

Abstract
To evaluate the mechanisms and consequences of chromosomal aberrations in colorectal cancer (CRC), we used a combination of spectral karyotyping, array comparative genomic hybridization (aCGH), and array-based global gene expression profiling on 31 primary carcinomas and 15 established cell lines. Importantly, aCGH showed that the genomic profiles of primary tumors are recapitulated in the cell lines. We revealed a preponderance of chromosome breakpoints at sites of copy number variants (CNVs) in the CRC cell lines, a novel mechanism of DNA breakage in cancer. The integration of gene expression and aCGH led to the identification of 157 genes localized within high-level copy number changes whose transcriptional deregulation was significantly affected across all of the samples, thereby suggesting that these genes play a functional role in CRC. Genomic amplification at 8q24 was the most recurrent event and led to the overexpression of MYC and FAM84B. Copy number dependent gene expression resulted in deregulation of known cancer genes such as APC, FGFR2, and ERBB2. The identification of only 36 genes whose localization near a breakpoint could account for their observed deregulated expression demonstrates that the major mechanism for transcriptional deregulation in CRC is genomic copy number changes resulting from chromosomal aberrations.
INTRODUCTION
Colorectal cancer (CRC) is among the most common malignancies in the Western World (Jemal et al., 2008). As a model for multistep carcinogenesis, colorectal neoplasia represents a genetic paradigm for cancer initiation and progression (Fearon and Vogelstein, 1990). Genomic copy number alterations (CNA) are a major characteristic of cancer cells and are extensively associated with progression of the disease. Numerous studies have revealed recurrent chromosomal gains and losses in CRC cells (Bardi et al., 1993; Ried et al., 1996; Douglas et al., 2004; Camps et al., 2006; Martin et al., 2007). Because gene expression changes associated with these genomic imbalances are ultimately responsible for the malignant phenotype, measuring the extent to which gene expression is affected by genomic insults is a powerful tool to identify putative cancer genes. This in turn may lead to the identification of cancer-specific molecular targets for therapeutic intervention.
The integrated application of high-throughput technologies to cancer cells generates an enormous wealth of knowledge. In particular, concerted analysis of the cancer genome using molecular karyotyping, high-resolution array-based CGH (aCGH), and global gene expression profiling builds a framework for the discovery of novel cancer genes in solid tumors. In addition, the identification of genomic amplifications and regions of high-level deletions is important for uncovering genes and biological pathways perturbed during tumorigenesis (Albertson, 2006; Myllykangas and Knuutila, 2006).
While primary colorectal carcinomas are ideal in that they truly represent the disease state, there are some aspects of tumor biology, such as the nature of the underlying chromosome aberrations, which we cannot currently interrogate in these samples. Using an approach similar to recent reports (Neve et al., 2006; Martin et al., 2007; Fix et al., 2008), we performed a combined high-throughput analysis of 31 primary colorectal tumors and 15 established CRC cell lines. The parallels we uncovered between primary tumors and cell lines provide a more thorough understanding of the nature of genomic alterations, the possible mechanism by which they are generated, their consequences on the transcriptome, and finally how the events in one sample can lead to genes and pathways generally affected in colorectal carcinogenesis.
MATERIALS AND METHODS
Cell Lines, DNA, and RNA Isolation
The following colorectal cancer cell lines were used in this study: DLD-1, HCT116, p53HCT116, SW48, and LoVo (near-diploid); SW480, SW837, HT-29, T84, Colo 201, Colo 320DM, LS411N, SK-CO-1, NCI-H508, and NCI-H716 (aneuploid). All of the cell lines were obtained from the ATCC (American Type Culture Collection) and cultured following their recommendations, except p53HCT116, a derivative of HCT116 with a homozygous disruption of TP53 (Bunz et al., 1998), which was kindly provided by Dr. Curtis C. Harris of the National Cancer Institute, NIH. Mismatch repair status was retrieved from the literature (Eshleman et al., 1998; Ghadimi et al., 2000; Abdel-Rahman et al., 2001).
DNA and RNA was extracted from the cell lines and primary tumors following standard procedures (http://www.riedlab.nci.nih.gov/protocols.asp). Nucleic acid quantification was determined using the Nanodrop ND-1000 UV-VIS spectrophotometer (Nanodrop, Rockland, DE) and RNA quality was assessed using the Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). Normal colon RNA isolated postmortem from five different donors without a history of colorectal cancer was purchased from Ambion (Applied Biosystems, Foster City, CA).
Array CGH and Gene Expression Microarrays
Oligonucleotide-based aCGH was performed according to the protocol provided by the manufacturer (Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis, protocol version 4.0, June 2006, Agilent Technologies, Santa Clara, CA), with minor modifications. Three micrograms of DNA from each cell line and tumor were labeled with Cy3 and combined with sex-matched commercially available pooled control DNA (Promega, Madison, WI) labeled with Cy5. Oligonucleotide-based Human Genome Microarrays (Agilent Technologies) containing 44K and 185K features, respectively, were used for hybridization.
One μg each of cell line or normal human colon RNA (Ambion, Austin, TX) and Universal Human Reference RNA (Stratagene, Cedar Creek, TX) were amplified and labeled with Cy3 and Cy5, respectively, using a T7 RNA Polymerase (Low RNA Input Fluorescent Linear Amplification Kit, Agilent) according to the manufacturer’s protocols, and hybridized to the 44K oligonucleotide-based Whole Human Genome Microarray (Agilent). Similarly, RNA from primary tumors and normal human colon were labeled with Cy3 and subjected to mono-channel hybridization onto 4 × 44K Whole Human Genome Microarray (Agilent).
Microarrays were washed and processed using an Agilent G2565BA scanner. Data were quality controled and extracted using Agilent Technologies’ Feature Extraction (version 9.1).
Data Analysis
Array CGH and gene expression analysis
The analyses of the microarray experiments were performed with in-house developed software based on R version 2.6.2 (http://www.R-project.org). DNA Copy package from Bioconductor (http://www.bioconductor.org) was used to analyze aCGH data. The data were smoothed using “smooth.CNA” function, with arguments smooth.region = 2, and smooth.SD.scale = 3, and followed by the generation of chromosome segments using Circular Binary Segmentation (CBS) (Olshen et al., 2004), using “segment” function with alpha = 0.02, undo.split = “sdundo” and undo.SD = 0.9. We centralized DNA copy number to the most common ploidy defined as the highest mode of the probability density function of sample versus reference log2 ratio across the total set of features in the array. Data were visualized using CGH Analytics™ (Agilent) and Nexus Copy Number (BioDiscovery, Inc.).
For the cell line dataset, gene expression data were obtained from 44K or 4 × 44K Agilent dual-channel arrays. Median per feature was used to summarize data when two or three technical replicates were available. The data were normalized using Linear & Lowess procedure in Agilent’s Feature Extraction software. Features for which signals were below background (as assessed by “gSurrogatedUsed” or “rSurrogatedUsed”) were forced to NA (not a number). We used the median measurement when more than one measurement was available per feature (i.e., median-summarization by array using “ProbeName”). The final cell line dataset contained 20 samples (15 cell lines, and five normal colon samples), and 40,380 features.
For the primary tumor dataset, gene expression data were obtained from 4 × 44K Agilent mono-channel arrays. We used the median measurement when more than one measurement was available per feature (i.e., median-summarization by array using “chr_coord”). Features for which signals were below background (as assessed by “gSurrogatedUsed”) were forced to zero. To compensate for any scanner distortion, we applied a 90 interpercentile range (90IPR) procedure to equalize the spread of Cy3 measurement per array (in log2 scale). The final dataset contained 28 samples (23 primary tumors, and five normal colon samples), and 40,365 features.
For the purpose of identifying features affected near breakpoint regions, outlier gene expression values were defined as having a >1.5-fold change relative to the next closest value among the remaining samples.
Processed microarray CGH and gene expression data are available as Supporting Information (Supporting Information Tables 1–3).
Determination of Breakpoints, Amplifications, and High-level Deletions
A breakpoint was defined as a shift between two adjacent CBS segments. As the genes between the features could not always be determined, we used the following criteria to determine which genes located at the breakpoints should be evaluated for changes in gene expression: (i) breakpoints spanning a distance of less than 250 kb, genes within a region ±150 kb from the midpoint between the aCGH features defining the breakpoint were assessed; (ii) for 250–300 kb breakpoint regions, genes within a 350-kb region were included; and (iii) breakpoints where the distance between the defining oligonucleotides was >300 kb, genes within ±25 kb of the ends were also examined.
Breakpoints were then mapped according to the hg17 build of the Database of Genomic Variants (http://projects.tcag.ca/variation/) to identify structural variants of the genome residing at these sites. The statistics of association of chromosomal breakpoints with CNV loci is the χ2 goodness of fit between the observed fraction of breakpoint in CNV loci (number of observed breakpoint in CNV loci/total observed breakpoints), and the fraction of expected breakpoints in CNV loci (total base-pair of CNV areas in array/total base-pair covered in array). The significance threshold for this statistical test is P value < α = 0.05 (two-sided).
