85 and 0.96, respectively. The former condition offers stable alignment around the working temperature 25 °C;41 the latter is the most popular choice in the studies employing PEG-hexanol media.42–44 Special care was taken to maintain the same alignment conditions throughout each series of samples. Toward this goal, we used the stock solutions containing 10% C12E5 and r = 0.85 (0.96) fraction of 1-hexanol. The anisotropic samples were prepared by mixing 150 μL aliquot of the stock solution with 150 μL aliquot of the protein solution. Each freshly prepared batch of protein was used to make two or three NMR samples, including some replicate samples. Every sample was allowed to equilibrate in the magnet for 2 hrs, before the residual quadrupolar coupling from 90% H2O – 10% D2O solvent, Δ(2H), was carefully measured. The 1HN-15N residual dipolar coupling data were collected using the IPAP-HSQC pulse sequence45, 46 on a Varian Inova 600 spectrometer equipped with a triple-resonance probe and z-axis gradient. Data sets for isotropic and partially aligned samples were collected as (192, 576) complex matrices with spectral widths of 1920 and 9000 Hz in nitrogen and proton dimensions, respectively. Each experiment took approximately 2 hours. After the data were collected, the residual 2H coupling was re-measured; the Δ(2H) drift never exceeded 0.2 Hz. The IPAP spectra were processed using the NMRPipe software package and peak positions were determined by means of the nlinLS fitting routine.47 In the case of tandem constructs, most of the peaks from the N-terminal domain are neatly overlapped with their counterpart peaks from the C-terminal domain. Hence, the RDC values measured for these residues represent the average of the N- and C-domain couplings. For a small number of sites that give rise to two resolved peaks, we choose to average the experimental RDCs on a pairwise basis (see Experimental Results). The experimental data from the tandem constructs represent, therefore, the average values (listed in Tabs. S2, S3). In analyzing the results, all datasets have been restricted to the core portion of the SH3 domain, residues 9–60.
Tab. 1.
Alignment parameters from a series of samples in r = 0.85 PEG/hexanol media. The results from replicate samples are shown in shaded columns.
| dSH3a | dSH3 | fSH3 | fSH3 (high salt)b | dSH3-sl-dSH3 | dSH3-sl-dSH3c | dSH3-ml-dSH3 | dSH3-ll-dSH3a | dSH3-ll-dSH3c | |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Aa (10−3) | 2.23 | 3.01 | −1.40 | −2.27 | −1.56 | −1.51 | 0.67 | 0.65 | 0.75 |
|
| |||||||||
| R | 0.62 | 0.40 | 0.37 | 0.39 | 0.37 | 0.36 | 0.65 | 0.27 | 0.29 |
|
| |||||||||
| {α, β, γ}d | 308° | 308° | 32° | 27° | 33° | 34° | 168° | 107° | 106° |
| 62° | 65° | 38° | 40° | 80° | 80° | 53° | 54° | 54° | |
| 81° | 80° | 320° | 325° | 63° | 63° | 159° | 155° | 155° | |
|
| |||||||||
| N | 29 | 25 | 46 | 27 | 42 | 42 | 39 | 45 | 45 |
|
| |||||||||
| Q | 0.25 | 0.30 | 0.22 | 0.21 | 0.22 | 0.21 | 0.27 | 0.26 | 0.27 |
|
| |||||||||
| Δ(2H) (Hz) | 24.9 | 21.8 | 24.7 | 25.8 | 23.3 | 23.1 | 16.9 | 23.5 | 24.4 |
|
| |||||||||
| GDO (10−3)e | 2.03f | 2.93 | 1.19f | 1.85 | 1.41f | 1.37 | 0.92f | 0.57f | 0.63 |
|
| |||||||||
| NSPg | 1.0 | 0.98 | 0.94 | 0.94 | −0.53 | −0.53 | −0.32 | 0.26 | 0.27 |
The RDC fits are shown in Fig. 6.
100 mM NaCl added to the buffer solution
The results included in Fig. 7a,b (green triangles)
Relative to the crystallographic coordinates 1U06. The rotation as defined in PALES is inverse of the rotation used by other programs.102, 103
Normalized toward Δ(2H) = 20 Hz
Values used to optimize the electrostatic alignment model
Relative to dSH3
Structural models
Each individual α-spectrin SH3 domain was modeled based on the crystallographic structure 1U06 (resolution 1.49 Å).39 Protons and several missing heavy atoms from the side chain of residue D48 were added to the structure and optimized using the facilities of CHARMM48 and XPLOR-NIH49 with the CHARMM22 force field. The resulting domain coordinates were used to build structures of tandem dSH3 and other constructs. The structure 1U06 contains five side chains that are modeled with two alternate conformations. In our main structural model all of them are placed in the conformation found in the chain A of the crystallographic structure. We have also tested other models, in particular the one where the ionizable E17 side chain is placed in conformation B. The energy of this alternative structure, as evaluated in implicit generalized Born solvent,50 was found to be within 6 kcal/mol from the all-A structure. The structure, labeled BAAAA, was therefore deemed suitable for the analyses. In addition, the NMR structure 1AEY38 was also used.
As an example of the SH3 tandem, let us consider the structural model for dSH3-ml-dSH3. The amino acid sequence for this construct is M1(E7…L61)D62-GGGGSGGGG-(E7…L61)D62. The bracketed portions of the sequence correspond to the structured portion of the domains, as represented by the crystallographic coordinates 1U06. The remaining part, comprising the 10-residue linker and single-residue termini, is modeled as a random coil. To generate the coordinates of these segments, we used the program TraDES (routine foldtraj, default input parameters).51 As an example, let us discuss the procedure for adding N-terminal residue M1. We begin by generating a random dipeptide with the sequence ME. This peptide is then `glued' onto the structure of the SH3 domain by overlaying the E residue of the peptide with E7 residue in the SH3 domain (specifically, N, Cα, and C' atoms are superimposed in this particular order). Finally, all redundant atoms are deleted. This includes all atoms from the peptide residue E, with the exception of HN. The equivalent procedure is used for the C-terminal residue D62. In this case, we begin with the random dipeptide LD, which is `glued' to the SH3 domain by overlaying the L residue of the peptide with L61 from the SH3 domain (C', Cα, and O atoms are superimposed). Subsequently, all atoms from the placeholder peptide residue L are deleted. The same prescriptions are applied to insert the ten-residue linker segment, DGGGGSGGGG.
The dSH3-ml-dSH3 model obtained in this fashion is subjected to a further round of MD refinement (1 ps at 100 K). The goal of the refinement is to relieve some of the strain caused by the `structure assembly' procedure. The relative position of the domains is preserved during this stage (for this purpose all atoms within the domains have been assigned an artificially large mass, 10 kDa). The refinement protocol is implemented using the torsional angle dynamics option in XPLOR NIH (IVM module52); the internal coordinates of the domains (all dihedral angles in the residues from 7 to 61, except for the angle φ in residue E7) are fixed and the planarity of peptide planes is enforced. This algorithm is used to produce the ensemble of 10,000 models for dSH3-ml-dSH3. Many of these models display severe steric clashes that cannot be resolved by our limited refinement procedure. To weed out these structures, we evaluate the energies of each model and analyze the resulting energy distribution as discussed below.
Fig. 1 shows a portion of the energy distribution for 10,000-strong ensemble of dSH3-ml-dSH3 models. The energies are evaluated using XPLOR-NIH/CHARMM22 in vacuo (the solvation term is inconsequential, as we seek to identify the structures with severe steric clashes). The low-energy part of the distribution follows the Gaussian curve (see inset in Fig. 1). This is to be expected, since the distribution arises from variability in the random-coil linker region. The Gaussian energy distribution for disordered protein systems can be predicted from general considerations and also has been derived from a number of theoretical models.53–55
Fig. 1.

Part of the energy distribution for 10,000-strong ensemble of dSH3-ml-dSH3 models (red circles) fitted with the Gaussian function (blue curve). The inset displays the expanded portion of the main graph; the dashed line corresponds to one standard deviation σ of the Gaussian curve.
In Fig. 1, those structures that comply with the Gaussian distribution constitute a bona fide conformational ensemble, whereas the high-energy structures outside the Gaussian distribution (appearing to the right in the plot) suffer from steric conflicts. To address this problem, we analyzed the part of the energy distribution to the right of the dashed line (corresponding to one standard deviation σ of the fitted Gaussian curve) and eliminated a fraction of structures that fall outside the curve. In this manner the original ensemble of 10,000 models was trimmed down to 2371 models. The retained structures are (i) free of significant steric clashes and (ii) have their two domains positioned according to the conformation of the (random-coil) linker, as generated by TraDES. Several randomly selected structures from the resulting reduced ensemble are shown in Fig. 2.
Fig. 2.

Five representative structures from the final dSH3-ml-dSH3 ensemble. The N-and C-terminal SH3 domains (residues 7–61, backbone coordinates same as in the crystallographic structure 1U06) are plotted in green and gold, respectively; the regions modeled as random coil are colored in red. The panel in the lower right corner shows the spatial distribution of the centers of mass of the C-terminal domain in the coordinate frame of the N-terminal domain (as plotted for the entire ensemble).
