

Abstract
Hepatitis C virus (HCV) has a high propensity to establish chronic infections. Failure of HCV-infected individuals to activate effective antiviral immune responses is at least in part related to HCV-induced impairment of dendritic cells (DCs) that play a central role in activating T cell responses. Although the impact of HCV on DC phenotype and function is likely to be more prominent in the liver, major HCV-induced alterations are detectable in peripheral blood DCs (pbDCs) that represent the most accessible source of DCs. These alterations include numerical reduction, impaired production of inflammatory cytokines and increased production of immunosuppressive IL10. These changes in DCs are relevant to our understanding the immune mechanisms underlying the propensity of HCV to establish persistent infection. Importantly, the non-invasive accessibility of pbDCs renders the analysis of these cells a convenient procedure that can be serially repeated in patient follow-up. Accordingly, the study of pbDCs in HCV-infected patients during conventional treatment with pegylated interferon and ribavirin indicated that restoration of normal plasmacytoid DC count may represent an additional mechanism contributing to the efficacy of the dual therapy. It also identified the pre-treatment levels of plasmacytoid DCs and IL10 as putative predictors of response to therapy. Treatment of chronic HCV infection is changing, as new generation direct-acting antiviral agents will soon be available for use in interferon-free therapeutic strategies. The phenotypic and functional analysis of pbDCs in this novel therapeutic setting will provide a valuable tool for investigating mechanisms underlying treatment efficacy and for identifying predictors of treatment response.
Keywords: Hepatitis C virus, Peripheral blood dendritic cells, Cytokines, Peg-interferon, Ribavirin
Core tip: Dendritic cells (DCs) are professional antigen-presenting cells that play a primary role in the activation and coordination of primary immune responses. In this review we will illustrate and discuss the emerging understanding of DC impairment occurring in patients with chronic hepatitis C virus (HCV) infection and the impact of therapy on DCs. Particular attention will be paid to HCV-induced alterations of DCs in the peripheral blood, as the non-invasive accessibility of these cells renders their analysis a convenient procedure that can be serially repeated in patient follow-up.
INTRODUCTION
One of the main features of the hepatitis C virus (HCV) is its high propensity to establish chronic infections that is related to impaired function and a decreased number of HCV-specific T cells[1,2]. Given the important role of dendritic cells (DCs) in activating T cell responses[3], DCs have become the target of investigation demonstrating that failure of HCV-infected individuals to activate effective antiviral immune responses is at least in part the result of HCV-induced impairment of DC function. Because of the difficulties in gaining access to liver DCs, although the liver is the primary site for HCV replication most studies addressing DC involvement in chronic HCV infection have focused on peripheral blood DCs (pbDCs). These studies provided useful information on the impact of HCV on DCs, the contribution of DCs to the pathogenesis of hepatitis C, the effects of therapy on DCs and DC features predictive of response to treatment.
CHRONIC INFECTION WITH HEPATITIS C VIRUS
Epidemiological aspects and natural history
HCV infection has a worldwide distribution and has been recognized as a major cause of chronic hepatitis, cirrhosis and end-stage liver disease. According to the 2013 World Health Organization report, about 150 million people are chronically infected by HCV worldwide and more than 350000 people die every year due to HCV-related complications[1].
Up to the 1990’s, the principal routes of HCV infection, were blood transfusion, unsafe injection procedures and intravenous drug use. Taken together these routes are estimated to be responsible for approximately 70% of chronic cases in developed countries. Currently screening of blood products for HCV by means of enzyme immunoassays (EIA) and nucleic acid testing has virtually eradicated transfusion-associated hepatitis C. Similarly, in the developed world, new HCV infections are infrequently related to unsafe medical or surgical procedures and spread among the community of people using intravenous drugs now accounts for the vast majority of incident cases. Other invasive behaviors, such as tattooing and acupuncture with unsafe materials are also implicated in occasional HCV transmissions. The risk of perinatal and of heterosexual transmission of HCV is low while male homosexual activity has become an important transmission route in Western Countries[4]. Conversely, the situation is quite different in resource-poor countries, where lack of public awareness and continuous use of unsafe medical tools still account for a considerable proportion of new HCV infections.
HCV is a positive strand RNA virus characterized by high sequence heterogeneity. Seven HCV genotypes, numbered 1 to 7, and a large number of subtypes have been described[5]. Genotypes and subtypes (which are identified by lowercase letters), differ among themselves by about 30% and 20% of their sequences respectively.
Acute hepatitis C is rarely severe, and symptoms occur in 10% to 50% of cases[4]. The incidence of acute HCV infection has decreased and it is about 1/100000 per year but this figure is probably underestimated because it mainly refers to symptomatic patients. Progression to chronic infection occurs in about 75% of cases but again, the estimate is very roughs mainly due to difficulties in the diagnosis of the acute infection[6] and possibly to difference related to the route of infection. For example, in a small outbreak of acute hepatitis C (genotype 2c) among subjects who took part in pharmacokinetics studies, chronicity occurred in less than 50% of subjects and some cases of very late clearance (up to 24 mo from the date of infection) were also described. In this latter case, unsafe procedures of blood sampling were supposed to be responsible for HCV infection[7].
Chronic hepatitis C is characterized by a variable degree of inflammation and with variable rates of fibrosis progression. Only exceptionally, HCV infection clear spontaneously in the chronic stage. Depending on the viral genotype, between 15% and 45% of infected cases will spontaneously resolve the infection within six months of infection without any treatment whilst the remaining proportion of cases will maintain the virus for years, with a risk of developing liver cirrhosis within 20 years up to 15%-30%[1]. On average, 20%-30% of patients develop cirrhosis over 20-30 years of infection[6]. The risk of developing hepatocellular carcinoma (HCC) in patients with HCV liver cirrhosis is approximately 1% to 5% per year. Patients with a diagnosis of HCC have a 33% probability of death during the first year of diagnosis[8]. Overall the standardized mortality rate in anti-HCV-positive persons ranges from 1.6 to 4.5 and was as high as 25 in a recent study from Scotland[9].
Hepatitis C progression to cirrhosis is highly variable depending on the presence of cofactors capable of accelerating the fibrotic process. Proven cofactors for fibrosis progression include older age at infection, male gender, chronic alcohol consumption, obesity, insulin resistance and type 2 diabetes, and immunosuppression (such as that occurring after solid organ transplantation and in untreated HIV infection)[10]. The clinical management of these cofactors represents a mainstay of HCV treatment, especially because many of these cofactors are associated with poor response to clinical treatment.
Clinical management
The primary goal of HCV therapy is to cure the infection, which is generally associated with the resolution of liver disease in patients with less advanced disease, that is, in patients without cirrhosis. However, patients with cirrhosis who have eradicated viral infection remain at risk of severe complications, mainly the development of HCC[11] and therefore need to continue active ultrasonographic follow up for early detection of HCC.
The infection is cured in more than 99% of patients who achieved a sustained virological response (SVR), defined as undetectable HCV RNA 24 wk after treatment completion[9]. Until recently, the only therapeutic option for chronic HCV infection was the combination of pegylated interferon (pegIFN) plus ribavirin (RBV), also known as dual therapy. With this regimen, patients infected with genotype 1 had SVR rates ranging from 40% in North America to 50% in Western Europe. Genotype 4 too, is characterized by low SVR rate. Higher SVR rates were achieved in patients infected with other HCV genotypes. For genotype 2, SVR rates accounted for about 80% while for genotypes 3, 5 and 6 the success rate appears to be lower. Both pegylated IFN molecules, pegylated IFNα2b (1.5 mcg/kg/wk) and pegylated IFNα2a (180 mcg/wk) can be used. Ribavirin should be given at a weight based daily dose (15 mg/kg) for genotypes 1, 4, 5 and 6 whereas, for genotype 2 and 3, a flat dose of 800 mg/d is indicated[9].
