

Abstract
Chronic alcohol consumption is one of the most common causes of the progression of alcoholic liver disease (ALD). In the past, alcohol-mediated hepatocyte injury was assumed to be a significantly major cause of ALD. However, a huge number of recent and brilliant studies have demonstrated that hepatic non-parenchymal cells including Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells and diverse types of lymphocytes play crucial roles in the pathogenesis of ALD by producing inflammatory mediators such as cytokines, oxidative stress, microRNA, and lipid-originated metabolites (retinoic acid and endocannabinoids) or by directly interacting with parenchymal cells (hepatocytes). Therefore, understanding the comprehensive roles of hepatic non-parenchymal cells during the development of ALD will provide new integrative directions for the treatment of ALD. This review will address the roles of non-parenchymal cells in alcoholic steatosis, inflammation, and liver fibrosis and might help us to discover possible therapeutic targets and treatments involving modulating the non-parenchymal cells in ALD.
Keywords: Alcoholic liver disease, Reactive oxygen stress, Endocannabinoid, NADPH oxidase
Core tip: Chronic alcohol consumption commonly causes chronic liver diseases including liver fibrosis and cirrhosis. According to recent studies, hepatic non-parenchymal cells including Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells and liver lymphocytes are important to modulate the pathogenesis of alcoholic liver disease (ALD) by producing inflammatory mediators or by interacting either hepatic parenchymal cells (hepatocytes) or non-parenchymal cells. Therefore, understanding the novel roles of hepatic non-parenchymal cells during the development of ALD is important and it will be considered as therapeutic targets for alcoholic liver diseases.
INTRODUCTION
Chronic alcohol abuse increases mortality rates worldwide due to alcoholic liver disease (ALD)[1]. ALD presents as a broad spectrum of liver disease, ranging from mild steatosis to more serious types of liver injury such as steatohepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma[2]. From a classical point of view, the liver has been considered a definite metabolic organ that is generally involved in diverse energy metabolisms of alcohol, glucose, lipid, and many significant molecules[3-5]. With excessive alcohol consumption, it is generally assumed that liver injury is due to the production of acetaldehyde-mediated adducts and oxidative stress causing compounds such as reactive oxygen species (ROS), which interfere with the metabolism of other energy sources[2,3]. These compounds are predominantly generated through the induction of cytochrome P450 2E1 (CYP2E1) and alcohol dehydrogenases (ADHs) in hepatocytes[2,3].
However, production of alcoholic metabolites and ROS is not enough to describe the entire pathogenesis of ALD. Interestingly, a line of evidence suggests that the liver seems to be another type of immunologic organ because of its enriched, innate, and adaptive immune cells, which play important roles in alcoholic immune responses against pathogens or pathogen associated molecular patterns derived from the gastrointestinal tracts, and danger signals from injured hepatocytes[6,7].
The liver is generally comprised of hepatocytes, which make up approximately two thirds of its total cell population (60%-70%), and non-parenchymal cells (30%-40%). The population of non-parenchymal cells includes liver sinusoidal endothelial cells (LSEC) (approximately 50%), Kupffer cells (approximately 20%), lymphocytes (approximately 25%), biliary cells (approximately 5%), and hepatic stellate cells (HSCs) (approximately 1%)[6,7]. Liver sinusoidal endothelial cells have a unique morphological phenotype called fenestrated endothelium. Normal morphology of LSEC is flat with a nucleus and organelles regularly and expresses CD45-CD146+ surface marker. The distal cytoplasm displayed as a lamina with many fenestrae, lacking the basement membrane underneath the endothelium[8]. Less than 100 nm nano-molecules are diffuse through the fenestrae, attributing a sieve function to LSEC[9]. Therefore, LSEC are responsible for the clearance of various macromolecules from the blood such as proteins, poly saccharides, lipids and nucleic acids[10]. Kupffer cells are known as liver resident macrophage and derived from circulating monocytes that arise from bone marrow progenitors[11]. The major role of Kupffer cells is to clear circulating endotoxin, microorganism, dead cells and debris. Endotoxins from gut activate Kupffer cells and produce pro-inflammatory cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α and transforming growth factor (TGF)-β. Liver lymphocytes are found scattered throughout the parenchyma and in the portal tract[7]. Liver is especially abundant in innate immune cells such as natural killer (NK), NKT, and γδ T cells; these comprise almost half of liver lymphocytes[6,7]. For example, the population of NK and NKT cells is approximately 30%-40% that of the total liver lymphocytes; γδ T cells account for 3% to 5% of these cells in mice and humans[6]. Infiltration of the liver by lymphocytes occurs in response to many insults such as autoimmune and toxic damage. The distribution of hepatic infiltration by lymphocytes varies depending on the inflammatory stimulus[12]. Hepatic stellate cells are situated in space of Disse between hepatocytes and the sinusoids. Hepatic stellate cells are generally quiescent in normal healthy livers (quiescent HSCs), however, they undergo morphological changes during liver injury. They gradually become activated (activated HSCs) and differentiate into myofibroblastic cells, characterized by a loss of vitamin A (retinol) and enhanced collagen expression[13,14].
Recently, cell-to-cell communication within the liver is a rising field to understand the pathogenesis of ALD[15]. For instance, HSCs and Kupffer cells have also been found to be involved in the pathogenesis of ALD via interaction with hepatic immune cells[16,17]. Therefore, the development of ALD is a sort of complex interaction between parenchymal (hepatocyte) and non-parenchymal cells.
In the present review, we summarize the novel specific roles of non-parenchymal cells in ALD with particular emphasis on alcoholic liver steatosis, inflammation, and fibrosis; we provide therapeutic strategies for curing ALD.