In contrast to single copy number gains which might result in small changes of the aCGH ratios, segments with a log2 ratio >1 and that differed in copy number from at least one adjacent segment by more than 1 (log2 ratio) were considered high-level, focal amplifications. High-level deletions were defined as CBS segments >100 kb with a log2 ratio <−1. In both analyses, segments encompassed within CNVs were discarded.
RESULTS
Genomic Profiling
To identify sites of CNAs, high-resolution aCGH was performed on 31 primary colon carcinomas (Camps et al., 2008) and 15 commonly used CRC cell lines. A total of 271 genomic imbalances, including whole chromosomal aneuploidies, were detected in the cell lines. Between two and five imbalances occurred in each of the five microsatellite unstable (MSI+), near-diploid cell lines, and from 14 to 34 in the 10 microsatellite stable (MSI−), aneuploid cell lines. Although the cell lines contained on average more CNAs than the primary tumors (18 versus 12.6), a remarkable consistency was observed with respect to the affected regions (Fig. 1). Low-level gains of chromosome arms 7, 8q, 11p, 13, 20q, and X occurred in greater than 25% for both cell lines and primary tumors. Similarly, low-level common losses were detected for chromosome arms 1p, 4q, 5q, 8p, 17p, 18, and 21. We therefore conclude that in general the cell lines have retained and mirror those chromosomal aberrations characteristic of primary colorectal carcinomas.
Figure 1.
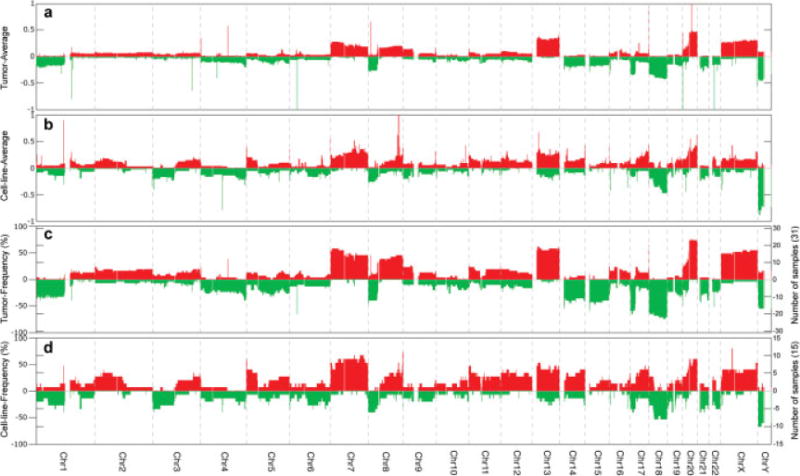
Comparison of genomic imbalances by array CGH analyses of human colorectal cancer cell lines and primary tumors. The average of copy number gains and losses for the 31 primary tumor (A) and for the 15 cell lines (B) is plotted as a function of genome location. Frequency distributions of increases or decreases in genome copy number changes are indicated for the primary tumors (C) and the cell lines (D). On the left hand Y-axis frequencies of gains and losses are represented as a percentage. On the right hand Y-axis the frequencies are displayed as a function of the total number of cases.
We and others have previously demonstrated a direct correlation between cancer specific genomic imbalances and the transcriptome in several primary tumor types (Monni et al., 2001; Pollack et al., 2002; Grade et al., 2006). We were therefore curious whether such a correlation was maintained in the CRC cell lines. As illustrated in Figure 2A, a positive correlation (r = 0.66) between the CNA segments and the expression level of the encompassed genes was observed. This overall positive correlation is depicted at the whole genome level for individual samples in Figure 2B.
Figure 2.
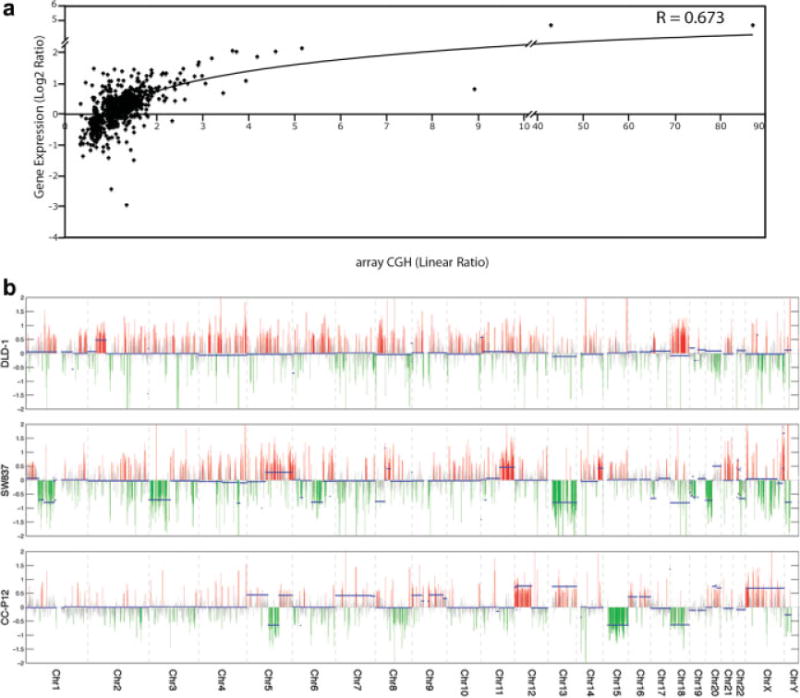
Correlation of genomic copy number changes with levels of gene expression. (A) Correlation of all of the CBS segments with their resident gene expression levels for 15 colorectal cancer cell lines. (B) Genome-transcriptome correlation plots for individual cell lines (DLD-1 and SW837) and primary tumor (CC-P12). Genomic copy number changes are indicated with solid blue bars and gene expression levels are indicated in red (overexpression) and green (underexpression) as a function of log2 ratio between the sample and five normal colon mucosa.
Mapping of High-Level Genomic Imbalances
Regions of the genome that undergo focal, high-level copy number gains are likely to contain oncogenes. Using aCGH, we identified 26 amplicons in the cell lines and 11 regions of amplification in the primary tumors (Table 1). The amplicons ranged in size from 50 kb to 27.22 Mb, with the average being 4.56 Mb. Cytogenetically, homogenously staining regions (hsr) accounted for four amplicons, double minutes (dmin) representing six different regions of high genome amplification were present in three cell lines, and nine amplifications were located near sites of chromosomal translocations. The level of amplification ranged from 2.368 to an astonishing 87-fold increase in genomic copy number. All of the amplicons occurred in MSI- cell lines. Four regions were independently amplified in multiple cell lines (chr6:42,008,700–42,937,190, chr8:125, 620,117–128,955,220, chr12:24,174,625–27,444,930, and chr13:27,392,825–27,439,502). Most notable was chromosome band 8q24, which was affected in four different cell lines (Fig. 3). While chromosomes 6, 8, 13, 17 and 20 contained amplicons in both the cell lines and primary tumors, shared amplified regions occurred on chromosomes 6 and 13 (Table 1).
TABLE 1.
