

Abstract
Thioredoxin 1 (Trx1) is a antioxidant protein that regulates protein disulfide bond reduction, transnitrosylation, denitrosylation and other redox post-translational modifications. In order to better understand how Trx1 modulates downstream protective cellular signaling events following cardiac ischemia, we conducted an expression proteomics study of left ventricles (LVs) after thoracic aortic constriction stress treatment of transgenic mice with cardiac-specific over-expression of Trx1, an animal model that has been proven to withstand more stress than its non-transgenic littermates. Although previous redox post-translational modifications proteomics studies found that several cellular protein networks are regulated by Trx1-mediated disulfide reduction and transnitrosylation, we found that Trx1 regulates the expression of a limited number of proteins. Among the proteins found to be upregulated in this study was SET and MYND domain-containing protein 1 (SMYD1), a lysine methyltransferase highly expressed in cardiac and other muscle tissues and an important regulator of cardiac development. The observation of SMYD1 induction by Trx1 following thoracic aortic constriction stress is consistent with the retrograde fetal gene cardiac protection hypothesis. The results presented here suggest for the first time that, in addition to being a master redox regulator of protein disulfide bonds and nitrosation, Trx1 may also modulate lysine methylation, a non-redox post-translational modification, via the regulation of SMYD1 expression. Such crosstalk between redox signaling and a non-redox PTM regulation may provide novel insights into the functions of Trx1 that are independent from its immediate function as a protein reductase.
Keywords: Thioredoxin, Methylation, iTRAQ, Mass spectrometry
1. Introduction
Cardiac hypertrophy is the enlargement of the ventricles in the heart, a tissue where compensatory growth is associated with cardiac dysfunctions [1]. Accumulating evidence suggests that oxidative stress plays an important role in the pathogenesis of cardiac hypertrophy [2, 3]. Although reactive oxygen species (ROS) such as superoxide anions, hydrogen peroxide, and hydroxyl radicals may be essential regulators of cellular signal transduction pathways, including the induction of hypertrophy, excess ROS levels will overwhelm the cellular antioxidant capacity, resulting in cellular damage and heart diseases [4,5]. In order to counteract elevated ROS levels and organize a cellular response to oxidative stress, cells employ a host of antioxidant mechanisms, including superoxide dismutase, catalase, thioredoxins (Trx), and glutathione [2, 6,7] to maintain an internal redox balance.
Trx is a highly conserved and widely expressed redox-regulating protein family [3,8]. In mammals, the Trx family consists of at least three members: Trx1, Trx2, and Sp-Trx [9]. Trx1 is localized in the cytosol and can be translocated into the nucleus upon stimulation. Trx2 is located in mitochondria, and Sp-Trx is located exclusively in spermatozoa [10,11]. Trx1 is a 12 kDa multifunctional protein involved in protein reduction, cell growth, death, cancer, cardiac diseases, and tissue development [12–18]. Within the human Trx1 catalytic center, Cys32 and 35 are crucial to its reductive activities [3,9]. Trx1 has been shown to regulate redox-dependent transcription, translation and protein turnover. Our earlier studies have reported that Trx1 acts as a negative regulator in cardiac hypertrophy and exhibits protective functions in the heart [3,10,19]. However, the mechanisms governing Trx1 cardiac hypertrophy inhibition are still unknown. In addition to a general antioxidant function, Trx1 is also involved in regulation of different redox post-translational modifications (PTMs), such as reduction of specific disulfide bonds, transnitrosylation, and denitrosylation [12,20–22]. Using isotope-coded affinity tags (ICAT), we were able to identify 78 redox-sensitive cysteines that are putative targets of Trx1. This result indicated that Trx1 may be involved in coordinating cellular functions related to cardiac energy production and utilization networks [23]. Upon the disulfide bond formation between Cys32 and Cys35, Trx1 can be nitrosylated at Cys73 and transnitrosylate target proteins. Using a biotin switch method coupled with a global proteomics approach, we identified 47 novel Trx1 transnitrosylated target proteins [21].
To elucidate the cardiac function of Trx1, RNA microarray analysis has been applied to delineate the differential gene expression profiles in hearts from transgenic mice with cardiac-specific over-expression of Trx1 (Tg-Trx1). The up-regulated genes in Tg-Trx1 hearts are involved in both mitochondrial oxidative phosphorylation and the tricarboxylic acid cycle [10]. However, the changes in mRNA levels may not always reflect the protein level changes. In order to identify proteomic changes in the LV of Tg-Trx1 transgenic mice, we used a 4-plex isobaric tag for relative and absolute quantitation, the iTRAQ-based method, which permits multiplex protein quantitation of multiple biological samples in a single experiment. Compared to other proteomic techniques, iTRAQ provides higher throughput and better peptide identification [24–28]. Following thoracic aortic constriction (TAC) stress, we identified expression changes for a limited number of proteins between Tg-Trx1 and the non-transgenic control animals, including SET and MYND domain-containing protein 1 (SMYD1).
The SMYD family of lysine methyltransferases is defined by each having a SET domain that is split into two segments by a MYND domain, followed by a cysteine-rich post-SET domain [29]. The SET domain is responsible for the methylation of lysine residues, whereas the MYND domain facilitates protein–protein interactions that may underlie methylation specificity [30–32]. SMYDs have been shown to play critical roles in the regulation of gene expression and DNA damage repair [33]. SMYD1 was initially identified as a histone methyltransferase that targets lysine 4 of histone 3 [34]. Other studies indicate that SMYD1 is important for both cardiac and skeletal muscle development and that a SMYD1 gene knocked out in mice could hinder the differentiation of cardiomyocytes and cause fetal mortality [29,35]. Based on the fact that SMYD1 is a methyltransferase, repeat analysis of iTRAQ data with a focus on lysine methylation revealed that Trx1 induction of the SMYD1 is positively correlated with the elevation of lysine methylation among selected target proteins, some of which are key players in modulating chromatin structure and gene expression. The induction of SMYD1 by Trx1 and subsequent protein lysine methylation is consistent with observations that the induction of fetal cardiac gene expression during oxidative stress may be important for cardioprotection.