NON-PARENCHYMAL CELLS IN ALCOHOLIC STEATOSIS AND INFLAMMATION OF LIVER
Hepatic steatosis is the most common response of the liver to acute binge and chronic alcohol consumption. If alcohol consumption is not stopped, hepatic steatosis subsequently progresses into inflammation. Thus steatosis and inflammation are important events in the initiation of alcoholic liver disease. It is generally believed that fat accumulation in hepatocytes is a consequence of imbalanced fat metabolism, such as up-regulated fat synthesis by sterol regulatory element-binding protein 1c (SREBP1c) and suppressed lipid oxidation by inhibited activation of AMP-activated protein kinase (AMPK)[2].
Contribution of activated Kupffer cell in development of hepatic steatosis and inflammation
Kupffer cells are mainly involved in the development of alcoholic steatosis in liver[18,19]. Enhanced gut permeabilization by alcohol consumption allows an increased uptake of lipopolysaccharide (LPS) in portal circulation[20,21], the delivered LPS in turn activates Kupffer cells via the toll-like receptor 4 (TLR4) signaling pathway, consequently leading to the production of pro-inflammatory mediators such as TNF-α, IL-1β, IL-6, and ROS[2,18,22]. It has been reported that TNF-α has the potential to increase the expression and maturation of SREBP1c in the liver of mice and human hepatocytes[23,24]. Furthermore, a recent report demonstrated that alcohol-mediated infiltration of macrophages into adipose tissue decreased the amount of adiponectin (known as an anti-steatosis peptide hormone that responses via up-regulation of AMPK activity) production of adipocytes, leading to alcoholic liver steatosis[25]. Therefore, Kupffer cells/macrophages might contribute to the development of alcoholic liver steatosis by down-regulating adiponectin-mediated activation of AMPK in hepatocytes. In contrast, IL-6 production by Kupffer cells/macrophages ameliorates alcohol-mediated hepatic steatosis by activating a signal transducer and activator of transcription 3 (STAT3) and inhibiting SREBP1c gene expression in hepatocytes[26-28].
If alcohol consumption is continued, alcoholic steatosis progresses into more severe types of liver disease such as hepatitis, in which many types of hepatic cells participate in the initiation of inflammation. As described above, one of the important factors in the progression to alcoholic hepatitis is increased LPS concentration in the portal blood stream. Alcohol increases levels of microRNA (miR)-212 in the gut epithelial cells that down-regulate the tight junction, Zonula occludens-1, inducing gut leakage by disruption of gut integrity and permeability[21]. Thereby elevated LPS activates TLR4 of the Kupffer cells to produce inflammatory mediators. Among these mediators, TNF-α plays the most important role not only in the development of steatosis but also in inflammatory responses in alcohol-induced liver injury[29]. In addition, ROS produced by NADPH oxidase (NOX) in Kupffer cells further enhances alcohol-mediated liver injury by stimulating the production of inflammatory mediators[30,31]. Moreover, chronic and binge ethanol drinking activates the NLRP3 (Nucleotide-binding domain and Leucine rich Repeat containing family, Pyrin domain containing 3) inflammasome in the Kupffer cells, inducing mature IL-1β release in ALD[32]. ROS has been considered one of several important factors in the maturation of IL-1β via NLRP3 in macrophages; LPS/TLR4 might be related with NOX-mediated ROS production in pulmonary endothelial cells, indicating a possible link between alcohol-mediated ROS production and the maturation of IL-1β in Kupffer cells[33,34]. However, there has as yet been no report on whether NOX-mediated ROS production regulates maturation of inflammasome-mediated IL-1β in alcoholic hepatitis. In the past decade, very important studies on micro RNAs have been performed and their regulatory functions against messenger RNAs have been reported. MicroRNAs including miR-125b, miR-146a, and miR-155 regulate inflammatory responses such as nuclear factor kappa B (NF-κB) activation and TNF-α production of LPS in macrophages and Kupffer cells[35,36]. However, chronic alcohol treatment increases the miR-155 level and TNF-α production in the Kupffer cells of mice[36]. Similarly, in isolated human HSCs, LPS treatment has been found to increase inflammatory cytokines (TNF- α and IL-1β) and adhesion molecules (ICAM-1 and VCAM-1) in NF-κB and JNK-dependent manners[37].
HSC activation and its retinol metabolism in alcoholic hepatic steatosis
In our previous study, we showed that chronic alcohol consumption increased production of 2-arachidonoylglycerol (2-AG), an endocannabinoid, in HSCs and that it enhanced fat accumulation in hepatocytes by suppressing the activity of AMPK while increasing the expression of SREBP1c and fatty acid synthase (FAS) through CB1R in hepatocytes[38]. Therefore, HSCs are regarded as a crucial factor in the endocannabinoid-dependent lipogenesis through paracrine activation of hepatic CB1 receptors in alcohol-induced steatosis. In contrast, chronic alcohol feeding (5% liquid ethanol diet for 8 wk) to mice does not induce liver fibrosis, although increased CYP2E1 expression and loss of retinol lipid droplets are detected in the liver and HSCs, respectively (Figure 1A). These changes might be due to NK cell killing of activated HSCs (Figure 1B). Previous interesting studies have demonstrated that NK cells can kill activated HSCs in a NKG2D-retinoic acid early inducible gene 1 (RAE1)-dependent manner[39-41]. During activation of HSCs, retinol lipid droplets are metabolized into retinoic acid such as all-trans retinoic acid or 9-cis retinoic acid, leading to induction of RAE1 through retinoic acid receptors[40]. In addition, freshly isolated HSCs from ethanol-fed mice showed enhanced RAE1 expression and were susceptible to NK cell killing (Figure 2). However, the mechanism of increased retinol metabolism and RAE1 expression in HSCs from ethanol-fed mice is not yet clearly understood. Recent studies have reported that class III ADH (ADH3) is the most important enzyme for retinol metabolism in HSCs, and that ADH3 might be involved in alcohol metabolism in the liver[42,43], suggesting that increased retinol metabolism in HSCs is due to the activation of ADH3 by alcohol. In rat HSCs, alcohol-mediated protein adducts such as malondialdehyde-acetaldehyde could increase chemokines such as monocyte chemoattractant protein (MCP)-1 and macrophage inflammatory protein (MIP)-2, contributing to hepatic inflammation in ALD[44].