The same procedure as described above has been used to build the models for other chimera proteins, as well as fSH3 and dSH3 (the former carries a 6-residue unstructured N-terminus and a single-residue C-terminus, the latter contains two single-residue termini).
Alignment parameters
Protein alignment in liquid-crystalline media is traditionally characterized by the degree of alignment Aa, rhombicity R, and three Euler angles that specify the orientation of the principal axes of the alignment tensor relative to the molecular frame.56 These parameters, however, are not necessarily best suited for drawing a comparison between different aligned samples (which becomes especially obvious when Euler angles are compared). For a suitable alternative we turn to the five-dimensional vector space spanned by the irreducible components of the Saupe alignment tensor.57–59 In this linear space the scalar product between the two vectors formed from the Saupe matrix elements can be defined as:
| (1) |
where Sij are the elements of the 3×3 Saupe matrices associated with the rigid molecule or molecular fragment (e.g. SH3 domain within the tandem construct). sample1 and sample2 designations refer to samples involving different alignment media or different protein constructs, that share the same fragment (SH3 domain). Note that both Ssample1 and Ssample2 should be expressed in the same coordinate frame, e.g. the SH3 pdb frame. Using the definition in Eq. (1), one can introduce the Generalized Degree of Order (GDO)60 that characterizes the magnitude of alignment for a given sample:
| (2) |
Furthermore, if the scalar product Eq. (1) is normalized, then it can be viewed as a generalized measure for relative orientation of the two alignment tensors:
| (3) |
The values of NSP close to 1.0 indicate that the two alignment tensors differ only by a scaling factor, whereas the values around 0.0 suggest that the alignment frames are `orthogonal'. Finally, the (Ssample1 | Ssample2) = −1 can be described as `antiparallel' alignment – the rhombicities and alignment frames are in this case identical, while the degrees of alignment Aa have opposite signs. This situation is observed in stretched vs. compressed gels,61, 62 conventional vs. `flipped' bicelles,63, 64 and other similar systems.65 Although NSP defined by Eq. (3) depends also on rhombicity R, it is mainly a measure of alignment axes orientation.
The definition of the Saupe tensor used here58 differs by a factor of 2 from the one used elsewhere.66 The definitions of the GDO and NSP, however, are unambiguous. The numeric results for Saupe matrices, as required for evaluating Eqs. (2) and (3), are reported by the program PALES,67, 68 both in the case of the experimentally determined (fitted) and predicted alignment tensors. Finally, the quality factor is defined as:69, 70
| (4) |
PALES predictions
The alignment mechanism in PEG/hexanol media is commonly assumed to be steric.41, 71–74 To model this media we used the PALES procedure originally developed for DMPC/DHPC bicelles.75 The thickness of the planar bilayer was set to 28.6 Å.76 It is worth mentioning that bicelle calculations in PALES have been programmed for DMPC/DHPC, so that the code implicitly accounts for the presence of 5 mM free DHPC in the solvent.77, 78 To correct for this small contribution and, in addition, account for 0.3 wt% of free hexanol in solution76 one should use a slightly altered value of the liquid crystal concentration. For instance, in the PALES calculations aimed at 5% PEG, r = 0.85 media the effective liquid crystal concentration should be set to 65 mg/ml. The order parameter of the PEG/hexanol bicelle was assumed to be the same (0.8) as for DMPC/DHPC.
We have also attempted to predict the alignment parameters under the assumption that an electrostatic mechanism plays a significant role. The corresponding PALES calculations involve several additional variables: charge density on the surface of the bilayer σ (treated as adjustable parameter), sample pH (actual value 3.5; also treated as adjustable parameter), and ionic strength of the solution (17 mM, corresponding to 20 mM citrate buffer at pH 3.5). To determine the charges on α-spc SH3, we have used the program PROPKA, which calculates the pKa values of ionizable groups.79, 80 Since there is significant structural variability in tandem constructs and fSH3, we have run PROPKA on each individual conformer from the structural ensemble. The results (i.e. the coordinates of a given conformer plus the charges calculated for this particular conformer) were then used as input for PALES calculations. In principle, more sophisticated methods for calculating charges can be used81, 82 and ultimately the pKa values can be determined experimentally.83, 84 However, such effort seems unwarranted given that the models used for prediction of electrostatic alignment remain relatively crude.85–87 Of note, we have found a programming error in the PALES module responsible for computations of electrostatic alignment (kindly confirmed by M. Zweckstetter). Specifically, natural logarithm is mistakenly used in the Henderson-Hasselbalch equation instead of the base 10 logarithm. As a result, given the set of pKa (either default or user-supplied values) the program fails to correctly calculate point charges at a given pH. The problem can be circumvented by using a table of user-supplied point charges as the PALES input.
The final step is the same for all PALES-based calculations (involving both steric and electrostatic mechanisms). Dipolar couplings predicted for the individual conformers are averaged, resulting in a single set of simulated RDCs. When drawing comparison with the experimental data from the tandem constructs, we further average the computed couplings from the N- and C-terminal domains (in accordance with our experimental procedure, see above). Note that such averaging procedure is fully justified so long as both domains have the same (invariant) internal structure. Indeed, RDCs by definition represent the average between many molecular orientations. The resulting dataset is subsequently restricted to the core portion of the α-spc SH3 domain, residues 9-60, and fitted with the (protonated) structure 1U06. In the situation when residues 9-60 are represented by one and the same set of coordinates throughout the course of the calculations, the residual χ2 from such fitting procedure is strictly zero. The alignment parameters extracted in this fashion are then used to calculate the theoretical values of the GDO and NSP, Eqs. (2) and (3). Note that the averaging of simulated RDCs with respect to the N- and C-terminal domains has an effect on the resulting GDO and NSP values.
Theory: general outline
Let us consider a generalized two-domain protein, where a pair of globular, near-spherical domains is connected via a flexible structureless linker. In the case when the linker is short, i.e. consists of no more than several residues, the protein has a dumbbell shape. Furthermore, a short linker allows for little motional freedom so that domain-domain motion is highly restricted. To a good approximation, the protein can be described as a rigid body with highly elongated shape. Being placed in a steric alignment media, this protein should efficiently align and display a high GDO value, Eq. (2). The long axis of the alignment tensor should roughly coincide with the line connecting the centers of mass of the two domains. Generally speaking, the alignment PAS in such a two-domain construct has no relation to the PAS of the individual (near-spherical) domain. Therefore the NSP between the two respective tensors, Eq. (3), should deviate from 1.0 (unless by coincidence).
Let us now consider the opposite situation, where the flexible linker is very long. In this case the two domains move almost freely and no longer sense each other's presence. Their GDOs, therefore, should approach that of an individual domain. Given that the shape of each domain is nearly spherical, the GDO should drop to a low value. Likewise, the PAS orientation should generally approach that found in the individual domain. Consequently, the scalar product between the two alignment tensors – the one from the domain which belongs to the tandem and the one from the isolated domain – should converge toward 1.0.
Note that neither GDO, nor NSP are expected to show a quantitative convergence toward the single-domain limit. Indeed, the behavior of a tethered domain is similar, but not identical, to that of the isolated domain. In particular, if the shape of the domain is very close to spherical then even a small alteration of the shape (such as caused by attachment of the linker) can lead to a significant change in alignment. Therefore, the convergence toward the single-domain limit should be rather viewed as a broad trend.
To illustrate this expected behavior, we have undertaken a series of PALES simulations for a number of protein domains, Fig. 3. For these simulations we have chosen the domains of different size with close-to-spherical shape; if desired, the results can be extended to domains with elongated or flattened shapes. The coordinates of the domains have been used to generate tandem structures, which were subsequently processed by PALES. As expected, increasing the linker length from three residues to five, then to seven, etc. leads to pronounced decreases in the GDO values.
Fig. 3.

PALES simulations of steric alignment in a series of computer-generated tandem proteins. To generate structural ensembles, the two identical domains (represented by crystallographic or NMR coordinates and presumed to be fully rigid) were directly connected via glycine/serine linkers. The simulated couplings from all non-proline residues in the N-terminal domain were used to derive the alignment parameters. Other details of the structure generation and RDC simulations are as described in the Materials and Methods. Shown are the results for the tandem constructs of the disintegrin domain (structure 1MPZ,88 41 residue, radius of gyration 8.8 Å, magenta symbols), SH2 domain (extracted from structure 1A81,89 106 residues, 13.0 Å, blue), and nucleotide-binding domain (3GWI,90 170 residues, 15.3 Å, green). (a) Simulated GDO values for the domains which belong to the tandems (circles) and the corresponding isolated domains (dashed lines). (b) Simulated NSP values characterizing the mutual projection of the alignment tensors from (i) the domain which belongs to the tandem and (ii) the corresponding isolated domain. The spline curves are added to the graphs for visual guidance. To verify the convergence of the simulations, we have regenerated this plot using a different set of randomly generated structural ensembles. The results are shown in Fig. S1.