Although they do not target a specific HCV protein or nucleic acid, both pegIFN and RBV exert their antiviral activity through induction of various antiviral proteins and through different immunomodulating actions. Dual therapy has been largely used for all genotypes but it is characterized by both limited efficacy and poor tolerability[9,12] and these limitations have stimulated research to explore the feasibility of new therapeutic tools. HCV interferon-dependent therapies rely on host factors such as IL28B polymorphism, liver fibrosis stage, and prior peg/IFN-RBV history to predict treatment response[13,14].
Recently, improvement in our knowledge of the molecular biology of the HCV replication life cycle led to the discovery of several molecules that specifically block various viral proteins[15,16]. These compounds are called direct-acting antiviral agents (DAA) and target different nonstructural proteins involved in the HCV life cycle, including the NS3/4A protease, the NS5B polymerase and the NS5A protein. There are 4 classes of DAAs: NS3/4A protease inhibitors; NS5B nucleos(t)ide inhibitors; NS5B nonnucleos(t)ide inhibitors; NS5A inhibitors[17]. The first DAAs approved for the treatment of chronic HCV infection were the protease inhibitors (PI) telaprevir and boceprevir. Efficacy of these two first PI generation is limited to genotype 1, being genotype 1a less responsive than genotype 1b. The addition of PI to dual therapy significantly improved the efficacy of treatment for genotype 1 HCV infection but at the expenses of worsened side effect profile with the potential for severe adverse events, mainly hematological complications[18-21].
All naive patients with compensated disease due to HCV should be considered for therapy. Treatment should not be deferred in case of significant fibrosis (METAVIR score F3-F4). For genotype 1 the addition of telaprevir or boceprevir to the dual therapy is the approved standard of care. Duration of therapy ranges between 24 and 48 wk depending on the rapidity of viral clearance. However, these triple-therapy regimens are complexes, with variable viral monitoring and stopping rules. All patients with cirrhosis should be treated for 48 wk. Only selected patients with contraindication to boceprevir or telaprevir and/or with highly likelihood of SVR should be administered dual therapy[9].
The combination of pegIFN and RBV is the approved standard of care for chronic hepatitis C, genotypes 2, 3, 4, 5 and 6. Dual-treatment duration should be tailored to on-treatment virological response. The likelihood of SVR is directly proportional to the speed of HCV RNA disappearance. Treatment should be stopped at week 12 if the HCV RNA decrease is less than 2 log UI/mL and at week 24 if HCV RNA is still detectable[9]. In patients with rapid virological response (undetectable HCV RNA at week 4), duration of treatment should be 24 wk but, for genotype 1 and 4, 24 wk of therapy may be sufficient only in the presence of low viral load (< 400000 UI/mL) at baseline; the remaining patients should be treated for 48 wk. Patients with later virological response should be treated for 48 wk, as well[9].
The protease inhibitor, Simeprevir and the nucleos(t)ide polymerase inhibitor, Sofosbuvir have been the first DAAs approved for HCV treatment. They were shown to be safer and more effective compounds compared with interferon-based therapy. Subsequently several other DAAs have also been approved and many other compounds from all DAA families are still under evaluation. Therefore, interferon-free strategies for the therapy of HCV chronic infection will be available in the near future with many advantages in terms of tolerability. This is the reason why many doctors and patients are now choosing to defer treatment rather than to proceed with dual or triple therapy.
IMMUNE EFFECTOR MECHANISMS IN THE CONTROL OF HCV INFECTION
The role of T cells
The mechanisms causing the high propensity of HCV to establish chronic infections are not clearly defined, but it is believed that alterations of the anti-viral immunity play a central role in determining the clinical outcome of infection. Indeed, the presence of strong and multi-specific T cell mediated antiviral responses is a hallmark of positive outcome of infection. It has been previously shown that a higher proportion of subjects who spontaneously cleared the HCV infection in the past have T cell proliferative responses to HCV peptides in comparison with patients with established chronic infection[22,23]. Furthermore, the magnitude of these responses was also greater in past resolvers[22]. In addition to different antigen-specific T cell proliferation profiles, different clinical outcomes of the HCV infections also correlate with different profiles of antigen-specific T cell cytokine production. T cells producing high amounts of IFNγ in response to multiple HCV antigens can be detected (1) in subjects with acute HCV infections as they progress to spontaneously clear the virus but not in subjects with acute HCV infection who become chronic[24]; (2) in subjects with past resolved HCV infection but not in patients with established chronic infection[23]; (3) in subjects with chronic HCV infection who respond to IFNα plus Ribavirin and pharmacologically clear the virus but not in those who do not respond[25]; and also (4) in chronic HCV treatment responders with faster response kinetics (fast responders) but only to a lesser extent in slow responders[26]. Furthermore, the presence of virus-specific T cell mediated responses can be observed even in the absence of anti-HCV antibodies in subjects who cleared the infection years before[23,27] and also in HCV-negative children born from HCV-positive mothers[28], where detectable HCV-specific T cells show increased production of IFNγ and reduced production of IL10 in response to HCV peptides in comparison to T cells obtained from the infected mothers, revealing a potential use of antigen-specific T cell detection as a powerful diagnostic marker to identify cases of viral exposure even in the absence of antibodies detectable with currently available methods.
The role of IFNγ and other cytokines
IFNγ is the most powerful T cell-derived antiviral factor and inducer of pro-inflammatory responses (reviewed in[29,30]). Amongst its functions, IFNγ boosts the cytotoxic activities of CD8+ T cells and NK cells, favours the differentiation of Th1 cells, enhances the killing activities mediated by phagocytes of the innate immunity, upregulates the expression of both class I and class II MHC molecules on by-stander cells and also stimulates activated B cells to perform antibody isotype switch towards IgG classes that are favourable for opsonisation and phagocytosis, complement activation and antibody-dependent cell cytotoxicity. Furthermore, besides its immune activities IFNγ can-like the other interferons, alpha, beta and lambda-act directly on infected cells, inducing intracellular genetic programs of direct antiviral response involving the expression of several interferon-stimulated genes that directly target viral metabolites suppressing viral replication. Infected cells can therefore be purged of the virus without undergoing immune-mediated killing, and by-stander cells can achieve an antiviral state that renders them refractory to infection. The production of IFNγ is facilitated by innate pro-inflammatory cytokines such as IL12, IL15 and IL18, which are mainly produced by activated antigen-presenting cells.
IL10 and other inhibitory pathways
If the production of IFNγ develops hand-in-hand with and favours the control of the infection, the production of anti-inflammatory IL10 follows different kinetics. In contrast with the pro-inflammatory and antiviral role of IFNγ, IL10 is one of the strongest endogenous immune suppressing agents physiologically produced by multiple cell subsets of both the innate and the adaptive immune system, including antigen-presenting cells (monocytes, dendritic cells) and subsets of antigen-specific T cells (reviewed in[31,32]). The physiological role of IL10 is to shut down no longer required immune responses, thus preventing continuous immune activation and immune-mediated tissue damage. Functionally speaking, IL10 contrasts several functions exerted by IFNγ, effectively suppressing antigen processing and presentation by antigen-presenting cells and T-cell-mediated antiviral functions, both direct and indirect. IL10 can suppress the production of almost all pro-inflammatory cytokines and chemokines[32], and can direct the differentiation of anergic T cells from naïve precursors, thus contributing to the development of impaired T-cell responses[33-35].