Figure 1.
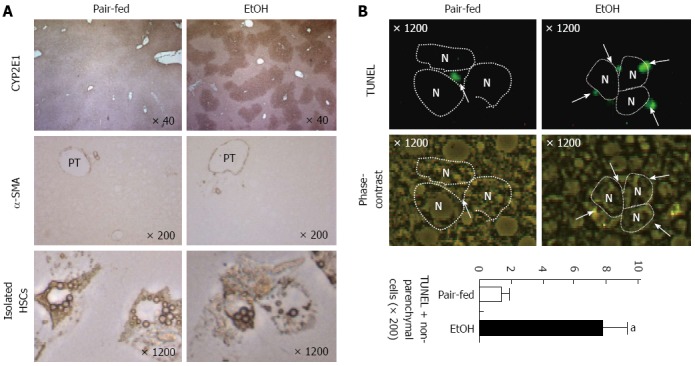
Chronic ethanol consumption increases CYP2E1 expression in hepatocytes and apoptosis of activated hepatic stellate cells. Mice were fed with 5% liquid ethanol (EtOH) or isocaloric control (Pair-fed) diet for 8 wk. A: Liver tissues were stained with antibodies of CYP2E1 and α-smooth muscle actin (α-SMA). Hepatic stellate cells (HSCs) were freshly isolated from pair-fed and ethanol-fed mice; B: Liver tissues were stained with TUNEL and apoptotic bodies of non-parenchymal cells were counted. PT: Portal triad; N: Nucleus of hepatocyte. aP < 0.05 in comparison with the corresponding control.
Figure 2.
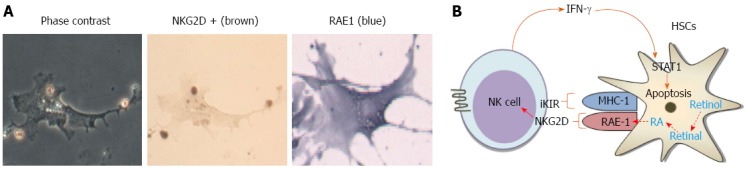
Freshly isolated hepatic stellate cells from liquid ethanol-fed mice were susceptible to natural killer cell killing. A: Mice were fed with 5% liquid ethanol diet for 8 wk. Hepatic stellate cells (HSCs) were freshly isolated from ethanol-fed mice and then co-cultured with natural killer (NK) cells. NK cells were stained with NKG2D antibody and HSCs were stained with RAE1; B: Schematic overview of NKT cell cytotoxicity against activated HSCs via interferon (IFN)-γ-dependent manner.
Dysfunction of LSEC and role of liver NKT cells during alcohol-mediated liver injury
LSEC is affected in various pathological conditions. During liver injury, LSEC lose their fenestration in a process called “capillarization” or “defenestration” due to the formation of extracellular matrix resulting in LSEC dysfunction. In acute or chronic alcohol exposure of mice, LSEC and the hepatic sinusoid undergo structural and functional alternations. Especially, the impairment of scavenging function of LSEC was observed in a dose- and time-dependent manner in acute alcohol consumption model[15]. Furthermore, the number of fenestrae was significantly decreased and the formation of the basement membrane of LSEC occurred gradually after chronic alcohol consumption[8,45]. Therefore, alcohol consumption is associated with changes in LSEC scavenging function and structure that precede the appearance of detectable pathological alterations in hepatocytes and the pathological alternations of the LSEC and possibly other non-parenchymal cells (e.g. HSCs) leading to “capillarization” of the hepatic sinusoid. Consequently, alcohol consumption inhibits the exchange between the sinusoidal plasma and parenchymal cells, ultimately producing liver parenchymal cell injury[15].
In liver lymphocytes, hepatic NKT cells also play important roles in liver injury by producing inflammatory cytokines[32]. Hepatic NKT cells activated by Kupffer cells are recruited in alcoholic steatohepatitis of mice in NLRP3 inflammasome- and IL-1β-dependent manners, resulting in the increased expression of interferon-γ (IFN-γ) and TNF-α in NKT cells[32]. In human patients with alcoholic liver disease, CD8 and CD4 T cells were infiltrated in the liver, that infiltration was related with inflammation and regeneration[46]. In addition, chronic alcohol consumption decreases hepatic NK cell activity [cytotoxicity against Yac-1 cells and polyinosinic-polycytidylic acid (poly I:C)-mediated activation], thereby accelerating murine cytomegalovirus-induced hepatitis and liver injury[47].
Taken together, alcoholic hepatic steatosis and inflammation form a mixture of metabolic and immunologic responses between parenchymal cells and non-parenchymal cells. Especially, recent papers have focused on and highlighted Kupffer cell-mediated inflammatory responses and de novo lipogenesis in hepatocytes by 2-AG production of HSCs (Figure 3)[2,48-50].
Figure 3.