Eventually, each curve approaches a plateau which is close to, but distinct from the GDO of the isolated domain (dashed horizontal lines in Fig. 3a). The shift between the asymptotic GDO value observed in the tandem and the GDO of the free domain is not unexpected – the domain which is a part of the tandem senses the presence of the linker. More specifically, it senses the presence of several linker residues which are closest to the attachment point. These residues tend to point in a certain specific direction (due to the finite stiffness of the polypeptide chain) and therefore effectively alter the shape of the domain. Conversely, remote part of the linker is thoroughly randomized and has no effect on the alignment of the domain. As a result, further lengthening of the linker does not change the simulated GDO.
A similar pattern is observed for the NSP parameter: as the linker length is increased, the NSP value becomes closer to 1.0 (Fig. 3b). The trend, however, is weak. As already pointed out, this can be attributed to the difference in the orientation of the alignment frame in the tethered domain and in the isolated domain. This difference persists even when the linker becomes very long.
It is interesting to discuss how the results scale with the size of the system. Generally, one would expect that for the smallest unit, disintegrin, the two domains become `uncoupled' and the GDO (NSP) plateau is reached already with a moderately long linker. Conversely, for the biggest unit, nucleotide-binding domain (NBD), this does not happen until the linker becomes much longer. A rough theoretical estimate can be readily obtained for the scaling properties of the two-domain constructs. The radii of gyration of the disintegrin and NBD domains are 8.8 and 15.3 Å, respectively. Let us accept that both domains are spherical. The average end-to-end distance in the random peptide chain, such as the computer-generated linker, scales with the number of residues as N0.6.91 From here we can estimate the `scaling factor' that relates the NBD tandem to disintegrin tandem. For example, the NBD tandem with 25-residue linker can be viewed as a `resized' copy of the disintegrin tandem with 10-residue linker. This is roughly consistent with the trends seen in Fig. 3a.
Generally speaking, N- and C-terminal domains in the tandem are inequivalent: one is attached to the linker through its C-terminal residues while the other through the N-terminal residues. In principle, this should lead to doubling of the spectral resonances. This situation is illustrated in Fig. 4a, which shows two domains connected in tandem. The scheme Fig. 4a suggests that some of the amide signals from N- and C-domains should be resolved with respect to their chemical shift. Furthermore, most of the signals should be distinguishable with respect to dipolar couplings. For instance, the equivalent residues from N- and C-terminal domains (blue circles) sense somewhat different chemical environment: one is located next to the fused terminus, while the other is close to the free terminus. Hence one can expect to observe two distinct HSQC peaks from these sites. Moreover, the orientation of the 15N-1HN vectors at the two sites (blue arrows) relative to the long axis of the alignment tensor (dashed line) is also different. Hence the residual dipolar couplings for these two sites should be different as well. In fact, it is anticipated that most of the N- and C-terminal residues can be differentiated on the basis of their dipolar couplings. For instance, another pair of equivalent residues (red circles) illustrates the case where chemical shifts are likely to be degenerate, but RDCs are very different. Fig. 4b represents a `softer' version of the same scenario. When N- and C-termini of each domain are positioned close to each other, the RDCs from the two domains may turn out to be similar. Apparently, this scenario holds true for the α-spc SH3 tandem (see Experimental Results). Of the simulated constructs, see Fig. 3, NBD tandem illustrates the situation where the couplings from the two domains are reasonably close.
Fig. 4.
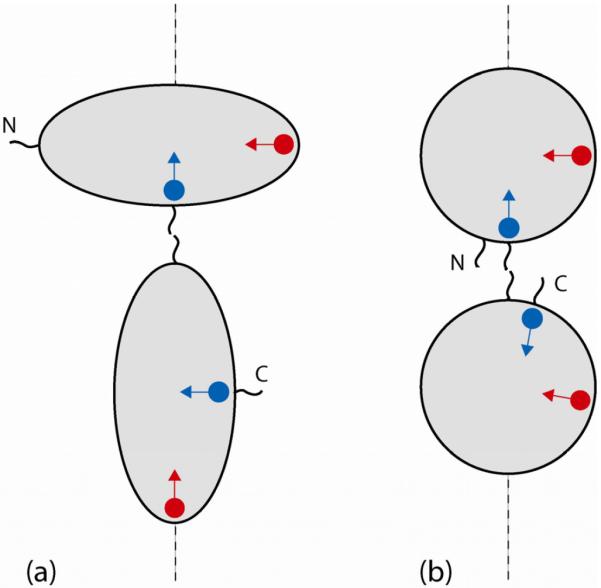
Schematic structure of tandem domains with resident spins. Blue circles represent 15N spins belonging to i-th residue in the N- and C-terminal domains (assuming that residue numbering is the same in the two domains); blue arrows represent the corresponding 15N-1HN vectors. Red circles/arrows refer to the j-th residue. The dashed line corresponds to the long axis of the steric alignment tensor.
Results
RDC data
The spectra of the dSH3 tandems turn out to be very similar to the spectrum of the isolated dSH3. For example, Fig. 5 shows the spectrum of the two-domain construct with long disordered linker superimposed on the spectrum of the single domain (spectral peaks colored red and blue, respectively). The resonances from multiple glycine residues of the random-coil GG(GGSGG)5GG sequence are all grouped in the area of the spectrum where they do not interfere with intra-domain peaks (outlined by the green contour). Most of the peaks from N- and C-terminal domains overlap. Furthermore, they overlap or fall very close to the peaks from the isolated dSH3 domain (cf. red and blue contours in Fig. 5), so that the assignments38 can be transferred in a straightforward fashion. The transfer of resonance assignment from a single domain to a multidomain assembly is presently a common practice.92–96 The lack of resonance shifts between the isolated domain and the tandem confirms the absence of specific domain-domain interactions (along the same lines, 15N relaxation study has not found any evidence of protein aggregation in α-spc SH397).
Fig. 5.

HSQC spectra of the isolated dSH3 domain (blue) and dSH3-ll-dSH3 tandem (red). The dSH3 resonances are labeled except for Trp, Asn, and Gln side-chain correlations. The elliptical contours outline the resonances from the glycine-rich linker and from the residue V9, which demonstrates peak doubling effect.
As discussed above, a small number of residues that are located close to the linker attachment point give rise to a pair of resolved peaks, corresponding to the sites in the N- and C-terminal domains (one example of this behavior is residue V9, outlined by the grey contour in the spectral map Fig. 5). The effect is most pronounced in the short-linker construct, dSH3-sl-dSH3, where the two domains are spatially close. In this sample, 18 residues show the peak doubling effect, with half of them fully resolved. In particular, the peaks from terminal residues 7, 8 and 61, 62 are strongly affected. Inspection of the crystallographic structure shows that these two segments are connected: there is a backbone-backbone hydrogen bond between residues 8 and 61. Thus all of the four residues prove to be in the immediate vicinity of the linker region, resembling the arrangement shown in Fig. 4b. The split peaks associated with residues 7, 8, and 61, 62 show large chemical-shift separations, which makes it impossible to assign most of them. Furthermore, it is likely that the conformation of these residues in dSH3-sl-dSH3 differs from that seen in the crystallographic structure of fSH3. Considering these complications we choose to restrict the data analysis to the core region of the domains, residues 9–60. This restriction has been applied to all α-spc SH3 constructs investigated experimentally and via simulations.
A separate question arises as to whether the resonances from N- and C-terminal domains can be distinguished on the basis of residual dipolar couplings. In principle, one may expect that a pair of peaks that is completely overlapped in HSQC spectrum would become resolved in IPAP-HSQC (due to potentially large differences in RDCs, on the order of tens of Hz; see Fig. 4a and surrounding discussion). In reality, however, we have not found any examples of such behavior, despite high resolution of the IPAP-HSQC spectra. Indeed, it appears that RDCs in the two domains are similar. In a handful of cases where HSQC peaks are resolved, the determined RDCs turn out to be approximately equal. For example, in the case of V9, Fig. 5, the difference between the two measured RDCs does not exceed 0.5 Hz. In the experiments involving dSH3-sl-dSH3, a few RDCs that have been measured separately for the sites in the N- and C-domains differ by no more than several Hz. At this level, RDCs alone cannot produce resolved spectral peaks. In summary, it appears that tandem dSH3 constructs behave as indicated in the scheme Fig. 4b: the N- and C-termini of each domain are aligned and together form a linker attachment point; the RDCs in the two domains are similar.
Given that only few residues allow for separate measurements of RDCs associated with N- and C-terminal domains and the measured values prove to be very similar, we choose to average such couplings and add the results to the bigger dataset consisting of the average RDCs. For instance, in the case of dSH3-ll-dSH3 we have measured 44 couplings that inherently represent the average values, plus two distinct couplings from the residue V9. After these two couplings are averaged, the final data set consists of 45 entries (pertaining to the region 9–60 in the dSH3 domain). The same approach has been applied to the other samples (Tabs. S2, S3).
