

Abstract
Objective
To assess the effects of proteasome inhibition on the development of burn-induced hypermetabolism.
Methods
Rats underwent 30–40% TBSA scald burn or sham injury. The proteasome inhibitor bortezomib (0.1 mg/kg) or vehicle (n=10) was administered i.p. 3×weekly starting at 2h (Early-Bortezomib, n=20) or 48h (Late-Bortezomib, n=13) post-burn. Body weights were determined weekly. Resting energy expenditures (REE) were measured at days 0 (baseline), 7, 14, 21 and 42 post-burn. At day 42, blood and pectoral muscle were harvested. Routine blood chemistry parameters were analyzed. Proteasome content, proteasome peptidase activities and ubiquitin-protein conjugates were measured in muscle extracts.
Results
As compared with sham-vehicle treated animals, specific proteasome activities were increased after burn and vehicle treatment. Bortezomib treatment inhibited proteasome activities and increased ubiquitin-protein conjugates after sham and burn injury. Bortezomib treatment did not affect REE after sham procedure. REE significantly increased by 47% within 7 days and remained elevated until day 42 after burn and vehicle treatment. After Early-Bortezomib treatment, burn-induced increases in REE were delayed and significantly reduced by 42% at day 42, as compared with vehicle treatment. With Late-Bortezomib treatment, burn-induced increases in REE were also delayed but not attenuated at day 42. Mortality was 20% with vehicle, 65% (median survival time: 1.875 days) with Early-Bortezomib and 25% with Late-Bortezomib treatment after burns (p<0.05 Early-Bortezomib vs. vehicle and Late-Bortezomib).
Conclusions
Proteasome inhibition delays development of burn-induced hypermetabolism. Although proteasome inhibition early after burn injury reduces the hypermetabolic response, it significantly increases early burn-associated mortality.
Keywords: Bortezomib, resting energy expenditure, hypermetabolism, mortality, ubiquitin-protein conjugates, skeletal muscle
Introduction
Severe thermal injury induces hypermetabolism, which persists for months to years after complete wound closure (1–5). The hallmark of the post-burn hypermetabolic response in patients is increased energy expenditure with profound protein loss. Burn-induced increases in resting energy expenditure (REE) are correlated with the degree and size of the burn injury and can reach almost 200% of normal (2, 6–8). Because skeletal muscle accounts for the majority of body cell dry weight and body protein, muscle catabolism dominates this response and is associated with increased morbidity, prolonged recovery periods and potential mortality (1, 2, 9, 10). Although anabolic compounds seem to attenuate consequences of burn-induced hypermetabolism(11–14), causative pharmacological approaches that limit the development and duration of hypermetabolism are not available. Such drugs, however, are desirable as they may provide the opportunity to reduce muscle cachexia and improve outcomes from severe burn injuries.
Whereas the exact mechanisms leading to hypermetabolism and muscle catabolism after burns are not well understood, studies on muscle protein turnover suggested that exaggerated protein degradation is the primary effector of muscle catabolism (15).
The ubiquitin-proteasome pathway of protein degradation (UPP) is the major non-lysosomal proteolytic pathway in all eukaryotic cells (16). The UPP is involved in the regulation of a multitude of biological processes, including protein turnover, and has been shown to contribute to the pathophysiology of various diseases (17). Increased proteolysis via the UPP is thought to be a principal cause for muscle wasting in various catabolic conditions, including cancer cachexia, sepsis and burns (16, 18). Selective proteasome inhibitors have been developed, and bortezomib, a reversible proteasome inhibitor, and carfilzomib, an irreversible proteasome inhibitor, have been approved by the US Food and Drug Administration (FDA) for the treatment of multiple myeloma and mantle cell lymphoma (19, 20). The effects of proteasome inhibition on the development of hypermetabolism after burns, however, are unknown. Thus, it was the aim of the present study to determine how proteasome inhibition affects the development of burn-induced hypermetabolism in a rat scald burn model. To inhibit the proteasome, we employed the reversible FDA approved proteasome inhibitor bortezomib and utilized measurements of REE throughout a six week period as a physiologically relevant read-out for burn-induced hypermetabolism.
Materials and Methods
Animal Protocol
All procedures were performed according to National Institutes of Health (NIH) Guidelines for Use of Laboratory Animals and approved by the Loyola Institutional Animal Care and Use Committee (IACUC) and the Department of Defense Animal Care and Use Review Office (ACURO). Male Sprague Dawley rats (325–375g body weight, Harlan, Indianapolis, IN) were anesthetized with 2.5% isoflurane, shaved and placed into a template that exposes a dorsal body area corresponding to 30–40% of their total body surface area. A full thickness burn was then induced by immersion of the dorsal skin into boiling water for 17 seconds (21). Sham animals were treated as described, except that their dorsal surface was immersed in tepid water. Animals were then resuscitated with crystalloid solution i.p. as per the Parkland formula (4 mL/kg per percent TBSA) over the first 48h after burn injury. The following experimental groups were performed:
Sham – vehicle treatment (n = 10). Animals underwent sham injury and 0.4 mL of 0.9% NaCl (=vehicle) was injected i.p. immediately following sham injury.
Sham – bortezomib treatment (n = 13). Animals underwent sham injury and 0.1 mg/kg of bortezomib in 0.4 mL of 0.9% NaCl was injected i.p. three times weekly, beginning at 2 h after sham injury
Burn – vehicle treatment (n = 10). Animals underwent burn injury and 0.4 mL 0.9% NaCl was injected i.p. three times weekly, beginning at 2h after burn injury.
Burn – Early Bortezomib (n = 20). Animals underwent burn injury and 0.1 mg/kg bortezomib in 0.4 mL 0.9% NaCl was injected i.p. three times weekly, beginning at 2h after burn injury.
Burn – Late Bortezomib (n = 13). Animals underwent burn injury and 0.1 mg/kg bortezomib in 0.4 mL 0.9% NaCl was injected i.p. three times weekly, beginning at 48h after burn injury.