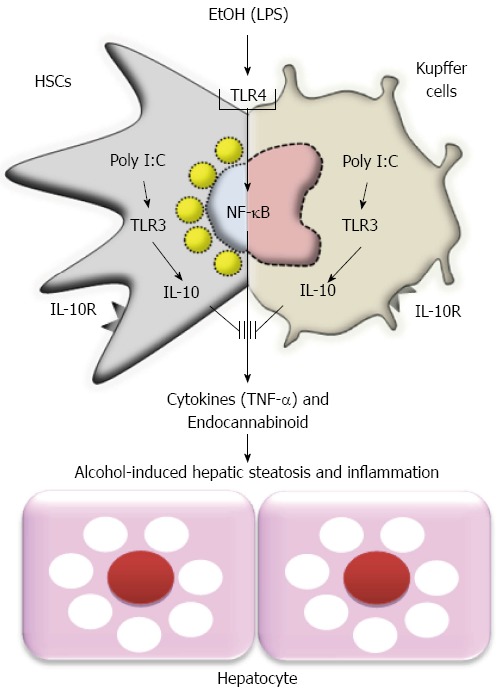
Model of ethanol-mediated hepatic steatosis and inflammation and preventive effects of toll-like receptor 3 activation in hepatic stellate cells and Kupffer cells against alcoholic liver injury. HSCs: Hepatic stellate cells; EtOH: Ethanol; TLR3: Toll-like receptor 3; IL: Interleukin; LPS: Lipopolysaccharide; TNF-α: Tumor necrosis factor α; NF-κB: Nuclear factor kappa B.
NON-PARENCHYMAL CELLS IN ALCOHOLIC FIBROSIS IN LIVER
Chronic alcohol consumption is one of the causes of liver fibrosis; the underlying mechanism of alcoholic liver fibrosis is not completely understood, although there are huge amounts of accumulated data in the literature. Generally, chronic alcohol drinking induces simple steatosis, but only 10%-40% of heavy drinkers progress from steatosis to alcoholic liver fibrosis[22,51]. In the past, it was believed that alcohol-mediated hepatocyte injury and the phagocytosis of hepatocyte apoptotic bodies in Kupffer cells and in HSCs predispose the liver to the production of collagen fibers, eventually leading to accelerated liver fibrosis[52-55]. However, recent lines of evidence have recommended a novel possibility that chronic alcohol consumption accelerates liver fibrosis by modulating hepatic non-parenchymal cells such as HSCs, Kupffer cells, and liver lymphocytes.
Interaction with activated HSCs and Kupffer cells during alcohol-mediated liver fibrosis
During HSC activation by various stimuli, one of several specific markers is the loss of intracellular retinol (vitamin A) lipid droplets. No convincing data have been reported to address the mechanism that links the activation of HSC to the loss of retinols. However, a recent interesting study has reported that the gene ADH3 is only expressed for retinol metabolism in HSCs of humans and mice; ADH3 expression is significantly increased in proportion to production of retinoic acid and collagen in HSCs in vitro[43]. Studies so far have been suggesting that alcohol-mediated activation of ADH3 might accelerate liver fibrosis by production of retinoic acid in HSCs. Furthermore, CYP2E1 plays important roles not only in alcohol metabolism but in ROS production in hepatocytes; alcohol consumption-mediated ROS production has been considered one of the major causes of the induction of liver fibrosis. In co-culture experiments of HSCs and HepG2 cells expressing CYP2E1, CYP2E1-derived ROS is responsible for the increase of collagen in HSCs[52]. In addition to ROS, the LPS/TLR4 signaling pathway makes HSCs more sensitive to alcohol and acetaldehyde in the production of collagen and IL-6[56]. By using reciprocal bone marrow transplantation between WT and TLR4 KO mice, TLR4-mediated fibrogenic responses such as increased expression of collagen and TGF-β1 were increased in HSCs during intragastric ethanol feeding to WT mice[57]. Consistently, in chemical and bile duct ligation-mediated liver fibrosis, TLR4 activation in HSCs induces down-regulation of Bambi, TGF-β pseudoreceptor, and leads to the sensitizing of HSCs to TGF-β signaling pathway, consequently enhancing liver fibrosis[58]. Thus, TLR4 has been considered one of several important targets for the treatment of liver fibrosis. In high fat diet and alcohol-induced liver fibrosis models, this combination leads synergistically to the development of liver fibrosis by TLR4 activation of non-parenchymal cells[59]. Furthermore, the direct impact of Kupffer cells on the activation of HSCs has been well recognized. In the co-culturing model of primary Kupffer cells and HSCs, Kupffer cells enhance the activation of HSCs by producing oxidative stress and IL-6[60]. Ethanol and arachidonic acid synergize to activate Kupffer cells and modulate the fibrogenic response via enhancing of the production of TNF-α and generation of NOX-mediated ROS, inducing activation of HSCs[61].
Interactions with activated HSCs and liver lymphocytes in the pathogenesis of alcohol-mediated liver fibrosis
Recent interesting studies have suggested that diverse types of lymphocytes are actively involved in the pathogenesis of liver fibrosis because the liver is normally enriched with innate and adaptive lymphocytes[6,7]. Among these lymphocytes, NK cells have anti-fibrotic effects, directly killing activated HSCs in NKG2D- and tumor necrosis factor-related apoptosis and inducing ligand (TRAIL)-dependent manners but not quiescent HSCs[41,62]. Moreover, NK cells can suppress liver fibrosis via production of IFN-γ, which can induce cell cycle arrest and apoptosis of HSCs in a STAT1-dependant manner and autocrine activation of NK cells[63,64]. However, lines of evidence have reported chronic alcohol consumption-mediated impairments of immune surveillance of lymphocytes against activated HSCs. Decreased cytotoxicity and number of liver NK cells against HSCs and tumor cells were observed in mice subject to chronic alcohol consumption[39,47]. Indeed, direct IFN-γ treatment failed to increase activation of NK cells or to suppress activated HSCs in chronically alcohol-fed mice, indicating no beneficial effects of IFN-γ in alcoholic liver fibrosis[39]. The suppressed NK cell activity and low susceptibility to IFN-γ in alcoholic liver fibrosis are due to the increased expression and production of TGF-β1 and SOCS1 by activated HSCs, respectively[39]. In line with these findings, patients with liver cirrhosis showed decreased NK cell numbers and reduced cytotoxic activity of T cells in peripheral blood[65]. Similarly, NKT cells can also suppress HSC activation by killing and IFN-γ production in the early stages of liver fibrosis[66]. In chronic alcohol consumption, the significantly increased number of NKT cells contributes in an inverse manner to accelerated alcoholic liver injury by up-regulating the NF-κB signaling pathway or by Fas and TNF-receptor-mediated hepatocyte apoptosis[67,68]. Nevertheless the effects of alcohol on NKT cells are still controversial. A recent interesting study reported that CD4 T cell-mediated IL-17A production promotes liver fibrosis by stimulating Kupffer cells and HSCs in chemical and bile duct ligation-mediated liver fibrosis models in mice[69]. In parallel with this report, CD4 T cells stimulated by ADH peptides were found to produce higher levels of IL-17 and IFN-γ in active drinkers, while IL-4 production was lower than that in abstinent patients, leading to the encouragement of liver fibrosis in patients with alcohol-related cirrhosis[70]. In addition, γδ T cells, a minor part of the population of liver lymphocytes, could ameliorate chemical and diet-induced liver fibrosis of mice by Fas ligand-mediated killing of HSCs[71]. However, there has been no report on the role of γδ T cells in alcoholic liver fibrosis. Therefore, in addition to information on stimuli from damaged hepatocytes, information on the diverse but specific roles of each of the non-parenchymal cells (HSCs, Kupffer cells, NK, NKT, CD4 T cells, and γδ T cells) is important in attempts to understand the pathogenesis of alcoholic liver fibrosis.