The RDC data obtained as described above have been fitted to the crystallographic coordinates 1U06, chain A (see Materials and Methods). Although the experimental RDCs from the tandem constructs represent the averages between the N- and C-terminal domains, they have been fitted in the standard fashion, same as the data from the isolated dSH3 and fSH3 domains. As already commented, this approach is fully legitimate, so long as the internal structure of the two domains is identical. Indeed, residual dipolar couplings represent, by definition, the average between many molecular orientations. The quality of the RDC fits for dSH3 and dSH3-ll-dSH3 is illustrated in Figs. 6a and 6b, respectively. In both cases the agreement between the experimental and fitted couplings is good, as evidenced by the Q factors of 0.25 and 0.26 (typical of a small globular protein represented by a crystallographic structure with 1.5 Å resolution).70
Fig. 6.

Fits of the experimental RDCs from (a) the isolated dSH3 domain and (b) dSH3-ll-dSH3 tandem with the structural model based on the coordinate set 1U06 (protonated, energy minimized). The fitting is limited to the core portion of the protein, residues 9–60, and does not include the data from N47 and D48 (grey circles in the plot). The total number of the fitted RDCs and the figure of merit Q are indicated in the graphs.
Two residues consistently show poor agreement in all fits – N47 and D48 (grey circles in Fig. 6). This discrepancy should be attributed to the local conformational variability at this particular site. In the crystal, N47 assumes an unusual conformation (ϕ ≈ 50°, ψ ≈ −110°)98 which has a significant destabilizing effect on the structure.99 A similarly unfavorable conformation is seen in solution structure (φ ≈ 60°, ψ ≈ −75°), except for one conformer.38 The built-in strain apparently triggers local motions at this site. Both N47 and D48 display sharply elevated B-factors and several of the side-chain atoms in D48 cannot be resolved by x-ray crystallography at all. D48 is also the only residue featuring exchange broadening in solution (Rex = 28 s−1 at 600 MHz).100 As it turns out, the RDC data from N47 and D48 can be accommodated using the existing structural models. Specifically, if these two residues are `grafted' from the NMR structure 1AEY onto the crystallographic structure 1U06, some of the resulting hybrid models lead to a satisfactory RDC fit.
Alignment
The alignment parameters derived from multiple SH3 samples are summarized in Tabs. 1, 2 and in Fig. 7. As already discussed, special care was taken to ensure that the alignment conditions are the same throughout each series of samples. In the case of r = 0.85 samples, the residual quadrupolar splittings Δ(2H) proved to be fairly uniform and reproducible (with the apparent exception of dSH3-ml-dSH3, see Tab. 1). For r = 0.96, the splittings were lower than expected and varied significantly from one sample to the other (see Tab. 2). This latter media appears to be only marginally stable at 25 °C.41 Generally, the dilute PEG/hexanol media used in NMR experiments falls close to the boundary between lamellar phase Lα and a `sponge phase' L3, which itself has a complex and variable morphology.101 In this situation, minor variations in the composition of the sample – possibly, the differences between the protein constructs themselves – may cause significant changes in the alignment. To account for these changes, we choose to normalize the GDO parameter according to the magnitude of Δ(2H), see Tab. 1. In doing so we assume that Δ(2H) reflects the aligning properties of the media not only with respect to water, but also to different α-spectrin SH3 constructs. This is an admittedly ad hoc approach, with only limited experimental justification.41 Note, however, that this issue is largely inconsequential in the case of r = 0.85 data.
Tab. 2.
Alignment parameters from a series of samples in r = 0.96 PEG/hexanol media. The conventions are the same as in Tab. 1
| dSH3 | fSH3 | dSH3-sl-dSH3 | dSH3-ml-dSH3 | dSH3-ll-dSH3 | |
|---|---|---|---|---|---|
|
| |||||
| Aa (10−3) | 2.23 | −0.78 | −1.50 | −0.78 | 0.41 |
|
| |||||
| R | 0.40 | 0.33 | 0.36 | 0.64 | 0.41 |
|
| |||||
| {α, β, γ} | 306° | 32° | 33° | 36° | 103° |
| 65° | 39° | 80° | 83° | 54° | |
| 79° | 321° | 63° | 64° | 154° | |
|
| |||||
| N | 43 | 48 | 41 | 40 | 45 |
|
| |||||
| Q | 0.25 | 0.21 | 0.22 | 0.26 | 0.25 |
|
| |||||
| Δ(2H) (Hz) | 17.1 | 12.3 | 20.3 | 15.0 | 11.0 |
|
| |||||
| GDO (10−3) | 2.76 | 1.32 | 1.54 | 1.18 | 0.80 |
|
| |||||
| NSP | 1.0 | 0.90 | −0.64 | −0.47 | 0.21 |
Fig. 7.

GDO and NSP alignment parameters in a series of constructs of α-spc SH3 domain. (a,b) Experimental data from dSH3-sl-dSH3, dSH3-ml-dSH3, and dSH3-lldSH3 tandem constructs (squares for the main series of samples, triangles for replicate samples), as well as dSH3 and fSH3 single-domain constructs (dashed and dotted horizontal lines, respectively). The data from r = 0.85 and 0.96 samples are indicated in green and red, respectively. Spline curves are added to guide the eye. The NSP parameter is calculated relative to the dSH3 construct; the corresponding value for the dSH3 itself is 1.0. (c,d) The corresponding PALES-based simulations using the steric alignment model which assumes planar bicelle bilayers with the thickness 28.6 Å.
The consistency of different sample preparations can be verified through measurements on replicate samples. The results from the duplicate dSH3-sl-dSH3 and dSH3-ll-dSH3 samples are in very good agreement with each other (see Tab. 1; also compare squares and rectangles in Fig. 7). Furthermore, the data taken at r = 0.85 and 0.96 are also in good agreement insofar as rhombicity and the orientation of the alignment frame are concerned. One apparent exception is dSH3-ml-dSH3, cf. Tabs. 1 and 2. This construct displays the rhombicity R approaching the maximum value of 2/3. Under these circumstances, the labeling of the principal axes of the alignment tensor becomes ambiguous, causing apparent differences in the Euler angles. In fact, the two data sets are highly consistent, as confirmed by the NSP of the respective alignment tensors, 0.998.
The only sample where reproducibility truly appears to be an issue is dSH3. While the orientation of the alignment frame is well reproduced, the rhombicity shows a significant amount of variation, Tabs. 1 and 2. Furthermore, the GDO value varies significantly from one preparation to the other. One thing is certain, however: the alignment of dSH3 is unexpectedly strong in all anisotropic samples, cf. Fig. 6 (also confirmed by the results from an additional dSH3 sample, not shown). In what follows, this observation is discussed in greater detail.
Fig. 7a provides the experimental verification of the concept presented in this paper, namely, that the tandem with a short linker should align strongly, whereas the tandem with a long linker should align more weakly (given that the shape of the domains is close to spherical, the linker is unstructured, and there is no significant domain-domain interactions). Furthermore, it appears that the simple steric alignment model, as implemented in PALES, in conjunction with the conformational ensembles generated with the help of TraDES, provide an adequate explanation for the experimental observations (compare squares/triangles in Fig. 7a and circles in Fig. 7c). Note that the PALES-based prediction algorithm does not involve any adjustable parameters (see Materials & Methods). The NSP data in Fig. 7b confirm our hypothesis that with increase in linker length the tandem domains begin to behave as independent entities, i.e. align similarly to isolated domains.
From inspection of Fig. 7 it becomes obvious, however, that the steric alignment model is only partially successful in reproducing the experimental data. While the GDO values for tandem constructs are predicted with reasonable accuracy, the predictions for isolated domains, dSH3 and fSH3, are off the mark (cf. dashed / dotted lines in Figs. 7a and 7c). The case of the dSH3 is particularly instructive. This domain represents a spheroid with modest anisotropy, I∥ / I⊥ = 0.83. Its structure is well defined: only one N-terminal and one C-terminal residue are classified as `unstructured'. Assuming the mechanism of alignment is steric, dSH3 should show only a modest degree of alignment. Yet the experimental GDO value is exceedingly high (see also Fig. 6a). Moreover, the experimentally determined alignment frame bears no similarity to the predicted one, as indicated by the low NSP value, −0.11. This leads us to suggest that the alignment mechanism in the PEG/hexanol media is, in fact, different from the simple steric model postulated in PALES.
To further investigate this possibility, we turn to the literature reports of RDC measurements in the PEG/hexanol media. For ubiquitin, the orientation of the alignment frame is predicted by PALES with reasonable accuracy, NSP = 0.78 – 0.86. However, the GDO values are overestimated by a factor 2.1 – 4.5.104, 105 Our own measurements on ubiquitin in PEG/hexanol media at pH 3.5 led to similar results, with GDO overestimated by 2.1 – 2.3 (data not shown). The agreement becomes even poorer if the results are scaled according to Δ(2H). The data from another small protein, GB3 domain, produce NSP = 0.94 and another poor GDO prediction, off by a factor 3.6.42 Taken together with our current data for α-spectrin SH3, these results suggest that PALES can both under- and overestimate the degree of alignment by a wide margin.
