

Abstract
The clinical utility of peptides is limited by their rapid degradation by endogenous proteases. Modification of the peptide backbone can generate functional analogues with enhanced proteolytic stability. Existing principles for the design of such oligomers have focused primarily on effective structural mimicry. A more robust strategy would incorporate a rational approach for engineering maximal proteolytic stability at minimal unnatural residue content. We report here the systematic comparison of the proteolytic resistance imparted by four backbone modifications commonly employed in the design of protease-stable analogues of peptides with complex folding patterns. The degree of protection is quantified as a function of modification type, position, and tandem substitution in the context of a long, unstructured host sequence and a canonical serine protease. These results promise to inform ongoing work to develop biostable mimics of increasingly complex peptides and proteins.
Keywords: peptides, peptidomimetics, foldamers, proteases, protein-protein interactions
Introduction
The involvement of proteins in human pathology is a significant research interest, and pharmaceuticals that target proteins are in high demand. While small molecules can effectively bind to pockets that natively recognize small ligands, disrupting protein-protein interactions (PPIs) involving extended interfaces remains a substantial challenge.[1] One solution to this problem is the use of larger peptide and protein based scaffolds, which have shown the unique ability to inhibit certain PPIs where small molecules have failed.[2] The promise of peptide therapeutics is attenuated in part by poor oral bioavailability, often necessitating administration by invasive and inconvenient parenteral methods.[3] A significant contributor to poor peptide bioavailability is rapid degradation by endogenous proteases, which can result in very short in vivo lifetimes.[3]
Alterations to the chemical connectivity of the l-α-peptide backbone can be a useful means to improve proteolytic stability, and targeted backbone modification in short peptides has a rich history in the field of peptidomimetics research.[4] More recent work has explored modified backbones in the context of larger oligomers, seeking to recreate complex functions of diverse bioactive peptides and proteins on protease-resistant scaffolds. Found at one end of the spectrum of such efforts is the introduction of one or two unnatural building blocks in a natural α-peptide to improve efficacy or stability.[5] This tactic offers the advantage of employing a biological sequence as the prototype and demonstrates that strategic placement of limited unnatural backbone content can lead to dramatically altered properties. At the other extreme are highly unnatural oligomeric backbones with protein-like folds and functions, often termed “foldamers.”[6] Foldamers comprised entirely of a single type of unnatural monomer are inert to proteases and can show interesting biological activities;[7] however, the design of completely unnatural sequences that effectively mimic natural peptides can be challenging.
Recent results suggest significant potential for oligomers in which ~20–30% of the α-residues in a bioactive sequence are replaced by some unnatural analogue.[8] Such backbone heterogeneity takes on another dimension when many classes of unnatural building blocks are incorporated alongside α-residues in a single chain.[9] An advantage of heterogeneous-backbone foldamers over homogeneous-backbone counterparts is the prospect of drawing from the wellspring of natural peptides for the design of unnatural analogues. In most examples of heterogeneous-backbone foldamer design, α-residue replacements are made in a manner guided primarily by structural considerations (e.g., maintaining local folding pattern and key interactions for receptor binding). While this can generate adequate proteolytic stability for biological applications, a more rational substitution approach would enable the construction of oligomers where proteolytic protection is considered alongside structural issues at the outset.
We report here a systematic examination of the proteolytic protection imparted by four of the most common modifications employed in the design of heterogeneous-backbone foldamers (Figure 1): d-α-residues, the Cα-methylated α-residue Aib, N-Me-α-residues, and β3-residues. Although each of these building blocks has been examined in isolation,[10] no prior report has sought to compare their effectiveness at shielding a substrate from proteolytic hydrolysis. Motivated by this gap in knowledge, we sought to address a number of open questions: (1) How do the variables of backbone modification type and position relative to a cleavage site affect protease efficiency? (2) What are the molecular mechanisms by which various modified backbones exert proteolytic protection? (3) How do multiple backbone modifications work in concert to protect a peptide from hydrolysis? Gaining a deeper understanding of these issues promises new design strategies for achieving maximal proteolytic protection in heterogeneous-backbone foldamers with minimal unnatural residue content.
Figure 1.
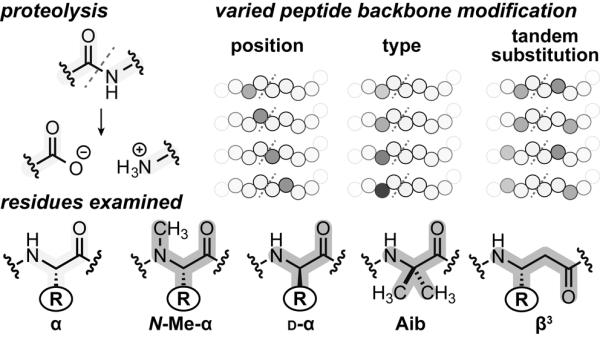
Chemical structures of the protease susceptible amide bond in a peptide, and the natural or unnatural residues examined in this work. Systematic backbone modification patterns varying unnatural residue types, sequence positions, and tandem substitutions.