Summary of the Amplicons and Candidate Target Genes Identified in Colon Primary Tumors and Colorectal Cancer Cell Lines
| ID | Sample | Cytoband | Starting bp | Ending bp | Size (Mb) | aCGH ratio | Amplification mechanism | GOI upregulated amplicon-specifica,b | GOI upregulated in tumors and cell linesa,c |
|---|---|---|---|---|---|---|---|---|---|
| Genomic amplification in primary tumors | |||||||||
| Amp-T1 | CC-P14 | 5q33.1-q33.2 | 147,325,084 | 152,440,343 | 5.12 | 1.917 | n.d. | FBXO38, GRPEL2, NDST1, SYNPO, RBM22, DCTN4, MST150, GM2A, SLC36A1 | SH3TC2, GRPEL2 |
| Amp-T2 | CC-P9 | 6p22.1-p21.33 | 27,775,301 | 31,010,736 | 3.24 | 2.223 | n.d. | HIST1H4J, HIST1H4K, ZNF193, ZNF187, ZNF452, RFP, GABBR1, ZNRD1, TRIM26,HCG18, TRIM39, RPP21, ABCF1, C6orf136, DHX16, MDC1, FLOT1, DDR1, GTF2H4, VARSL | HIST1H3H, HIST1H2AM, ZNF165, TRIM27, RNF39, HCG18, C6orf134, MDC1, IER3, SFTPG |
| Amp-T3 | CC-P14 | 6p21.1 | 41,451,467 | 42,008,700 | 0.56 | 1.187 | n.d. | – | BYSL |
| Amp-T4 | CC-P1 | 8p23.1 | 10,607,890 | 10,995,687 | 0.39 | 1.516 | n.d. | n.d. | – |
| Amp-T5 | CC-P72 | 8p22 | 12,927,635 | 13,469,526 | 0.54 | 1.002 | n.d. | – | – |
| Amp-T6 | CC-P47 | 13q12.13-q12.3 | 26,222,778 | 28,886,810 | 2.66 | 1.411 | n.d. | – | – |
| Amp-T7 | CC-P47 | 13q34 | 109,376,185 | 111,674,042 | 2.30 | 1.816 | n.d. | C13orf16 | C13orf29, ANKRD10 |
| Amp-T8 | CC-P65 | 16q11.2-q12.2 | 45,091,146 | 52,567,393 | 7.48 | 1.754 | n.d. | CHD9, RBL2 | SHCBP1, ORC6L, GPT2, NETO2, HEATR3, ADCY7, NKD1, KIAA1005 |
| Amp-T9 | CC-P45 | 16q22.1 | 67,552,540 | 68,408,029 | 0.86 | 1.203 | n.d. | – | HAS3, VPS4A, COG8, CYB5B, NQO1 |
| Amp-T10 | CC-P56 | 17q12q21.2 | 34,837,463 | 36,548,195 | 1.71 | 3.870 | n.d. | PNMT, PERLD1, ERBB2, C17orf37, GRB7, SMARCE1, KRT10, TMEM99, KRT12, KRT20, KRTAP3-2, KRTAP1-1 | GRB7, CDC6, TOP2A, TNS4, KRT23, KRTAP1-1 |
| Amp-T11 | CC-P16 | 20p12.1 | 13,421,057 | 16,034,785 | 2.61 | 1.130 | n.d. | – | – |
| Genomic amplification in cell lines | |||||||||
| Amp-CL1 | Colo 302DM | 1q21.1 | 143,456,705 | 145,971,637 | 2.51 | 1.244 | Translocation | PRKAB2 | – |
| Amp-CL2 | Colo 302DM | 1q21.1 | 146,628,218 | 147,584,714 | 0.95 | 2.567 | Translocation | CA14, TARSL1, ADAMTSL4, ENSA, GOLPH3L | TARSL1 |
| Amp-CL3 | Colo 302DM | 2q14.3-q21.1 | 127,150,203 | 130,993,283 | 3.84 | 1.613 | Translocation | – | PROC, POLR2D, UGCGL1 |
| Amp-CL4 | Colo 201 | 6p21.2-p21.1 | 38,005,714 | 42,937,190 | 4.93 | 2.197 | n.d. | BTBD9, C6orf64, UNC5CL, C6orf130, C6orf49, USP49, TRFP, TRERF1, TBCC, KIAA0240, RPL7L1 | BYSL |
| Amp-CL5 | Colo 201 | 6p12.2-p12.1 | 51,326,264 | 56,199,244 | 4.87 | 1.093 | n.d. | EFHC1, ICK | MCM3 |
| Amp-CL6 | Colo 201 | 6q12 | 64,228,424 | 68,244,706 | 4.02 | 1.258 | n.d. | – | – |
| Amp-CL7 | Colo 201 | 6q23.2-q23.3 | 133,461,261 | 138,461,571 | 4.52 | 2.231 | n.d. | ALDH8A1, HBS1L, AHI1 | FAM54A, MAP7, PEX7, IL20RA, PERP |
| Amp-CL8 | SK-CO-1 | 8p21.1-p12 | 29,037,499 | 31,277,948 | 2.21 | 1.532 | Translocation | LEPROTL1 | – |
| Amp-CL9 | SW837 | 8p12-p11.23 | 37,913,540 | 38,703,881 | 0.79 | 1.141 | Translocation | PPAPDC1B | EIF4EBP1 |
| Amp-CL10 | HT-29 | 8q23.3-q24.3 | 115,354,884 | 142,570,330 | 27.22 | 1.577 | hsr | ANXA13, ST3GAL1, SLC45A4 | MAL2, DCC1, MTBP, SNTB1, ATAD2, ZNF572, SQLE, TRIB1, FAM84B, MYC, EIF2C2 |
| Amp-CL11 | NCI-H716 | 8q24.12 | 121,090,646 | 121,513,421 | 0.42 | 5.853 | dmin | DEPDC6, COL14A1 | – |
| Amp-CL12 | NCI-H716 | 8q24.13-q24.21 | 125,620,117 | 128,955,220 | 3.34 | 5.418 | dmin | NDUFB9, MTSS1, ZNF572, SQLE, KIAA0196, NSMCE2, TRIB1 | ZNF572, SQLE, TRIB1, FAM84B, MYC |
| Amp-CL13 | SW480 | 8q24.13-q24.21 | 126,642,555 | 129,574,570 | 2.93 | 2.367 | Translocation | – | FAM84B, MYC |
| Amp-CL14 | Colo 302DM | 8q24.21 | 127,633,844 | 128,955,220 | 1.32 | 6.443 | dmin/hsr | – | FAM84B, MYC |
| Amp-CL15 | NCI-H716 | 10q26.13 | 123,231,641 | 123,590,573 | 0.36 | 5.070 | dmin | FGFR2 | ATE1 |
| Amp-CL16 | SW480 | 12p12.1-12p11.23 | 21,809,476 | 27,444,930 | 5.64 | 1.377 | Translocation | – | – |
| Amp-CL17 | SK-CO-1 | 12p12.1-12p11.23 | 24,174,625 | 27,717,940 | 3.54 | 1.366 | hsr | – | ARNTL2 |
| Amp-CL18 | Colo 302DM | 13q12.2 | 27,392,825 | 27,439,502 | 0.05 | 5.513 | dmin/hsr | – | – |
| Amp-CL19 | Colo 302DM | 13q22.1 | 72,036,581 | 73,638,705 | 1.54 | 1.728 | n.d. | – | FLJ22624, C13orf37 |
| Amp-CL20 | Colo 302DM | 13q32.2-q32.3 | 97,969,328 | 98,705,914 | 0.74 | 1.291 | n.d. | – | – |
| Amp-CL21 | NCI-H508 | 14q12-q13.1 | 27,552,070 | 32,568,740 | 4.57 | 2.518 | dmin | KIAA1333, STRN3, AP4S1, HECTD1, C14orf126, NUBPL, ARHGAP5 | – |
| Amp-CL22 | SK-CO-1 | 17q24.3-q25.3 | 64,743,167 | 77,119,105 | 12.38 | 1.419 | n.d. | – | SOX9, NAT9, FDXR, RECQL5, SFRS2, TK1, BIRC5, CBX8, CBX4, AZI1, SLC38A10, TMEM105, CCDC40 |
| Amp-CL23 | SK-CO-1 | 18q12.3-q21.2 | 40,535,628 | 46,962,763 | 6.43 | 1.535 | hsr | KIAA1632, SMAD2 | C18orf24 |
| Amp-CL24 | NCI-H716 | 20q13.2-q13.33 | 51,013,575 | 62,363,574 | 11.23 | 1.313 | Translocation | GNAS, GM632 | AURKA, CSTF1, RAE1, STX16, C20orf45, TAF4, SS18L1, CABLES2, C20orf20, C20orf59, YTHDF1, C20orf195, SAMD10 |
| Amp-CL25 | SK-CO-1 | 22q11.22-q12.1 | 21,514,768 | 24,977,835 | 3.46 | 1.784 | hsr | RAB36, CABIN1 | RTDR1, SMARCB1, ADRBK2 |
| Amp-CL26 | SW837 | Xq28 | 149,736,681 | 154,405,100 | 4.67 | 1.676 | Translocation | PASD1, GABRE, ZNF185, CXorf12, MECP2, RPL10, F8A1 | HMGB3, LOC203547, CSAG1, CSAG3, ZNF185, SNORA70, LAGE3, DKC1 |
n.d., Not determined.
Only RefSeq genes are indicated.
Genes of interest (GOI) that showed an expression value greater than twofold relative to the mucosa and at least a threefold separation from the remaining samples.
Genes of interest (GOI) that showed an average expression across all the cell lines and primary tumors higher than twofold (P < 0.05).
Figure 3.