The production of IL10 can be induced in combination with other immunological negative check points that share functional similarities, such as the programmed-death-1 (PD1) pathway. Several groups have shown that in patients with established chronic HCV infection HCV-specific T cells manifest with an immune exhausted phenotype[36-38] characterized by suppressed antigen-specific proliferation and cytokine production together with increased expression of PD1 and other T-cell activation markers that act as inhibitory immune check points, such as T-cell immunoglobulin mucin-3 (TIM3), lymphocyte attivation gene-3, cytotoxic T-lymphocyte-associated protein 4 and CD244[39]. Furthermore, antigen-specific T-cell exhaustion is not specific of chronic HCV infections only, but it can be observed during other chronic viral infections, including HBV[40-44] and HIV[45-47]. Monocytes expressing PD1 were shown to produce IL10 in response to PD1 engagement during HIV infections[48], with the consequence of suppressing HIV-specific immunity. Furthermore, in alcoholic liver disease the production of IL10 was correlated with reduced expression of IFNγ and increased expression of the immune inhibitors PD1 and TIM3 on several immune subsets in response to bacterial stimulation[49]. Several dysfunctions associated with immune exhaustion can be successfully reversed using blocking antibodies targeting these hyper-expressed immune inhibitory receptors and their respective ligands[45-47,50-52], and immunotherapeutic protocols using these antibodies are currently being evaluated in clinical studies for a plethora of medical conditions, including viral infections and cancer[53,54].
In the context of acute HCV infections, strong HCV-specific IL10 responses develop only during later stages of the acute infection in subjects who develop chronicity[24], possibly as a compensatory anti-inflammatory mechanism dedicated to suppress unwanted immune-mediated tissue damage in the infected livers, and are also detectable in subjects with established chronic infection and in treatment non-responders[23,25]. It has also been previously shown that there is an inverse correlation between IL10 production and the presence of HCV-specific T-cell proliferative responses in patients with established chronic HCV infection[22,23], supporting the hypothesis that effective endogenous anti-inflammatory and immune inhibiting responses take place in association with a poorer outcome of the infection[55].
The role of NK cells
Immunological events developing early on during the first phases of the acute infection are key in defining and shaping the subsequent adaptive responses. The innate immunological signatures in subjects with acute HCV infection differ in respect to their clinical outcome. Cytotoxic NK cells are proportionally predominant in comparison to CD56bright NK cells in acutely infected subjects who are spontaneously resolving the infection whilst this predominance is absent in patients progressing to chronicity[24,56,57]. Furthermore, the relocation of innate and adaptive immune cells into the infected liver may be compromised in subjects progressing to chronicity, because these patients produce higher concentrations of CXCL10 antagonist in comparison to those who spontaneously resolve the infection as a consequence of a higher dipeptidyl peptidase-4 (DPP4) activity present in their bloodstream[24,58-61]. The role of DPP4 in skewing the clinical outcome of HCV infections has also been demonstrated anecdotally on the basis of these publications, in a case report of a patient with diabetes on the background of chronic HCV infection who was treated with the DPP4 inhibitor sitagliptin (a commonly used anti-diabetic drug) before undergoing antiviral treatment. During treatment with sitagliptin alone, the patient experienced a 2-log reduction in HCV viral load, which could not be ascribed to any cause other than the DPP4 inhibitor[62].
DENDRITIC CELLS AND CHRONIC HCV INFECTION
DCs are professional antigen-presenting cells and have a primary role in the activation and coordination of primary immune responses. Different types of DCs exist through the body. They express different markers and home to different tissues; some re-circulate in the bloodstream[3,63-65]. Dendritic cells sense pathogen-associated molecular patterns of bacterial, fungal or viral origin (PAMPs) thanks to their expression of several innate receptors such as TLRs and RLRs[3,64]. Immature DCs are primarily sampling the immunological milieu of the tissue where they reside. However, upon activation immature DCs undergo a transformation process that includes upregulation of class I and class II MHC molecules and co-stimulatory molecules such as CD80 and CD86, production of type-1 and type-3 interferons (IFNα, IFNβ, IFNλ), production of pro-inflammatory cytokines IL12, IL15, IL18 (amongst others), production of tissue-protecting IL10, and radical changes in their chemokine receptor and adhesion molecule profile[3,63-65]. Activated mature DCs migrate to the lymphoid organs, where they interact with and activate both naïve and experienced T cells. Interactions between DCs and NK cells are also important for the modulation of the innate immunity (reviewed in[66]). The induction of a pro-inflammatory milieu locally also activates tissue-resident fibroblasts[67-69], and the production of new extracellular matrix (fibrogenesis) marks the beginning of the healing response and the deposition of scar tissue. In the liver, this is associated with the activation of hepatic stellate cells (HSCs), which favours the development of fibrosis and cirrhosis, hallmarks of chronic liver disease[70,71].
The idea that DC functions may be impaired during HCV infection has been evaluated in a multiplicity of studies, often with conflicting results depending on the experimental conditions and the cohorts of patients studied. However, on the overall, all these studies point at the non-equivocal conclusion that DCs do behave differently in patients with hepatitis C than they do in healthy subjects.
Liver dendritic cells in chronic HCV infection
As the liver is the primary site of HCV replication, it is conceivable that DCs would migrate into the infected tissues and that DC changes induced by the virus are prominent in the liver. Investigation of DCs in the liver of HCV-infected patients is indeed hampered by difficulties in gaining access to liver DCs. Therefore, our knowledge about liver DCs during HCV infection are limited. The few studies that investigated these cells demonstrated by either direct or indirect methods that DCs are enriched in the liver of HCV-infected patients[72-75] (reviewed in[76,77]). Contrasting results have been reported regarding the reciprocal distribution of DC subsets in the liver[73,78]. Notably, the demonstration that DCs in livers from patients with hepatitis C localize in areas of liver necrosis[79], together with the experimental observation that DCs and T cells are together recruited within HCV-infected human liver slices[80], strongly support a role for DCs in the activation of HCV-specific immune responses. The mechanisms underlying DC enrichment in the liver of HCV-infected individuals are still poorly defined. Beyond an increased local recruitment, the observation that in vitro the migration of DCs is strongly inhibited by the interaction of DCs with the viral protein HCV E2[81] may suggest that DC entrapment within the liver may contribute to the process.
Dendritic cells in the peripheral blood
pbDCs are the most accessible source of DCs. They can be divided into two main subsets: myeloid DCs (mDCs) and plasmacytoid DCs (pDCs)[3,63-65]. Neither of them express lineage-specific markers (CD3, CD14, CD16, CD19, CD20), but both of them express high levels of HLA-DR. mDCs are characterized by high expression of the integrin CD11c and the blood DC antigens (BDCA) 1 (CD1c) or 3 (CD141). pDCs do not express CD11c and BDCA1-3, but express high levels of the IL3 receptor (CD123), BDCA2 (CD303) and BDCA4 (CD304) instead[82]. Activated mDCs and pDCs have very distinct cytokine profiles. mDCs produce preferentially IL12 and IL10, whilst pDCs are the strongest producers of type-1 and type-3 interferons (IFNα, IFNλ)[3,63-65]. pDCs express high levels of TLR3, 7-9, and are therefore highly sensitive to viral nucleic acids, nucleobases and ribonucleosides. mDCs may play a stronger role as orchestrators of pro-inflammatory responses, but pDCs are certainly strongly involved in the development of anti-viral responses. mDCs and pDCs can be counted and characterized by flow cytometry directly performed on peripheral blood samples[83,84].