Results and Discussion
Host Peptide Sequence and Experimental Design
As outlined above, the primary goals at the outset of the present work were to gain new insights into the relationship between peptide backbone composition and proteolytic susceptibility, as well as the molecular origins of observed trends. To this end, we chose to carry out in vitro studies with an isolated sequence-specific protease rather than a promiscuous enzyme or whole serum. Chymotrypsin is the type example of the largest subfamily of serine proteases, which account for ~1/3 of total proteases in humans.[11] Its prevalence has led to a robust literature on structure, mechanism, and substrate recognition behavior (spanning well beyond the primary cleavage site).[11] While chymotrypsin substrate scope is well studied, the vast majority of that work has focused on short peptides, many of which contain unnatural leaving groups to facilitate spectroscopic analysis.
We sought a host sequence for backbone modification that was more analogous to what the enzyme might encounter in the context of a larger bioactive peptide or protein. The host peptide sequence is loosely based on a recently reported sequence motif selected by phage display for high inherent resistance to intestinal proteases, including chymotrypsin.[12] Changes were made to avoid complications from Cys side chains, minimize conformational rigidity, and eliminate large hydrophobic residues (Met, Phe) that might retain even slight susceptibility to chymotrypsin. We fused two of the modified motifs in a single chain and introduced a defined chymotrypsin-specific cleavage site (AY↓K)[11b] at the center to generate 21-residue α-peptide 1 (Figure 2). Peptide 1 is processed cleanly by chymotrypsin, resulting in two products observed by MALDI-TOF-MS from hydrolysis of the amide between residues Tyr11 and Lys12. Monitoring the reaction time course by HPLC reveals a single-phase exponential decay with a half-life of 8 min for 160 μM peptide in the presence of 50 nM chymotrypsin (Figure S2). Peptide 1 is highly resistant to cleavage by the enzyme outside the engineered hydrolysis site; the Tyr11→Ala mutant showed no apparent degradation up to 5 days under the same conditions. α-Peptide 1 lacks any measurable secondary structure by circular dichroism (CD) spectroscopy (Figure S1). This is an important feature as it enables the attribution of protection afforded by various modifications to changes in backbone chemical connectivity rather than changes in the folded state.
Figure 2.

A) Prototype α-peptide sequence with putative site of proteolysis (red hashed line) and corresponding residue nomenclature. Positions of unnatural residue substitution are highlighted gray. B) Normalized peptide half-lives as a function of substitution type and position. α➔Aib half-lives are normalized to the corresponding Ala mutants of peptide 1. All others are normalized to the half-life of peptide 1. Error bars represent the error in the fit of the full progress curve (SI). An asterisk over a bar indicates that proteolysis was observed outside of the putative site.
To explore the effect of backbone modification on proteolytic stability, we synthesized a library comprising 32 analogues of peptide 1 in which individual α-residues in the prototype were replaced by one of four unnatural analogues: N-Me-α-residues, d-α-residues, β3-residues, or the Cα-Me-α-residue Aib. These four classes of backbone modification were each scanned across eight positions surrounding the cleavage site (Arg7 to Ala14, P5 to P3' in the Schechter and Berger nomenclature).[13] In contrast to the other types of backbone modification, α→Aib substitution involves the loss of a side chain functional group. We therefore synthesized a set of Ala mutants of the parent sequence to isolate the effects of Cα-methylation from any potential change in proteolytic susceptibility resulting from the removal of a side chain.
Each of the peptides described above was synthesized by standard methods and purified by preparative reverse phase HPLC; the identity and purity of the final products were assessed by MALDI-TOF-MS and analytical HPLC, respectively. After synthesis and purification, each peptide was subjected to proteolysis under identical experimental conditions (50 nM chymotrypsin, 160 μM peptide, 50 mM TBS buffer pH 7.5) and the reactions were monitored over time by analytical HPLC and MALDI-TOF-MS. The resulting data set for single residue substitutions is summarized as a set of normalized half-life values for degradation as a function of backbone modification type and position (Figure 1B).
Impact of Backbone Modification Type and Position
Predictably, modification of the backbone at the primary specificity site P1 (i.e., Tyr11) resulted in significant protection (100–1000 fold increased half-life). The more intriguing aspects of the data were trends in the moderate changes in proteolytic susceptibility observed upon substitution near, but not at, the chymotrypsin cleavage site. Here both the magnitude and profile of protection differed for each backbone modification type. A summary of the key observations for each class of unnatural monomer is provided in the following section, along with interpretation of some results in terms of potential molecular mechanisms of protection.