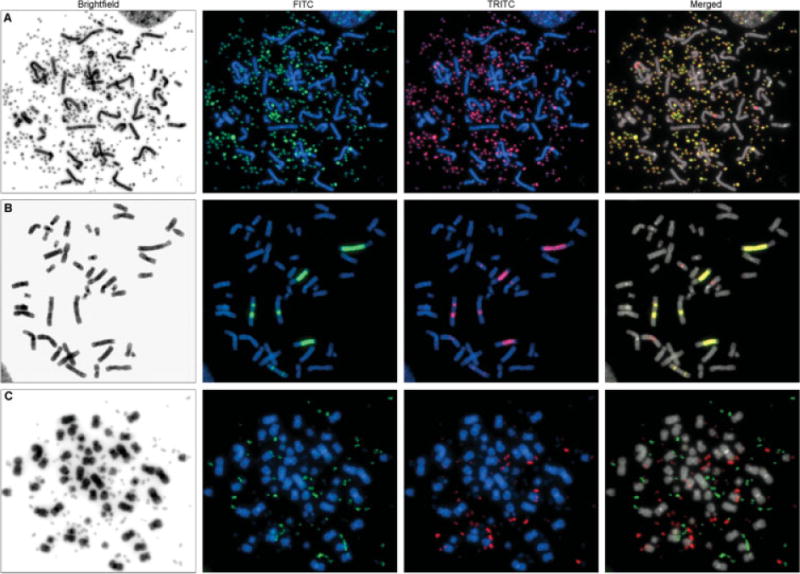
Chromosomal localization of amplified sequences in cell lines Colo 320DM and NCI-H716. Panels A and B show the coamplification of genomic material from chr8:127,633,844-128,955,220 and chr13:27,392,825–27,439,502 as dmin (A) and hsr (B), respectively, in Colo 320DM. Fluorescence in situ hybridization using BAC clones CTD-3056O22 (green) and RP11-153M24 (red) demonstrated the coamplification of target genes MYC and CDX2, respectively, at chromosome locations 8q24.21 and 13q12.2. Overexpression of both genes in this cell line compared to normal mucosa was confirmed by RT-PCR (data not shown). Panel C shows the presence of two distinct populations of dmin in NCI-H716; one is comprised of genomic material from chr8:125,620,117–128,955,220 and the other consists of chr10:123,231,641-123,590,573. Fluorescence in situ hybridization was performed using BAC clones CTD-3056O22 (green) at 8q24.21, and RP11-62L18 (red) at 10q26.13, containing MYC and FGFR2, respectively. Microarray data showed overexpression of these two genes in NCI-H716 compared with normal colon mucosa.
An increase in genomic copy number alone, however, is insufficient for the identification of biologically relevant cancer genes. We therefore combined the aCGH with gene expression data in an attempt to identify those genes within the amplicons that showed a concomitant increase in expression. This resulted in 101 genes whose altered expression was a direct consequence of a genomic amplification based on their up-regulation in the primary tumor or cell line containing the amplicon (Table 1). The increased expression of five (COL14A1, CA14, ADAMTSL4, SLC45A4, and FGFR2) and three (ZNF187, FLOT1, and SYNPO) of these genes in the cell lines and tumors, respectively, was clearly dependent on genomic amplification because the expression levels in the remaining cell lines were actually lower than in the mucosa.
Amplification is only one mechanism whereby the expression level of genes critical to tumorigenesis is increased. Genes mapping within amplicons were therefore evaluated for their average expression across all of the cell lines and primary tumors irrespective of amplification. We identified 98 genes for which gene expression levels, despite being the highest in the samples containing the amplicons, were greater than the normal mucosa across all of the remaining samples (Table 1). For example, MYC was co-amplified with FAM84B, a member of the smc DNA repair complex, in several cell lines. Both genes were also highly transcribed in the majority of the cell lines and primary tumors despite being present in only two copies, raising the possibility that these two genes may be regulated in concert. NCI-H716 contained two distinct populations of dmin; one was comprised of genomic material from chromosome 8, including MYC, and the other consisted of a small amplified region of chromosome 10 containing FGFR2 and ATE1 (Fig. 3C). In this example, FGFR2 displayed a marked overexpression restricted to NCI-H716, whereas ATE1 was up-regulated in most of the samples. Thus, while the vast majority of overexpressed genes are not amplified, identification of those genes that have on occasion been subjected to amplification is one approach for the discovery of potential oncogenes.
Array CGH also revealed focal, high-level copy number losses putatively containing tumor suppressor genes. Fifteen and 25 high-level deletions were identified in the primary tumors and the cell lines, respectively (Table 2). These ranged in size from 100 kb to 22 Mb. Although four genomic locations were found commonly deleted in more than one sample (chr8:11,003,785-11,578,419, chr9:9,099,692-9,455,092, chr9:21,795, 270-22,510,695, and chr20:13,996,399-14,401,156), no deletions occurred in both cell lines and tumors nor was any particular chromosome more prone to these genomic alterations. As was true for the amplifications, none of the near-diploid cell lines contained high-level deletions.
TABLE 2.
Summary of the High-Level Deletions and the Candidate Target Genes Identified in Colon Primary Tumors and Colorectal Cancer Cell Lines
| ID | Sample | Cytoband | Starting bp | Ending bp | Size (Mb) | aCGH ratio | GOI downregulated deletion-specifica,b | GOI downregulated in tumors and cell linesa,c |
|---|---|---|---|---|---|---|---|---|
| High-level deletions in primary tumors | ||||||||
| Del-T1 | CC-P42 | 4p15.33 | 12,548,512 | 13,046,159 | 0.50 | −1.2867 | – | – |
| Del-T2 | CC-P8 | 4q28.1 | 126,606,401 | 126,762,093 | 0.16 | −1.006 | n.d. | FAT4 |
| Del-T3 | CC-P44 | 4q31.1 | 140,982,969 | 141,128,006 | 0.15 | −1.1933 | – | – |
| Del-T4 | CC-P42 | 5q22.2-q23.1 | 112,151,633 | 115,510,011 | 3.36 | −1.2096 | APC | CDO1 |
| Del-T5 | CC-P8 | 5q31.1 | 130,896,505 | 131,005,829 | 0.11 | −2.3969 | n.d. | – |
| Del-T6 | CC-P44 | 5q33.3-q35.3 | 159,578,940 | 180,630,148 | 21.05 | −1.1383 | PDLIM7, ZFP2 | C1QTNF2, GABRG2, SLIT3, DOCK2, KCNMB1, DUSP1, CPEB4, HMP19, PDLIM7, COL23A1, ZFP2, ADAMTS2, LTC4S, GFPT2, FLT4, MGAT1 |
| Del-T7 | CC-P14 | 8p23.3-p23.1 | 11,227 | 11,578,419 | 11.57 | −1.1322 | – | MSRA, SOX7, BLK |
| Del-T8 | CC-P1 | 8p23.1 | 11,003,785 | 11,578,419 | 0.57 | −1.0436 | n.d. | BLK |
| Del-T9 | CC-P48 | 10q26.11 | 121,358,075 | 121,464,144 | 0.11 | −1.4319 | n.d. | – |
| Del-T10 | CC-P44 | 12q21.2 | 75,323,497 | 75,548,096 | 0.22 | −1.0238 | – | – |
| Del-T11 | CC-P44 | 12q24.21-q24.31 | 114,772,695 | 119,747,722 | 4.98 | −1.156 | TMEM118, PEBP1 | HSPB8 |
| Del-T12 | CC-P44 | 12q24.31q24.32 | 123,351,688 | 126,419,287 | 3.07 | −1.1336 | – | – |
| Del-T13 | CC-P4 | 15q23 | 69,430,835 | 70,118,249 | 0.69 | −1.1357 | – | – |
| Del-T14 | CC-P42 | 20p12.1 | 13,965,181 | 15,338,637 | 1.37 | −1.0451 | FLRT3 | – |
| Del-T15 | CC-P44 | 20p12.1 | 13,996,399 | 14,401,156 | 0.40 | −1.1556 | FLRT3 | – |
| High-level deletions in cell lines | ||||||||
| Del-CL1 | LS411N | 1p33 | 49,343,592 | 49,615,373 | 0.27 | −2.4855 | – | – |
| Del-CL2 | T84 | 2q37.3 | 240,010,644 | 242,125,259 | 2.11 | −1.0729 | CR607745, HDLBP | KIF1A, SNED1 |
| Del-CL3 | LS411N | 3p14.2 | 60,310,673 | 60,554,735 | 0.24 | −4.2547 | – | – |
| Del-CL4 | HT-29 | 3p12.3-p11.1 | 81,621,645 | 90,264,118 | 8.64 | −1.4442 | ZNF654 | VGLL3 |
| Del-CL5 | Colo 320DM | 3q12.3-q13.11 | 104,330,485 | 104,604,162 | 0.27 | −1.0195 | – | – |
| Del-CL6 | T84 | 4q32.3-q35.2 | 169,107,625 | 189,395,512 | 20.29 | −1.163 | – | PALLD, SCRG1, HAND2, GPM6A, VEGFC, STOX2, SLC25A4, PDLIM3, SORBS2, FAM149A, CYP4V2 |
| Del-CL7 | SK-CO-1 | 5q31.2 | 138,281,186 | 138,561,371 | 0.28 | −3.8254 | CTNNA1, SIL1 | – |
| Del-CL8 | LS411N | 6q22.33 | 128,394,913 | 128,921,979 | 0.53 | −1.4794 | – | – |
| Del-CL9 | LS411N | 6q24.3 | 147,927,941 | 148,273,805 | 0.35 | −2.7292 | – | – |
| Del-CL10 | Colo 320DM | 7q35 | 145,163,801 | 145,358,245 | 0.19 | −1.1363 | – | – |
| Del-CL11 | Colo 320DM | 7q35 | 146,165,285 | 146,515,514 | 0.35 | −1.172 | – | – |
| Del-CL12 | T84 | 9p24.3-p21.3 | 855,779 | 22,889,584 | 22.03 | −1.2521 | – | C9orf26, MPDZ, NFIB, BNC2, C9orf94, ADFP |