TREATMENT
Paradoxically, one set of epidemiological data reported that moderate alcoholic beverage consumption has a beneficial effect on longevity, while another showed that it contributed to the development of cancers of the mouth, pharynx, larynx, esophagus, breast and liver[72-74]. Similarly, a study reported that acute ethanol treatment inhibited production of TNF-α and IL-1β, while production of IL-10 and TGF-β was increased in human monocytes during bacterial stimulation in vitro[75]. In addition, alcohol treatment can facilitate the passive loading of glycolipid to the CD1d molecule in CD1d transfected HeLa cells[76]. Thus, additional mechanistic study should be undertaken to consider these discrepant effects of alcohol drinking.
In alcoholic patients, abstinence is the best therapy but it is practically difficult to achieve. Although liver transplantation might be an alternative choice, it is not always a suitable treatment because of donor shortage[77]. Therefore, numerous efforts have been conducted to create therapeutics for ALD. To reduce or inhibit inflammation and oxidative stress, various anti-inflammatory drugs, anti-oxidants and anti-fibrotics have been developed and their effectiveness has been demonstrated in animals and patients with ALD[78-81]. On the other hand, clinical studies of treatment with antioxidants have shown no beneficial effects of these substances for either patients with alcoholic hepatitis or those with alcoholic cirrhosis. To overcome these discrepancies between animal studies and clinical trials, therapeutic strategies should be reconstituted and made to target to multiple cells among non-parenchymal cells. For example, treatments for alcoholic steatohepatitis should be targeted simultaneously at HSCs and Kupffer cells, because the two types of cells can concurrently produce endocannabinoid (e.g., 2-AG) and inflammatory mediators (e.g., TNF-α and ROS). For alcoholic liver fibrosis, the simultaneous suppression of HSCs and Kupffer cells and activation of NK cells should be considered because of the pro-fibrotic cytokine production of HSCs and Kupffer cells and the cytotoxicity of NK cells against activated HSCs. Our recent study suggested that poly I:C-mediated activation of TLR3 in HSCs and Kupffer cells increased the production of IL-10, leading to the simultaneous amelioration of alcoholic steatosis and inflammation in 2-wk binge ethanol drinking mice (Figure 3)[49]. Moreover, inhibition of IL-1 receptor-mediated signaling could ameliorate alcoholic steatohepatitis by down-regulating TLR4-dependent inflammatory signaling[82]. Recently, bone marrow infusion therapy has been highlighted as a treatment of liver fibrosis[83]. However, chronic alcohol consumption stimulated the recruitment of TNF-α and TGF-β1, producing activated HSCs from the bone marrow to the liver[84], inducing a worse situation of alcoholic liver injury. Thus, in the field of ALD treatment, we need to more carefully arrange strategies that are capable of increasing NK cell activity while concurrently suppressing activation of HSCs and Kupffer cells. However, to prevent alcohol-mediated disease, no therapeutic strategy can outperform abstinence from the drinking of alcohol.
CONCLUSION
The present review has addressed the important roles of hepatic non-parenchymal cells in the pathogenesis of ALD. By producing inflammatory mediators (TNF-α, IL-1β, IL-6, IL-17, TGF-β, ROS, and miRNA) and lipid-originated metabolites (retinoic acid and endocannabinoids) in immune cells (Kupffer cells and lymphocytes), LSEC and HSCs, multiple types of non-parenchymal cells are involved in the development of alcoholic steatosis, inflammation, and fibrosis. Moreover, interactions among non-parenchymal cells play important roles in ALD. Thus, we should concurrently consider the various roles of diverse non-parenchymal cells as we search for therapeutics for ALD. In conclusion, dedicated and orchestrated studies for the understanding of the roles of non-parenchymal cells will help us to conquer ALD.
Footnotes
Supported by A grant from the Next-Generation BioGreen 21 Program, No. PJ009957; Rural Development Administration; and partially from the Korea Advanced Institute of Science and Technology Institute for the BioCentury, South Korea.