The frequency of pbDCs in the bloodstream is extremely low. This has hampered the study of these cell populations, as high concentrations of pbDCs can only be obtained starting from high volumes of peripheral blood. However, DCs can be successfully differentiated from peripheral blood monocytes stimulated with either IL4 or IFNα in the presence of GM-CSF (monocyte-derived DCs, moDCs)[85], and this has allowed researchers to bypass the issue of low availability in the peripheral blood and has granted them a useful in vitro model for the functional characterization of these cells.
Impairment of peripheral blood DCs in chronic HCV infection
Several studies investigated the impact of HCV infection on DCs by analyzing pbDCs. The main findings are illustrated in Figure 1, that recapitulates the current view on pbDCs involvement in HCV pathogenesis. In particular, most studies reported a numerical reduction of pbDCs in patients with chronic HCV infection, yet with some conflicting results on whether either mDCs or pDCs or both subsets are affected[22,73,86-92]. Although pDCs are undoubtedly specialized in antiviral defenses, the reduction of mDCs may be relevant to HCV infection as well, as mDCs through IL12 production and the subsequent polarization of Th1 responses may contribute to the activation of cellular immunity. However, the reason for pbDC reduction is still unclear. pbDC reduction has been reported to be associated with the degree of liver inflammation[93], possibly suggesting that the reduction of DCs in the peripheral blood may be due, at least in part, to an enhanced recruitment of these cells in the inflamed liver. pbDC reduction has also been reported to be more pronounced in patients infected with HCV genotype 2[22], but not correlated with the viral load[22,73,86,88,91], suggesting that multiple viral and non viral mechanisms may directly and indirectly contribute to the decrease of mDCs and pDCs in the circulation. Notably, our previous demonstration that the number of pbDCs is unaffected in healthy HCV-seropositive patients who underwent spontaneous resolution of their HCV infection[22] clearly indicate that active HCV infection is needed in order to determine a reduction of pbDCs.
Figure 1.
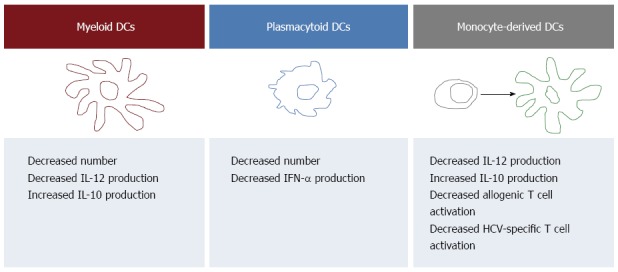
Summary of the hepatitis C virus-associated alterations of dendritic cells detectable in the peripheral blood. Patients with chronic hepatitis C undergo changes in the number and function of both myeloid dendritic cells (mDCs) and plasmacytoid DCs (pDCs) that may be relevant to the pathogenesis of chronic HCV infection. In particular, several Authors demonstrated that pDCs, which are specialized in antiviral immune responses, are decreased in HCV-infected patients and are endowed with a reduced ability to produce interferon (IFN). mDCs, that contribute to the activation of cellular immunity through the production of interleukin (IL)12, are decreased as well and characterized by reduced production of IL12 and increased production of the immunosuppressive cytokine IL10. These last functional defects are shared by monocyte-derived DCs that are also endowed with decreased ability to activate T cells. All references are reported in the text.
The functional assessment of pbDCs could also demonstrate that pbDCs from patients with chronic HCV infection are functionally impaired. Several studies demonstrated indeed that, upon stimulation with TLR ligands, viruses or interaction with T cells, both mDCs and pDCs from HCV-infected patients have impaired production of pro-inflammatory and anti-inflammatory cytokines compared with healthy controls. Due to the important role played by pDCs and IFN in antiviral immune responses, many Authors investigated the ability of pDCs to produce IFNα in response to distinct TLR ligands, with impaired IFN production observed in HCV-infected patients by some Authors[87,88,90,91], but not by others[89,94,95]. Moreover, mDCs from patients with chronic HCV infection were demonstrated to be characterized by impaired production of IL12, that in some cases was associated with impaired allostimulatory activity, and increased production of IL10[22,87,88,96,97]. All together, these observations indicate that, despite discrepancies among studies possibly due to differences in the patient cohorts and the experimental procedures, the analysis of circulating pbDCs can detect numerical and functional alterations of DCs occurring during HCV infection.
Similarly to pbDCs, also moDCs have been demonstrated to be impaired in HCV-infected patients, with reduced IL12 production, reduced allostimulatory activity and increased IL10 production reported in many studies but not confirmed in others, as reviewed elsewhere[92]. moDCs have been suggested to be less representative of in vivo functions compared with pbDCs, as discussed elsewhere[22]. Nevertheless, they have proven to be a very useful model for the study of DC functions. Yet, they have been largely used for studying the effects of cell culture-adapted strains of HCV on DCs and they provided insight into the molecular interactions of recombinant HCV proteins with DCs in vitro (reviewed in[98]). Although the mechanisms whereby HCV affects DC function still remain largely elusive, the effects of many viral components and proteins on DCs have been defined[99].
Effects of therapy with pegIFN plus RBV on peripheral blood DCs
As reported above, the analysis of pbDCs from patients with chronic HCV infection allows the detection of DC defects that can be relevant to the inadequate activation of immune responses leading to HCV persistence. Until recently the only therapeutic option for chronic HCV infection was the dual therapy with pegIFN plus RBV that achieved a sustained SVR only in part of the patients. For these reasons, several studies investigated the effects of dual therapy on pbDC number and functions according to virological response, in order to define the impact of treatment on DCs and to possibly identify correlates of clinical response. In most of the studies, the standard treatment with pegIFN plus RBV for 24 or 48 wk induced an increase of the number of pDCs in patients who achieved a SVR[100-102]. An early increase of pDCs during treatment was considered a correlate of favourable response in two studies[100,103]. pDC increase was accompanied by mDC increase in one study[102]. Only one study reported no variation in the number of pbDC subsets during treatment follow-up[104], but the reasons for this discrepancy are not evident. In this context, we can add our evidence supporting the impact of the dual therapy on pDCs. We analyzed pbDCs in a cohort of 14 HCV-infected patients (11 males, 3 females; mean age 49 years, range 29-68 years) undergone standard treatment with pegIFN and RBV for 24 or 48 wk depending on the viral genotype. pbDCs were analyzed before, after 1 mo of treatment, at the end of therapy and 6 mo after treatment completion. pbDCs analysis was performed by flow cytometry as previously described[22], in order to assess DC count, subset distribution, expression of activation/maturation markers (CD80, CD86, CD40, CD83), expression of molecules involved in HCV entry (CD81, DC-SIGN) and production of cytokines (IL12 and IL10). As shown in Figure 2, we observed that pegIFN and RBV treatment restored correct proportions of pDCs in SVR patients by inducing a rapid and progressive increase of these cells that was observed after 1 mo of treatment, further increased at the end of treatment and persisted after the end of treatment. SVR patients showed higher pDC levels than non-SVR at any time point. pegIFN and RBV treatment also induced a transient increase of IL12 production by pbDCs, that was observed after 1 mo of treatment but decreased thereafter, and that was more pronounced in SVR than non-SVR patients. The other pbDC parameters were not affected by the dual therapy. Because of the role played by chemokines and chemokine receptors in regulating DC migration, Mengshol and coll. also investigated the expression of CXCR3 and CXCR4 demonstrating that these molecules, upregulated on pbDCs at baseline, are normalized by pegIFN plus RBV, as well[101].
Figure 2.