In general, N-Me-α-residue incorporation provided the smallest degree of proteolytic protection among the unnatural monomer types examined. Only three positions clustered directly around the cleavage site (P1–P2′) gave rise to >10-fold improvement in stability relative to the prototype α-peptide. This result is not surprising, given that N-Me-α-residues are also most similar among the modified backbones examined to natural α-residues in folding propensity and structural properties. The positional dependence of N-Me-α-residue incorporation can be interpreted based on putative enzyme–substrate hydrogen bonds. Like most proteases, chymotrypsin recognizes substrates in an extended conformation, resulting in hydrogen bonds to alternating residues at sites P3, P1, and P2′ (Figure 3A).[11b] Since amide methylation replaces a hydrogen-bond donor with a potential steric clash, it would be expected to provide protection only when incorporated on the face of substrate that engages the enzyme binding pocket. This hypothesis is supported by the observation that α→N-Me-α replacement at P1 or P3 leads to moderate to strong proteolytic protection, while the same modification at P2 leads to no change in susceptibility relative to the parent α-peptide. The proteolytic resistance imparted by P1′ N-methylation is likely the result of a steric clash during formation of the acylenzyme intermediate.[14]
Figure 3.
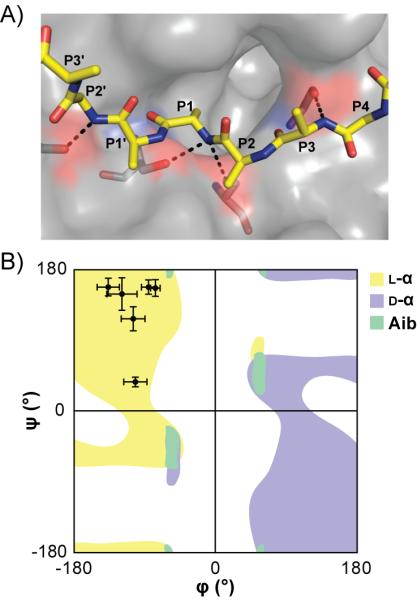
Proposed modes of protection for N-Me-residues (A), and d-/Aib residues (B). A) Chymotrypsin (surface) complexed with a representative non-covalent inhibitor (PDB: 1GL0). Amide protons of residues P3, P1, and P2' are hydrogen bonded (hashed black lines) to chymotrypsin. B) Overlayed Ramachandran plots of L-α-residues, d-α-residues, and Aib. The data points indicate average dihedral angles observed for P4-P2' residues from 11 published crystal structures of chymotrypsin in complex with substrate-like inhibitors (PDB: 1ACB, 1CGI, 1CGJ, 1CHO, 1GL0, 1GL1, 1HJA, 1N8O, 2Y6T, 3BG4, 3RU4).
Cα-methylation through incorporation of Aib provided protection over a wider range from the cleavage site relative to α→N-Me-α substitution; >10-fold protection was observed from P3-P2′ and near 10-fold protection at P4 and P2′. Interestingly, >2-fold protection was observed as far as from the cleavage site as position P5, where all other backbone modifications examined had no effect. The complete protection observed upon substitution at P1 is unsurprising, given the loss of the key Tyr side chain. In order to focus analysis of data from the other peptides in the Aib series on the contribution of backbone methylation, we normalized the half-life of each to the corresponding Ala mutant of prototype α-peptide 1. The strong protection conferred by Cα-methylation at P1′ likely results from a steric clash around the scissile amide bond; however, the behavior of the remaining Aib mutants is more readily interpreted as a result of altered conformational preferences.
Compared to l-α-residues, Aib has a more constrained range of accessible backbone conformations, favoring α-helical secondary structure.[15] An induced folded structure in the substrate would certainly alter proteolytic processing; however, analyses of the Aib series by CD indicated no measurable change to the random coil signature observed for the prototype peptide (Figure S1). We reasoned that Aib, although not inducing a particular folded state, may be altering the denatured ensemble to disfavor chymotrypsin recognition of the substrate. Support for this hypothesis can be found in the analysis of 11 published crystal structures of the enzyme in complex with inhibitors that mimic natural substrate recognition.[16] A Ramachandran plot comparing the average backbone conformation observed at substrate positions P4-P2′ to a previously calculated profile for Aib shows that the conformation necessary for effective recognition by the enzyme falls in a disallowed region for the unnatural residue (Figure 3B).[17]
Among the backbone modification types examined, alteration of Cα stereochemistry through single incorporation of d-α-residues resulted in the greatest degree of protection over the widest range. >10-fold increases in half-life were observed after incorporation of a d-residue at any of six positions surrounding the cleavage site (P4-P2′). Known features of chymotrypsin stereospecificity explain some aspects of the data;[18] however, the dramatic protection resulting from d-residue incorporation at remote sites was surprising. Literature precedent suggests the S3 site of chymotrypsin is not particularly stereospecific, but these conclusions were based on analysis of short 4–5 residue substrates. Our results show that, in a more native-like extended oligomer, d-residues impart strong protection as far from the cleavage site as P4. The impact of d-residue incorporation on proteolytic susceptibility can be most readily interpreted in terms of an altered ensemble constituting the denatured state. As with Aib, the typical substrate conformation at residues P4-P2′ falls in the disallowed region of the d-residue Ramachandran plot (Figure 3B).