| Del-CL13 | NCI-H716 | 9p23 | 9,099,692 | 9,455,092 | 0.36 | −4.6694 | – | – |
| Del-CL14 | Colo 320DM | 9p21.3 | 21,795,270 | 22,510,695 | 0.72 | −1.1371 | – | – |
| Del-CL15 | NCI-H716 | 10q22.1 | 71,458,068 | 73,219,377 | 1.76 | −1.1721 | AMID, KIAA1274, SGPL1 | C10orf54 |
| Del-CL16 | NCI-H716 | 11p11.12-q13.1 | 51,244,499 | 63,148,801 | 11.90 | −1.3467 | MED19, TMEM138, HEAB, STX3, HRASLS2 | SLC43A3, SERPING1, YPEL4, MS4A6A, MS4A7, NYD-SP21, MS4A1, ZP1, SLC15A3, PGA5, CYBASC3, RAB3IL1, AHNAK, ROM1, GNG3, RARRES3 |
| Del-CL17 | SK-CO-1 | 11q24.3-q25 | 129,864,067 | 132,103,390 | 2.24 | −1.4579 | SNX19 | – |
| Del-CL18 | Colo 201 | 13q34 | 112,869,272 | 114,077,063 | 1.21 | −1.1494 | – | – |
| Del-CL19 | Colo 320DM | 15q21.3 | 55,065,891 | 55,315,120 | 0.25 | −1.0653 | – | – |
| Del-CL20 | SK-CO-1 | 16q22.3-q23.1 | 73,148,905 | 74,268,040 | 1.12 | −1.3298 | TMEM170A | – |
| Del-CL21 | LS411N | 16q23.1 | 77,023,863 | 77,153,748 | 0.13 | −2.5688 | – | – |
| Del-CL22 | LS411N | 16q23.1 | 77,168,499 | 77,370,336 | 0.20 | −1.7107 | – | – |
| Del-CL23 | NCI-H508 | 20p12.1 | 14,517,126 | 14,634,135 | 0.12 | −1.3008 | – | – |
| Del-CL24 | NCI-H508 | 20p12.1 | 14,966,270 | 15,113,421 | 0.15 | −5.3923 | – | – |
| Del-CL25 | NCI-H508 | 20p12.1 | 15,121,525 | 15,225,287 | 0.10 | −3.2493 | – | – |
n.d., Not determined.
Only RefSeq genes are indicated.
Genes of interest (GOI) that showed an expression value smaller than 0.5-fold relative to the mucosa and at least a threefold separation from the remaining samples.
Genes of interest (GOI) that showed an average expression across all the cell lines and primary tumors lower than 0.5-fold (P < 0.05).
Genes found to be specifically down-regulated in samples carrying high-level deletions are indicated in Table 2. In particular, SGPL1, HEAB, MED19, TMEM138, PCID2, ADPRHL1, and TMEM170A in the cell lines, and TRIAP1 in primary tumors were exclusively transcriptionally repressed in those samples with the high-level deletion, attesting to the causative effect of their loss on gene expression levels. Fifty-nine genes mapping within regions of high-level deletion in some samples were likewise deregulated in the remaining samples independent of a genomic loss (Table 2). BLK, present in two different high-level deletions, and FAT4 were the only genes found within a microdeletion (<1 Mb) and commonly down-regulated across all of the samples, suggesting a role in tumor suppression.
One of the endeavers of global gene expresison analysis is to demonstrate the interconnection of differentially expressed genes through their involvement in common biological pathways or cellular processes which could then potentially be targeted therapeutically. Such is the case for some of those genes mapping within amplicons and high-level deletions whose gene expression deregulation was on average more than twofold higher in all of the samples compared with normal mucosa (P < 0.05). Ingenuity Pathway Analysis (Ingenuity Systems) assigned these genes into the cancer, gastrointestinal disease, genetic disorder, and cell cycle biofunctions (P < 1.0E-4). As seen in Figure 4, there is an interrelatedness between these genes, as all of the genes contained in this network converge on the well known oncogene MYC. Thus, the colorectal cancer cells are simultaneously using a multipronged approach to target the activities of a central “hub” protein involved in many aspects of cellular biology and thus essential for tumor growth.
Figure 4.
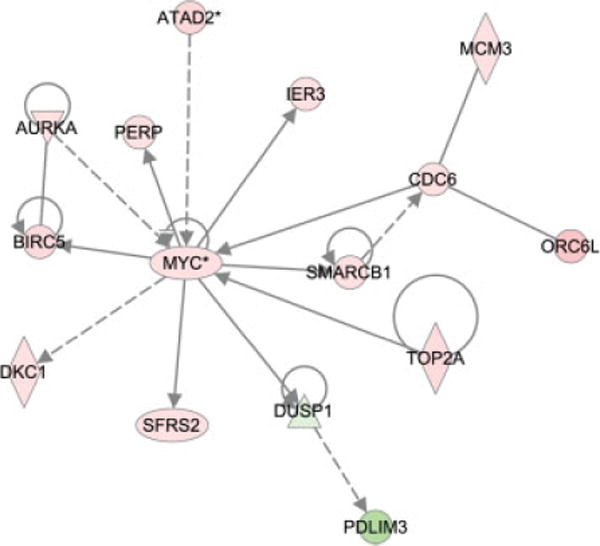
Network of genes located within high-level copy number changes and deregulated across all of the cell lines and tumors. Ingenuity pathway analysis was used to assess the potential interconnection between genes representing the most significantly affected cellular functions. Red, increased expression; green, decreased expression.
Consequences of Chromosomal Breakpoints on Gene Expression
To understand the mechanism by which genomic imbalances arose, we used spectral karyotyping (SKY) to characterize chromosomal aberrations occurring in the 15 CRC cell lines. A total of 87% of the genomic imbalances detected by aCGH correlated with cytogenetically detectable chromosome aberrations elucidated by SKY, thus enabling identification of the molecular events responsible for the observed genomic imbalances. This was particularly informative with respect to the recurrent breakpoints (Supporting Information Table 4). The complete karyotypes of these cell lines will be published elsewhere (Knutsen et al., submitted) and can be retrieved at http://www.ncbi.nlm.nih.gov/projects/sky/.
Sixteen microdeletions and six microduplications flanking sites of copy number alterations were identified in the CRC cell lines with aCGH (Supporting Information Table 5). Analysis of the corresponding breakpoint assessed by SKY enabled us to determine the nature of the chromosomal aberration occurring at the site of these submicroscopic genomic alterations, possibly caused as a consequence of a breakage-fusion-bridge event (Gisselsson et al., 2000). As illustrated in Supporting Information Figure 1, a subtle deletion of chromosome 4 maps to the fusion site in the der(4)t(4;17) in HCT116. Subsequently, this rearrangement underwent a further recombination with chromosome 18 [der(18)t(17;18)t(4;17)]. Although previous examples of this have been described, they involved a single locus-specific analysis in each study (Yoshimoto et al., 2007; Alsop et al., 2008; Li et al., 2008).
Structural reorganization of chromosomes can affect either the expression of genes or their biological functions via premature truncation or fusion events. We identified 1,645 array features mapping within the vicinity of 333 CBS-determined breakpoint regions in the 15 cell lines, of which 75% (n = 1,235) had intensity ratios that could be analyzed. We then identified the features mapping to these breakpoints whose expression was an outlier value (see Materials and Methods). Ninety-nine such features occurred in cell lines containing the breakpoint, 65 (5.27%) with the highest expression and 34 (2.75%) with the lowest expression. Another 534 features occurred in cell lines without the breakpoint. This was statistically significant compared to what would be expected by chance (1.56% and 1.53%, highest and lowest respectively, P < 2.2E-16). After looking closely through the 59 breakpoint regions, eight were regions of amplification and 12 were within deletions. The validity of some breakpoints was difficult to evaluate whereas others mapped near the centromeric repeats, where it was not possible to define narrowly the breakpoint due to the absence of features in the array. In the end, we identified only 36 features that mapped to genes whose altered gene expression could reasonably have been the direct result of a chromosomal break (Table 3). Some of them, namely FOXA2, MRPS35, LOC341346, SRCRB4D, C21orf63, TEMEM98, and WASF3 were deregulated across all of the cell lines and/or tumors (P < 0.05), indicating that chromosome breakage might be one, but not the only, mechanism affecting the expression of these genes.