2. Materials and methods
2.1. Materials
Triethylammonium bicarbonate (TEAB), Na2CO3, protease inhibitor cocktail, and trifluoroacetic acid (TFA) were purchased from Sigma (St. Louis, MO). Tris(2-carboxyethyl)-phosphine (TCEP), methyl methanethiosulfonate (MMTS), and iTRAQ reagents were purchased from AB Sciex (Foster City, CA). Trypsin was purchased from Promega Corp. (Madison, WI). PepClean C18 spin columns were purchased from Thermo Scientific (Rockford, IL). Acetonitrile (ACN) and water were purchased from J. T. Baker Inc., (Center Valley, PA).
2.2. Transgenic mice
Tg-Trx1 mice were generated on an FVB background using the α-myosin heavy chain promoter (courtesy of J. Robbins, University of Cincinnati, Cincinnati, OH) to achieve cardiac-specific expression [2]. Transgenic mice were treated with TAC to induce oxidative stress. In brief, the mice were anesthetized with a mixture of ketamine (0.065 mg/g), xylazine (0.013 mg/g), and acepromazine (0.002 mg/g) and were mechanically ventilated. The left chest was opened at the second intercostal space. TAC was performed by ligation of the transverse thoracic aorta between the innominate artery and the left common carotid artery with a 28-gauge needle using a 7–0 braided polyester suture. A sham operation was performed without constricting the aorta. Two weeks after the ligation, mice were euthanized by exposure to CO2. The LV were dissected out, rinsed thoroughly with saline to remove blood, and then stored at −80 °C until use. All animal handling protocols were performed according to the Institutional Animal Care and Use Committee at the Rutgers University.
2.3. Protein extraction and iTRAQ labeling
The iTRAQ experimental procedure is similar to the previous publication [23]. Briefly, twenty milligrams of LV tissues from two control and two Tg-Trx1 mice were each homogenized by a tissue homogenizer (Omni International, Marietta, GA) in 450 μL lysis buffer that contained 25 mM TEAB, 20 mM Na2CO3, and 2 μL of protease inhibitor cocktail. After centrifugation at 19,000 × g for 30 min, the protein concentrations were determined by a Bradford assay using a Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Fifty micrograms of each protein sample were subjected to trypsin digestion prior to labeling with the 4-plex iTRAQ reagent (AB Sciex). The peptides from Non-Tg mouse hearts were labeled with iTRAQ tags 114 and 115, and the peptides from the Tg-Trx1 mouse hearts were labeled with iTRAQ tags 116 and 117. The labeled peptides were combined and dried in a SpeedVac prior to strong cation exchange (SCX) fractionation.
2.4. 2D-LC fractionation
The iTRAQ-labeled peptides were re-suspended in Buffer A, which contained 10 mM KH2PO4 and 20% ACN (pH 3.0), and was fractioned by SCX using a polysulfoethyl A column (4.6 × 200 mm, 5 μm, 300 Å, Poly LC, Columbia, MD, USA) on a BioCAD™ Perfusion Chromatography System (AB Sciex) at a constant flow rate of 1 mL/min. The composition of Buffer B was 10 mM KH2PO4, 20% ACN, and 600 mM KCl (pH 3.0). The peptides were separated using a 60-min linear gradient. Thirty-five fractions were collected. Following PepClean C18 spin column desalting (Pierce, Rockford, IL, USA), each peptide fraction from SCX was further separated on a reversed-phase C18 capillary column (0.75 × 150 mm, 3 μm, 100 Å, Dionex, Sunnyvale, CA, USA) on an Ultimate LC system coupled with a Probot spotting device (Dionex) as described previously [36]. The peptides eluted from the reversed-phase LC were mixed with a MALDI matrix (5 mg/mL α-cyano-4-hydroxycinnamic acid in 60% ACN, 0.1% TFA, 5 mM ammonium monobasicphosphate, 50 fmol/μL each of glu-fibrinogen peptide (m/z 1570.677), and adrenocorticotrophin hormone fragments 18–39 (m/z 2465.199) and were spotted onto the stainless steel MALDI plates for MS/MS analysis.
2.5. Mass spectrometry analysis
The peptides spotted on MALDI plates were analyzed by a 4800 MALDI TOF/TOF analyzer (AB Sciex) in a plate-wide data-dependent analysis manner. The ten most intense ions within a mass range of m/z 800–3500 were chosen for MS/MS analysis. CID was used for peptide fragmentation with a collision energy of 1 keV and a collision gas pressure of 5 × 10−7 Torr. Glu-fibrinogen peptide (m/z 1570.677) and adrenocorticotrophin hormone fragments 18–39 (m/z 2465.199) were used as internal mass calibration standards to achieve accurate precursor mass measurements.
2.6. MS data analysis and protein quantitation
The peak lists of the MS/MS spectra were generated using TS2Mascot software and saved as a MGF file format. Protein identification was performed using a local MASCOT search engine (v. 2.3) on a Proteome Discoverer platform (V. 1.3, Thermo Scientific). Database searching was restricted to mouse sequences in the UniRef database (51,551 entries, downloaded in September, 2014). Trypsin was selected as a cleavage enzyme with one miss cleavage. The precursor ions mass tolerance was 50 ppm and MS/MS fragment ions mass tolerance was 0.5 Da. iTRAQ-labeled N-termini, lysine, and cysteine methanethiolation were selected as fixed modifications, while methionine oxidation and iTRAQ-labeled tyrosines were considered as variable modifications. The decoy database containing both forward and reverse sequences was used to evaluate the false discovery rate (FDR). Proteins were considered as confidently identified if they contained at least one peptide with a confidence interval value (C. I. value) greater than 95% and less than 1% FDR. Proteins that shared identical peptides were grouped to reduce redundancy. Only unique peptides were used for protein identification and quantification.