This shortcoming seems to be specific to the PEG/hexanol media. In the case of DMPC/DHPC bicelles, the steric PALES model fares much better. For ubiquitin, NSP = 0.96 – 0.97 and the predicted GDO value is only 1.3 times higher than the experimental value. For GB3 domain, NSP = 0.90 and the predicted GDO is essentially exact. Clearly, it would have been desirable to repeat our experiments in the DMPC/DHPC bicelles. Unfortunately, fSH3 and especially dSH3 constructs tend to precipitate at pH 6.5–7.0 required by this alignment media.106 This situation is not atypical, which is one the reasons for the increasing popularity of the PEG/hexanol media.
Alignment mechanisms
Unexpectedly, the results Fig. 7a indicate that nearly-spherical dSH3 and fSH3 domains align stronger than the markedly anisotropic dSH3-sl-dSH3 construct. This result appears paradoxical only in the light of the simple steric model involving planar bicelles. Alternatively, if the morphology of the media is complex, then a steric mechanism can, in principle, lead to an arbitrary alignment tensor. Indeed, assume for a moment that anisotropic media contains cavities that can (transiently) accommodate certain proteins. Assume further that the cavities are shaped such that they form `lock and key' pair with a given protein. Clearly, the resulting alignment tensor will be different from the one that is predicted by PALES.
In fact, recent studies showed that the topology of the most used liquid-crystal media is far more complex than previously believed. The DMPC/DHPC bicelles, which were originally described as large disks, have been instead visualized as “two-dimensional networks of flattened, highly branched, cylindrical micelles and lamellar sheets perforated by large irregular holes.”107 Similar observations have been made with regard to the PEG-hexanol media. Early on it has been noted that the Lα bilayers display collective wave-like modulation (undulation).108, 109 Gaemers and Bax pointed out that dilute conditions used in NMR experiments bring the system close to the transitions region where the lamellar phase Lα coexists with a `sponge phase' L3.101 Their diffusion data further suggest that the motion of probe molecules between the oriented bilayers (i.e. along the z-axis in the NMR tube) is significantly obstructed. These obstructions may arise from the `bridges' between the adjacent bilayers, the presence of small globular structures (e.g. micelles) in the interbilayer space, etc. None of this potentially complex topology is taken into consideration in the PALES model – which may explain its apparent lack of accuracy, Fig. 7.
From a more general perspective, the failure of the PALES can be attributed to site-specific interactions between the proteins and the media. Generally, PEG-based media are protein-friendly in a sense that there is little protein adsorption. Nevertheless, specific interactions involving PEG-hexanol have been observed for short peptides,110 N-terminal domain of the enzyme I from bacterial phosphotransferase system111 and two cytoplasmic domains of chitobiose transporter.112 There is no doubt that many more cases remain undocumented (dismissed as unsuccessful attempts to prepare aligned samples).
PEG doped with hexanol forms a bilayer where the surface is lined with hydroxyl headgroups. The interactions of the proteins with such bilayers have been modeled in several influential theoretical studies.113, 114 All-atom force field simulations have been also reported.115–117 These simulations clearly identify the preferred orientation of the protein (lysozyme) on a surface of a PEG bilayer. Of note, the binding interface appears to be formed by charged / polar residues interacting with PEG oxygens at or near the surface of the bilayer.116 Of course, the binding is only weakly specific since many sites on the protein surface can form such favorable contacts. Note that electrostatic effects play a prominent role in molecular alignment even when the media is not charged – it is sufficient that the media is polar. These aspects has been extensively discussed in the small-molecule studies.118–122
To elucidate a possible role of electrostatics in PEG/hexanol media, we have prepared an additional sample of fSH3 containing 100 mM NaCl. If the alignment is purely steric, as is commonly held, then one would not expect to observe any significant changes upon addition of NaCl. In reality, however, the GDO parameter jumps from 1.19·10−3 to 1.85·10−3, see Tab. 1. This result is unexpected and, at a first glance, counterintuitive. Indeed, if electrostatic interactions are responsible, then addition of salt should screen out these interactions and thus lower the degree of alignment. In fact, a number of plausible explanations can be suggested for this finding. The increase in GDO may result from the interplay between the electrostatic and steric mechanisms. Furthermore, it may also occur in the context of purely electrostatic alignment. As described by Wu and co-workers,87 excessive electrostatic repulsion drives the solute away from the charged media and into the bulk, where it cannot effectively align. Adding NaCl to the solution reverses this process, thus increasing the degree of alignment. Details of alignment notwithstanding, the observed dependence of the GDO on solution ionic strength suggests that an electrostatic mechanism plays a role.
To further probe the relevance of electrostatic interactions, we have undertaken an additional series of PALES simulations. In doing so we did not aim to quantitatively model the alignment of α-spc SH3 in PEG/hexanol media. Instead, the intention was to explore the broad trends associated with electrostatic alignment. Unlike the steric model, which has no adjustable parameters, the electrostatic model contains several such quantities. As described in the Materials & Methods section, the ionic strength of the solution was set to the actual experimental value, 17 mM. The other two parameters, pH and the surface charge density of the medium σ, were optimized in an ad hoc fashion to obtain the best possible agreement with the five experimental GDO values (listed in the legend of Tab. 1). The optimization was conducted by means of the grid-search in the space of two parameters. Calculations at each point on the grid involved many thousand PALES jobs (addressing five conformational ensembles, each comprised of several thousand conformers).
The optimized electrostatic model produced the predictions for GDO and NSP as shown in Fig. 8. The main result of this simulation is that it is indeed possible to recreate the situation where dSH3 and fSH3 display a higher degree of alignment than tandem constructs, as observed experimentally (see Fig. 7a). Aside from that, the agreement between Figs. 7a,b and Fig. 8 is unimpressive and does not support any far-reaching conclusions.
Fig. 8.

The GDO and NSP alignment parameters for α-spc SH3 constructs as obtained from PALES-based electrostatic simulations. The model assumes planar bicelle bilayers with the thickness 28.6 Å, surface charge density σ = 0.05 e/nm2, and effective pH 4.8.
The optimized model parameters are σ = 0.05 e/nm2, effective pH 4.8. The latter value is significantly higher than the actual experimental pH, 3.5. It should be noted, however, that our modeling of electrostatic interactions is associated with significant uncertainty. Specifically, PROPKA program predicts pKa values with the typical error of 0.5 – 1 unit and maximum error of up to 2 units.79 Furthermore, the predictions of electrostatic alignment are highly sensitive to fine details of the charge distribution. For instance, the predicted alignment parameters may change in response to a conformational jump of a single charged side chain. To demonstrate this effect, we regenerated the SH3 conformational ensembles using the BAAAA structure of the SH3 domain, i.e. a structure where the single side chain, E17, is placed in an alternative conformation (see Materials & Methods). This minor structural modification led to significant decrease in the predicted GDO of the fSH3 sample, from 4.12·10−3 to 3.80·10−3.
The obtained effective charge density, σ = 0.05 e/nm2, can be questioned as well. On one hand, this value is relatively low in comparison, for example, with Pf1 phage, −0.47 e/nm2. In principle, surface charge may arise from adsorption of ions on the interface between water and non-ionic surfactants.122–126 On the other hand, it appears that the charge density on the surface of a C12E5 bilayer in water is much lower than 0.05 e/nm2 and negative,124, 126 even in the presence of a small amount of ionic surfactant.127, 128 Finally, it is worth noting that the effect of NaCl on PEG/hexanol alignment is system-specific. For instance, we have not observed any significant impact of salt on alignment of ubiquitin. We conclude that the present PALES-based electrostatic model demonstrates the feasibility of the scenario in which dSH3 and fSH3 align more efficiently than the tandem constructs. Beyond that, we do not attach any particular significance to the results shown in Fig. 8.
One can envisage that more sophisticated prediction tools will be developed in future, where a fully atomistic approach is extended to the liquid crystal media. It can also be expected that MD-based methods will eventually emerge. The example of such developments have been cited above.116, 117, 121, 122, 129–131 The progress in this area, however, is limited by our relatively poor knowledge of the liquid crystal structure and dynamics.
Conclusion
In this study we focus on proteins comprised of two modular domains connected through a random-coil linker (α-spc SH3 domain connected through glycine-serine linker). When the linker is short, the tandem SH3 construct represents a dumbbell-shaped molecule with relatively little domain-domain mobility. Being placed in steric alignment media, such as PEG/hexanol, this molecule is expected to display a high degree of alignment. As the length of the linker is increased, the domains become effectively uncoupled and start behaving as independent entities. Consequently, the degree of alignment observed in each of the tandem domains approaches that of the isolated SH3 domain.
In this study we attempted to quantify the dependence of the alignment parameters (amplitude and orientation) on the length of the linker. The form of the corresponding transition curves depends on the details of the system, e.g. the shape of the individual domains and the linker attachment points. In the case of electrostatic alignment, the trend can be easily reversed, i.e. the degree of alignment may increase with lengthening of the linker. The transition phenomenon itself, however, is sufficiently general.