TABLE 3.
Expression of Genes Mapping at Breakpoints
| Breakpoint | Oligonucleotide/gene name | Cell line | Chr:Mapping position | Expression |
|---|---|---|---|---|
| 1 | MCF2L | Colo 201 | 13:112,800,332-112,800,391 | Increased |
| 2 | AK056384 | Colo 320DM | 21:33,027,032-33,027,091 | Increased |
| 3 | MSRB3 | Colo 320DM | 12:63,966,606-63,966,665 | Increased |
| 4 | RPL34 | Colo 320DM | 4:109,903,909-109,903,968 | Increased |
| 5 | AGXT2L1 | Colo 320DM | 4:110,020,929-110,020,870 | Increased |
| 6 | A_24_P200962 | Colo 320DM | 7:120,210,671-120,210,730 | Increased |
| 7 | FLJ21986 | Colo 320DM | 7:120,362,418-120,362,477 | Increased |
| 8 | FLJ39609 | NCI-H716 | 1:893,632-893,573 | Increased |
| 9 | C20orf56 | NCI-H716 | 20:22,489,440-22,489,381 | Increased |
| 10 | FOXA2b | NCI-H716 | 20:22,509,943-22,509,884 | Increased |
| 11 | AL096727 | NCI-H716 | 20:25,702,745-25,702,686 | Increased |
| 12 | PCOLCE | NCI-H716 | 7:99,848,718-99,848,777 | Increased |
| 13 | ACTL6B | NCI-H716 | 7:99,889,787-99,889,728 | Increased |
| 14 | PYGB | HT-29 | 20:25,226,326-25,226,385 | Increased |
| 15 | FLJ43826 | HT-29 | 17:34,462,723-34,462,782 | Increased |
| 16 | GGTL4 | SK-CO-1 | 22:21,313,375-21,313,434 | Increased |
| 17 | MRPS35a,b | SK-CO-1 | 12:27,800,373-27,800,432 | Increased |
| 18 | CR749704 | SK-CO-1 | 8:58,304,783-58,304,841 | Increased |
| 19 | ACTG1 | SK-CO-1 | 17:77,091,659-77,091,609 | Increased |
| 20 | LOC341346a | SW480 | 12:27,546,306-27,546,365 | Increased |
| 21 | SRCRB4Da,b | SW480 | 7:75,663,359-75,663,300 | Increased |
| 22 | THC2317822 | SW480 | 5:93,765,126-93,765,067 | Increased |
| 23 | AF118067 | SW837 | 17:20,855,929-20,855,988 | Increased |
| 24 | PTGER3 | SW837 | 1:71,030,382-71,030,323 | Increased |
| 25 | RPS12 | Colo 201 | 6:133,180,332-133,180,391 | Decreased |
| 26 | C21orf63b | Colo 320DM | 21:32,751,887-32,761,921 | Decreased |
| 27 | HAP1 | Colo 320DM | 17:37,132,425-37,132,418 | Decreased |
| 28 | PLEKHN1 | NCI-H716 | 1:950,367-950,426 | Decreased |
| 29 | GINS1 | HT-29 | 20:25,346,791-25,353,865 | Decreased |
| 30 | TMEM98a | HT-29 | 17:28,292,241-28,292,300 | Decreased |
| 31 | KPNA2 | HT-29 | 17:63,473,120-63,473,179 | Decreased |
| 32 | ARHGEF7 | HT-29 | 13:110,745,422-110,745,481 | Decreased |
| 33 | WASF3a,b | SK-CO-1 | 13:26,160,694-26,160,753 | Decreased |
| 34 | BAHCC1 | SK-CO-1 | 17:77,047,529-77,047,588 | Decreased |
| 35 | BC047380 | SW837 | 22:24,176,324-24,177,801 | Decreased |
| 36 | LRRC40 | SW837 | 1:70,322,640-70,322,581 | Decreased |
Deregulation of this gene in the same direction as the sample with the breakpoint was observed across all the CRC cell lines (P < 0.05).
Deregulation of this gene in the same direction as the sample with the breakpoint was observed across all the primary colorectal tumors (P < 0.05).
Structural Variants of the Genome Colocalize with Chromosomal Breakpoints
The total number of DNA breakpoints that we identified by aCGH was 333, ranging from one to six in the near-diploid and from 11 to 50 in the aneuploid CRC cell lines. In agreement with our previous results in primary tumors (Camps et al., 2008), 45.9% of the breakpoints in the CRC cell lines occurred within sites of known structural variants of the genome (P < 1.0E-11), either CNVs or segmental duplications (Fig. 5 and Supporting Information Table 6). As for the microdeletions and microduplications associated with chromosomal breakpoints, five spanned a CNV and the other 12 contained a CNV at one end of the imbalance. Interestingly, 51% of the amplicons contained a structural variant at one or both ends, suggesting that these features are not only involved in DNA double strand breaks that result in chromosomal translocations, but that these breaks might result in the generation of high-level copy number gains more frequently than expected by chance (P < 0.0005). In contrast, only 32% (P = 0.3) of high-level deletions contained a structural variant in at least one end of the deletion. We then interrogated the distribution of CNVs in each chromosome aberration detected by SKY. Results indicated that 52.5% of the genomic rearrangements involved a structural variant for at least one partner of the chromosome marker. Of these, 24.5% contain structural variants in both ends of the partners that originate the chromosome aberration.
Figure 5.
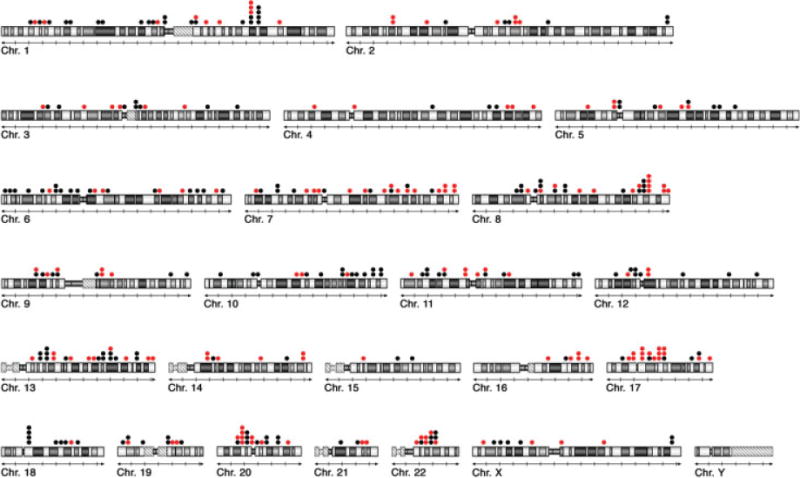
Prevalence of chromosomal breakpoints at sites of structural variants of the human genome. A total of 333 breakpoints were mapped and their coordinates compared with the physical position of CNVs and SDs annotated in the Database of Genomic Varaints (http://projects.tcag.ca/variation). Red dots (n = 151) indicate the location of breakpoints that coincide with sites of structural variants of the human genome.
Because CNVs occur with the same frequency at breakpoints in the primary CRC tumors and the CRC cell lines, we examined the extent to which the breakpoints were shared among the samples. We identified 710 breakpoints in the 15 CRC cell lines and 31 primary colon carcinomas, of which 45 occurred in two or more samples (Supporting Information Table 4). A total of 237 annotated CNVs mapped to breakpoints (n = 309), of which 15 were shared among the tumors, seven were located within breaks in two or more cell lines, and nine resided within breakpoint regions found in both the tumors and the cell lines. Thus, 13% of the CNVs mapped within regions of the genome where changes in copy number occurred in multiple samples.
DISCUSSION
This study represents a systematic and comprehensive integration of SKY, aCGH, and gene expression data of colorectal cancer. While our data are in general agreement with previously published cytogenetic and molecular cytogenetic analyses (Abdel-Rahman et al., 2001; Roschke et al., 2003; Camps et al., 2004; Kleivi et al., 2004), the fine mapping of breakpoints, identification of subtle regions of amplification and high-level deletions, refinement of the composition of dmin and hsr, and determining their consequences at the gene expression level are an important advancement for identifying relevant tumor-related events and gene loci involvement. Our results corroborate the finding that the main consequence of chromosomal aneuploidy in cancer is to affect the average expression of all genes, rather than a select few, within the regions of copy number alteration (Monni et al., 2001; Pollack et al., 2002; Grade et al., 2006). Analysis of genes localized within focal amplifications and deletions, however, demonstrated a tendency toward the deregulation of specific genes. Thus, our analysis resulted in the identification of several putative oncogenes and tumor suppressor genes for which an association with colorectal cancer has hitherto not been described. Furthermore, the expression of genes mapping near breakpoints was significantly affected. However, we did not find recurrent breakpoints in the majority of the samples. We therefore conclude that in contrast to what is observed in hematologic malignancies where recurrent breakpoints are common (Mitelman et al., 2004), breakpoints do not represent a frequent mechanism to deregulate gene expression in colorectal tumorigenesis.