Conflict-of-interest statement: The authors have no conflict of interest to report.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 27, 2015
First decision: September 9, 2015
Article in press: December 14, 2015
P- Reviewer: Han JL, Seth D S- Editor: Gong ZM L- Editor: A E- Editor: Wang CH
References
- 1.O’Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 2.Purohit V, Gao B, Song BJ. Molecular mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res. 2009;33:191–205. doi: 10.1111/j.1530-0277.2008.00827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63:49–63. doi: 10.1079/pns2003327. [DOI] [PubMed] [Google Scholar]
- 4.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56:952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 5.Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4:177–197. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao B, Jeong WI, Tian Z. Liver: An organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 7.Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- 8.Wang BY, Ju XH, Fu BY, Zhang J, Cao YX. Effects of ethanol on liver sinusoidal endothelial cells-fenestrae of rats. Hepatobiliary Pancreat Dis Int. 2005;4:422–426. [PubMed] [Google Scholar]
- 9.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1:1. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elvevold K, Smedsrød B, Martinez I. The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol. 2008;294:G391–G400. doi: 10.1152/ajpgi.00167.2007. [DOI] [PubMed] [Google Scholar]
- 11.Gale RP, Sparkes RS, Golde DW. Bone marrow origin of hepatic macrophages (Kupffer cells) in humans. Science. 1978;201:937–938. doi: 10.1126/science.356266. [DOI] [PubMed] [Google Scholar]
- 12.Lalor PF, Shields P, Grant A, Adams DH. Recruitment of lymphocytes to the human liver. Immunol Cell Biol. 2002;80:52–64. doi: 10.1046/j.1440-1711.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- 13.Friedman SL, Rockey DC, Bissell DM. Hepatic fibrosis 2006: report of the Third AASLD Single Topic Conference. Hepatology. 2007;45:242–249. doi: 10.1002/hep.21459. [DOI] [PubMed] [Google Scholar]
- 14.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarphie G, D’Souza NB, Van Thiel DH, Hill D, McClain CJ, Deaciuc IV. Dose- and time-dependent effects of ethanol on functional and structural aspects of the liver sinusoid in the mouse. Alcohol Clin Exp Res. 1997;21:1128–1136. [PubMed] [Google Scholar]
- 16.Yi HS, Jeong WI. Interaction of hepatic stellate cells with diverse types of immune cells: foe or friend? J Gastroenterol Hepatol. 2013;28 Suppl 1:99–104. doi: 10.1111/jgh.12017. [DOI] [PubMed] [Google Scholar]
- 17.Cohen JI, Nagy LE. Pathogenesis of alcoholic liver disease: interactions between parenchymal and non-parenchymal cells. J Dig Dis. 2011;12:3–9. doi: 10.1111/j.1751-2980.2010.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bird GL, Sheron N, Goka AK, Alexander GJ, Williams RS. Increased plasma tumor necrosis factor in severe alcoholic hepatitis. Ann Intern Med. 1990;112:917–920. doi: 10.7326/0003-4819-112-12-917. [DOI] [PubMed] [Google Scholar]
- 19.Lin HZ, Yang SQ, Zeldin G, Diehl AM. Chronic ethanol consumption induces the production of tumor necrosis factor-alpha and related cytokines in liver and adipose tissue. Alcohol Clin Exp Res. 1998;22:231S–237S. doi: 10.1097/00000374-199805001-00004. [DOI] [PubMed] [Google Scholar]
- 20.Nagy LE. Recent insights into the role of the innate immune system in the development of alcoholic liver disease. Exp Biol Med (Maywood) 2003;228:882–890. doi: 10.1177/153537020322800803. [DOI] [PubMed] [Google Scholar]
- 21.Tang Y, Banan A, Forsyth CB, Fields JZ, Lau CK, Zhang LJ, Keshavarzian A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol Clin Exp Res. 2008;32:355–364. doi: 10.1111/j.1530-0277.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- 22.Jeong WI, Gao B. Innate immunity and alcoholic liver fibrosis. J Gastroenterol Hepatol. 2008;23 Suppl 1:S112–S118. doi: 10.1111/j.1440-1746.2007.05274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawler JF Jr, Yin M, Diehl AM, Roberts E, Chatterjee S. Tumor necrosis factor-alpha stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J Biol Chem. 1998;273:5053–5059. doi: 10.1074/jbc.273.9.5053. [DOI] [PubMed] [Google Scholar]
- 24.Endo M, Masaki T, Seike M, Yoshimatsu H. TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c) Exp Biol Med (Maywood) 2007;232:614–621. [PubMed] [Google Scholar]
- 25.Kang L, Sebastian BM, Pritchard MT, Pratt BT, Previs SF, Nagy LE. Chronic ethanol-induced insulin resistance is associated with macrophage infiltration into adipose tissue and altered expression of adipocytokines. Alcohol Clin Exp Res. 2007;31:1581–1588. doi: 10.1111/j.1530-0277.2007.00452.x. [DOI] [PubMed] [Google Scholar]
- 26.El-Assal O, Hong F, Kim WH, Radaeva S, Gao B. IL-6-deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol Immunol. 2004;1:205–211. [PubMed] [Google Scholar]
- 27.Hong F, Radaeva S, Pan HN, Tian Z, Veech R, Gao B. Interleukin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease. Hepatology. 2004;40:933–941. doi: 10.1002/hep.20400. [DOI] [PubMed] [Google Scholar]
- 28.Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, Osei-Hyiaman D, Moh A, Fu XY, Pacher P, et al. Cell type-dependent pro- and anti-inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology. 2008;134:1148–1158. doi: 10.1053/j.gastro.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 30.Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol. 2006;79:1348–1356. doi: 10.1189/jlb.1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Minicis S, Brenner DA. Oxidative stress in alcoholic liver disease: role of NADPH oxidase complex. J Gastroenterol Hepatol. 2008;23 Suppl 1:S98–S103. doi: 10.1111/j.1440-1746.2007.05277.x. [DOI] [PubMed] [Google Scholar]