To translate the proposed intuitive model into quantitative predictions, we generated a series of conformational ensembles representing α-spc SH3 constructs with different linker lengths (the disordered linkers and terminal sequences were built using the program TraDES). These ensembles were used to predict the parameters of steric alignment (program PALES). To test the validity of the theoretical predictions, we prepared three tandem constructs of α-spc SH3 and two variants of the isolated SH3 domain. These chimeras were used to measure backbone 1HN-15N RDCs in two series of PEG/hexanol samples (r = 0.85 and 0.96). It was found that the alignment of tandem proteins is in line with expectations and agrees reasonably well with the results from PALES-based simulations. The isolated domains, however, displayed much stronger alignment than expected. Furthermore, the degree of alignment turned out to be sensitive to the ionic strength of the solution, which is generally not expected to be the case for a well-behaved steric alignment media.
Several factors may potentially contribute to this situation. First, the topology of the liquid crystal media is known to be more complex than envisaged in the simple PALES model (i.e. planar bilayers). Furthermore, there is a possibility that the phase equilibrium of the liquid crystal media changes in response to addition of protein or salt (these changes may not necessarily be fully reflected in Δ(2H)).77 It is also likely that protein alignment in PEG/hexanol significantly depends on (weak) site-specific interactions. In particular, an electrostatic mechanism appears to play a role, visibly affecting the alignment of isolated domains.
In summary, it seems that PEG/hexanol bears little resemblance to an idealized alignment media envisioned in PALES. One would be well-advised to keep this in mind, especially in the context of studies where PALES is used to simulate RDC data from disordered proteins dissolved in PEG-based media.132–135 Indeed, if we cannot reliably predict the alignment of globular proteins, the predictions should be even less reliable for disordered proteins. In this latter case, all residues can potentially form site-specific contacts with the media and, furthermore, these contacts may significantly skew the original conformational equilibrium (for those protein molecules that are in the vicinity of the media and give rise to observable RDCs).
In conclusion, we have demonstrated that alignment parameters obtained in the orienting media (liquid crystals, gels, etc.) can be used to characterize the degree of domain coupling in modular multidomain proteins. The chimera protein investigated in this work is comprised of two non-interacting domains connected by a variable-length random linker. It can thus be described as an example of the system with minimal domain coupling. This example provides an important point of reference for future studies of the proteins with substantial domain coupling, either through domain-domain interactions (e.g. Crk adaptor protein,2 Syk kinase25) or through a structured linker (e.g. calmodulin,136 troponin137). In the case of popular PEG/hexanol media, the usefulness of this approach is presently limited by lack of precise information about the morphology of the oriented phase and the details of alignment mechanism. We expect that the DMPC/DHPC media offers a better chance of success, although it is far more restrictive in terms of sample conditions. Further progress in this area depends on development of more accurate alignment prediction tools. Ultimately, such tools should be based on all-atom MD simulations focusing on the interactions between the protein and the alignment media.
Supplementary Material
Acknowledgements
This work has been funded through NSF grant MCB-044563. We thank Ryan Muir for his help with early versions of the structure-generating scripts. We are also grateful to Etti Harms, Nina Gorenstein, and Yi Xue for their advice on different aspects of experimental work.
References
- (1).Cesareni G, Gimona M, Sudol M, Yaffe M, editors. Modular Protein Domains. Wiley-VCH: Weinheim; 2005. [Google Scholar]
- (2).Kobashigawa Y, Sakai M, Naito M, Yokochi M, Kumeta H, Makino Y, Ogura K, Tanaka S, Inagaki F. Nat. Struct. Mol. Biol. 2007;14:503. doi: 10.1038/nsmb1241. [DOI] [PubMed] [Google Scholar]
- (3).Defilippi P, Di Stefano P, Cabodi S. Trends Cell Biol. 2006;16:257. doi: 10.1016/j.tcb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- (4).Sharma A, Mayer BJ. BMC Cell Biol. 2008;9 doi: 10.1186/1471-2121-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Feller SM. Oncogene. 2001;20:6348. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- (6).Birge RB, Kalodimos C, Inagaki F, Tanaka S. Cell Commun. Signal. 2009;7:7. doi: 10.1186/1478-811X-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. Nature. 1998;394:337. doi: 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- (8).van den Berghe N, Cool RH, Horn G, Wittinghofer A. Oncogene. 1997;15:845. doi: 10.1038/sj.onc.1201407. [DOI] [PubMed] [Google Scholar]
- (9).Brugnera E, Haney L, Grimsley C, Lu MJ, Walk SF, Tosello-Trampont AC, Macara IG, Madhani H, Fink GR, Ravichandran KS. Nat. Cell Biol. 2002;4:574. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- (10).Girardin SE, Yaniv M. EMBO J. 2001;20:3437. doi: 10.1093/emboj/20.13.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Antoku S, Saksela K, Rivera GM, Mayer BJ. J. Cell. Sci. 2008;121:3071. doi: 10.1242/jcs.031575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shishido T, Akagi T, Chalmers A, Maeda M, Terada T, Georgescu MM, Hanafusa H. Genes Cells. 2001;6:431. doi: 10.1046/j.1365-2443.2001.00431.x. [DOI] [PubMed] [Google Scholar]
- (13).Linghu H, Tsuda M, Makino Y, Sakai M, Watanabe T, Ichihara S, Sawa H, Nagashima K, Mochizuki N, Tanaka S. Oncogene. 2006;25:3547. doi: 10.1038/sj.onc.1209398. [DOI] [PubMed] [Google Scholar]
- (14).Nishihara H, Tanaka S, Tsuda M, Oikawa S, Maeda M, Shimizu M, Shinomiya H, Tanigami A, Sawa H, Nagashima K. Cancer Lett. 2002;180:55. doi: 10.1016/s0304-3835(01)00763-7. [DOI] [PubMed] [Google Scholar]
- (15).Miller CT, Chen G, Gharib TG, Wang H, Thomas DG, Misek DE, Giordano TJ, Yee J, Orringer MB, Hanash SM, Beer DG. Oncogene. 2003;22:7950. doi: 10.1038/sj.onc.1206529. [DOI] [PubMed] [Google Scholar]
- (16).Takino T, Nakada M, Miyamori H, Yamashita J, Yamada KM, Sato H. Cancer Res. 2003;63:2335. [PubMed] [Google Scholar]
- (17).Watanabe T, Tsuda M, Makino Y, Ichihara S, Sawa H, Minami A, Mochizuki N, Nagashima K, Tanaka S. Mol. Cancer Res. 2006;4:499. doi: 10.1158/1541-7786.MCR-05-0141. [DOI] [PubMed] [Google Scholar]
- (18).Ogawa S, Toyoshima H, Kozutsumi H, Hagiwara K, Sakai R, Tanaka T, Hirano N, Mano H, Yazaki Y, Hirai H. Oncogene. 1994;9:1669. [PubMed] [Google Scholar]
- (19).Akakura S, Kar B, Singh S, Cho L, Tibrewal N, Sanokawa-Akakura R, Reichman C, Ravichandran KS, Birge RB. J. Cell. Physiol. 2005;204:344. doi: 10.1002/jcp.20288. [DOI] [PubMed] [Google Scholar]
- (20).Muralidharan V, Dutta K, Cho J, Vila-Perello M, Raleigh DP, Cowburn D, Muir TW. Biochemistry. 2006;45:8874. doi: 10.1021/bi060590z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sarkar P, Reichman C, Saleh T, Birge RB, Kalodimos CG. Mol. Cell. 2007;25:413. doi: 10.1016/j.molcel.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Donaldson LW, Gish G, Pawson T, Kay LE, Forman-Kay JD. Proc. Natl. Acad. Sci. USA. 2002;99:14053. doi: 10.1073/pnas.212518799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cipres A, Abassi YA, Vuori K. Cell. Signal. 2007;19:1662. doi: 10.1016/j.cellsig.2007.02.011. [DOI] [PubMed] [Google Scholar]