The dose of bortezomib was chosen based on its toxicity profile during long term administration in previous studies (22, 23). On day 42 after sham procedure or burn injury, animals were euthanized (isoflurane inhalation, bilateral pneumothorax), and pectoral muscle and blood were harvested. We selected pectoral muscle, a red muscle (24), as an uninjured skeletal muscle remote from the dorsal burn injury to exclude possible effects of direct thermal injury and to be able to compare results from the present study with our previous observations within the same time interval (25).
Muscle biopsies were snap frozen in liquid nitrogen and stored at −70°C until further processing. Blood was used for blood gas analyses and plasma preparation. Plasma was stored at −70°C until further analyses were performed.
Resting Energy Expenditure (REE)
REE was measured using indirect calorimetry, as described (25, 26). Respiratory gas exchange was measured in an open-circuit respirometer (Columbus Instruments, OH). Rats were placed in a plexiglas metabolic chamber (4 L capacity). Air inlets and outlets contained columns of calcium sulfate to dry both inlet and expired air. Airflow rate was monitored continuously for 2 min per rat per cycle for 6 cycles (total of 12 min), and oxygen consumption and CO2 production were calculated by multiplying the rate of airflow by changes in O2 and CO2 concentrations of air entering and exiting the chamber. From these values, the differences in O2 intake (DO2), CO2 (DCO2) output and REE were calculated using the Oxymax software (Columbus Instruments, OH). REE was measured at baseline (day 0) and on days 7, 14, 21 and 42 after burn injury or sham procedure, respectively.
Blood gas analyses
Arterial blood was used for measurements of pH, pCO2, pO2, hemoglobin, sodium, glucose and lactate using a blood gas analyzer (Stat Profile pHOx Plus L, Nova Biomedical).
Blood Chemistry
Plasma samples were assayed for total protein, cholesterol, glucose, sodium, creatinine, blood urea nitrogen (BUN), gamma-glutamyltranspeptidase (GGT), alanine aminotransferase (ALT), and alkaline phosphatase (ALP), using a veterinary blood chemistry analyzer (DRI-CHEM 7000 Chemistry Analyzer, Heska).
Complete blood counts (CBC)
Heparinized blood samples were used for the analyses of CBC on a veterinary hematology analyzer (HemaTrue, Heska).
Preparation of tissue extracts
Snap frozen pectoral muscles were homogenized in 1/10 phosphate buffered saline, pH 7.4 (1:5 weight/volume), centrifuged (16,600×g, 4°C, 30min) and supernatants (=extracts) aliquoted, as described (27, 28). Protein concentrations in the tissue extracts were determined using the DC protein assay (Bio-Rad, Hercules, CA). All measurements in tissue extracts were standardized to total protein content.
Proteasome peptidase activities
Proteasome peptidase activities were measured employing the fluorogenic chymotryptic-like peptide substrate N-Suc-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Suc-LLVY-AMC, Biomol), as described (27, 29, 30). Reaction mixtures contained 10 mM Tris/HCl, pH 7.5, 2 mM ATP, 5 mM MgCl2, 200 µM peptide substrate and 50 µg of muscle extract protein. Mixtures were incubated for 40 minutes at 37°C. Ethanol (2:1; volume: volume) was added, mixtures placed on ice for 10 minutes and centrifuged at 16000 × g, 5°C for 6 minutes. Supernatants were transferred into microplates (Corning, Acton, MA) and free 7-amino-4-methylcoumarincleaved from the substrates measured in a microplate reader (Synergy 2, Biotek, λexcitation/emission=340/440 nm) against standard curves of 7-amino-4-methylcoumarin (Sigma). To differentiate the proteasome from other peptidase activities, the epoxomicin (specific proteasome inhibitor) sensitive proportion was determined by addition of 7 µM epoxomicin (Boston Biochem) to the mixtures (31). All enzyme assays were performed immediately after preparation of the muscle extracts to prevent proteasome inactivation by freeze-thawing. Enzyme time progression curves showed linearity for 40 min. Proteasome peptidase activity (total peptidase activity minus peptidase activity in the presence of epoxomicin) is expressed as 7-amino-4-methylcoumarin released from the peptide substrate (relative fluorescence units; RFU) per 40 min and per mg of extract protein. Specific proteasome activity is expressed as RFU/40 min/ng 20S proteasome because the 20S core particle contains the catalytic sites in free 20S particles and within the 26S proteasome complex.
20S proteasome enzyme linked immunosorbent assay (ELISA)
20S proteasome content in muscle extracts was quantified by ELISA, as described in detail previously(29). In brief, microtiter plates (NuncMaxisorb, NalgeNunc International, Rochester, NY) were coated with anti-20S subunit α6 (PW8100, Biomol, Plymouth Meeting, PA) diluted in phosphate buffer saline (PBS), pH 7.4 over-night at 4°C. After coating plates were blocked with PBS, 1% bovine serum albumin (Sigma, PBS-BSA). Standard curves were prepared employing highly purified 20S (PW8270; Biomol) diluted in PBS-BSA. 100 μL of standards and samples diluted 1:5 in PBS-BSA were placed in the wells and incubated for two hours. After washing the plates, the anti-20S “core subunits” (α5,α7, β1, β5, β5i, β7) (PW8155, Biomol) was added to the wells and incubated for 1 hour at room temperature. The plates were washed again and HRP labeled anti-rabbit (GE Healthcare, Piscataway, NJ) was added. After 1 hour of incubation, plates were washed again and the bound antibodies were detected using tetramethylbenzidine (TMB, Sigma-Aldrich, St. Louis, MO). The reaction was stopped by addition of 100 µL 2NHCL and the optical densities (OD) were determined at 450/540 nm in a microplate reader (Synergy 2, Biotek Instruments, Winooski, VT). The 20S proteasome ELISA detects free 20S proteasomes and 20S proteasomes within the 26S proteasome complex (29). The lower detection limit was 1.5 ng/mL 20S proteasome.