Scaffold Q+ software (V. 1.3) was used to quantify the proteins. The iTRAQ reporter ion cluster areas were corrected for isotopic carryover. The average protein expression ratios between Tg-Trx1 and the wild type groups were calculated as the mean of the unique peptides of the protein. In this study, two biological replicates of the iTRAQ-labeled sample were analyzed and a corresponding student’s t-test was performed. Proteins with a p value less than or equal to 0.05 in the t-test and ratios ≥1.20-fold increased or ≤0.8-fold decreased were considered as differentially expressed based on our previously determined analytical variations of our system [37,38].
2.7. Cell culture and molecular biology
Cell culture and transfections were performed as previously described [21]. Briefly, a human Trx1 gene inserted into the shuttle vector pDC316 with Flag tag at the N-termini was constructed. HeLa cells were cultured at 37 °C in 5% CO2 atmosphere. Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) was used. Cells were transiently transfected with either the Trx1 plasmid or an empty pDC316 vector using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen, Grand Island, NY, USA.). Forty-eight hours after transfection, the cells were harvested via centrifugation at 500 ×g for 5 min and washed with phosphate-buffered saline (PBS) prior to Western blotting.
2.8. Western blotting
Proteins extracted from HeLa cells (20 μg) or the LV from three control and three Tg-Trx1 mice (30 μg each) were used for Western blotting. In brief, proteins were separated by 10% or 15% SDS-PAGE gels and transferred onto nitrocellulose membranes (Bio-Rad Hercules, CA, USA). The membranes were blocked with 5% milk and probed with primary antibodies against Trx1 (Abcam Inc., Cambridge, MA, USA, ab1754, 1:5000), or SET and MYND domain-containing proteins 1, 2, 3 and 5 (Abcam Inc., Cambridge, MA, USA, SMYD1 (ab49327), SMYD2 (ab108217), SMYD3 (ab187149), SMYD5 (ab137622), −1:2000) overnight, followed by a 1 h incubation with an HRP-conjugated secondary antibody. The signals were detected using the ECL chemiluminescence method (Perkin-Elmer, Boston, MA, USA). The densities of the bands were determined using Quantity One software (v.4.3.1, Bio-Rad, Hercules, CA, USA). GAPDH antibody Western blotting was used to verify equal protein loading of the blots. The data were expressed as mean ± SEM. To compare two independent groups, we used Student’s unpaired t-test. P < 0.05 was taken as a minimal level of significance.
For 2D electrophoresis gel (2DE) and 2D Western blotting, we followed a similar protocol as previously described [21]. Briefly, proteins from Tg-Trx1 and control mice LVs were extracted and dissolved in the 2DE buffer (7 M urea, 2 M thiourea, 4% CHAPS, 65 mM DTT, 0.2% BioLyte, pH 3–10, and 0.01% bromophenol blue). In the first dimension, IPG strips (11 cm, pH 3–10 non-linear; Bio-Rad) were used for protein isoelectric focusing (IEF). After rehydration at 50 V for 12 h, the proteins were focused using the following program: at 250 V for 0.5 h, then the voltage was ramped to 8000 V in 3 h, followed by a 6 h focusing at 8000 V. After a 15 min disulfide reduction with DTT (2%, w/v) followed by a 15 min alkylation with iodoacetamide (2.5%, w/v), the IPG strips were equilibrated in a buffer containing 6 M urea, 375 mM Tris–HCl (pH 8.8), 2% SDS, and 20% glycerol for 15 min. In the second dimension separation, the proteins were separated using 12.5% SDS-PAGE gels. After fixing with 40% methanol and 10% acetic acid for 30 min, the gels were stained with SYPRO Ruby dye and scanned using a Typhoon 9400 imager (GE Healthcare). For 2D Western blotting, the proteins were separated by 2DE as described above and transferred onto nitrocellulose membranes. The membranes were blocked with 5% milk, probed with an anti-lysine methylation antibody (Abcam, 1:2,500), and visualized with an ECL substrate (PerkinElmer Life Science, Waltham, MA).
3. Results and discussion
3.1. Differentially expressed proteins in Tg-Trx1 mice
The iTRAQ experiment was performed in a 4-plex fashion with proteins from two non-transgenic control animals labeled with iTRAQ reagents 114 and 115, and two Tg-Trx1 samples labeled with iTRAQ reagents 116 and 117. In total, 4040 unique peptide sequences were identified, which corresponded to 647 proteins (Supplementary Table 1). To achieve confident protein identification and quantification, we only quantified the proteins that contained at least two unique peptides with a C. I. value ≥95% and a FDR less than 1%. Based on our previously developed bioinformatics workflow [37], 24 proteins were differentially expressed in Tg-Trx1 mouse hearts (Table 1). Twenty-two proteins were up-regulated, whereas two proteins were downregulated. Fig. 1 shows two representative MS/MS spectra corresponding to the peptides derived from Trx1 and SMYD1 and their iTRAQ quantifications, indicating their increased expression in the LVs of Tg-Trx1 mice following TAC stress.
Table 1.