- (24).Peterson ME, Long EO. Immunity. 2008;29:578. doi: 10.1016/j.immuni.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang YJ, Oh H, Burton RA, Burgner JW, Geahlen RL, Post CB. Proc. Natl. Acad. Sci. USA. 2008;105:11760. doi: 10.1073/pnas.0708583105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bae SH, Dyson HJ, Wright PE. J. Am. Chem. Soc. 2009;131:6814. doi: 10.1021/ja809687r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Walsh JD, Meier K, Ishima R, Gronenborn AM. Biophys. J. 2010;99:2636. doi: 10.1016/j.bpj.2010.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chen K, Tjandra N. J. Am. Chem. Soc. 2008;130:12745. doi: 10.1021/ja803557t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wong V, Case DA, Szabo A. Proc. Natl. Acad. Sci. USA. 2009;106:11016. doi: 10.1073/pnas.0809994106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).de la Torre JG, Huertas ML, Carrasco B. Biophys. J. 2000;78:719. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bertini I, Del Bianco C, Gelis I, Katsaros N, Luchinat C, Parigi G, Peana M, Provenzani A, Zoroddu MA. Proc. Natl. Acad. Sci. USA. 2004;101:6841. doi: 10.1073/pnas.0308641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bertini I, Gupta YK, Luchinat C, Parigi G, Peana M, Sgheri L, Yuan J. J. Am. Chem. Soc. 2007;129:12786. doi: 10.1021/ja0726613. [DOI] [PubMed] [Google Scholar]
- (33).Ikegami T, Verdier L, Sakhaii P, Grimme S, Pescatore B, Saxena K, Fiebig KM, Griesinger C. J. Biomol. NMR. 2004;29:339. doi: 10.1023/B:JNMR.0000032611.72827.de. [DOI] [PubMed] [Google Scholar]
- (34).Rodriguez-Castaneda F, Haberz P, Leonov A, Griesinger C. Magn. Reson. Chem. 2006;44:S10. doi: 10.1002/mrc.1811. [DOI] [PubMed] [Google Scholar]
- (35).Keizers PHJ, Desreux JF, Overhand M, Ubbink M. J. Am. Chem. Soc. 2007;129:9292. doi: 10.1021/ja0725201. [DOI] [PubMed] [Google Scholar]
- (36).Musacchio A, Noble M, Pauptit R, Wierenga R, Saraste M. Nature. 1992;359:851. doi: 10.1038/359851a0. [DOI] [PubMed] [Google Scholar]
- (37).Huston JS, Levinson D, Mudgetthunter M, Tai MS, Novotny J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, Oppermann H. Proc. Natl. Acad. Sci. USA. 1988;85:5879. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Blanco FJ, Ortiz AR, Serrano L. J. Biomol. NMR. 1997;9:347. doi: 10.1023/a:1018330122908. [DOI] [PubMed] [Google Scholar]
- (39).Chevelkov V, Faelber K, Diehl A, Heinemann U, Oschkinat H, Reif B. J. Biomol. NMR. 2005;31:295. doi: 10.1007/s10858-005-1718-z. [DOI] [PubMed] [Google Scholar]
- (40).Lazar GA, Desjarlais JR, Handel TM. Protein Sci. 1997;6:1167. doi: 10.1002/pro.5560060605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rückert M, Otting G. J. Am. Chem. Soc. 2000;122:7793. [Google Scholar]
- (42).Ulmer TS, Ramirez BE, Delaglio F, Bax A. J. Am. Chem. Soc. 2003;125:9179. doi: 10.1021/ja0350684. [DOI] [PubMed] [Google Scholar]
- (43).Bewley CA. Structure. 2001;9:931. doi: 10.1016/s0969-2126(01)00653-0. [DOI] [PubMed] [Google Scholar]
- (44).Braddock DT, Baber JL, Levens D, Clore GM. EMBO J. 2002;21:3476. doi: 10.1093/emboj/cdf352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Yang DW, Nagayama K. J. Magn. Reson. Ser. A. 1996;118:117. [Google Scholar]
- (46).Ottiger M, Delaglio F, Bax A. J. Magn. Reson. 1998;131:373. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- (47).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J. Biomol. NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- (48).Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J. Comput. Chem. 2009;30:1545. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. J. Magn. Reson. 2003;160:65. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- (50).Dominy BN, Brooks CL. J. Phys. Chem. B. 1999;103:3765. [Google Scholar]
- (51).Feldman HJ, Hogue CWV. Proteins: Struct. Funct. Genet. 2000;39:112. [PubMed] [Google Scholar]
- (52).Schwieters CD, Clore GM. J. Magn. Reson. 2001;152:288. doi: 10.1006/jmre.2001.2413. [DOI] [PubMed] [Google Scholar]
- (53).Bryngelson JD, Wolynes PG. Proc. Natl. Acad. Sci. USA. 1987;84:7524. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Shakhnovich EI, Gutin AM. Biophys. Chem. 1989;34:187. doi: 10.1016/0301-4622(89)80058-4. [DOI] [PubMed] [Google Scholar]
- (55).Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Proteins: Struct. Funct. Genet. 1995;21:167. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- (56).Tjandra N, Bax A. Science. 1997;278:1111. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- (57).Moltke S, Grzesiek S. J. Biomol. NMR. 1999;15:77. doi: 10.1023/A:1008309630377. [DOI] [PubMed] [Google Scholar]
- (58).Sass J, Cordier F, Hoffmann A, Rogowski M, Cousin A, Omichinski JG, Lowen H, Grzesiek S. J. Am. Chem. Soc. 1999;121:2047. [Google Scholar]
- (59).Prestegard JH, Al-Hashimi HM, Tolman JR. Quart. Rev. Biophys. 2000;33:371. doi: 10.1017/s0033583500003656. [DOI] [PubMed] [Google Scholar]
- (60).Tolman JR, Al-Hashimi HM, Kay LE, Prestegard JH. J. Am. Chem. Soc. 2001;123:1416. doi: 10.1021/ja002500y. [DOI] [PubMed] [Google Scholar]
- (61).Sass HJ, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. J. Biomol. NMR. 2000;18:303. doi: 10.1023/a:1026703605147. [DOI] [PubMed] [Google Scholar]
- (62).Mohana-Borges R, Goto NK, Kroon GJA, Dyson HJ, Wright PE. J. Mol. Biol. 2004;340:1131. doi: 10.1016/j.jmb.2004.05.022. [DOI] [PubMed] [Google Scholar]
- (63).Opella SJ, De Angelis AA. Nat. Protoc. 2007;2:2332. doi: 10.1038/nprot.2007.329. [DOI] [PubMed] [Google Scholar]
- (64).Koenig BW, Hu JS, Ottiger M, Bose S, Hendler RW, Bax A. J. Am. Chem. Soc. 1999;121:1385. [Google Scholar]
- (65).Denisov AY, Kloser E, Gray DG, Mittermaier AK. J. Biomol. NMR. 2010;47:195. doi: 10.1007/s10858-010-9423-y. [DOI] [PubMed] [Google Scholar]
- (66).Bax A, Kontaxis G, Tjandra N. Method Enzymol. 2001;339:127. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- (67).Zweckstetter M, Bax A. J. Am. Chem. Soc. 2000;122:3791. [Google Scholar]
- (68).Zweckstetter M. Nat. Protoc. 2008;3:679. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- (69).Cornilescu G, Marquardt JL, Ottiger M, Bax A. J. Am. Chem. Soc. 1998;120:6836. [Google Scholar]
- (70).Bax A. Protein Sci. 2003;12:1. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Zhang YB, Zuiderweg ERP. Proc. Natl. Acad. Sci. USA. 2004;101:10272. doi: 10.1073/pnas.0401313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Fischer D, Geyer A. Magn. Reson. Chem. 2005;43:893. doi: 10.1002/mrc.1653. [DOI] [PubMed] [Google Scholar]
- (73).Maltsev AS, Ahmed AH, Fenwick MK, Jane DE, Oswald RE. Biochemistry. 2008;47:10600. doi: 10.1021/bi800843c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Berlin K, O'Leary DP, Fushman D. J. Magn. Reson. 2009;201:25. doi: 10.1016/j.jmr.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Bax A, Tjandra N. J. Biomol. NMR. 1997;10:289. doi: 10.1023/a:1018308717741. [DOI] [PubMed] [Google Scholar]
- (76).Freyssingeas É , Nallet F, Roux D. Langmuir. 1996;12:6028. [Google Scholar]
- (77).Ottiger M, Bax A. J. Biomol. NMR. 1998;12:361. doi: 10.1023/a:1008366116644. [DOI] [PubMed] [Google Scholar]
- (78).Simon K, Xu J, Kim C, Skrynnikov NR. J. Biomol. NMR. 2005;33:83. doi: 10.1007/s10858-005-2601-7. [DOI] [PubMed] [Google Scholar]