The comparison of cell lines and primary tumors in this study shows that CRC cell lines maintain genomic imbalances identified in primary colon tumors with a high fidelity (Fig. 1). The number of CNAs, including high-level copy number changes, was nearly 40% higher in the cell lines, most of which occurring in the mismatch repair proficient, aneuploid lines. In addition, our data showed that primary tumors tended to contain more whole chromosome arm alterations, whereas smaller chromosomal regions were predominantly involved in structural rearrangements in the cell lines, reflected also on the wide spread distribution of the chromosomal breakpoints along the genome (Fig. 5). Thus, either culture conditions compared to the tumor microenvironment and/or the developmental “age” of the cell lines resulted in the accumulation of a higher level of genomic instability.
Global genomic examination of these cell lines corroborated our recent observation that chromosomal breakpoints in primary tumors occur preferentially at sites of structural variants of the human genome (Camps et al., 2008). Subsequently, this phenomenon has also been shown in mantle cell lymphoma (Bea et al., 2009). Two specific examples are the genomic amplifications involving chromosome bands 8q24.1–24.3 and 12p11.23–12.1 that occurred in multiple cell lines. The boundaries of these amplicons were not identical in each of the cell lines, but the clustering of breakpoints and the ensuing amplification indicate that these genomic regions are unstable and prone to chromosomal breaks. Interestingly, five of the 12 breakpoints leading to these two amplifications occurred at sites of CNVs. Thus, we conclude that CNVs not only appear to promote double strand breaks that lead to chromosomal translocations, but are also significantly (P < 0.0005) involved in the mechanism that leads to localized high-level copy number amplifications. Such an association was not observed for deletion events. Because the frequency of CNV-associated breaks is not altered by the increased accumulation of genomic aberrations in the cell lines, we conclude that this CNV-specific instability remains active in these samples perhaps as a potential mechanism to generate CNAs.
A direct link between genes affected by either high-level amplification or loss-of-heterozygosity and tumorigenesis has clearly been demonstrated in solid tumors and has in some instances provided targets for therapeutic intervention (Clark and Cookson, 2008; Prat and Baselga, 2008). Applying this approach, we identified 37 amplicons within the 46 samples analyzed, of which only four were observed in more than one sample. BYSL, MYC, FAM84B, SEQL, and TRIB1 were recurrently amplified and overexpressed. Interestingly, several genes mapping within amplicons were significantly overexpressed in the cell lines and tumors irrespective of their copy number; however, those samples with an amplicon generally had higher expression, suggesting that the transcription of these genes was increased as a direct consequence of the change in gene dosage (Table 1). BYSL, a gene involved in ribosome biogenesis and cell growth, maps within the amplified region chr6:41,451,467-42,008,700 in Colo 201 and the primary tumor CC-P14. Overexpression of this gene has previously been described in several human cancer cell lines (Miyoshi et al., 2007), in diffuse large B-cell lymphoma (Kasugai et al., 2005), and in primary gastric cancer (Tsukamoto et al., 2008). KIAA1333 and C14orf126 within the amplification at chromosome 14 in NCI-H508 were also overexpressed in all of the colorectal cell lines, and their expression was further enhanced more than threefold in NCI-H508, again reflecting an amplicon-specific effect on gene expression. The high expression level of KIAA1333 in some of the primary tumors further supports its oncogenic potential.
A number of amplified regions, conversely, did not contain any genes with increased expression across the samples. While it is formally possible that increased copy number of these genomic regions does not convey any advantage to the cancer cell, the potential exists for alterations in other genomic elements such as non-coding RNAs. Two such examples are chr8:10,607,890-10,995,687 and chr12: 21,809,476-27,444,930 in CC-P1 and SW480, respectively, which contain known miRNAs.
A similar approach using high-level genomic deletions as a means to detect putative colon cancer tumor suppressor genes resulted in the identification of BLK and FAT4. Although these genes demonstrated the lowest expression in those samples harboring the genomic deletion, they were systematically down-regulated in all of the samples relative to the normal mucosa. FAT4, involved in kidney development (Saburi et al., 2008), has recently been proposed to be a tumor suppressor gene as its transcriptional repression in the non-tumorigenic mammary epithelial cell line NOG8 induced tumorigenesis (Qi et al., 2009). We suggest that FAT4 might be one of the candidate genes that lead to the selection of the common genomic loss of 4q in later stages of colorectal cancer (Arribas et al., 1999; Knösel et al., 2004).
Regions of copy number alteration may in fact harbor multiple genes whose altered expression is part of the etiology. One such example is the invariable coamplification of FAM84B with MYC, which occurred independently in three different cell lines (Table 1). Both of these genes displayed increased expression levels that were directly correlated with gene dosage. Although further functional analyses are required to determine whether an interaction exists between the biological actions of these two proteins, our data at the least support a model in which multiple overexpressed genes contained within an amplicon may contribute to the oncogenic phenotype. Examples of this phenomenon have been demonstrated in several tumor types (Guan et al., 1994; Squire et al., 1995; Huang et al., 2006; Kendall et al., 2007), but this is to our knowledge the first description of its occurrence in colorectal cancer.
In conclusion, we carried out the integration of molecular cytogenetics, genome-wide gene copy number, and expression microarray profiling of colorectal cancer cell lines and primary colon adenocarcinomas, and further applied statistical analysis to identify putiative target genes that are deregulated in association with high-level copy number changes. A comprehensive comparison of the aberration patterns between cell lines and primary tumors supports the usage of in vitro models to assess further functional genomics. Investigation of clinical significance and biological validation studies should be conducted to elucidate the mechanism of action of the target genes.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Yidong Chen for discussion and generating the visual outputs of the aCGH and gene expression data and Dr. Michael Erdos for use of NHGRI instrumentation. The authors also thank Buddy Chen and Joseph Cheng for IT and editorial assistance.