Western blots
Western blotting with anti-ubiquitin (Life Sensors, Malvern, PA) was performed as described (27, 28, 32). Anti-glyceraldehyde 3-phosphate dehydrogenase (Anti-GAPDH; Applied Biosciences, Foster City, CA) in combination with HRP labeled anti-mouse (GE Healthcare, Burr Ridge, IL) were used as protein loading control. Chemiluminescence signals were detected with a Chemidoc imaging system (BioRad, Hercules, CA).
Power analysis, data analyses and statistics
The number of animals in each group was chosen based on a power analysis that was calculated with the software program StatMate (GraphPad Software). Based on our previous observations in this animal model (25), we estimated a standard deviation of 15% for REE as the primary outcome variable and assumed that mortality after burn injury will be below 30%. Thus, with a sample size of at least n = 10 animals in each group, we assumed that 7–8 animals will survive the entire observation period. We calculated that a sample size of n=7 provides a power of 0.8 to detect a 24.5% difference in REE between groups on a two-tailed p<0.05 level. As we observed unexpected mortality in the Burn – Early Bortezomib group, we increased the sample size accordingly to be able to analyze REE with n=7 in this group.
Data are presented as mean ± SEM. Data were analyzed with Student’s t test, one-way analysis of variance (ANOVA) with Holm-Sidak’s multiple comparisons test or two-way ANOVA with Tukey'smultiple comparisons test to correct for multiple testing, as appropriate. Survival was plotted using the Kaplan-Meier method and survival between groups was compared with the Log-rank test. Statistical analyses were calculated with the GraphPad Prism program (GraphPad Software). A two-tailed p<0.05 was considered significant.
Results
Bortezomib treatment after sham procedure
The body weights and REE of animals after sham procedure and vehicle or bortezomib treatment are shown in Fig. 1A/B. There were no statistically significant differences between the sham groups at baseline (day 0). Body weights increased continuously throughout the observation period with vehicle and bortezomib treatment (Fig. 1A). With bortezomib, however, weight gain was less pronounced than with vehicle treatment (p<0.05 vs. vehicle on days 7–42). REE remained constant throughout the observation period with vehicle and bortezomib treatment (Fig. 1B). Furthermore, there were no statistically significant differences in blood gas analyses or in any of the routine blood chemistry parameters between the groups (Table 1). All animals after vehicle treatment and 12 out of 13 animals after bortezomib treatment survived the entire observation period (p>0.05 vehicle vs. bortezomib). Post-mortem necropsy of the single animal that died on day 3 after sham procedure and bortezomib treatment did not reveal any macroscopic abnormality of the internal organs.
Figure 1.
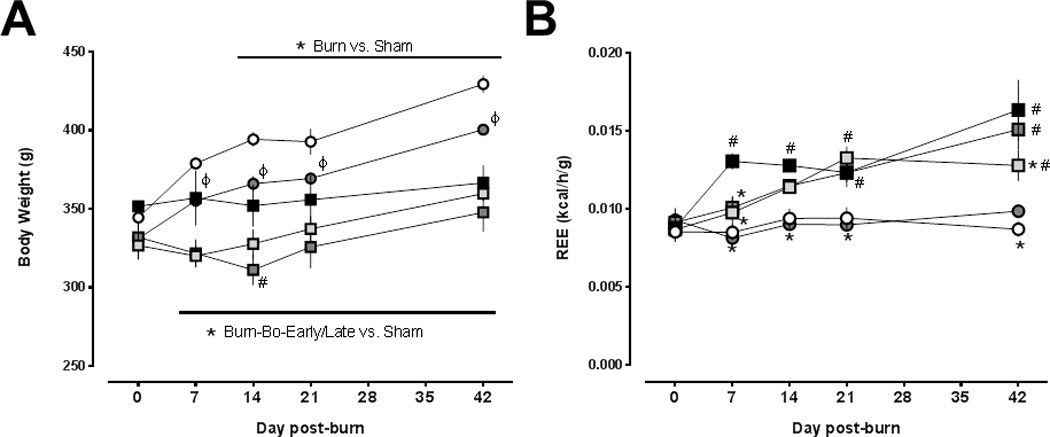
(A) Body weights (g) after sham procedure and burn injury. Open circles: vehicle treatment – sham procedure (n=10). Grey circles: bortezomib treatment – sham procedure (n=12–13). Black squares: burn, vehicle treatment (n=8–10). Light grey squares: burn, early bortezomib treatment (n=7–20). Dark grey squares: burn, late bortezomib treatment (n=9–13). Data are mean ± SEM. ϕ p<0.05 sham procedure and vehicle treatment vs. sham procedure and bortezomib treatment. *: p<0.05 burn vs. sham procedure and vehicle treatment. #: p<0.05 burn and vehicle treatment vs. burn and late bortezomib treatment. (B) Resting energy expenditures (REE, kcal/h/g) after sham procedure and burn injury. Same symbols and sample sizes as in (A). Data are mean ± SEM. *: p<0.05 vs. burn and vehicle treatment. #: p<0.05 vs. sham procedure and vehicle treatment.
Table 1.
Routine laboratory parameters at day 42.