Peptides containing α→β3 substitutions showed a moderate degree of protection from degradation by chymotrypsin. Substitution adjacent to the site of hydrolysis (P1 or P1′) resulted in only a 100-fold improvement in proteolytic stability (compared to >1000-fold for Aib), and the range of >10-fold protection only extended over four residues (P3-P1′). Although the extra methylene unit relative to an α-residue leads to additional conformational flexibility, β3-residues have been shown capable of recreating protein-like helix, sheet, loop and turn secondary structures when incorporated into α/β-peptides.[8a] Thus, the majority of proteolytic protection afforded by α→β3 residue substitution is not likely the result of the inability of the modified peptide to adopt a native-like conformation. The extra backbone methylene unit may interfere sterically by changing the spacing of side chains distributed along the substrate. Alternatively, β3-residue incorporation may disrupt chymotrypsin-substrate interactions by inverting the display of side chains along the extended strand.[19]
Impact of Tandem Backbone Modifications
The above data provided insights into the role of backbone modification type and position in determining susceptibility of a given amide bond to hydrolysis by chymotrypsin; however, open questions remained. In the design of heterogeneous-backbone foldamers that mimic larger peptides and proteins, there are many potential cleavage sites, the identities of which are often not known a priori. Efforts to generate stable analogues are characterized by two opposing trends. The proteolytic stability of a backbone is generally correlated with its unnatural content; completely unnatural backbones (e.g., β-peptides, d-α-peptides) are inert, and the stability of heterogeneous backbones will improve with increasing unnatural content. An opposite trend is often seen in terms of activity; the more a backbone is modified, the less likely it is to maintain a native-like folding pattern and function. When the entire peptide backbone is considered protease susceptible, evenly spaced modification along a sequence is the most efficient means to achieve maximal biostability with minimal chemical alteration. Thus, we sought to determine the optimal density of modification and how it varies with the type of unnatural monomer employed.
We synthesized and characterized a series of additional peptides bearing two substitutions flanking the cleavage site. Comparing their degradation rates to the corresponding singly substituted analogues provided insights into synergistic effects of backbone modification on proteolytic protection. Due to the prevalence of bioactive α/β-peptides in the recent heterogeneous-backbone foldamer literature,[8b] we focused attention initially on tandem β3-residue substitutions. Five peptides were prepared and characterized, each bearing two α→β3 substitutions separated by two to six α-residues surrounding the cleavage site (Figure 4). In most of the series, multiple α→β3 substitutions were synergistic (i.e., the observed half-life was greater than that expected, assuming that the activation energies of orthogonal proteolysis reactions are additive). Tandem mutants bearing β-residues separated by two, three, or four α-residues (βα↓αβ, βαα↓αβ, βααα↓αβ) showed a degree of protection that was comparable to β3-residue substitution directly at the cleavage site. This observation suggests there may be a maximum achievable stability conferred by a particular type backbone modification. Increasing the spacing between substitutions α-residues (βααα↓ααβ) resulted a half-life that was lower, albeit still greater than predicted from the single substitutions. Interestingly, incorporation of two β3-residues separated by six α-residues (βαααα↓ααβ) led to a tandem mutant that was significantly less stable than any of the single β3 substitutions and even more susceptible to chymotrypsin than the host α-peptide. This observation is somewhat surprising and suggests that incorporation of β3-residues in certain patterns may actually promote conformations that facilitate proteolysis. Together, these results demonstrate that combined backbone modifications can have positive or negative synergistic effects, and the correlation between unnatural residue density and proteolytic stability is not necessarily direct.
Figure 4.
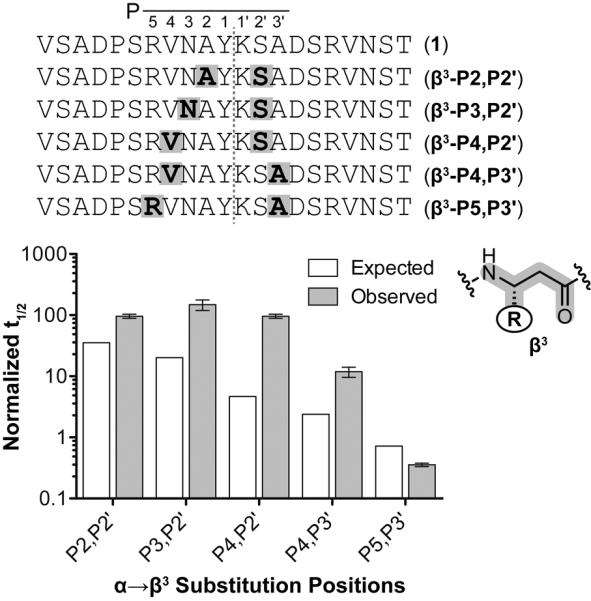
Prototype α-peptide and tandem β3-residue substituted sequences with putative site of proteolysis (red hashed line) and corresponding residue nomenclature. Positions of β3-residue substitution are highlighted cyan. Normalized peptide half-lives as a function of tandem β3-residue substitution position. Half-lives were normalized to the half-life of peptide 1. Error bars represent the error in the fit of the full progress curve (SI).