Supported by: Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Abdel-Rahman WM, Katsura K, Rens W, Gorman PA, Sheer D, Bicknell D, Bodmer WF, Arends MJ, Wyllie AH, Edwards PA. Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement. Proc Natl Acad Sci USA. 2001;98:2538–2543. doi: 10.1073/pnas.041603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson DG. Gene amplification in cancer. Trends Genet. 2006;22:447–455. doi: 10.1016/j.tig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Alsop AE, Taylor K, Zhang J, Gabra H, Paige AJ, Edwards PA. Homozygous deletions may be markers of nearby heterozygous mutations: The complex deletion at FRA16D in the HCT116 colon cancer cell line removes exons of WWOX. Genes Chromosomes Cancer. 2008;47:437–447. doi: 10.1002/gcc.20548. [DOI] [PubMed] [Google Scholar]
- Arribas R, Risques RA, Gonzalez-Garcia I, Masramon L, Aiza G, Ribas M, Capella G, Peinado MA. Tracking recurrent quantitative genomic alterations in colorectal cancer: Allelic losses in chromosome 4 correlate with tumor aggressiveness. Lab Invest. 1999;79:111–122. [PubMed] [Google Scholar]
- Bardi G, Johansson B, Pandis N, Mandahl N, Bak-Jensen E, Lindstrom C, Tornqvist A, Frederiksen H, Andren-Sandberg A, Mitelman F, Heim S. Cytogenetic analysis of 52 colorectal carcinomas—Non-random aberration pattern and correlation with pathologic parameters. Int J Cancer. 1993;55:422–428. doi: 10.1002/ijc.2910550317. [DOI] [PubMed] [Google Scholar]
- Bea S, Salaverria I, Armengol L, Pinyol M, Fernandez V, Hartmann EM, Jares P, Amador V, Hernandez L, Navarro A, Ott G, Rosenwald A, Estivill X, Campo E. Uniparental disomies, homozygous deletions, amplifications and target genes in mantle cell lymphoma revealed by integrative high-resolution whole genome profiling. Blood. 2009;113:3059–3069. doi: 10.1182/blood-2008-07-170183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Camps J, Armengol G, del Rey J, Lozano JJ, Vauhkonen H, Prat E, Egozcue J, Sumoy L, Knuutila S, Miro R. Genome-wide differences between microsatellite stable and unstable colorectal tumors. Carcinogenesis. 2006;27:419–428. doi: 10.1093/carcin/bgi244. [DOI] [PubMed] [Google Scholar]
- Camps J, Grade M, Nguyen QT, Hormann P, Becker S, Hummon AB, Rodriguez V, Chandrasekharappa S, Chen Y, Difilippantonio MJ, Becker H, Ghadimi BM, Ried T. Chromosomal breakpoints in primary colon cancer cluster at sites of structural variants in the genome. Cancer Res. 2008;68:1284–1295. doi: 10.1158/0008-5472.CAN-07-2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps J, Morales C, Prat E, Ribas M, Capella G, Egozcue J, Peinado MA, Miro R. Genetic evolution in colon cancer KM12 cells and metastatic derivates. Int J Cancer. 2004;110:869–874. doi: 10.1002/ijc.20195. [DOI] [PubMed] [Google Scholar]
- Clark PE, Cookson MS. The von Hippel-Lindau gene: Turning discovery into therapy. Cancer. 2008;113:1768–1778. doi: 10.1002/cncr.23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas EJ, Fiegler H, Rowan A, Halford S, Bicknell DC, Bodmer W, Tomlinson IP, Carter NP. Array comparative genomic hybridization analysis of colorectal cancer cell lines and primary carcinomas. Cancer Res. 2004;64:4817–4825. doi: 10.1158/0008-5472.CAN-04-0328. [DOI] [PubMed] [Google Scholar]
- Eshleman JR, Casey G, Kochera ME, Sedwick WD, Swinler SE, Veigl ML, Willson JK, Schwartz S, Markowitz SD. Chromosome number and structure both are markedly stable in RER colorectal cancers and are not destabilized by mutation of p53. Oncogene. 1998;17:719–725. doi: 10.1038/sj.onc.1201986. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Fix A, Lucchesi C, Ribeiro A, Lequin D, Pierron G, Schleiermacher G, Delattre O, Janoueix-Lerosey I. Characterization of amplicons in neuroblastoma: High-resolution mapping using DNA microarrays, relationship with outcome, and identification of overexpressed genes. Genes Chromosomes Cancer. 2008;47:819–834. doi: 10.1002/gcc.20583. [DOI] [PubMed] [Google Scholar]
- Ghadimi BM, Sackett DL, Difilippantonio MJ, Schrock E, Neumann T, Jauho A, Auer G, Ried T. Centrosome amplification and instability occurs exclusively in aneuploid, but not in diploid colorectal cancer cell lines, and correlates with numerical chromosomal aberrations. Genes Chromosomes Cancer. 2000;27:183–190. [PMC free article] [PubMed] [Google Scholar]
- Gisselsson D, Pettersson L, Hoglund M, Heidenblad M, Gorunova L, Wiegant J, Mertens F, Dal Cin P, Mitelman F, Mandahl N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci USA. 2000;97:5357–5362. doi: 10.1073/pnas.090013497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grade M, Ghadimi BM, Varma S, Simon R, Wangsa D, Barenboim-Stapleton L, Liersch T, Becker H, Ried T, Difilippantonio MJ. Aneuploidy-dependent massive deregulation of the cellular transcriptome and apparent divergence of the Wnt/beta-catenin signaling pathway in human rectal carcinomas. Cancer Res. 2006;66:267–282. doi: 10.1158/0008-5472.CAN-05-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XY, Meltzer PS, Dalton WS, Trent JM. Identification of cryptic sites of DNA sequence amplification in human breast cancer by chromosome microdissection. Nat Genet. 1994;8:155–161. doi: 10.1038/ng1094-155. [DOI] [PubMed] [Google Scholar]
- Huang XP, Rong TH, Wang JY, Tang YQ, Li BJ, Xu DR, Zhao MQ, Zhang LJ, Fang Y, Su XD, Liang QW. Negative implication of C-MYC as an amplification target in esophageal cancer. Cancer Genet Cytogenet. 2006;165:20–24. doi: 10.1016/j.cancergencyto.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Kasugai Y, Tagawa H, Kameoka Y, Morishima Y, Nakamura S, Seto M. Identification of CCND3 and BYSL as candidate targets for the 6p21 amplification in diffuse large B-cell lymphoma. Clin Cancer Res. 2005;11:8265–8272. doi: 10.1158/1078-0432.CCR-05-1028. [DOI] [PubMed] [Google Scholar]
- Kendall J, Liu Q, Bakleh A, Krasnitz A, Nguyen KC, Lakshmi B, Gerald WL, Powers S, Mu D. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci USA. 2007;104:16663–16668. doi: 10.1073/pnas.0708286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleivi K, Teixeira MR, Eknaes M, Diep CB, Jakobsen KS, Hamelin R, Lothe RA. Genome signatures of colon carcinoma cell lines. Cancer Genet Cytogenet. 2004;155:119–131. doi: 10.1016/j.cancergencyto.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Knösel T, Schluns K, Stein U, Schwabe H, Schlag PM, Dietel M, Petersen I. Chromosomal alterations during lymphatic and liver metastasis formation of colorectal cancer. Neoplasia. 2004;6:23–28. doi: 10.1016/s1476-5586(04)80050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MM, Nimmakayalu MA, Mercer D, Andersson HC, Emanuel BS. Characterization of a cryptic 3.3 Mb deletion in a patient with a “balanced t(15;22) translocation” using high density oligo array CGH and gene expression arrays. Am J Med Genet A. 2008;146:368–375. doi: 10.1002/ajmg.a.32116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ES, Tonon G, Sinha R, Xiao Y, Feng B, Kimmelman AC, Protopopov A, Ivanova E, Brennan C, Montgomery K, Kucherlapati R, Bailey G, Redston M, Chin L, DePinho RA. Common and distinct genomic events in sporadic colorectal cancer and diverse cancer types. Cancer Res. 2007;67:10736–10743. doi: 10.1158/0008-5472.CAN-07-2742. [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet. 2004;36:331–334. doi: 10.1038/ng1335. [DOI] [PubMed] [Google Scholar]
- Miyoshi M, Okajima T, Matsuda T, Fukuda MN, Nadano D. Bystin in human cancer cells: Intracellular localization and function in ribosome biogenesis. Biochem J. 2007;404:373–381. doi: 10.1042/BJ20061597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monni O, Barlund M, Mousses S, Kononen J, Sauter G, Heiskanen M, Paavola P, Avela K, Chen Y, Bittner ML, Kallioniemi A. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc Natl Acad Sci USA. 2001;98:5711–5716. doi: 10.1073/pnas.091582298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllykangas S, Knuutila S. Manifestation, mechanisms and mysteries of gene amplifications. Cancer Lett. 2006;232:79–89. doi: 10.1016/j.canlet.2005.07.045. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Baselga J. The role of hormonal therapy in the management of hormonal-receptor-positive breast cancer with co-expression of HER2. Nat Clin Pract Oncol. 2008;5:531–542. doi: 10.1038/ncponc1179. [DOI] [PubMed] [Google Scholar]
- Qi C, Zhu YT, Hu L, Zhu YJ. Identification of Fat4 as a candidate tumor suppressor gene in breast cancers. Int J Cancer. 2009;124:793–798. doi: 10.1002/ijc.23775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried T, Knutzen R, Steinbeck R, Blegen H, Schrock E, Heselmeyer K, du Manoir S, Auer G. Comparative genomic hybridization reveals a specific pattern of chromosomal gains and losses during the genesis of colorectal tumors. Genes Chromosomes Cancer. 1996;15:234–245. doi: 10.1002/(SICI)1098-2264(199604)15:4<234::AID-GCC5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Roschke AV, Tonon G, Gehlhaus KS, McTyre N, Bussey KJ, Lababidi S, Scudiero DA, Weinstein JN, Kirsch IR. Karyotypic complexity of the NCI-60 drug-screening panel. Cancer Res. 2003;63:8634–8647. [PubMed] [Google Scholar]
- Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat Genet. 2008;40:1010–1015. doi: 10.1038/ng.179. [DOI] [PubMed] [Google Scholar]
- Squire JA, Thorner PS, Weitzman S, Maggi JD, Dirks P, Doyle J, Hale M, Godbout R. Co-amplification of MYCN and a DEAD box gene (DDX1) in primary neuroblastoma. Oncogene. 1995;10:1417–1422. [PubMed] [Google Scholar]
- Tsukamoto Y, Uchida T, Karnan S, Noguchi T, Nguyen LT, Tanigawa M, Takeuchi I, Matsuura K, Hijiya N, Nakada C, Kishida T, Kawahara K, Ito H, Murakami K, Fujioka T, Seto M, Moriyama M. Genome-wide analysis of DNA copy number alterations and gene expression in gastric cancer. J Pathol. 2008;216:471–482. doi: 10.1002/path.2424. [DOI] [PubMed] [Google Scholar]
- Yoshimoto M, Ludkovski O, Bayani J, Graham C, Zielenska M, Squire JA. Microdeletion and concurrent translocation associated with a complex TMPRSS2:ERG prostate cancer gene fusion. Genes Chromosomes Cancer. 2007;46:861–863. doi: 10.1002/gcc.20470. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.