| Sham | Burn | ||||
|---|---|---|---|---|---|
| Analyte | Vehicle | Bo | Vehicle | Early-Bo | Late-Bo |
| Total protein (mg/dL) | 2.14 ± 0.73 | 3.05 ± 1.09 | 4.68 ± 1.41 | 4.56 ± 1.33 | 4.32 ± 1.16 |
| Cholesterol (mg/dL) | 215.70 ± 13.45 | 198.60 ± 46.25 | 172.70 ± 47.76 | 203.71 ± 19.96 | 211.00 ± 12.65 |
| Glucose (mg/dL) | 221.70 ± 40.08 | 243.70 ± 54.86 | 259.00 ± 58.07 | 205.60 ± 18.34 | 231.10 ± 56.29 |
| Na (mM) | 142.30 ± 2.08 | 141.70 ± 1.76 | 143.50 ± 2.34 | 141.90 ± 2.53 | 142.80 ± 1.49 |
| Creatinine (mg/dL) | 0.22 ± 0.04 | 0.22 ± 0.06 | 0.20 ± 0.00 | 0.21 ± 0.04 | 0.20 ± 0.00 |
| BUN (mg/dL) | 19.00 ± 2.99 | 20.65 ± 4.34 | 19.44 ± 2.19 | 23.50 ± 4.07 | 21.83 ± 3.02 |
| GGT (U/L) | 189.30 ± 60.37 | 215.90 ± 81.85 | 196.30 ± 91.29 | 247.40 ± 28.45 | 257.29 ± 42.92 |
| ALT (U/L) | 101.80 ± 22.12 | 103.80 ± 21.15 | 76.13 ± 23.58 | 114.86 ± 26.19 | 108.86 ± 32.27 |
| ALP (U/L) | 482.60 ± 38.66 | 464.60 ± 44.96 | 416.60 ± 176.10 | 539.83 ± 80.74 | 535.71 ± 84.15 |
| WBC (103/mL) | 12.29 ± 2.93 | 12.69 ± 3.13 | 14.79 ± 6.13 | 14.19 ± 1.94 | 17.34 ± 1.82 |
| HGB (g/dL) | 13.76 ± 1.10 | 13.55 ± 1.26 | 13.64 ± 1.14 | 12.66 ± 0.36 | 12.71 ± 0.72 |
| HCT (%) | 39.03 ± 2.30 | 37.56 ± 4.29 | 37.41 ± 3.31 | 34.83 ± 2.79 | 34.87 ± 1.60 |
| PLT (103/mL) | 334.10 ± 183.50 | 453.10 ± 189.30 | 618.40 ± 170.90 | 485.90 ± 341.80 | 519.90 ± 239.10 |
| pH | 7.39 ± 0.20 | 7.37 ± 0.11 | 7.34 ± 0.13 | 7.37 ± 0.12 | 7.34 ± 0.14 |
| PO2 (mmHg) | 427.80 ± 204.60 | 364.10 ± 207.30 | 530.30 ± 96.98 | 388.40 ± 257.20 | 441.90 ± 151.00 |
| HCO3 (mmol/L) | 33.80 ± 2.30 | 34.54 ± 1.80 | 33.80 ± 2.30 | 33.29 ± 3.20 | 32.76 ± 2.30 |
| BE (mEq/L) | 8.67 ± 4.38 | 9.17 ± 1.95 | 7.90 ± 2.98 | 7.84 ± 4.66 | 6.46 ± 3.16 |
Bo: Bortezomib. BUN: blood urea nitrogen. GGT:gamma-glutamyltranspeptidase. ALT: alanine aminotransferase. ALP: alkaline phosphatase. WBC: white blood cell count. HGB: hemoglobin. HCT: hematocrit. PLT: platelets. BE: base excess.
There were no statistically significant differences among the groups.
To confirm that bortezomib treatment resulted in inhibition of the proteasome, we then measured proteasome peptidase activities and content in muscle extracts at day 42 (Fig. 2A–C). While there were no statistically significant differences between vehicle and bortezomib treated animals in proteasome peptidase activities and content per mg of muscle extract protein (Fig. 2A/B), bortezomib treatment significantly reduced the specific proteasome peptidase activity expressed as activity per ng of 20S proteasome (Fig. 2C). Consistent with inhibition of the specific proteasome activity, we observed that ubiquitin-protein conjugates, the physiological substrates of the proteasome, were increased in muscle extracts after bortezomib treatment (Fig. 2D).
Figure 2.
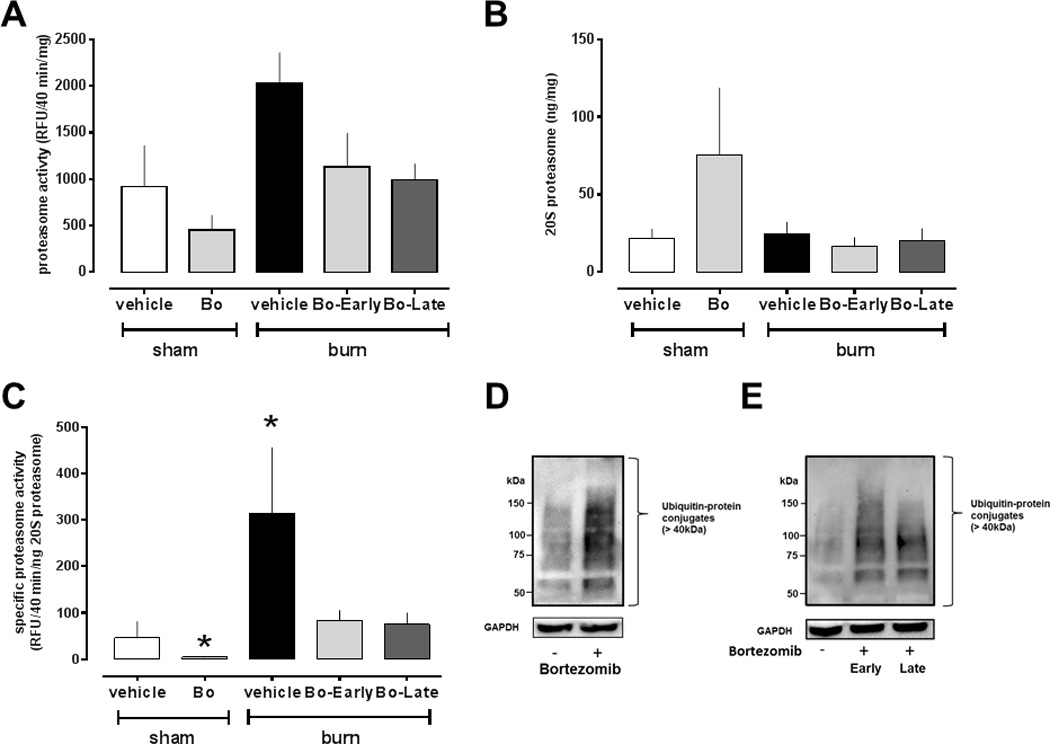
Proteasome peptidase activities, proteasome content and ubiquitin-protein conjugates in skeletal muscle extracts after sham procedure and burn injury (day 42). Sham procedure and vehicle treatment, n = 10. Sham procedure and bortezomib (Bo) treatment, n = 7. Burn and vehicle treatment, n=6. Burn, early Bo treatment, n = 6. Burn, late Bo treatment, n=6. (A) Proteasome peptidase activity (RFU/40 min/mg). (B) 20S proteasome concentration (ng 20S proteasome/mg protein). (C) Specific proteasome activity (RFU/40min/ng 20S proteasome). *: p<0.05 vs. sham procedure and vehicle treatment. (D) Top: Western blot analysis of ubiquitin-protein conjugates (>40kDa) in skeletal muscle extracts from uninjured animals after vehicle (−) and bortezomib (+) treatment. Bottom: Blots were re-probed with anti-GAPDH as a protein loading control. Migration positions of molecular mass standards are indicated on the left. (E) Top: Western blot analysis of ubiquitin-protein conjugates (>40kDa) in skeletal muscle extracts from animals after burn and vehicle (−) or early and late bortezomib (+) treatment. Bottom: Blots were re-probed with anti-GAPDH as a protein loading control. Migration positions of molecular mass standards are indicated on the left.