The trends observed in the tandem β3-residue mutants prompted us to investigate potential synergistic effects of incorporating the other monomer types. Thus, d-α, Aib, and N-Me-α residues were incorporated in combination in a fixed pattern (Xααα↓ααX) where β3-residue incorporation led to only a modest improvement in proteolytic stability (Figure 5). Backbone modifications that alter local folding propensity (d-α, Cα-Me-α) had strong positive synergistic effects on proteolytic stability when combined in a single chain. In contrast, combining N-Me-α residues led to a degree of protection no different than predicted from the corresponding single substitutions. This corroborates our hypothesis that N-Me-α-residues confer proteolytic resistance by disrupting specific enzyme– substrate interactions rather than by affecting local folding.
Figure 5.
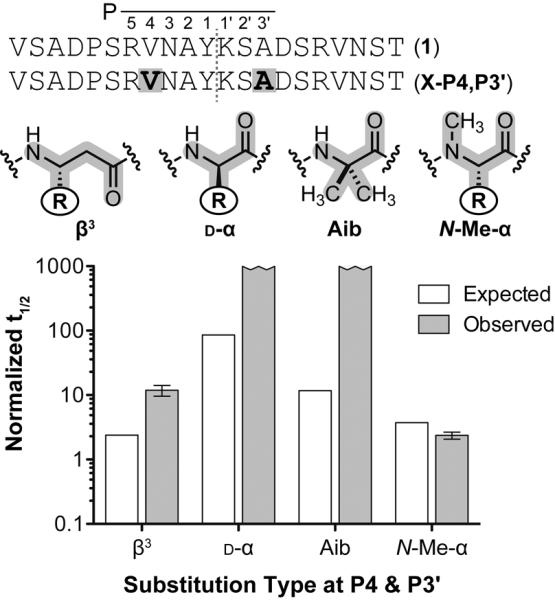
Prototype α-peptide and various tandem substituted sequences with putative site of proteolysis (red hashed line) and corresponding residue nomenclature. Positions of unnatural substitution are highlighted gray. Normalized peptide half-lives as a function of unnatural residue substitution type at sequence positions P4 and P3'. The half-life of the tandem Aib substituted peptide is normalized to the corresponding Ala mutants. All others are normalized to the half-life of peptide 1. Error bars represent the error in the fit of the full progress curve (SI).
Conclusions
In summary, we have reported here a comparison of the proteolytic protection afforded by four common building blocks used in heterogeneous-backbone foldamers. Systematic examination of the variables of backbone modification type and position in the context of a long unstructured host α-peptide and the canonical serine protease chymotrypsin reveal some clear trends. In terms of isolated α-residue replacement, the degree of protection imparted as a function of modification type follows as d-α > Cα-Me-α > β3 > N-Me-α. Differences are most apparent at sites 2–4 residues removed from the scissle amide bond. The modes by which the unnatural residues exert protection fall under two broad categories: (1) interference with specific enzyme-substrate contacts (local effects), and (2) alteration of the denatured ensemble of the unstructured host peptide (global effects).
d-α and Cα-Me-α residues likely impart proteolytic stability by shifting the denatured ensemble away from conformers effectively recognized by the enzyme. As a result, the protection they exert from proteolysis is large in magnitude, wide-ranging, and synergistic when substitutions are combined in a single chain. N-Me-α-residues appear to influence proteolytic susceptibility primarily through disruption of discrete enzyme–substrate contacts, making their effects modest in magnitude, short-range, and simply additive when combined. Among the residue types examined, the behavior of β3-residues was most complex. The protection afforded was modest in magnitude yet somewhat wide-ranging, and the molecular origins are difficult to unambiguously define. The effects of combined backbone α→β3 replacements can be synergistic; however, the density of β3-residues is not directly correlated to proteolytic resistance.
Although the scope of the present findings is limited by involving only a single protease, they generate a number of clear hypotheses for future work on the development of agents that recreate natural peptides and protein function on biostable scaffolds. Considerations for effective structural mimicry are now well defined for many systems, and we can begin to develop design principles for achieving maximal proteolytic stability at minimal unnatural backbone content. For example, both Aib and β3-residues can be readily incorporated into peptide and protein helices. β3-Residues have the functional advantage of retaining native side chains, but our data suggest Aib may impart a greater degree of proteolytic protection at lower substitution density. Taken together, these observations suggest that chiral Cα-methylated amino acids bearing protein-derived side chains may be superior to both Aib and β3-residues as building blocks in heterogeneous-backbone foldamers. As another example of a new design insight, the folding propensity of d-α-residues differs greatly from their l-α counterparts, but they are quite effective turn inducers. Given the far-ranging proteolytic protection provided by even limited backbone stereochemical alteration, our data suggest d-residues as an ideal backbone modification in situations where they would be structurally accommodated. It is our hope that these and other aspects of the present study will inform ongoing work on the design of heterogeneous-backbone foldamers and prompt further investigation into the mechanisms by which unnatural amino acids impart proteolytic stability. Areas of particular interest include how backbone alteration influences the structure of the denatured ensemble[20] as well as the detailed kinetics (i.e., kcat, Km) for the enzymatic degradation of modified substrates.