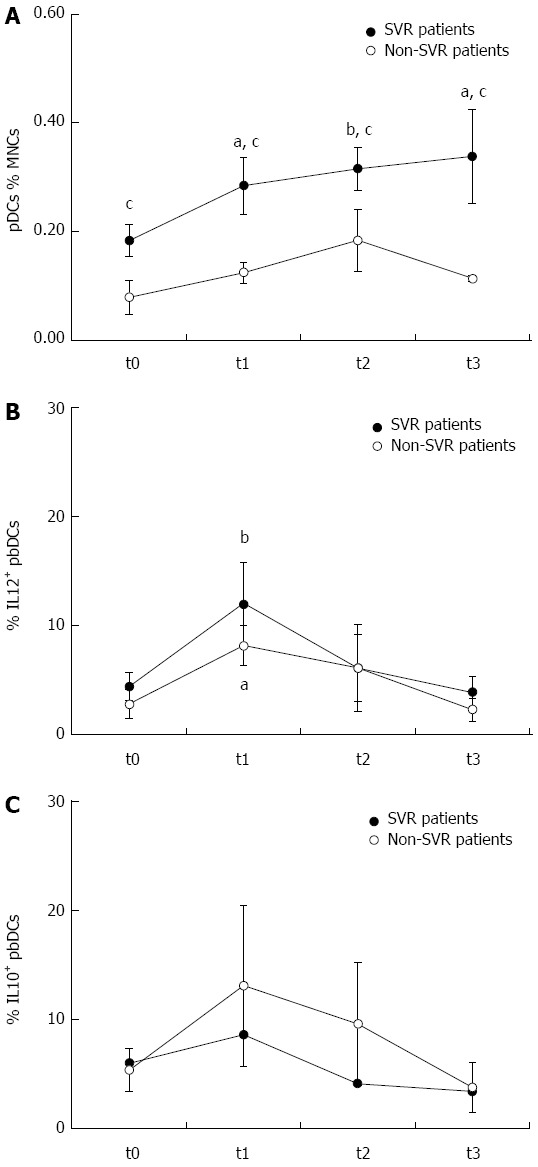
Effects of dual therapy with pegylated interferon and ribavirin on peripheral blood dendritic cells. The treatment induced a rapid, progressive and persistent increase of plasmacytoid dendritic cells (pDCs) in sustained virological response (SVR) but not in non-SVR patients. The frequency of pDCs was higher in SVR than non-SVR patients at all the time points. pegIFN and RBV treatment also induced a transient increase of peripheral blood DC production of interleukin (IL)12, that was observed after 1 mo of treatment but decreased thereafter, and that was more pronounced in SVR than non-SVR patients. IL10 production showed a similar trend and tended to be higher in non-SVR patients. aP < 0.05 and bP < 0.01, respectively, any time vs t0 within each group, as assessed by Wilcoxon signed rank test; cP < 0.05, SVR vs non-SVR, as assessed by Mann-Whitney test. t0: Before treatment; t1: After 1 mo of treatment; t2: At the end of treatment; t3: 6 mo after treatment completion.
The effects of therapy have also been assessed on moDCs, in order to investigate the mechanisms responsible for the impaired stimulation of T cells by DCs from HCV-infected patients. In an elegant cross-over study involving chronic HCV patients undergoing anti-viral treatment with pegIFN and RBV[105], peripheral blood DCs and T cells were independently purified from the same patient at two time points: baseline (BL), with high serum HCV viral load, and treatment week 12 (TW12), with pharmacologically reduced serum HCV viral load. Co-cultures of autologous T cells and DCs were prepared using all four possible combinations from each subject (BL DCs plus BL T cells or TW12 T cells, and TW12 DCs plus BL T cells or TW12 T cells) and DC-induced T cell responses were measured in order to ascertain the role of DCs or T cells in the context of altered HCV-specific responses. The study showed that the measurable reduction in T-cell responses was not dependent on the T cells used, but it was always only the use of DCs obtained at baseline, with high viral presence, which was associated with low T cell immunity. Conversely, DCs obtained at the later time point, after pharmacological viral clearance, were able to stimulate with similar higher efficiency both BL T cells and TW12 T cells.
Peripheral blood DCs as predictors of response to treatment
Predicting the outcome of a treatment that is administered to a patient is a goal for both patients and physicians. Accordingly, the identification of pbDC parameters measured at baseline or at the beginning of treatment that may predict SVR or non-SVR was a primary aim of the studies that analyzed pbDCs in HCV-infected patients treated with pegIFN and RBV. Notably pDCs, whose increase represented the most relevant treatment-induced pbDC modification, also resulted good predictors of response in two studies, where higher levels of pDCs during the first weeks of treatment were associated with SVR[100,103]. High levels of pDCs at baseline was a predictor of SVR in our cohort of patients, as well, independent from other clinical features including viral genotype and viral load (Figure 2). An increased pbDC production of IL-10 at baseline has been reported as non-SVR predictor by Liang and coll[102], and confirmed in our cohort of patients.
CONCLUSION
Although the impact of HCV on DC phenotype and function is likely to be more prominent in the liver as it is primarily infected by the virus, nevertheless several HCV-induced alterations are detectable in pbDCs that represent the most accessible source of DCs. These alterations include numerical reduction, impaired production of IL12 and IFNα, impaired allostimulatory activity and increased production of immunosuppressive IL10. Despite somehow controversial because variably observed among different cohorts of patients, these HCV-associated DC features are relevant to our understanding the immune mechanisms underlying the propensity of HCV to establish persistent infection. Importantly, the non-invasive accessibility of pbDCs renders the analysis of these cells a convenient procedure that can be serially repeated in patient follow-up. Accordingly, the study of pbDCs in HCV-infected patients during conventional treatment with pegIFN and RBV indicated that restoration of normal pDC count may represent a further novel mechanism contributing to the efficacy of the dual therapy. It also identified pDCs and IL10 as putative predictors of response to therapy. Treatment of chronic HCV infection is changing, as new generation direct-acting antivirals will soon be available for use in interferon-free therapeutic strategies. The phenotypic and functional analysis of pbDCs in this novel therapeutic setting will provide a valuable tool for investigating mechanisms underlying treatment efficacy and for identifying predictors of treatment response.