Bortezomib treatment after burn injury
The survival curves for all animals after burn injury are shown in Fig. 3. There was 20% mortality with vehicle treatment. Late bortezomib treatment resulted in 25% mortality (p>0.05 vs. vehicle treatment). Early bortezomib treatment, however, resulted in 65% mortality (median survival time: 1.875 days; Hazard ratio early bortezomib/vehicle (Mantel-Haenszel): 3.75 (95% confidence interval: 1.3 – 10.7); p = 0.0084 vs. vehicle treatment). Post-mortem necropsy of animals that died did not reveal macroscopic abnormalities or signs of infection. All surviving animals recovered from the burn injury and showed normal feeding and drinking behavior within seven days.
Figure 3.
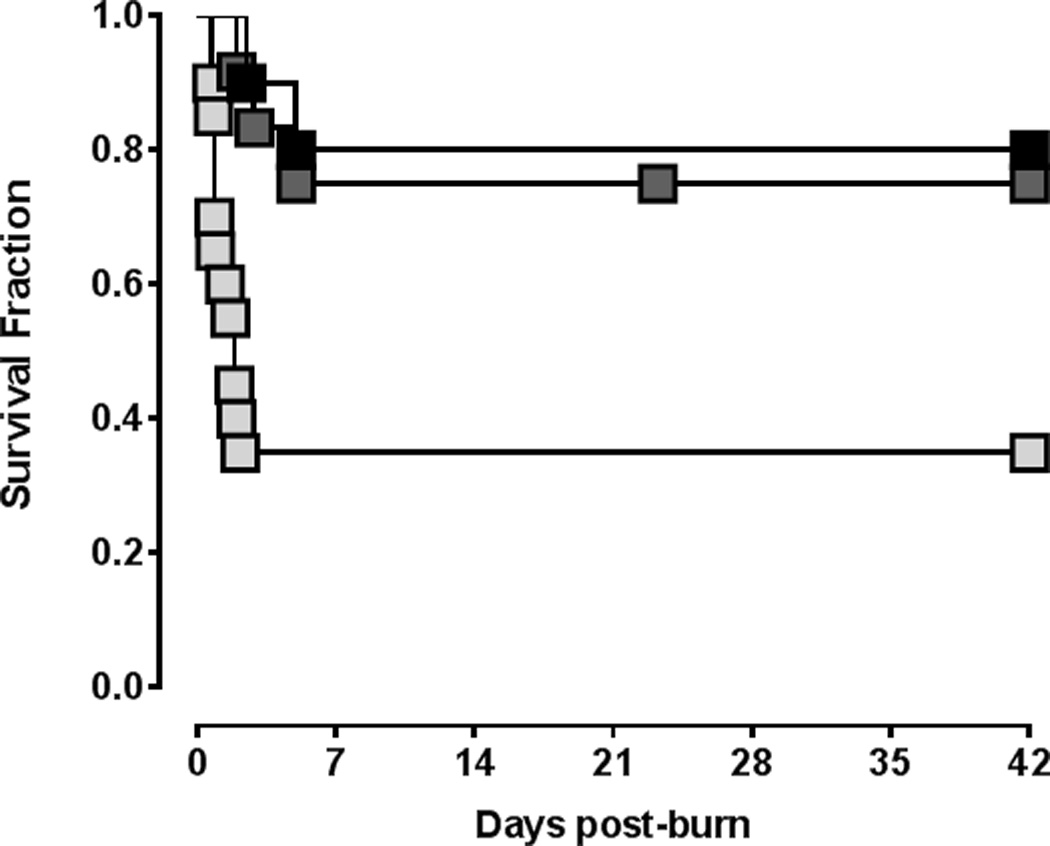
Survival after burn injury. Black squares: vehicle treatment (n=10). Light grey squares: Early bortezomib treatment (n=20). Dark grey squares: Late bortezomib treatment (n=13).
The body weights and REE of animals after burn injury and vehicle or early and late bortezomib treatment are shown in Fig. 1A/B. In all animals after burn injury, body weights remained constant and were significantly lower than after sham procedure on days 7 – 42. Except on post-burn day 14, where body weights were significantly lower with late bortezomib treatment than with vehicle treatment, there were no significant differences in body weights among animals after burn injury and vehicle or early and late bortezomib treatment (Fig. 1A).