- 32.Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou R, Bai L, Lian Z, Wei H, Sun R, et al. Invariant NKT cells promote alcohol-induced steatohepatitis through interleukin-1β in mice. J Hepatol. 2015;62:1311–1318. doi: 10.1016/j.jhep.2014.12.027. [DOI] [PubMed] [Google Scholar]
- 33.Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest. 2013;123:887–902. doi: 10.1172/JCI65647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 36.Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, Szabo G. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286:1436–1444. doi: 10.1074/jbc.M110.145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- 38.Jeong WI, Osei-Hyiaman D, Park O, Liu J, Bátkai S, Mukhopadhyay P, Horiguchi N, Harvey-White J, Marsicano G, Lutz B, et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7:227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 39.Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. 2008;134:248–258. doi: 10.1053/j.gastro.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radaeva S, Wang L, Radaev S, Jeong WI, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293:G809–G816. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]
- 41.Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 42.Haseba T, Ohno Y. A new view of alcohol metabolism and alcoholism--role of the high-Km Class III alcohol dehydrogenase (ADH3) Int J Environ Res Public Health. 2010;7:1076–1092. doi: 10.3390/ijerph7031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yi HS, Lee YS, Byun JS, Seo W, Jeong JM, Park O, Duester G, Haseba T, Kim SC, Park KG, et al. Alcohol dehydrogenase III exacerbates liver fibrosis by enhancing stellate cell activation and suppressing natural killer cells in mice. Hepatology. 2014;60:1044–1053. doi: 10.1002/hep.27137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kharbanda KK, Todero SL, Shubert KA, Sorrell MF, Tuma DJ. Malondialdehyde-acetaldehyde-protein adducts increase secretion of chemokines by rat hepatic stellate cells. Alcohol. 2001;25:123–128. doi: 10.1016/s0741-8329(01)00174-4. [DOI] [PubMed] [Google Scholar]
- 45.Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–959. doi: 10.1083/jcb.140.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chedid A, Mendenhall CL, Moritz TE, French SW, Chen TS, Morgan TR, Roselle GA, Nemchausky BA, Tamburro CH, Schiff ER. Cell-mediated hepatic injury in alcoholic liver disease. Veterans Affairs Cooperative Study Group 275. Gastroenterology. 1993;105:254–266. doi: 10.1016/0016-5085(93)90034-a. [DOI] [PubMed] [Google Scholar]
- 47.Pan HN, Sun R, Jaruga B, Hong F, Kim WH, Gao B. Chronic ethanol consumption inhibits hepatic natural killer cell activity and accelerates murine cytomegalovirus-induced hepatitis. Alcohol Clin Exp Res. 2006;30:1615–1623. doi: 10.1111/j.1530-0277.2006.00194.x. [DOI] [PubMed] [Google Scholar]
- 48.Tilg H, Moschen AR, Kaneider NC. Pathways of liver injury in alcoholic liver disease. J Hepatol. 2011;55:1159–1161. doi: 10.1016/j.jhep.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 49.Byun JS, Suh YG, Yi HS, Lee YS, Jeong WI. Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating Kupffer cells and stellate cells to produce interleukin-10 in mice. J Hepatol. 2013;58:342–349. doi: 10.1016/j.jhep.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 50.Suh YG, Jeong WI. Hepatic stellate cells and innate immunity in alcoholic liver disease. World J Gastroenterol. 2011;17:2543–2551. doi: 10.3748/wjg.v17.i20.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol. 2010;105:14–32; quiz 33. doi: 10.1038/ajg.2009.593. [DOI] [PubMed] [Google Scholar]
- 52.Nieto N, Friedman SL, Cederbaum AI. Cytochrome P450 2E1-derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. J Biol Chem. 2002;277:9853–9864. doi: 10.1074/jbc.M110506200. [DOI] [PubMed] [Google Scholar]
- 53.Svegliati-Baroni G, Inagaki Y, Rincon-Sanchez AR, Else C, Saccomanno S, Benedetti A, Ramirez F, Rojkind M. Early response of alpha2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-beta independent. Hepatology. 2005;42:343–352. doi: 10.1002/hep.20798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, Gores GJ. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188–1198. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 55.Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, Torok NJ. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 56.Quiroz SC, Bucio L, Souza V, Hernández E, González E, Gómez-Quiroz L, Kershenobich D, Vargas-Vorackova F, Gutiérrez-Ruiz MC. Effect of endotoxin pretreatment on hepatic stellate cell response to ethanol and acetaldehyde. J Gastroenterol Hepatol. 2001;16:1267–1273. doi: 10.1046/j.1440-1746.2001.02619.x. [DOI] [PubMed] [Google Scholar]
- 57.Inokuchi S, Tsukamoto H, Park E, Liu ZX, Brenner DA, Seki E. Toll-like receptor 4 mediates alcohol-induced steatohepatitis through bone marrow-derived and endogenous liver cells in mice. Alcohol Clin Exp Res. 2011;35:1509–1518. doi: 10.1111/j.1530-0277.2011.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 59.Gäbele E, Dostert K, Dorn C, Patsenker E, Stickel F, Hellerbrand C. A new model of interactive effects of alcohol and high-fat diet on hepatic fibrosis. Alcohol Clin Exp Res. 2011;35:1361–1367. doi: 10.1111/j.1530-0277.2011.01472.x. [DOI] [PubMed] [Google Scholar]
- 60.Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology. 2006;44:1487–1501. doi: 10.1002/hep.21427. [DOI] [PubMed] [Google Scholar]