- (79).Li H, Robertson AD, Jensen JH. Proteins. 2005;61:704. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- (80).Bas DC, Rogers DM, Jensen JH. Proteins. 2008;73:765. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- (81).Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A. Nucleic Acids Res. 2005;33:W368. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Khandogin J, Brooks CL. Biochemistry. 2006;45:9363. doi: 10.1021/bi060706r. [DOI] [PubMed] [Google Scholar]
- (83).Tollinger M, Forman-Kay JD, Kay LE. J. Am. Chem. Soc. 2002;124:5714. doi: 10.1021/ja020066p. [DOI] [PubMed] [Google Scholar]
- (84).Andre I, Linse S, Mulder FAA. J. Am. Chem. Soc. 2007;129:15805. doi: 10.1021/ja0721824. [DOI] [PubMed] [Google Scholar]
- (85).Zweckstetter M, Hummer G, Bax A. Biophys. J. 2004;86:3444. doi: 10.1529/biophysj.103.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Zweckstetter M. Eur. Biophys. J. 2006;35:170. doi: 10.1007/s00249-005-0018-6. [DOI] [PubMed] [Google Scholar]
- (87).Wu B, Petersen M, Girard F, Tessari M, Wijmenga SS. J. Biomol. NMR. 2006;35:103. doi: 10.1007/s10858-006-9004-2. [DOI] [PubMed] [Google Scholar]
- (88).Moreno-Murciano MP, Monleon D, Marcinkiewcz C, Calvete JJ, Celda B. J. Mol. Biol. 2003;329:135. doi: 10.1016/s0022-2836(03)00371-1. [DOI] [PubMed] [Google Scholar]
- (89).Futterer K, Wong J, Grucza RA, Chan AC, Waksman G. J. Mol. Biol. 1998;281:523. doi: 10.1006/jmbi.1998.1964. [DOI] [PubMed] [Google Scholar]
- (90).Hakansson KO. Acta Crystallogr. D. 2009;65:1181. doi: 10.1107/S090744490903306X. [DOI] [PubMed] [Google Scholar]
- (91).Fitzkee NC, Rose GD. Proc. Natl. Acad. Sci. USA. 2004;101:12497. doi: 10.1073/pnas.0404236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Tzakos AG, Grace CRR, Lukavsky PJ, Riek R. Annu. Rev. Bioph. Biom. 2006;35:319. doi: 10.1146/annurev.biophys.35.040405.102034. [DOI] [PubMed] [Google Scholar]
- (93).Gelis I, Bonvin AMJJ, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos CG. Cell. 2007;131:756. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Sprangers R, Kay LE. Nature. 2007;445:618. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- (95).Heikkinen O, Permi P, Koskela H, Ylanne J, Kilpelainen I. Biomol. NMR Assign. 2009;3:53. doi: 10.1007/s12104-008-9140-6. [DOI] [PubMed] [Google Scholar]
- (96).Brosey CA, Chagot ME, Ehrhardt M, Pretto DI, Weiner BE, Chazin WJ. J. Am. Chem. Soc. 2009;131:6346. doi: 10.1021/ja9013634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Chevelkov V, Zhuravleva AV, Xue Y, Reif B, Skrynnikov NR. J. Am. Chem. Soc. 2007;129:12594. doi: 10.1021/ja073234s. [DOI] [PubMed] [Google Scholar]
- (98).Vega MC, Martínez JC, Serrano L. Protein Sci. 2000;9:2322. doi: 10.1110/ps.9.12.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Martinez JC, Pisabarro MT, Serrano L. Nat. Struct. Biol. 1998;5:721. doi: 10.1038/1418. [DOI] [PubMed] [Google Scholar]
- (100).Chevelkov V, Xue Y, Linser R, Skrynnikov NR, Reif B. J. Am. Chem. Soc. 2010;132:5015. doi: 10.1021/ja100645k. [DOI] [PubMed] [Google Scholar]
- (101).Gaemers S, Bax A. J. Am. Chem. Soc. 2001;123:12343. doi: 10.1021/ja011967l. [DOI] [PubMed] [Google Scholar]
- (102).Skrynnikov NR, Goto NK, Yang DW, Choy WY, Tolman JR, Mueller GA, Kay LE. J. Mol. Biol. 2000;295:1265. doi: 10.1006/jmbi.1999.3430. [DOI] [PubMed] [Google Scholar]
- (103).Valafar H, Prestegard JH. J. Magn. Reson. 2004;167:228. doi: 10.1016/j.jmr.2003.12.012. [DOI] [PubMed] [Google Scholar]
- (104).Lakomek NA, Carlomagno T, Becker S, Griesinger C, Meiler J. J. Biomol. NMR. 2006;34:101. doi: 10.1007/s10858-005-5686-0. [DOI] [PubMed] [Google Scholar]
- (105).Briggman KB, Tolman JR. J. Am. Chem. Soc. 2003;125:10164. doi: 10.1021/ja035904+. [DOI] [PubMed] [Google Scholar]
- (106).van Rossum BJ, Castellani F, Pauli J, Rehbein K, Hollander J, de Groot HJM, Oschkinat H. J. Biomol. NMR. 2003;25:217. doi: 10.1023/a:1022819921584. [DOI] [PubMed] [Google Scholar]
- (107).van Dam L, Karlsson G, Edwards K. Langmuir. 2006;22:3280. doi: 10.1021/la052988m. [DOI] [PubMed] [Google Scholar]
- (108).Strey R, Schomacker R, Roux D, Nallet F, Olsson U. J. Chem. Soc. Faraday Trans. 1990;86:2253. [Google Scholar]
- (109).Jonstromer M, Strey R. J. Phys. Chem. 1992;96:5993. [Google Scholar]
- (110).Ohnishi S, Shortle D. Proteins: Struct. Funct. Genet. 2003;50:546. doi: 10.1002/prot.10327. [DOI] [PubMed] [Google Scholar]
- (111).Suh JY, Cai ML, Clore GM. J. Biol. Chem. 2008;283:18980. doi: 10.1074/jbc.M802211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Jung YS, Cai ML, Clore GM. J. Biol. Chem. 2010;285:4173. doi: 10.1074/jbc.M109.080937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Jeon SI, Lee JH, Andrade JD, De Gennes PG. J Colloid. Interf. Sci. 1991;142:149. [Google Scholar]
- (114).McPherson T, Kidane A, Szleifer I, Park K. Langmuir. 1998;14:176. [Google Scholar]
- (115).Lim K, Herron JN. Molecular simulation of protein-PEG interaction. In: Harris JM, editor. Poly(ethylene glycol): biotechnical and biomedical applications. Plenum Press; New York: 1992. [Google Scholar]
- (116).Zheng J, Li LY, Chen SF, Jiang SY. Langmuir. 2004;20:8931. doi: 10.1021/la036345n. [DOI] [PubMed] [Google Scholar]
- (117).Zheng J, Li LY, Tsao HK, Sheng YJ, Chen SF, Jiang SY. Biophys. J. 2005;89:158. doi: 10.1529/biophysj.105.059428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Emsley JW, Palke WE, Shilstone GN. Liq. Cryst. 1991;9:643. [Google Scholar]
- (119).Terzis AF, Photinos DJ. Mol. Phys. 1994;83:847. [Google Scholar]
- (120).Syvitski RT, Burnell EE. J. Chem. Phys. 2000;113:3452. [Google Scholar]
- (121).Dingemans T, Photinos DJ, Samulski ET, Terzis AF, Wutz C. J. Chem. Phys. 2003;118:7046. [Google Scholar]
- (122).Wardle KE, Carlson E, Henderson D, Rowley RL. J. Chem. Phys. 2004;120:7681. doi: 10.1063/1.1669374. [DOI] [PubMed] [Google Scholar]
- (123).Elworthy PH, Rogers JA, Florence AT. J Colloid. Interf. Sci. 1971;35:23. [Google Scholar]
- (124).Balzer D. Langmuir. 1993;9:3375. [Google Scholar]
- (125).Karraker KA, Radke CJ. Adv. Colloid. Interface Sci. 2002;96:231. doi: 10.1016/s0001-8686(01)00083-5. [DOI] [PubMed] [Google Scholar]
- (126).Stubenrauch C, von Klitzing R. J. Phys.: Condens. Matt. 2003;15:R1197. [Google Scholar]
- (127).Schomacker R, Strey R. J. Phys. Chem. 1994;98:3908. [Google Scholar]
- (128).von Berlepsch H, de Vries R. Eur. Phys. J. E. 2000;1:141. [Google Scholar]
- (129).Zangi R, Engberts JBFN. J. Am. Chem. Soc. 2005;127:2272. doi: 10.1021/ja044426f. [DOI] [PubMed] [Google Scholar]
- (130).Polyansky AA, Volynsky PE, Nolde DE, Arseniev AS, Efremov RG. J. Phys. Chem. B. 2005;109:15052. doi: 10.1021/jp0510185. [DOI] [PubMed] [Google Scholar]
- (131).Aroulanda C, Celebre G, De Luca G, Longeri M. J. Phys. Chem. B. 2006;110:10485. doi: 10.1021/jp061345s. [DOI] [PubMed] [Google Scholar]
- (132).Alexandrescu AT, Kammerer RA. Protein Sci. 2003;12:2132. doi: 10.1110/ps.03164403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Bernado P, Blanchard L, Timmins P, Marion D, Ruigrok RWH, Blackledge M. Proc. Natl. Acad. Sci. USA. 2005;102:17002. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Jensen MR, Houben K, Lescop E, Blanchard L, Ruigrok RWH, Blackledge M. J. Am. Chem. Soc. 2008;130:8055. doi: 10.1021/ja801332d. [DOI] [PubMed] [Google Scholar]
- (135).Pinheiro AS, Marsh JA, Forman-Kay JD, Peti W. J. Am. Chem. Soc. 2011;133:73. doi: 10.1021/ja107810r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Ikura M, Kay LE, Krinks M, Bax A. Biochemistry. 1991;30:5498. doi: 10.1021/bi00236a024. [DOI] [PubMed] [Google Scholar]
- (137).Slupsky CM, Kay CM, Reinach FC, Smillie LB, Sykes BD. Biochemistry. 1995;34:7365. doi: 10.1021/bi00022a009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.