Footnotes
Conflict-of-interest statement: The authors have no conflict of interest to report.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 12, 2015
First decision: August 26, 2015
Article in press: November 13, 2015
P- Reviewer: Navas-Martin S, Shimizu Y S- Editor: Gong ZM L- Editor: A E- Editor: Zhang DN
References
- 1.World Health Organization. Hepatitis C factsheet No. 164. Updated July. 2013. Available from: http://www.who.int/mediacentre/factsheets/fs164/en/ [Google Scholar]
- 2.Chang KM, Thimme R, Melpolder JJ, Oldach D, Pemberton J, Moorhead-Loudis J, McHutchison JG, Alter HJ, Chisari FV. Differential CD4(+) and CD8(+) T-cell responsiveness in hepatitis C virus infection. Hepatology. 2001;33:267–276. doi: 10.1053/jhep.2001.21162. [DOI] [PubMed] [Google Scholar]
- 3.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van de Laar TJ, Matthews GV, Prins M, Danta M. Acute hepatitis C in HIV-infected men who have sex with men: an emerging sexually transmitted infection. AIDS. 2010;24:1799–1812. doi: 10.1097/QAD.0b013e32833c11a5. [DOI] [PubMed] [Google Scholar]
- 5.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esteban JI, Sauleda S, Quer J. The changing epidemiology of hepatitis C virus infection in Europe. J Hepatol. 2008;48:148–162. doi: 10.1016/j.jhep.2007.07.033. [DOI] [PubMed] [Google Scholar]
- 7.Larghi A, Zuin M, Crosignani A, Ribero ML, Pipia C, Battezzati PM, Binelli G, Donato F, Zanetti AR, Podda M, et al. Outcome of an outbreak of acute hepatitis C among healthy volunteers participating in pharmacokinetics studies. Hepatology. 2002;36:993–1000. doi: 10.1053/jhep.2002.36129. [DOI] [PubMed] [Google Scholar]
- 8.John-Baptiste A, Krahn M, Heathcote J, Laporte A, Tomlinson G. The natural history of hepatitis C infection acquired through injection drug use: meta-analysis and meta-regression. J Hepatol. 2010;53:245–251. doi: 10.1016/j.jhep.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 9.European Association for Study of Liver. EASL Clinical Practice Guidelines: management of hepatitis C virus infection. J Hepatol. 2014;60:392–420. doi: 10.1016/j.jhep.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Grebely J, Dore GJ. What is killing people with hepatitis C virus infection? Semin Liver Dis. 2011;31:331–339. doi: 10.1055/s-0031-1297922. [DOI] [PubMed] [Google Scholar]
- 11.Bruno S, Crosignani A, Facciotto C, Rossi S, Roffi L, Redaelli A, de Franchis R, Almasio PL, Maisonneuve P. Sustained virologic response prevents the development of esophageal varices in compensated, Child-Pugh class A hepatitis C virus-induced cirrhosis. A 12-year prospective follow-up study. Hepatology. 2010;51:2069–2076. doi: 10.1002/hep.23528. [DOI] [PubMed] [Google Scholar]
- 12.McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J, Nyberg LM, Lee WM, Ghalib RH, Schiff ER, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med. 2009;361:580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- 13.Pawlotsky JM. Treatment of chronic hepatitis C: current and future. Curr Top Microbiol Immunol. 2013;369:321–342. doi: 10.1007/978-3-642-27340-7_13. [DOI] [PubMed] [Google Scholar]
- 14.Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int. 2013;33 Suppl 1:93–104. doi: 10.1111/liv.12076. [DOI] [PubMed] [Google Scholar]
- 15.Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology. 2007;132:1979–1998. doi: 10.1053/j.gastro.2007.03.116. [DOI] [PubMed] [Google Scholar]
- 16.Soriano V, Vispo E, Poveda E, Labarga P, Martin-Carbonero L, Fernandez-Montero JV, Barreiro P. Directly acting antivirals against hepatitis C virus. J Antimicrob Chemother. 2011;66:1673–1686. doi: 10.1093/jac/dkr215. [DOI] [PubMed] [Google Scholar]
- 17.Poveda E, Wyles DL, Mena A, Pedreira JD, Castro-Iglesias A, Cachay E. Update on hepatitis C virus resistance to direct-acting antiviral agents. Antiviral Res. 2014;108:181–191. doi: 10.1016/j.antiviral.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 18.Poordad F, McCone J, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD, Sings HL, Boparai N, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1207–1217. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 21.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, et al. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364:2417–2428. doi: 10.1056/NEJMoa1013086. [DOI] [PubMed] [Google Scholar]
- 22.Della Bella S, Crosignani A, Riva A, Presicce P, Benetti A, Longhi R, Podda M, Villa ML. Decrease and dysfunction of dendritic cells correlate with impaired hepatitis C virus-specific CD4+ T-cell proliferation in patients with hepatitis C virus infection. Immunology. 2007;121:283–292. doi: 10.1111/j.1365-2567.2007.02577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cramp ME, Carucci P, Rossol S, Chokshi S, Maertens G, Williams R, Naoumov NV. Hepatitis C virus (HCV) specific immune responses in anti-HCV positive patients without hepatitis C viraemia. Gut. 1999;44:424–429. doi: 10.1136/gut.44.3.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riva A, Laird M, Casrouge A, Ambrozaitis A, Williams R, Naoumov NV, Albert ML, Chokshi S. Truncated CXCL10 is associated with failure to achieve spontaneous clearance of acute hepatitis C infection. Hepatology. 2014;60:487–496. doi: 10.1002/hep.27139. [DOI] [PubMed] [Google Scholar]
- 25.Cramp ME, Rossol S, Chokshi S, Carucci P, Williams R, Naoumov NV. Hepatitis C virus-specific T-cell reactivity during interferon and ribavirin treatment in chronic hepatitis C. Gastroenterology. 2000;118:346–355. doi: 10.1016/s0016-5085(00)70217-4. [DOI] [PubMed] [Google Scholar]
- 26.Tang KH, Herrmann E, Cooksley H, Tatman N, Chokshi S, Williams R, Zeuzem S, Naoumov NV. Relationship between early HCV kinetics and T-cell reactivity in chronic hepatitis C genotype 1 during peginterferon and ribavirin therapy. J Hepatol. 2005;43:776–782. doi: 10.1016/j.jhep.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 27.Takaki A, Wiese M, Maertens G, Depla E, Seifert U, Liebetrau A, Miller JL, Manns MP, Rehermann B. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med. 2000;6:578–582. doi: 10.1038/75063. [DOI] [PubMed] [Google Scholar]
- 28.Della Bella S, Riva A, Tanzi E, Nicola S, Amendola A, Vecchi L, Nebbia G, Longhi R, Zanetti AR, Villa ML. Hepatitis C virus-specific reactivity of CD4+-lymphocytes in children born from HCV-infected women. J Hepatol. 2005;43:394–402. doi: 10.1016/j.jhep.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 29.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 30.Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol. 1993;11:571–611. doi: 10.1146/annurev.iy.11.040193.003035. [DOI] [PubMed] [Google Scholar]
- 31.Moore KW, O’Garra A, de Waal Malefyt R, Vieira P, Mosmann TR. Interleukin-10. Annu Rev Immunol. 1993;11:165–190. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- 32.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 33.Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol. 2002;129:263–276. doi: 10.1159/000067596. [DOI] [PubMed] [Google Scholar]
- 34.Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood. 2002;99:2468–2476. doi: 10.1182/blood.v99.7.2468. [DOI] [PubMed] [Google Scholar]
- 35.Kapsenberg ML. Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol. 2003;3:984–993. doi: 10.1038/nri1246. [DOI] [PubMed] [Google Scholar]