Differentially expressed proteins in Tg-Trx1 mice.
| Protein name | Acc. no. | Gene name | M. W. | Unique peptides | Ratio* (KO/Ctrl) | T-test |
|---|---|---|---|---|---|---|
| Up-regulated proteins | ||||||
| NADH dehydrogenase 1 beta subcomplex subunit 4 | Q9CQC7 | NDUFB4 | 15 kDa | 3 | 1.6 | 0.04 |
| Hexokinase-2 | E9Q5B5 | HK2 | 99 kDa | 6 | 1.6 | 0.02 |
| Thioredoxin | P10639 | TXN | 12 kDa | 3 | 1.4 | 0.00 |
| Eukaryotic translation initiation factor 3 subunit B | Q8JZQ9 | EIF3B | 91 kDa | 4 | 1.4 | 0.02 |
| Heterogeneous nuclear ribonucleoprotein H | O35737 | HNRNPH1 | 49 kDa | 2 | 1.4 | 0.05 |
| Acylpyruvase FAHD1 | Q8R0F8 | FAHD1 | 25 kDa | 2 | 1.3 | 0.03 |
| Actin-related protein 2/3 complex subunit 2 | Q9CVB6 | ARPC2 | 34 kDa | 2 | 1.3 | 0.02 |
| 2-oxoisovalerate dehydrogenase subunit alpha | P50136 | BCKDHA | 50 kDa | 3 | 1.3 | 0.02 |
| 14-3-3 protein beta/alpha | Q9CQV8 | YWHAB | 28 kDa | 5 | 1.3 | 0.02 |
| Glutathione S-transferase omega-1 | O09131 | GST01 | 27 kDa | 5 | 1.2 | 0.05 |
| Reticulon-4-interacting protein 1 | Q924D0 | RTN4IP1 | 43 kDa | 2 | 1.2 | 0.05 |
| Cytochrome b-c1 complex subunit 6 | P99028 | UQCRHL | 10 kDa | 5 | 1.2 | 0.05 |
| Pyruvate dehydrogenase (acetyl-transferring) kinase isozyme 1 | Q8BFP9 | PDK1 | 49 kDa | 5 | 1.2 | 0.05 |
| Cytosolic 5′-nucleotidase 3A | Q9D020 | NT5C3A | 37 kDa | 2 | 1.2 | 0.05 |
| Serpin | F8WIV2 | SERPINB6 | 45 kDa | 6 | 1.2 | 0.04 |
| 40S ribosomal protein S3a | D3Z6C3 | RPS3A2 | 30 kDa | 3 | 1.2 | 0.04 |
| Peptidyl-prolyl cis-trans isomerase FKBP1A | P26883 | FKBP1A | 12 kDa | 2 | 1.2 | 0.04 |
| Isoform M1 of pyruvate kinase PKM | P52480-2 | PKM | 58 kDa | 23 | 1.2 | 0.04 |
| Short-chain specific acyl-CoA dehydrogenase | Q07417 | ACADS | 45 kDa | 10 | 1.2 | 0.03 |
| Histone-lysine N-methyltransferase SMYD1 | P97443 | SMYD1 | 56 kDa | 4 | 1.2 | 0.03 |
| Tubulin alpha-4A chain | P68368 | TUBA4A | 50 kDa | 11 | 1.2 | 0.03 |
| 40S ribosomal protein S10 | P63325 | RPS10 | 19 kDa | 4 | 1.2 | 0.03 |
| Down-regulated proteins | ||||||
| Glutaredoxin-related protein 5 | Q80Y14 | GLRX5 | 16 kDa | 2 | 0.8 | 0.04 |
| Myosin-6 | Q02566 | MYH6 | 224 kDa | 93 | 0.7 | 0.01 |
Proteins are sorted by iTRAQ ratios. All protein identification and quantification information are shown in Supplementary Table 1.
Fig. 1.
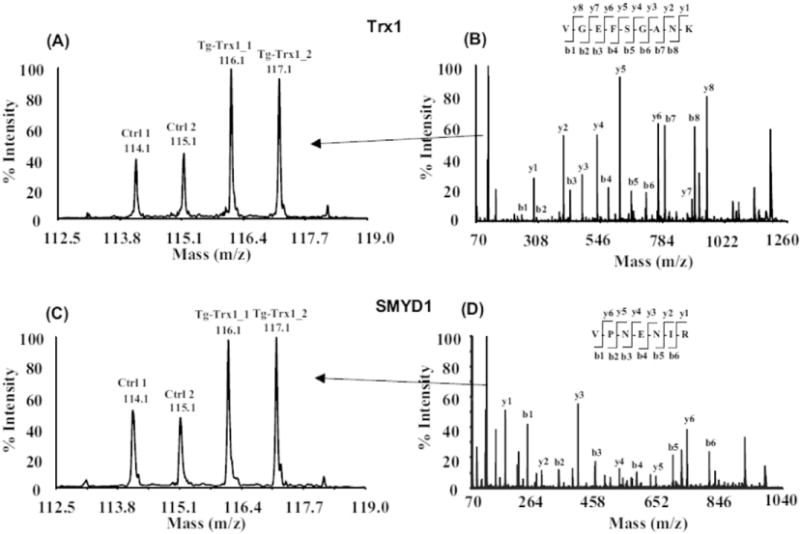
MS/MS spectra and iTRAQ quantifications of selected peptides derived from Trx1 and SMYD1. Proteins from two control and two Tg-Trx1 samples were extracted, reduced, alkylated and digested by trypsin. The resulting peptides were labeled with iTRAQ reagents and mixed together. The iTRAQ-labeled peptides were separated by 2D-LC and subjected to tandem mass spectrometry analysis. Protein and peptide quantitation are based on iTRAQ reporter ions (A, C) and the identification is based on the observation of continuous b- and y-series of ions (B, D).