Experimental Section
Materials
2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU), Fmoc-D-Val-OH, Fmoc-D-Lys(Boc)-OH, Fmoc-D-Arg(Pbf)-OH, Fmoc-β3-HAsn(Trt)-OH, Fmoc-β3-HArg(Pbf)-OH and Fmoc-N-Me-Asn(Trt)-OH were purchased from AAPPTec LLC. Fmoc-D-Asn(Trt)-OH, Fmoc-D-Ala-OH, Fmoc-D-Tyr(tBu)-OH, Fmoc-D-Ser(tBu)-OH, Fmoc-β3-HVal-OH, Fmoc-β3-HAla-OH, Fmoc-β3-HTyr(tBu)-OH, Fmoc-β3-HLys(Boc)-OH, Fmoc-β3-HSer(tBu)-OH Fmoc-N-Me-Val-OH, Fmoc-N-Me-Ala-OH, Fmoc-N-Me-Tyr(tBu)-OH, and Fmoc-N-Me-Ser(tBu)-OH were purchased from Chem-Impex International, Inc. Fmoc-N-Me-Arg(Pbf)-OH was purchased from ChemPep. NovaPEG Rink Amide Resin and Fmoc-protected α-amino acids were purchased from Novabiochem. Fmoc-α-Me-Ala-OH and Fmoc-N-Me-Lys(Boc)-OH, N,N-diisopropylethylamine (DIEA) and bovine α-chymotrypsin were purchased from Sigma-Aldrich. All other reagents were purchased from Acros Organics, Fisher Scientific, JT Baker, and Sigma-Aldrich.
Peptide Synthesis
Peptides were synthesized using microwave-assisted (CEM MARS) Fmoc solid-phase synthesis techniques. All peptides were prepared as the C-terminal carboxamide using NovaPEG Rink Amide resin (0.025 mmol scale). Fmoc protected amino acids (0.1 mmol) were coupled to the growing peptide chain with HCTU (0.1 mmol) and DIEA (0.15 mmol) in NMP (1 mL) for 2 minutes after a 1.5 minute ramp to 90 °C. Double couplings were carried out at hindered amine nucleophiles (i.e. proline or N-methyl residues). Fmoc groups were removed with 4-methylpiperidine in DMF (20% v/v) for 2 minutes after a 2 minute ramp up to 80 °C. Resin was washed three times with DMF after each reaction. After the final deprotection, resin was rinsed three times each with DMF, dichloromethane, and methanol, and then dried. Peptides were cleaved from resin using a solution of TFA/EDT/H2O/TIS (94%/2.5%/2.5%/1%) for 3 hours. After precipitation in cold ethyl ether, the solutions were centrifuged and the pelleted solids were dissolved in mixtures of 0.1% TFA in H2O and 0.1% TFA in acetonitrile.
Peptides were purified by preparative RP-HPLC on a Phenomenex Luna C18 column using gradients between 0.1% TFA in H2O and 0.1% TFA in acetonitrile. Peptide identity and purity were determined by mass spectrometry (Voyager DE Pro MALDI-TOF) and analytical RP-HPLC, respectively. Peptide stock concentrations were quantified by UV spectroscopy (ε280 = 1280 M−1 cm−1). Stock concentrations for peptides lacking a chromophoric residue (Y11A, Y11Aib) were estimated by mass.
Proteolysis reactions
The concentrations of bovine α-chymotrypsin stock solutions were quantified by UV spectroscopy (ε280 = 51,240 M−1 cm−1). Aliquots of 0.25 μM chymotrypsin in TBS (50 mM Tris, 150 mM NaCl, pH 7.5) were made and frozen. Each proteolysis reaction was initiated by the addition of chymotrypsin from a freshly thawed aliquot described above to a 200 μM solution of peptide in water to a final sample composition of 160 μM peptide, 50 nM chymotrypsin, 50 mM Tris, 150 mM NaCl, pH 7.5. For each time point, a 50 μL portion of the reaction was removed and quenched with 0.5% (v/v) trifluoroacetic acid (40 μL).
Quenched samples were analyzed by analytical RP-HPLC (90 μL injection), and the undigested peptide remaining quantified by integration of the corresponding chromatogram. MALDI-MS was also performed at each time point to map the sites of enzymatic hydrolysis. A plot of percent peptide remaining vs. time was generated, and the data fit to a one-phase exponential decay using GraphPad Prism to generate a calculated half-life. Each data point is representative of 2–3 independent experiments. Some of the hybrid peptides exhibited digestion profiles that plateaued at a non-zero value. This has been seen before in proteolysis of backbone-modified peptides,[5a, 21] and we attribute it to enzyme inactivation.[22] While enzyme inactivation is a form of proteolytic protection, the focus of the present study is on inherent susceptibility of a particular backbone. Thus, we treated the data for samples with non-zero baselines using only early time points before product inhibition was apparent and a constrained baseline for the exponential decay based on complete peptide degradation. Assuming that, under different circumstances, the initial rate of proteolysis could be sustained, this treatment of the data provides the most conservative estimate of the degree of protection that could be afforded by a particular backbone modification. Full progress curves and digestion product analyses for all peptides can be found in the supplementary information.