- 36.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81:9249–9258. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, Hanson HL, Steinberg JP, Masopust D, Wherry EJ, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81:2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. 2010;129:474–481. doi: 10.1111/j.1365-2567.2010.03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans A, Riva A, Cooksley H, Phillips S, Puranik S, Nathwani A, Brett S, Chokshi S, Naoumov NV. Programmed death 1 expression during antiviral treatment of chronic hepatitis B: Impact of hepatitis B e-antigen seroconversion. Hepatology. 2008;48:759–769. doi: 10.1002/hep.22419. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe T, Bertoletti A, Tanoto TA. PD-1/PD-L1 pathway and T-cell exhaustion in chronic hepatitis virus infection. J Viral Hepat. 2010;17:453–458. doi: 10.1111/j.1365-2893.2010.01313.x. [DOI] [PubMed] [Google Scholar]
- 42.Tzeng HT, Tsai HF, Liao HJ, Lin YJ, Chen L, Chen PJ, Hsu PN. PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PLoS One. 2012;7:e39179. doi: 10.1371/journal.pone.0039179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, Cheng Y, Rosenberg W, Dusheiko G, Gilson R, ChinAleong J, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS One. 2012;7:e47648. doi: 10.1371/journal.pone.0047648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye B, Liu X, Li X, Kong H, Tian L, Chen Y. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis. 2015;6:e1694. doi: 10.1038/cddis.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 46.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 47.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 48.Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, Hill BJ, Noto A, Ancuta P, Peretz Y, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med. 2010;16:452–459. doi: 10.1038/nm.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Markwick LJ, Riva A, Ryan JM, Cooksley H, Palma E, Tranah TH, Manakkat Vijay GK, Vergis N, Thursz M, Evans A, et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology. 2015;148:590–602.e10. doi: 10.1053/j.gastro.2014.11.041. [DOI] [PubMed] [Google Scholar]
- 50.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, Schacker T, Roederer M, Douek DC, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Porichis F, Hart MG, Zupkosky J, Barblu L, Kwon DS, McMullen A, Brennan T, Ahmed R, Freeman GJ, Kavanagh DG, et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J Virol. 2014;88:2508–2518. doi: 10.1128/JVI.02034-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Golden-Mason L, Madrigal-Estebas L, McGrath E, Conroy MJ, Ryan EJ, Hegarty JE, O’Farrelly C, Doherty DG. Altered natural killer cell subset distributions in resolved and persistent hepatitis C virus infection following single source exposure. Gut. 2008;57:1121–1128. doi: 10.1136/gut.2007.130963. [DOI] [PubMed] [Google Scholar]
- 57.Meier UC, Owen RE, Taylor E, Worth A, Naoumov N, Willberg C, Tang K, Newton P, Pellegrino P, Williams I, et al. Shared alterations in NK cell frequency, phenotype, and function in chronic human immunodeficiency virus and hepatitis C virus infections. J Virol. 2005;79:12365–12374. doi: 10.1128/JVI.79.19.12365-12374.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ragab D, Laird M, Duffy D, Casrouge A, Mamdouh R, Abass A, Shenawy DE, Shebl AM, Elkashef WF, Zalata KR, et al. CXCL10 antagonism and plasma sDPPIV correlate with increasing liver disease in chronic HCV genotype 4 infected patients. Cytokine. 2013;63:105–112. doi: 10.1016/j.cyto.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 59.Casrouge A, Bisiaux A, Stephen L, Schmolz M, Mapes J, Pfister C, Pol S, Mallet V, Albert ML. Discrimination of agonist and antagonist forms of CXCL10 in biological samples. Clin Exp Immunol. 2012;167:137–148. doi: 10.1111/j.1365-2249.2011.04488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Casrouge A, Decalf J, Ahloulay M, Lababidi C, Mansour H, Vallet-Pichard A, Mallet V, Mottez E, Mapes J, Fontanet A, et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest. 2011;121:308–317. doi: 10.1172/JCI40594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Proost P, Schutyser E, Menten P, Struyf S, Wuyts A, Opdenakker G, Detheux M, Parmentier M, Durinx C, Lambeir AM, et al. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood. 2001;98:3554–3561. doi: 10.1182/blood.v98.13.3554. [DOI] [PubMed] [Google Scholar]
- 62.Yanai H. Dipeptidyl peptidase-4 inhibitor sitagliptin significantly reduced hepatitis C virus replication in a diabetic patient with chronic hepatitis C virus infection. Hepatobiliary Pancreat Dis Int. 2014;13:556. doi: 10.1016/s1499-3872(14)60308-8. [DOI] [PubMed] [Google Scholar]
- 63.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 64.Boltjes A, van Wijk F. Human dendritic cell functional specialization in steady-state and inflammation. Front Immunol. 2014;5:131. doi: 10.3389/fimmu.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collin M, McGovern N, Haniffa M. Human dendritic cell subsets. Immunology. 2013;140:22–30. doi: 10.1111/imm.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferlazzo G, Morandi B. Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front Immunol. 2014;5:159. doi: 10.3389/fimmu.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buckley CD. Why does chronic inflammation persist: An unexpected role for fibroblasts. Immunol Lett. 2011;138:12–14. doi: 10.1016/j.imlet.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. 2014;5:123. doi: 10.3389/fphar.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu R, Zhang Z, Wang FS. Liver fibrosis: mechanisms of immune-mediated liver injury. Cell Mol Immunol. 2012;9:296–301. doi: 10.1038/cmi.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dooley S, ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res. 2012;347:245–256. doi: 10.1007/s00441-011-1246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gallè MB, DeFranco RM, Kerjaschki D, Romanelli RG, Montalto P, Gentilini P, Pinzani M, Romagnoli P. Ordered array of dendritic cells and CD8+ lymphocytes in portal infiltrates in chronic hepatitis C. Histopathology. 2001;39:373–381. doi: 10.1046/j.1365-2559.2001.01241.x. [DOI] [PubMed] [Google Scholar]
- 73.Wertheimer AM, Bakke A, Rosen HR. Direct enumeration and functional assessment of circulating dendritic cells in patients with liver disease. Hepatology. 2004;40:335–345. doi: 10.1002/hep.20306. [DOI] [PubMed] [Google Scholar]
- 74.Dolganiuc A, Norkina O, Kodys K, Catalano D, Bakis G, Marshall C, Mandrekar P, Szabo G. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology. 2007;133:1627–1636. doi: 10.1053/j.gastro.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ryan EJ, Dring M, Ryan CM, McNulty C, Stevenson NJ, Lawless MW, Crowe J, Nolan N, Hegarty JE, O’Farrelly C. Variant in CD209 promoter is associated with severity of liver disease in chronic hepatitis C virus infection. Hum Immunol. 2010;71:829–832. doi: 10.1016/j.humimm.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 76.Dolganiuc A, Szabo G. Dendritic cells in hepatitis C infection: can they (help) win the battle? J Gastroenterol. 2011;46:432–447. doi: 10.1007/s00535-011-0377-y. [DOI] [PubMed] [Google Scholar]
- 77.Zhou Y, Zhang Y, Yao Z, Moorman JP, Jia Z. Dendritic cell-based immunity and vaccination against hepatitis C virus infection. Immunology. 2012;136:385–396. doi: 10.1111/j.1365-2567.2012.03590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lai WK, Curbishley SM, Goddard S, Alabraba E, Shaw J, Youster J, McKeating J, Adams DH. Hepatitis C is associated with perturbation of intrahepatic myeloid and plasmacytoid dendritic cell function. J Hepatol. 2007;47:338–347. doi: 10.1016/j.jhep.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 79.Tanimoto K, Akbar SM, Michitaka K, Horiike N, Onji M. Antigen-presenting cells at the liver tissue in patients with chronic viral liver diseases: CD83-positive mature dendritic cells at the vicinity of focal and confluent necrosis. Hepatol Res. 2001;21:117–125. doi: 10.1016/s1386-6346(01)00084-5. [DOI] [PubMed] [Google Scholar]