Several of the proteins identified by this method, such as myosin 6, NADH dehydrogenase 1, 14–3–3 proteins, and serpin have previously been shown to be cardioprotective. Myosin 6 (MYH6) is an actin-activated Mg2+ ATPase that is required for vesicle transport, cell migration, mitosis, myofibril formation and assembly. Mutations in the MYH6 gene have been implicated in atrial septal defects and hypertrophic dilated cardiomyopathy [39]. MYH6 downregulation in Tg-Trx1 mice suggests that Trx1 may exert cardioprotective effects by regulating myofibril architecture.
Among the other proteins induced in Tg-Trx1 mice, 14–3–3 proteins are adaptor proteins that bind to serine/threonine phosphorylated proteins and mediate phosphorylation signaling in various cellular processes such as cell cycle control, apoptosis, and mitosis. Cardiac specific expression of a dominate negative-14–3–3 protein in mice increased apoptotic cell death under stress such as cardiac hypertrophy, diabetic cardiomyopathy, and heart failure, suggesting that 14–3–3 proteins are essential for cardioprotection during stress to the heart [40].
Serpins are serine-protease inhibitors that control various signaling pathways that are important for regulating cell survival and cell death. A recombinant serpin, LEX032, has been shown to reduce the infarct size in a mouse ischemia/reperfusion model by inhibiting neutrophil accumulation and thus, preventing superoxide release from the neutrophils [41,42]. Another study by Ma et al. showed that antithrombin, a serpin, exhibited anti-inflammatory and cardioprotection activity during ischemia/reperfusion injury by activating the AMPK signaling pathway [43]. Upregulation of these proteins in Tg-Trx1 mice during TAC suggests that new pathways, such as myofibril organization, may be regulated by Trx1 during cardiac stress and will require further studies to understand their exact mechanisms.
3.2. Validation of iTRAQ results using Western blotting
To validate the protein expression results obtained from iTRAQ analysis, we selected two differentially expressed proteins, Trx1 and SMYD1, for Western blotting validation among a larger number of independent animals. GAPDH was chosen as a control for sample loading. Fig. 2A shows the increase of both Trx1 (~12 KDa) and SMYD1 (~56 KDa) in Tg-Trxs tissues; Trx1 and SMYD1 had a 4.1- and 1.5-fold increase in Tg-Trx1 mice compared to the control animals, respectively (Fig. 2B). Although the exact fold changes in Western blotting are different from the ones obtained from the iTRAQ analysis (1.4- and 1.3-fold increases), the trend of the protein changes were the same.
Fig. 2.
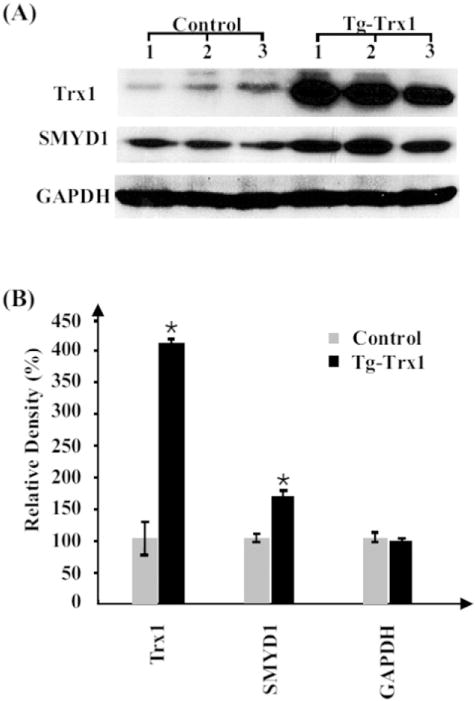
Western blotting validation of iTRAQ results. (A) Protein (30 μg) from three control and three Tg-Trx1 samples were blotted with specific antibodies. (B) The relative densities of the Western blots were plotted to indicate the increase of both Trx1 and SMYD1 in Tg-Trx1 mice, *p < 0.05. GAPDH was used to adjust sample equal loading.
SMYD1, a lysine methyl transferase, is essential for both cardiomyogenesis and myofibril organization [44]. It interacts with several cytoplasmic and nuclear protein complexes and likely regulates their activities via methylation [35,45].
3.3. Upregulation of lysine methylation in Tg-Trx1 mice
Recent studies have shown that epigenetic regulations such as protein methylation, histone methylation and acetylation, chromatin remodeling, and gene regulation by non-coding RNAs (e.g., miRNAs) play a key role during heart failure. For example, lysine methylation of KMT4/H3K79 has been implicated in maintaining cardiac structural integrity and preserving cardiac function [46]. Because the cardioprotective Tg-Trx1 mice showed an increase in SMYD1, a protein that contains a SET domain and can thus methylate lysine residues, we wanted to determine the protein methylation status in Tg-Trx1 mice. We performed a 2DE analysis of the proteins obtained from both control and Tg-Trx1 mice. Similar to iTRAQ expression profiling, the global protein expressions of both the Tg-Trx1 and control group samples were very similar, as shown by 2DE analysis (Fig. 3A and B). However, 2D Western blotting with an antibody that recognizes methylated lysines showed a different pattern between the two samples (Fig. 3C and D), indicating selective changes of lysine methylation among proteins in Tg-Trx1 tissues.
Fig. 3.