There were no differences in REE among the groups at baseline (day 0). As compared with animals after sham procedure and vehicle treatment, REE increased significantly by 48 ± 6% on day 7 and remained elevated until day 42 after burn and vehicle treatment (p<0.05 vs. sham-vehicle on days 7 – 42; Fig. 1B). After burn and early bortezomib treatment, increases in REE were delayed until post-burn day 21 (p>0.05 vs. sham-vehicle on days 7 and 14; p<0.05 vs. burn-vehicle on day 7). Furthermore, early bortezomib treatment after burns significantly reduced REE on day 42, as compared with animals after burn and vehicle treatment (increase in REE: burn-vehicle - 86 ± 22% of day 0; burn-early bortezomib – 47 ± 11% of day 0). With late bortezomib treatment after burns, increases in REE were also delayed until day 21. Late bortezomibtreatment, however, did not reduce the magnitude of the increase in REE on day 42 (increase in REE on day 42: 65 ± 14% of day 0; p>0.05 vs. burn-vehicle), as compared with animals after burn and vehicle treatment.
There were no significant differences in any laboratory parameters among the groups on post-burn day 42 (Table 1). Furthermore, the dorsal burn wounds were indistinguishable by gross examination and eschar separation occurred at comparable time points in animals with vehicle or early and late bortezomib treatment. The quantification of proteasome peptidase activities and content in muscle extracts after burn injury and vehicle or bortezomib treatment are shown in Fig 2. There were no significant differences in proteasome peptidase activities and content per mg of total muscle protein among the groups (Fig. 2A/B). Quantification of specific proteasome activities in muscle extracts revealed a significant increase in animals after burn injury and vehicle treatment, as compared with animals after sham procedure and vehicle treatment. The burn-induced increase in specific proteasome peptidase activities could be prevented with early and late bortezomib treatment (Fig. 2C). As observed after sham procedure, early and late bortezomib treatment increased the content of ubiquitin-protein conjugates in muscle extracts (Fig. 2E).
Discussion
In the present study, we provide an initial assessment of the effects of proteasome inhibition on the development of hypermetabolism after burn injury. There are several new findings from the present study. First, specific proteasome peptidase activities in skeletal muscle are significantly increased six weeks after burn injury. Second, proteasome inhibition delays burn-induced increases in REE. Third, while proteasome inhibition early after burn injury delays and attenuates burn-induced increases in REE, it significantly increases early burn-associated mortality.
The persistent increases in REE during the six week observation period and the lack of weight gain in animals after burn injury document that our model was able to induce hypermetabolism within a clinically relevant time frame. Furthermore, the magnitude of the increase in REE that we observed in animals compares well with increases in REE that have been measured with indirect calorimetry in patients with similar burn sizes (33, 34).
Although the UPP has been suggested to be involved in the pathophysiology of burn-induced hypermetabolism and muscle cachexia, information on the regulation of the skeletal muscle proteasome after burn injury is sparse. While increases in skeletal muscle proteasome activities per mg of total protein have been observed within 7 days after burns in rats by others (35, 36), evidence for persistent proteasome activation during documented burn-induced hypermetabolism and within clinically relevant time periods is lacking. We have previously described that skeletal muscle proteasome peptidase activities per mg of protein were not significantly altered within a six week post-burn time period (25), which is consistent with the findings of the present study. Normalization of proteasome peptidase activities to proteasome content in the present study, however, revealed significantly increased specific proteasome activities in skeletal muscle extracts from animals with documented post-burn hypermetabolism. Thus, our findings now provide initial biochemical evidence for persistent activation of the skeletal muscle proteasome after burn injury, which justifies studies on the role of the proteasome as a drug target during burn-induced hypermetabolism.
As documented by enzyme activity measurements and analyses of endogenous intracellular protein substrates of the proteasome, bortezomib treatment reduced skeletal muscle proteasome activity after sham and burn injury. Although there were no significant differences in survival after sham procedure with and without bortezomib treatment, one of the animals after bortezomib treatment died. Preclinical toxicity studies showed that the maximum safe level of proteasome inhibition by bortezomib is approximately 90% and that proteasome inhibition beyond this threshold results in severe gastrointestinal toxicities and lethal hemodynamic consequences (37–39). In our study, bortezomib treatment after sham injury inhibited specific proteasome activity on average by 86%. Therefore, it is possible that the safety threshold level for bortezomib was exceeded in the single non-surviving animal after sham procedure. After burn injury, bortezomib treatment inhibited specific proteasome activities by 73–76%, as compared with vehicle treatment. This degree of inhibition resulted in specific proteasome activities that were comparable with normal activities in animals after sham procedure, suggesting that intrinsic drug toxicity is unlikely to account for the observed mortality with early bortezomib treatment after burn injury. This assumption is further supported by the finding that late bortezomib treatment was not associated with increased mortality after burns, when compared with vehicle treated animals.
The acute response to burn injury is traditionally referred to as the ebb or shock phase, which is characterized by a hypodynamic-hypometabolic state and lasts for 12–24 h after burn (40, 41). The ebb phase is then followed by the flow phase, which persists for months to years after burns and is characterized by hypermetabolism and net protein loss. Therefore, our findings suggest that adequate proteasome function during the ebb phase after burns is essential for survival.
Besides the roles of the UPP in the regulation of protein turnover, it is also involved in the regulation of a multitude of other essential cellular functions, including a broad variety of intracellular signaling pathways and regulation of inflammation (42). Thus, the identification of the predominant mechanisms through which proteasome inhibition alters REE and induces mortality after burns in particular, and by which proteasome inhibition influences any other cellular function in normal and pathological conditions in general, remains a challenge. Proteasome inhibitors are known to have profound anti-inflammatory and immune suppressive actions (43, 44). Thus, it may be speculated that inhibiting the acute inflammatory response during the ebb phase after burns contributed to the observed mortality with early bortezomib treatment.
As documented by measurements of REE, hypermetabolism developed within 7 days post-burn and persisted during the six week observation period. Whereas early and late bortezomib treatment delayed the increase in REE, only early bortezomib treatment significantly affected the magnitude of the burn-induced hypermetabolic response. These findings have several implications. Our data imply that the UPP contributes to post-burn hypermetabolism and document that pharmacological modulation of a major proteolytic pathway can reduce increases in REE. Furthermore, these findings suggest that molecular events during the initial phase after burns determine the magnitude of the hypermetabolic response weeks after injury.