Circular Dichroism
Solutions of 40 μM peptide in 20 mM sodium phosphate buffer (pH 6.9) were subjected to CD scans using an Olis DSM17 circular dichroism spectrophotometer. Scans were performed at 25 °C from λ 200 – 260 nm in 1 nm increments with a 2 nm bandwidth. The Savitzky-Golay method was used to smooth the data.[23]
Supplementary Material
Acknowledgements
Funding for this work was provided by the National Institutes of Health (R01GM107161).
References
- [1].a) Arkin MR, Tang Y, Wells James A. Chem. Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guo W, Wisniewski JA, Ji H. Bioorg. Med. Chem. Lett. 2014;24:2546–2554. doi: 10.1016/j.bmcl.2014.03.095. [DOI] [PubMed] [Google Scholar]
- [2].Nevola L, Giralt E. Chem. Commun. 2015;51:3302–3315. doi: 10.1039/c4cc08565e. [DOI] [PubMed] [Google Scholar]
- [3].a) Werle M, Bernkop-Schnürch A. Amino Acids. 2006;30:351–367. doi: 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]; b) Nestor JJ., Jr Curr. Med. Chem. 2009;16:4399–4418. doi: 10.2174/092986709789712907. [DOI] [PubMed] [Google Scholar]; c) Stevenson CL. Curr. Pharm. Biotechnol. 2009;10:122–137. doi: 10.2174/138920109787048634. [DOI] [PubMed] [Google Scholar]; d) Rink R, Arkema-Meter A, Baudoin I, Post E, Kuipers A, Nelemans SA, Akanbi MHJ, Moll GN. J. Pharmacol. Toxicol. Methods. 2010;61:210–218. doi: 10.1016/j.vascn.2010.02.010. [DOI] [PubMed] [Google Scholar]; e) Bellmann-Sickert K, Beck-Sickinger AG. Trends Pharmacol. Sci. 2010;31:434–441. doi: 10.1016/j.tips.2010.06.003. [DOI] [PubMed] [Google Scholar]; f) Weinstock MT, Francis JN, Redman JS, Kay MS. Pept. Sci. 2012;98:431–442. doi: 10.1002/bip.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Diao L, Meibohm B. Clin. Pharmacokinet. 2013;52:855–868. doi: 10.1007/s40262-013-0079-0. [DOI] [PubMed] [Google Scholar]
- [4].Avan I, Hall CD, Katritzky AR. Chem. Soc. Rev. 2014;43:3575–3594. doi: 10.1039/c3cs60384a. [DOI] [PubMed] [Google Scholar]
- [5].a) Fiacco SV, Roberts RW. ChemBioChem. 2008;9:2200–2203. doi: 10.1002/cbic.200800208. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gaston F, Granados GC, Madurga S, Rabanal F, Lakhdar-Ghazal F, Giralt E, Bahraoui E. ChemMedChem. 2009;4:570–581. doi: 10.1002/cmdc.200800390. [DOI] [PubMed] [Google Scholar]; c) Dong JZ, Shen Y, Zhang J, Tsomaia N, Mierke DF, Taylor JE. Diabetes Obes. Metab. 2011;13:19–25. doi: 10.1111/j.1463-1326.2010.01313.x. [DOI] [PubMed] [Google Scholar]
- [6].Gellman SH. Acc. Chem. Res. 1998;31:173–180. [Google Scholar]
- [7].a) Bautista AD, Craig CJ, Harker EA, Schepartz A. Curr. Opin. Chem. Biol. 2007;11:685–692. doi: 10.1016/j.cbpa.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Goodman CM, Choi S, Shandler S, DeGrado WF. Nat. Chem. Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Seebach D, Gardiner J. Acc. Chem. Res. 2008;41:1366–1375. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]; d) Guichard G, Huc I. Chem. Commun. 2011;47:5933–5941. doi: 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]; e) Horne WS. Expert Opin. Drug Discov. 2011;6:1247–1262. doi: 10.1517/17460441.2011.632002. [DOI] [PubMed] [Google Scholar]
- [8].a) Reinert ZE, Horne WS. Org. Biomol. Chem. 2014;12:8796–8802. doi: 10.1039/c4ob01769b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Werner HM, Horne WS. Curr. Opin. Chem. Biol. 2015;28:75–82. doi: 10.1016/j.cbpa.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Reinert ZE, Lengyel GA, Horne WS. J. Am. Chem. Soc. 2013;135:12528–12531. doi: 10.1021/ja405422v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Tugyi R, Uray K, Iván D, Fellinger E, Perkins A, Hudecz F. Proc. Natl. Acad. Sci. USA. 2005;102:413–418. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wada S.-i., Tsuda H, Okada T, Urata H. Bioorg. Med. Chem. Lett. 2011;21:5688–5691. doi: 10.1016/j.bmcl.2011.08.030. [DOI] [PubMed] [Google Scholar]; c) Chatterjee J, Rechenmacher F, Kessler H. Angew. Chem. Int. Ed. 2013;52:254–269. doi: 10.1002/anie.201205674. [DOI] [PubMed] [Google Scholar]; d) Cabrele C, Martinek TA, Reiser O, Berlicki Ł. J. Med. Chem. 2014 doi: 10.1021/jm5010896. [DOI] [PubMed] [Google Scholar]