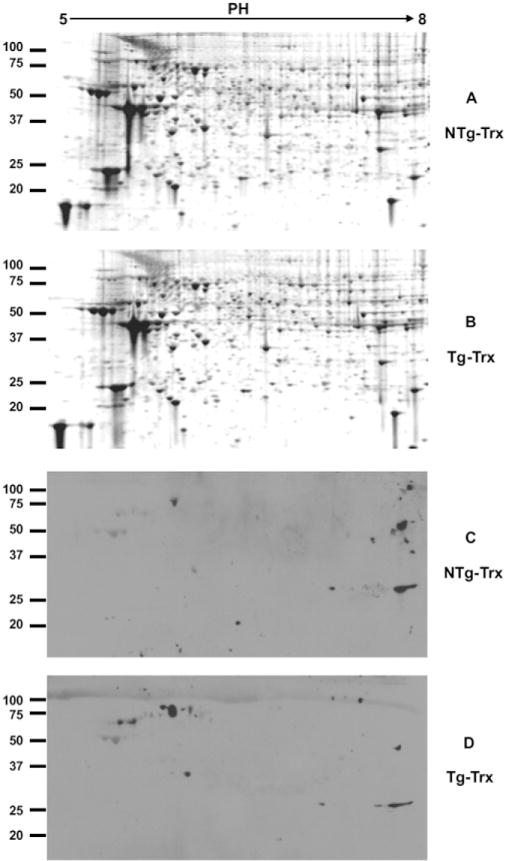
2DE and 2D Western blotting of total protein and their lysine methylation status. Proteins from both Tg-Trx1 and control mice hearts were extracted and separated by 2DE. Gel images were acquired on a Typhoon 9400 imager (GE Healthcare) after SYPRO Ruby staining (A and B). Protein lysine methylation was analyzed by 2D Western blotting using an anti-lysine methylation antibody (C and D).
3.4. Regulation of SMYD proteins by Trx1 also occurs in other cell types
SMYD proteins are defined by having a SET domain separated into two segments by a MYND domain. Five SET-domain containing proteins (SMYD1, 2, 3, 4 and 5) contain highly conserved sequences in their MYND domains, specifically a zinc finger motif that facilities the interaction between SMYD and target proteins [45]. Within the SMYD family, SMYD1, 2 and 3 share conserved cysteine residues in their zinc finger motifs [47] (Supplementary Fig. S1). To determine whether the induction of SMYD1 by Trx1 is specific in cardiac cells or is a general mechanism for Trx1 to exert regulations of cellular functions, we over-expressed Trx1 in HeLa cells and found that it significantly promoted the expression of SMYD1, 2, 3 and 5 (Fig. 4) suggesting that Trx1 induction of SMYD methyl transferases may be a general mechanism in regulation (we were unable to determine if SMYD4 was induced because a SMYD4 specific antibody was not available at the time of the study).
Fig. 4.
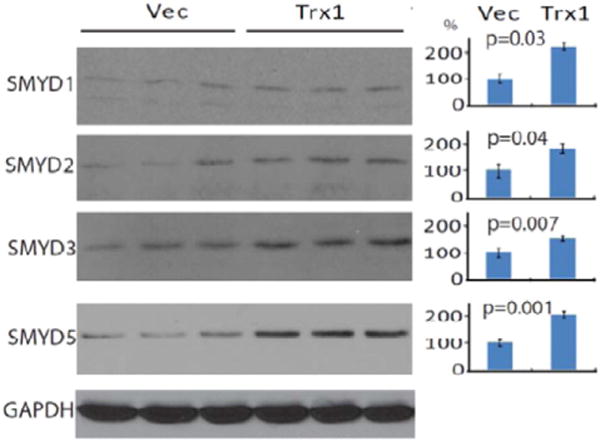
Western blotting of SMYD1, 2, 3, and 5 in Trx1 overexpressed HeLa cells. HeLa cells were transiently transfected with a human Trx1 plasmid or with an empty pDC316 vector for 48 h, after which the cells were harvested and washed with PBS. Proteins were extracted for Western blotting using anti-SMYD1, 2, 3, and 5 antibodies.
4. Conclusions
Earlier studies in cardiovascular biology indicated that when under stress, failing hearts reactivate certain fetal genes and revert to fetal metabolic patterns via the down regulation of adult gene transcripts rather than upregulating fetal genes [48]. Fetal gene over representation in stressed adult hearts may be an adaptation to a variety of pathophysiologic conditions including hypoxia, ischemia, and hypertrophy. Whether such adaptation is beneficial as an effective long-term stress response mechanism in the heart remains to be determined. The current study reveals that Trx1 may aid fetal gene upregulation following TAC stress via upregulating SMYD1 and the related family of methyl-transferases. The precise role of such non-redox regulation will be revealed upon detailed biological studies. Overall, this study uncovers that as a master redox regulator, Trx1 not only modulates cellular function via regulating redox PTMs, but it also exerts novel regulatory functions by modulating SMYD1 and protein methylation. Such regulation may be evolutionarily conserved and occur in different cell types.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbapap.2015.09.006.
Supplementary Material
Acknowledgments
The project described was supported by a grant from the NJ Health Foundation and the instrument used is supported by a grant (P30NS046593) from the National Institute of Neurological Disorders and Stroke. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke or the National Institutes of Health.
Abbreviations
- C. I.
confidence interval
- FDR
false discovery rate
- GAPDH
glyceraldehyde-3-phophate dehydrogenase
- ICAT
isotope-coded affinity tags
- iTRAQ
isobaric tags for relative and absolute quantitation
- MMTS
methyl methanethiosulfonate
- PTMs
post-translational modifications
- ROS
reactive oxygen species
- SMYD
SET and MYND domain-containing protein
- SCX
strong cation exchange
- TCEP
Tris(2-carboxyethyl)-phosphine
- TEAB
Triethylammonium bicarbonate buffer
- Tg-Trx1
transgenic mice with a cardiac-specific over-expression of Trx1
- Trx1
Thioredoxin 1
- TAC
thoracic aortic constriction
- TFA
trifluoroacetic acid
- LV
left ventricle
- FVB
Friend virus B-type
- IEF
isoelectric focusing
Footnotes
Conflict of interest
The authors report no conflict of interest.