During recent years several lines of evidence have suggested that the proteasome is also intricately involved in the regulation of lipid and carbohydrate metabolism and itself controlled by metabolic factors, such as O-linked N-acetylglucosamine(45–47). Thus, it appears possible that early proteasome inhibition during the ebb phase was able to attenuate activation of other molecular effectors that contribute to and ultimately determine the magnitude of the post-burn hypermetabolism, whereas such cascades were already activated with delayed proteasome inhibition that was initiated during the flow phase. The exact molecular mechanisms through which proteasome inhibition modulates REE and also induces mortality after burn injury, however, remain to be determined.
In conclusion, our findings provide evidence for the involvement of the proteasome in the pathophysiology of burn-induced hypermetabolism and demonstrate that proteasome inhibition can delay and attenuate post-burn increases in REE. The therapeutic window during which proteasome inhibition reduces the magnitude of the hypermetabolic response, however, appears to be limited to the ebb phase after burns, during which proteasome inhibition significantly increases burn-associated mortality. Our findings highlight the risks of inhibiting an essential proteolytic pathway early after burn injury and suggest that global proteasome inhibition does not provide a clinically feasible approach to reduce burn-induced hypermetabolism.
Acknowledgments
This research was made possible by a grant that was awarded and administered by the U.S. Army Medical Research & Materiel Command (USAMRMC) and the Telemedicine & Advanced Technology Research Center (TATRC), at Fort Detrick, MD, under Contract Number W81XWH1110559. The views, opinions and/or findings contained in this research are those of the author(s) and do not necessarily reflect the views of the Department of Defense and should not be construed as an official DoD/Army position, policy or decision unless so designated by other documentation. No official endorsement should be made. This work was also supported in part by National Institutes of Health Grant NIHT32GM008750 and the Dr. Ralph and Marian Falk Medical Research Trust.
Footnotes
This research has been presented, in part, at the 37th Annual Conference on Shock, Charlotte, North Carolina, June 2014, and at the 17th Congress of the International Society for Burn Injuries, Sydney, Australia, October 2014.
References
- 1.Pereira C, Murphy K, Jeschke M, Herndon DN. Post burn muscle wasting and the effects of treatments. Int J Biochem Cell Biol. 2005;37(10):1948–1961. doi: 10.1016/j.biocel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Hart DW, et al. Determinants of skeletal muscle catabolism after severe burn. Ann Surg. 2000;232(4):455–465. doi: 10.1097/00000658-200010000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia de Lorenzo y Mateos A, Ortiz Leyba C, Sanchez SM. Guidelines for specialized nutritional and metabolic support in the critically-ill patient: update. Consensus SEMICYUC-SENPE: critically-ill burnt patient. NutrHosp. 2011;26(Suppl 2):59–62. doi: 10.1590/S0212-16112011000800013. [DOI] [PubMed] [Google Scholar]
- 4.Williams FN, Herndon DN, Jeschke MG. The hypermetabolic response to burn injury and interventions to modify this response. Clin Plast Surg. 2009;36(4):583–596. doi: 10.1016/j.cps.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter C, Hurren NM, Herndon DN, Borsheim E. Whole body and skeletal muscle protein turnover in recovery from burns. Int J Burns Trauma. 2013;3(1):9–17. [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuda T, et al. The effect of burn wound size on resting energy expenditure. J Trauma. 1987;27(2):115–118. doi: 10.1097/00005373-198702000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Jeschke MG, et al. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS One. 2011;6(7):e21245. doi: 10.1371/journal.pone.0021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilmore DW, Aulick LH, Mason AD, Pruitt BA., Jr Influence of the burn wound on local and systemic responses to injury. Ann Surg. 1977;186(4):444–458. doi: 10.1097/00000658-197710000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter C, Herndon DN, Sidossis LS, Borsheim E. The impact of severe burns on skeletal muscle mitochondrial function. Burns. 2013;39(6):1039–1047. doi: 10.1016/j.burns.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majetschak M, Waydhas C. Infection, bacteremia, sepsis and the sepsis syndrome-metabolic alterations, hypermetabolism, and cellular alterations. In: Baue AE, Faist E, Fry DE, editors. Multiple organ failure: pathophysiology, prevention, and therapy. New York: Springer; 2000. pp. 101–107. [Google Scholar]
- 11.Tuvdendorj D, et al. Long-term oxandrolone treatment increases muscle protein net deposition via improving amino acid utilization in pediatric patients 6 months after burn injury. Surgery. 2011;149(5):645–653. doi: 10.1016/j.surg.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hart DW, et al. Anabolic effects of oxandrolone after severe burn. Ann Surg. 2001;233(4):556–564. doi: 10.1097/00000658-200104000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herndon DN, et al. Muscle protein catabolism after severe burn: effects of IGF-1/IGFBP-3 treatment. Ann Surg. 1999;229(5):713–720. doi: 10.1097/00000658-199905000-00014. discussion 720–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer NA, Barrow RE, Herndon DN. Combined insulin-like growth factor-1 and growth hormone improves weight loss and wound healing in burned rats. J Trauma. 1996;41(6):1008–1012. doi: 10.1097/00005373-199612000-00011. [DOI] [PubMed] [Google Scholar]
- 15.Biolo G, et al. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. J Clin Endocrinol Metab. 2002;87(7):3378–3384. doi: 10.1210/jcem.87.7.8699. [DOI] [PubMed] [Google Scholar]
- 16.Hershko A, Ciechanover A. The ubiquitin system for protein degradation. Annu Rev Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- 17.Ciechanover A. Intracellular protein degradation from a vague idea through the lysosome and the ubiquitin-proteasome system and on to human diseases and drug targeting: Nobel Lecture, December 8, 2004. Ann N Y AcadSci. 2007;1116:1–28. doi: 10.1196/annals.1402.078. [DOI] [PubMed] [Google Scholar]