- [11].a) Southan C. J. Pept. Sci. 2000;6:453–458. doi: 10.1002/1099-1387(200009)6:9<453::AID-PSC284>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]; b) Hedstrom L. Chem. Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]; c) Gráf L, Szilágyi L, Venekei I. In: Handbook of Proteolytic Enzymes. Salvesen NDR, editor. Academic Press; 2013. pp. 2626–2633. [Google Scholar]
- [12].Baeriswyl V, Heinis C. Prot. Eng. Des. Sel. 2013;26:81–89. doi: 10.1093/protein/gzs085. [DOI] [PubMed] [Google Scholar]
- [13].Berger A, Schechter I. Philos. Trans. R. Soc. London, Ser. B. 1970;257:249–264. doi: 10.1098/rstb.1970.0024. [DOI] [PubMed] [Google Scholar]
- [14].Bizzozero SA, Dutler H. Bioorg. Chem. 1981;10:46–62. [Google Scholar]
- [15].Wang D, Friedmann M, Gattin Z, Jaun B, van Gunsteren WF. Helv. Chim. Acta. 2010;93:1513–1531. [Google Scholar]
- [16].a) Fujinaga M, Sielecki AR, Read RJ, Ardelt W, Laskowski M, Jr, James MNG. J. Mol. Biol. 1987;195:397–418. doi: 10.1016/0022-2836(87)90659-0. [DOI] [PubMed] [Google Scholar]; b) Hecht HJ, Szardenings M, Collins J, Schomburg D. J. Mol. Biol. 1991;220:711–722. doi: 10.1016/0022-2836(91)90112-j. [DOI] [PubMed] [Google Scholar]; c) Frigerio F, Coda A, Pugliese L, Lionetti C, Menegatti E, Amiconi G, Schnebli HP, Ascenzi P, Bolognesi M. J. Mol. Biol. 1992;225:107–123. doi: 10.1016/0022-2836(92)91029-o. [DOI] [PubMed] [Google Scholar]; d) Roussel A, Mathieu M, Dobbs A, Luu B, Cambillau C, Kellenberger C. J. Biol. Chem. 2001;276:38893–38898. doi: 10.1074/jbc.M105707200. [DOI] [PubMed] [Google Scholar]; e) Kim H, Chu TTT, Kim DY, Kim DR, Nguyen CMT, Choi J, Lee J-R, Hahn M-J, Kim KK. J. Mol. Biol. 2008;376:184–192. doi: 10.1016/j.jmb.2007.11.089. [DOI] [PubMed] [Google Scholar]; f) Clark EA, Walker N, Ford DC, Cooper IA, Oyston PCF, Acharya KR. J. Biol. Chem. 2011;286:24015–24022. doi: 10.1074/jbc.M111.225730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Venkatraman J, Shankaramma SC, Balaram P. Chem. Rev. 2001;101:3131–3152. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]; b) Schweitzer-Stenner R, Gonzales W, Bourne GT, Feng JA, Marshall GR. J. Am. Chem. Soc. 2007;129:13095–13109. doi: 10.1021/ja0738430. [DOI] [PubMed] [Google Scholar]
- [18].a) Wilcox PE, Segal DM, Powers JC, Cohen GH, Davies DR. Biochemistry. 1971;10:3728–3738. doi: 10.1021/bi00796a014. [DOI] [PubMed] [Google Scholar]; b) Segal DM. Biochemistry. 1972;11:349–356. doi: 10.1021/bi00753a007. [DOI] [PubMed] [Google Scholar]
- [19].Lengyel GA, Horne WS. J. Am. Chem. Soc. 2012;134:15906–15913. doi: 10.1021/ja306311r. [DOI] [PubMed] [Google Scholar]
- [20].Reinert ZE, Horne WS. Chem. Sci. 2014;5:3325–3330. doi: 10.1039/C4SC01094A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Adessi C, Frossard M-J, Boissard C, Fraga S, Bieler S, Ruckle T, Vilbois F, Robinson SM, Mutter M, Banks WA, Soto C. J. Biol. Chem. 2003;278:13905–13911. doi: 10.1074/jbc.M211976200. [DOI] [PubMed] [Google Scholar]; b) Banerjee R, Basu G, Roy S, Chène P. J. Pept. Res. 2002;60:88–94. doi: 10.1034/j.1399-3011.2002.201005.x. [DOI] [PubMed] [Google Scholar]; c) Charpentier B, Durieux C, Pelaprat D, Dor A, Reibaud M, Blanchard J-C, Roques BP. Peptides. 1988;9:835–841. doi: 10.1016/0196-9781(88)90130-1. [DOI] [PubMed] [Google Scholar]
- [22].Selwyn MJ. Biochim. Biophys. Acta. 1965;105:193–195. doi: 10.1016/s0926-6593(65)80190-4. [DOI] [PubMed] [Google Scholar]
- [23].Savitzky A, Golay MJE. Anal. Chem. 1964;36:1627–1639. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.