- 80.Nascimbeni M, Bourdoncle P, Penna C, Saunier B. Recruitment and interaction of human dendritic and T cells in autologous liver slices experimentally infected with HCV produced in cell culture. J Immunol Methods. 2012;378:51–55. doi: 10.1016/j.jim.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 81.Nattermann J, Zimmermann H, Iwan A, von Lilienfeld-Toal M, Leifeld L, Nischalke HD, Langhans B, Sauerbruch T, Spengler U. Hepatitis C virus E2 and CD81 interaction may be associated with altered trafficking of dendritic cells in chronic hepatitis C. Hepatology. 2006;44:945–954. doi: 10.1002/hep.21350. [DOI] [PubMed] [Google Scholar]
- 82.Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, Buck DW, Schmitz J. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J Immunol. 2000;165:6037–6046. doi: 10.4049/jimmunol.165.11.6037. [DOI] [PubMed] [Google Scholar]
- 83.Giannelli S, Taddeo A, Presicce P, Villa ML, Della Bella S. A six-color flow cytometric assay for the analysis of peripheral blood dendritic cells. Cytometry B Clin Cytom. 2008;74:349–355. doi: 10.1002/cyto.b.20434. [DOI] [PubMed] [Google Scholar]
- 84.Della Bella S, Giannelli S, Taddeo A, Presicce P, Villa ML. Application of six-color flow cytometry for the assessment of dendritic cell responses in whole blood assays. J Immunol Methods. 2008;339:153–164. doi: 10.1016/j.jim.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 85.Della Bella S, Nicola S, Riva A, Biasin M, Clerici M, Villa ML. Functional repertoire of dendritic cells generated in granulocyte macrophage-colony stimulating factor and interferon-alpha. J Leukoc Biol. 2004;75:106–116. doi: 10.1189/jlb.0403154. [DOI] [PubMed] [Google Scholar]
- 86.Kunitani H, Shimizu Y, Murata H, Higuchi K, Watanabe A. Phenotypic analysis of circulating and intrahepatic dendritic cell subsets in patients with chronic liver diseases. J Hepatol. 2002;36:734–741. doi: 10.1016/s0168-8278(02)00062-4. [DOI] [PubMed] [Google Scholar]
- 87.Anthony DD, Yonkers NL, Post AB, Asaad R, Heinzel FP, Lederman MM, Lehmann PV, Valdez H. Selective impairments in dendritic cell-associated function distinguish hepatitis C virus and HIV infection. J Immunol. 2004;172:4907–4916. doi: 10.4049/jimmunol.172.8.4907. [DOI] [PubMed] [Google Scholar]
- 88.Kanto T, Inoue M, Miyatake H, Sato A, Sakakibara M, Yakushijin T, Oki C, Itose I, Hiramatsu N, Takehara T, et al. Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J Infect Dis. 2004;190:1919–1926. doi: 10.1086/425425. [DOI] [PubMed] [Google Scholar]
- 89.Longman RS, Talal AH, Jacobson IM, Rice CM, Albert ML. Normal functional capacity in circulating myeloid and plasmacytoid dendritic cells in patients with chronic hepatitis C. J Infect Dis. 2005;192:497–503. doi: 10.1086/431523. [DOI] [PubMed] [Google Scholar]
- 90.Murakami H, Akbar SM, Matsui H, Horiike N, Onji M. Decreased interferon-alpha production and impaired T helper 1 polarization by dendritic cells from patients with chronic hepatitis C. Clin Exp Immunol. 2004;137:559–565. doi: 10.1111/j.1365-2249.2004.02550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szabo G, Dolganiuc A. Subversion of plasmacytoid and myeloid dendritic cell functions in chronic HCV infection. Immunobiology. 2005;210:237–247. doi: 10.1016/j.imbio.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 92.Ryan EJ, O’Farrelly C. The affect of chronic hepatitis C infection on dendritic cell function: a summary of the experimental evidence. J Viral Hepat. 2011;18:601–607. doi: 10.1111/j.1365-2893.2011.01453.x. [DOI] [PubMed] [Google Scholar]
- 93.Ulsenheimer A, Gerlach JT, Jung MC, Gruener N, Wächtler M, Backmund M, Santantonio T, Schraut W, Heeg MH, Schirren CA, et al. Plasmacytoid dendritic cells in acute and chronic hepatitis C virus infection. Hepatology. 2005;41:643–651. doi: 10.1002/hep.20592. [DOI] [PubMed] [Google Scholar]
- 94.Piccioli D, Tavarini S, Nuti S, Colombatto P, Brunetto M, Bonino F, Ciccorossi P, Zorat F, Pozzato G, Comar C, et al. Comparable functions of plasmacytoid and monocyte-derived dendritic cells in chronic hepatitis C patients and healthy donors. J Hepatol. 2005;42:61–67. doi: 10.1016/j.jhep.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 95.Goutagny N, Vieux C, Decullier E, Ligeoix B, Epstein A, Trepo C, Couzigou P, Inchauspe G, Bain C. Quantification and functional analysis of plasmacytoid dendritic cells in patients with chronic hepatitis C virus infection. J Infect Dis. 2004;189:1646–1655. doi: 10.1086/383248. [DOI] [PubMed] [Google Scholar]
- 96.Kanto T, Inoue M, Miyazaki M, Itose I, Miyatake H, Sakakibara M, Yakushijin T, Kaimori A, Oki C, Hiramatsu N, et al. Impaired function of dendritic cells circulating in patients infected with hepatitis C virus who have persistently normal alanine aminotransferase levels. Intervirology. 2006;49:58–63. doi: 10.1159/000087264. [DOI] [PubMed] [Google Scholar]
- 97.Tsubouchi E, Akbar SM, Murakami H, Horiike N, Onji M. Isolation and functional analysis of circulating dendritic cells from hepatitis C virus (HCV) RNA-positive and HCV RNA-negative patients with chronic hepatitis C: role of antiviral therapy. Clin Exp Immunol. 2004;137:417–423. doi: 10.1111/j.1365-2249.2004.02544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pachiadakis I, Pollara G, Chain BM, Naoumov NV. Is hepatitis C virus infection of dendritic cells a mechanism facilitating viral persistence? Lancet Infect Dis. 2005;5:296–304. doi: 10.1016/S1473-3099(05)70114-6. [DOI] [PubMed] [Google Scholar]
- 99.Saha B, Szabo G. Innate immune cell networking in hepatitis C virus infection. J Leukoc Biol. 2014;96:757–766. doi: 10.1189/jlb.4MR0314-141R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kanto T, Inoue M, Oze T, Miyazaki M, Sakakibara M, Kakita N, Matsubara T, Higashitani K, Hagiwara H, Iio S, et al. Dynamics of regulatory T cells and plasmacytoid dendritic cells as immune markers for virological response in pegylated interferon-α and ribavirin therapy for chronic hepatitis C patients. J Gastroenterol. 2012;47:169–178. doi: 10.1007/s00535-011-0466-y. [DOI] [PubMed] [Google Scholar]
- 101.Mengshol JA, Golden-Mason L, Castelblanco N, Im KA, Dillon SM, Wilson CC, Rosen HR. Impaired plasmacytoid dendritic cell maturation and differential chemotaxis in chronic hepatitis C virus: associations with antiviral treatment outcomes. Gut. 2009;58:964–973. doi: 10.1136/gut.2008.168948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liang CC, Liu CH, Lin YL, Liu CJ, Chiang BL, Kao JH. Functional impairment of dendritic cells in patients infected with hepatitis C virus genotype 1 who failed peginterferon plus ribavirin therapy. J Med Virol. 2011;83:1212–1220. doi: 10.1002/jmv.22096. [DOI] [PubMed] [Google Scholar]
- 103.Itose I, Kanto T, Inoue M, Miyazaki M, Miyatake H, Sakakibara M, Yakushijin T, Oze T, Hiramatsu N, Takehara T, et al. Involvement of dendritic cell frequency and function in virological relapse in pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C patients. J Med Virol. 2007;79:511–521. doi: 10.1002/jmv.20809. [DOI] [PubMed] [Google Scholar]
- 104.Hammond T, Lee S, Watson MW, Flexman JP, Cheng W, Price P. Decreased IFNγ production correlates with diminished production of cytokines by dendritic cells in patients infected with hepatitis C virus and receiving therapy. J Viral Hepat. 2011;18:482–492. doi: 10.1111/j.1365-2893.2010.01331.x. [DOI] [PubMed] [Google Scholar]
- 105.Pachiadakis I, Chokshi S, Cooksley H, Farmakiotis D, Sarrazin C, Zeuzem S, Michalak TI, Naoumov NV. Early viraemia clearance during antiviral therapy of chronic hepatitis C improves dendritic cell functions. Clin Immunol. 2009;131:415–425. doi: 10.1016/j.clim.2009.02.001. [DOI] [PubMed] [Google Scholar]