- 18.Jagoe RT, Goldberg AL. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr Opin Clin Nutr Metab Care. 2001;4(3):183–190. doi: 10.1097/00075197-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Twombly R. First proteasome inhibitor approved for multiple myeloma. J Natl Cancer Inst. 2003;95(12):845. doi: 10.1093/jnci/95.12.845. [DOI] [PubMed] [Google Scholar]
- 20.Herndon TM, et al. U.s. Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013;19(17):4559–4563. doi: 10.1158/1078-0432.CCR-13-0755. [DOI] [PubMed] [Google Scholar]
- 21.Walker HL, Mason AD., Jr A standard animal burn. J Trauma. 1968;8(6):1049–1051. doi: 10.1097/00005373-196811000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Cavaletti G, et al. Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp Neurol. 2007;204(1):317–325. doi: 10.1016/j.expneurol.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Nowis D, et al. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am J Pathol. 2010;176(6):2658–2668. doi: 10.2353/ajpath.2010.090690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanchaer MC, Van Wijhe M. Isozymes of lactic dehydrogenase in skeletal muscle. Am J Physiol. 1962;202:827–829. doi: 10.1152/ajplegacy.1962.202.5.827. [DOI] [PubMed] [Google Scholar]
- 25.Wong YM, et al. Activities of nonlysosomal proteolytic systems in skeletal and cardiac muscle during burn-induced hypermetabolism. J Burn Care Res. 2014;35(4):319–327. doi: 10.1097/BCR.0000000000000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer NA, Barrow RE, Herndon DN. Combined insulin-like growth factor-1 and growth hormone improves weight loss and wound healing in burned rats. J Trauma. 1996;41(6):1008–1012. doi: 10.1097/00005373-199612000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Majetschak M, et al. Cardiac proteasome dysfunction during cold ischemic storage and reperfusion in a murine heart transplantation model. Biochem Biophys Res Commun. 2008;365(4):882–888. doi: 10.1016/j.bbrc.2007.11.092. [DOI] [PubMed] [Google Scholar]
- 28.Geng Q, et al. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem Biophys Res Commun. 2009;390(4):1136–1141. doi: 10.1016/j.bbrc.2009.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majetschak M, Sorell LT. Immunological methods to quantify and characterize proteasome complexes: development and application. J Immunol Methods. 2008;334(1–2):91–103. doi: 10.1016/j.jim.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Patel MB, Majetschak M. Distribution and interrelationship of ubiquitin proteasome pathway component activities and ubiquitin pools in various porcine tissues. Physiol Res. 2007;56(3):341–350. doi: 10.33549/physiolres.931005. [DOI] [PubMed] [Google Scholar]
- 31.Meng L, et al. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci U S A. 1999;96(18):10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker TA, et al. Prolongation of myocardial viability by proteasome inhibition during hypothermic organ preservation. Biochem Biophys Res Commun. 2010;401(4):548–553. doi: 10.1016/j.bbrc.2010.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda T, et al. The effect of burn wound size on resting energy expenditure. J Trauma. 1987;27(2):115–118. doi: 10.1097/00005373-198702000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Khorram-Sefat R, Behrendt W, Heiden A, Hettich R. Long-term measurements of energy expenditure in severe burn injury. World J Surg. 1999;23(2):115–122. doi: 10.1007/pl00013172. [DOI] [PubMed] [Google Scholar]
- 35.Fang CH, et al. Burn injury upregulates the activity and gene expression of the 20 S proteasome in rat skeletal muscle. Clin Sci. 2000;99(3):181–187. [PubMed] [Google Scholar]
- 36.Ni B, et al. Interleukin-1 up-regulates the expression and activity of 26S proteasome in burned rat. Burns. 2007;33(5):621–627. doi: 10.1016/j.burns.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 37.Hamilton AL, et al. Proteasome inhibition with bortezomib (PS-341): a phase I study with pharmacodynamic end points using a day 1 and day 4 schedule in a 14-day cycle. J Clin Oncol. 2005;23(25):6107–6116. doi: 10.1200/JCO.2005.01.136. [DOI] [PubMed] [Google Scholar]
- 38.Adams J, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59(11):2615–2622. [PubMed] [Google Scholar]
- 39.Adams J. Development of the proteasome inhibitor PS-341. Oncologist. 2002;7(1):9–16. doi: 10.1634/theoncologist.7-1-9. [DOI] [PubMed] [Google Scholar]
- 40.Wolfe RR. Review: acute versus chronic response to burn injury. Circ Shock. 1981;8(1):105–115. [PubMed] [Google Scholar]
- 41.Tredget EE, Yu YM. The metabolic effects of thermal injury. World J Surg. 1992;16(1):68–79. doi: 10.1007/BF02067117. [DOI] [PubMed] [Google Scholar]
- 42.Reinstein E. Immunologic aspects of protein degradation by the ubiquitin-proteasome system. Isr Med Assoc J. 2004;6(7):420–424. [PubMed] [Google Scholar]
- 43.Mohty M, Brissot E, Savani BN, Gaugler B. Effects of bortezomib on the immune system: a focus on immune regulation. Biol Blood Marrow Transplant. 2013;19(10):1416–1420. doi: 10.1016/j.bbmt.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 44.Nencioni A, Grunebach F, Patrone F, Ballestrero A, Brossart P. The proteasome and its inhibitors in immune regulation and immune disorders. Crit Rev Immunol. 2006;26(6):487–498. doi: 10.1615/critrevimmunol.v26.i6.20. [DOI] [PubMed] [Google Scholar]
- 45.Ronnebaum SM, Patterson C, Schisler JC. Minireview: Hey U(PS): Metabolic and Proteolytic Homeostasis Linked via AMPK and the Ubiquitin Proteasome System. Mol Endocrinol. 2014;28(10):1602–1615. doi: 10.1210/me.2014-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharpe LJ, Cook EC, Zelcer N, Brown AJ. The UPS and downs of cholesterol homeostasis. Trends BiochemSci. 2014;39(11):527–535. doi: 10.1016/j.tibs.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Zhang F, Paterson AJ, Huang P, Wang K, Kudlow JE. Metabolic control of proteasome function. Physiology. 2007;22:373–379. doi: 10.1152/physiol.00026.2007. [DOI] [PubMed] [Google Scholar]