References
- 1.Marian AJ. Genetic determinants of cardiac hypertrophy. Curr Opin Cardiol. 2008;23:199–205. doi: 10.1097/HCO.0b013e3282fc27d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamamoto M, Yang G, Hong C, Liu J, Holle E, et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest. 2003;112:1395–1406. doi: 10.1172/JCI17700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshioka J, Schulze PC, Cupesi M, Sylvan JD, MacGillivray C, et al. Thioredoxin-interacting protein controls cardiac hypertrophy through regulation of thioredoxin activity. Circulation. 2004;109:2581–2586. doi: 10.1161/01.CIR.0000129771.32215.44. [DOI] [PubMed] [Google Scholar]
- 4.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 5.Adiga IK, Nair RR. Multiple signaling pathways coordinately mediate reactive oxygen species dependent cardiomyocyte hypertrophy. Cell Biochem Funct. 2008;26:346–351. doi: 10.1002/cbf.1449. [DOI] [PubMed] [Google Scholar]
- 6.Hardt SE, Sadoshima J. Negative regulators of cardiac hypertrophy. Cardiovasc Res. 2004;63:500–509. doi: 10.1016/j.cardiores.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res. 2015;116:531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collet JF, Messens J. Structure, function, and mechanism of thioredoxin proteins. Antioxid Redox Signal. 2010;13:1205–1216. doi: 10.1089/ars.2010.3114. [DOI] [PubMed] [Google Scholar]
- 9.Ago T, Sadoshima J. Thioredoxin and ventricular remodeling. J Mol Cell Cardiol. 2006;41:762–773. doi: 10.1016/j.yjmcc.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ago T, Yeh I, Yamamoto M, Schinke-Braun M, Brown JA, et al. Thioredoxin1 upregulates mitochondrial proteins related to oxidative phosphorylation and TCA cycle in the heart. Antioxid Redox Signal. 2006;8:1635–1650. doi: 10.1089/ars.2006.8.1635. [DOI] [PubMed] [Google Scholar]
- 11.Miranda-Vizuete A, Ljung J, Damdimopoulos AE, Gustafsson JA, Oko R, et al. Characterization of sptrx, a novel member of the thioredoxin family specifically expressed in human spermatozoa. J Biol Chem. 2001;276:31567–31574. doi: 10.1074/jbc.M101760200. [DOI] [PubMed] [Google Scholar]
- 12.Weichsel A, Brailey JL, Montfort WR. Buried S-nitrosocysteine revealed in crystal structures of human thioredoxin. Biochemistry. 2007;46:1219–1227. doi: 10.1021/bi061878r. [DOI] [PubMed] [Google Scholar]
- 13.Lu J, Holmgren A. Thioredoxin system in cell death progression. Antioxid Redox Signal. 2012;17:1738–1747. doi: 10.1089/ars.2012.4650. [DOI] [PubMed] [Google Scholar]
- 14.Yoshioka J, Schreiter ER, Lee RT. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxid Redox Signal. 2006;8:2143–2151. doi: 10.1089/ars.2006.8.2143. [DOI] [PubMed] [Google Scholar]
- 15.Powis G, Kirkpatrick DL. Thioredoxin signaling as a target for cancer therapy. Curr Opin Pharmacol. 2007;7:392–397. doi: 10.1016/j.coph.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi-Miura M, Shioji K, Hoshino Y, Masutani H, Nakamura H, et al. Oxygen sensing and redox signaling: the role of thioredoxin in embryonic development and cardiac diseases. Am J Physiol Heart Circ Physiol. 2007;292:H2040–H2050. doi: 10.1152/ajpheart.01316.2006. [DOI] [PubMed] [Google Scholar]
- 17.Shioji K, Nakamura H, Masutani H, Yodoi J. Redox regulation by thioredoxin in cardiovascular diseases. Antioxid Redox Signal. 2003;5:795–802. doi: 10.1089/152308603770380106. [DOI] [PubMed] [Google Scholar]
- 18.Dunn LL, Buckle AM, Cooke JP, Ng MK. The emerging role of the thioredoxin system in angiogenesis. Arterioscler Thromb Vasc Biol. 2010;30:2089–2098. doi: 10.1161/ATVBAHA.110.209643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ago T, Sadoshima J. Thioredoxin1 as a negative regulator of cardiac hypertrophy. Antioxid Redox Signal. 2007;9:679–687. doi: 10.1089/ars.2007.1529. [DOI] [PubMed] [Google Scholar]
- 20.Wu C, Parrott AM, Fu C, Liu T, Marino SM, et al. Thioredoxin 1-mediated post-translational modifications: reduction, transnitrosylation, denitrosylation, and related proteomics methodologies. Antioxid Redox Signal. 2011;15:2565–2604. doi: 10.1089/ars.2010.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C, Liu T, Chen W, Oka S, Fu C, et al. Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol Cell Proteomics. 2010;9:2262–2275. doi: 10.1074/mcp.M110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sengupta R, Holmgren A. Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antioxid Redox Signal. 2013;18:259–269. doi: 10.1089/ars.2012.4716. [DOI] [PubMed] [Google Scholar]
- 23.Fu C, Wu C, Liu T, Ago T, Zhai P, et al. Elucidation of thioredoxin target protein networks in mouse. Mol Cell Proteomics. 2009;8:1674–1687. doi: 10.1074/mcp.M800580-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gagne JP, Ethier C, Gagne P, Mercier G, Bonicalzi ME, et al. Comparative proteome analysis of human epithelial ovarian cancer. Proteome Sci. 2007;5:16. doi: 10.1186/1477-5956-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hergenroeder G, Redell JB, Moore AN, Dubinsky WP, Funk RT, et al. Identification of serum biomarkers in brain-injured adults: potential for predicting elevated intracranial pressure. J Neurotrauma. 2008;25:79–93. doi: 10.1089/neu.2007.0386. [DOI] [PubMed] [Google Scholar]