- 61.Cubero FJ, Nieto N. Ethanol and arachidonic acid synergize to activate Kupffer cells and modulate the fibrogenic response via tumor necrosis factor alpha, reduced glutathione, and transforming growth factor beta-dependent mechanisms. Hepatology. 2008;48:2027–2039. doi: 10.1002/hep.22592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taimr P, Higuchi H, Kocova E, Rippe RA, Friedman S, Gores GJ. Activated stellate cells express the TRAIL receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis. Hepatology. 2003;37:87–95. doi: 10.1053/jhep.2003.50002. [DOI] [PubMed] [Google Scholar]
- 63.Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441–1451. doi: 10.1002/hep.21419. [DOI] [PubMed] [Google Scholar]
- 64.Baroni GS, D’Ambrosio L, Curto P, Casini A, Mancini R, Jezequel AM, Benedetti A. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology. 1996;23:1189–1199. doi: 10.1002/hep.510230538. [DOI] [PubMed] [Google Scholar]
- 65.Laso FJ, Almeida J, Torres E, Vaquero JM, Marcos M, Orfao A. Chronic alcohol consumption is associated with an increased cytotoxic profile of circulating lymphocytes that may be related with the development of liver injury. Alcohol Clin Exp Res. 2010;34:876–885. doi: 10.1111/j.1530-0277.2010.01160.x. [DOI] [PubMed] [Google Scholar]
- 66.Park O, Jeong WI, Wang L, Wang H, Lian ZX, Gershwin ME, Gao B. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology. 2009;49:1683–1694. doi: 10.1002/hep.22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaruga B, Hong F, Kim WH, Sun R, Fan S, Gao B. Chronic alcohol consumption accelerates liver injury in T cell-mediated hepatitis: alcohol disregulation of NF-kappaB and STAT3 signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2004;287:G471–G479. doi: 10.1152/ajpgi.00018.2004. [DOI] [PubMed] [Google Scholar]
- 68.Minagawa M, Deng Q, Liu ZX, Tsukamoto H, Dennert G. Activated natural killer T cells induce liver injury by Fas and tumor necrosis factor-alpha during alcohol consumption. Gastroenterology. 2004;126:1387–1399. doi: 10.1053/j.gastro.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 69.Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765–76.e1-3. doi: 10.1053/j.gastro.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin F, Taylor NJ, Su H, Huang X, Hussain MJ, Abeles RD, Blackmore L, Zhou Y, Ikbal MM, Heaton N, et al. Alcohol dehydrogenase-specific T-cell responses are associated with alcohol consumption in patients with alcohol-related cirrhosis. Hepatology. 2013;58:314–324. doi: 10.1002/hep.26334. [DOI] [PubMed] [Google Scholar]
- 71.Hammerich L, Bangen JM, Govaere O, Zimmermann HW, Gassler N, Huss S, Liedtke C, Prinz I, Lira SA, Luedde T, et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology. 2014;59:630–642. doi: 10.1002/hep.26697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klatsky AL, Friedman GD. Alcohol and longevity. Am J Public Health. 1995;85:16–18. doi: 10.2105/ajph.85.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Longnecker MP. Alcohol consumption and risk of cancer in humans: an overview. Alcohol. 1995;12:87–96. doi: 10.1016/0741-8329(94)00088-3. [DOI] [PubMed] [Google Scholar]
- 74.Willett WC, Stampfer MJ, Colditz GA, Rosner BA, Hennekens CH, Speizer FE. Moderate alcohol consumption and the risk of breast cancer. N Engl J Med. 1987;316:1174–1180. doi: 10.1056/NEJM198705073161902. [DOI] [PubMed] [Google Scholar]
- 75.Szabo G, Mandrekar P, Girouard L, Catalano D. Regulation of human monocyte functions by acute ethanol treatment: decreased tumor necrosis factor-alpha, interleukin-1 beta and elevated interleukin-10, and transforming growth factor-beta production. Alcohol Clin Exp Res. 1996;20:900–907. doi: 10.1111/j.1530-0277.1996.tb05269.x. [DOI] [PubMed] [Google Scholar]
- 76.Buschard K, Hansen AK, Jensen K, Lindenbergh-Kortleve DJ, de Ruiter LF, Krohn TC, Hufeldt MR, Vogensen FK, Aasted B, Osterbye T, et al. Alcohol facilitates CD1d loading, subsequent activation of NKT cells, and reduces the incidence of diabetes in NOD mice. PLoS One. 2011;6:e17931. doi: 10.1371/journal.pone.0017931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Williams R. Global challenges in liver disease. Hepatology. 2006;44:521–526. doi: 10.1002/hep.21347. [DOI] [PubMed] [Google Scholar]
- 78.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 79.Spahr L, Rubbia-Brandt L, Pugin J, Giostra E, Frossard JL, Borisch B, Hadengue A. Rapid changes in alcoholic hepatitis histology under steroids: correlation with soluble intercellular adhesion molecule-1 in hepatic venous blood. J Hepatol. 2001;35:582–589. doi: 10.1016/s0168-8278(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 80.Taïeb J, Mathurin P, Elbim C, Cluzel P, Arce-Vicioso M, Bernard B, Opolon P, Gougerot-Pocidalo MA, Poynard T, Chollet-Martin S. Blood neutrophil functions and cytokine release in severe alcoholic hepatitis: effect of corticosteroids. J Hepatol. 2000;32:579–586. doi: 10.1016/s0168-8278(00)80219-6. [DOI] [PubMed] [Google Scholar]
- 81.Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119:1637–1648. doi: 10.1053/gast.2000.20189. [DOI] [PubMed] [Google Scholar]
- 82.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suh YG, Kim JK, Byun JS, Yi HS, Lee YS, Eun HS, Kim SY, Han KH, Lee KS, Duester G, et al. CD11b(+) Gr1(+) bone marrow cells ameliorate liver fibrosis by producing interleukin-10 in mice. Hepatology. 2012;56:1902–1912. doi: 10.1002/hep.25817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fujimiya T, Liu J, Kojima H, Shirafuji S, Kimura H, Fujimiya M. Pathological roles of bone marrow-derived stellate cells in a mouse model of alcohol-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2009;297:G451–G460. doi: 10.1152/ajpgi.00055.2009. [DOI] [PubMed] [Google Scholar]