- 26.Ralhan R, Desouza LV, Matta A, Chandra Tripathi S, Ghanny S, et al. Discovery and verification of head-and-neck cancer biomarkers by differential protein expression analysis using iTRAQ-labeling and multidimensional liquid chromatography and tandem mass spectrometry. Mol Cell Proteomics. 2008 doi: 10.1074/mcp.M700500-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu T, Donahue KC, Hu J, Kurnellas MP, Grant JE, et al. Identification of differentially expressed proteins in experimental autoimmune encephalomyelitis (EAE) by proteomic analysis of the spinal cord. J Proteome Res. 2007;6:2565–2575. doi: 10.1021/pr070012k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenzweig D, Smith D, Myler PJ, Olafson RW, Zilberstein D. Post-translational modification of cellular proteins during Leishmania donovani differentiation. Proteomics. 2008 doi: 10.1002/pmic.200701043. [DOI] [PubMed] [Google Scholar]
- 29.Gottlieb PD, Pierce SA, Sims RJ, Yamagishi H, Weihe EK, et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat Genet. 2002;31:25–32. doi: 10.1038/ng866. [DOI] [PubMed] [Google Scholar]
- 30.Trievel RC, Beach BM, Dirk LM, Houtz RL, Hurley JH. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell. 2002;111:91–103. doi: 10.1016/s0092-8674(02)01000-0. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, et al. Structure of the neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 2002;111:117–127. doi: 10.1016/s0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foreman KW, Brown M, Park F, Emtage S, Harriss J, et al. Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS One. 2011;6:e22290. doi: 10.1371/journal.pone.0022290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abu-Farha M, Lanouette S, Elisma F, Tremblay V, Butson J, et al. Proteomic analyses of the SMYD family interactomes identify HSP90 as a novel target for SMYD2. J Mol Cell Biol. 2011;3:301–308. doi: 10.1093/jmcb/mjr025. [DOI] [PubMed] [Google Scholar]
- 34.Tan X, Rotllant J, Li H, De Deyne P, Du SJ. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc Natl Acad Sci U S A. 2006;103:2713–2718. doi: 10.1073/pnas.0509503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Just S, Meder B, Berger IM, Etard C, Trano N, et al. The myosin-interacting protein SMYD1 is essential for sarcomere organization. J Cell Sci. 2011;124:3127–3136. doi: 10.1242/jcs.084772. [DOI] [PubMed] [Google Scholar]
- 36.Liu T, D’Mello V, Deng L, Hu J, Ricardo M, et al. A multiplexed proteomics approach to differentiate neurite outgrowth patterns. J Neurosci Methods. 2006;158:22–29. doi: 10.1016/j.jneumeth.2006.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu J, Qian J, Borisov O, Pan S, Li Y, et al. Optimized proteomic analysis of a mouse model of cerebellar dysfunction using amine-specific isobaric tags. Proteomics. 2006;6:4321–4334. doi: 10.1002/pmic.200600026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen SS, Raval A, Johnson AJ, Hertlein E, Liu TH, et al. Epigenetic changes during disease progression in a murine model of human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2009;106:13433–13438. doi: 10.1073/pnas.0906455106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Granados-Riveron JT, Ghosh TK, Pope M, Bu’Lock F, Thornborough C, et al. Alpha-cardiac myosin heavy chain (MYH6) mutations affecting myofibril formation are associated with congenital heart defects. Hum Mol Genet. 2010;19:4007–4016. doi: 10.1093/hmg/ddq315. [DOI] [PubMed] [Google Scholar]
- 40.Sari FR, Watanabe K, Widyantoro B, Thandavarayan RA, Harima M, et al. Partial inactivation of cardiac 14–3–3 protein in vivo elicits endoplasmic reticulum stress (ERS) and activates ERS-initiated apoptosis in ERS-induced mice. Cell Physiol Biochem. 2010;26:167–178. doi: 10.1159/000320548. [DOI] [PubMed] [Google Scholar]
- 41.Murohara T, Guo JP, Lefer AM. Cardioprotection by a novel recombinant serine protease inhibitor in myocardial ischemia and reperfusion injury. J Pharmacol Exp Ther. 1995;274:1246–1253. [PubMed] [Google Scholar]
- 42.Delyani JA, Murohara T, Lefer AM. Novel recombinant serpin, LEX-032, attenuates myocardial reperfusion injury in cats. Am J Physiol. 1996;270:H881–H887. doi: 10.1152/ajpheart.1996.270.3.H881. [DOI] [PubMed] [Google Scholar]
- 43.Ma Y, Wang J, Gao J, Yang H, Wang Y, et al. Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury. Thromb Haemost. 2014;113 doi: 10.1160/TH14-04-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao J, Li J, Li BJ, Yagil E, Zhang J, et al. Expression and functional characterization of Smyd1a in myofibril organization of skeletal muscles. PLoS One. 2014;9:e86808. doi: 10.1371/journal.pone.0086808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leinhart K, Brown M. SET/MYND lysine methyltransferases regulate gene transcription and protein activity. Genes (Basel) 2011;2:210–218. doi: 10.3390/genes2010210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sirinupong N, Brunzelle J, Ye J, Pirzada A, Nico L, et al. Crystal structure of cardiac-specific histone methyltransferase SmyD1 reveals unusual active site architecture. J Biol Chem. 2010;285:40635–40644. doi: 10.1074/jbc.M110.168187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, et al. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.