

Abstract
Hepatic steatosis defined as lipid accumulation in hepatocytes is very frequently found in adults and obese adolescents in the Western World. Etiologically, obesity and associated insulin resistance or excess alcohol intake are the most frequent causes of hepatic steatosis. However, steatosis also often occurs with chronic hepatitis C virus (HCV) infection and is also found in rare but potentially life-threatening liver diseases of pregnancy. Clinical significance and outcome of hepatic triglyceride accumulation are highly dependent on etiology and histological pattern of steatosis. This review summarizes current concepts of pathophysiology of common causes of hepatic steatosis, including non-alcoholic fatty liver disease (NAFLD), alcoholic fatty liver disease, chronic HCV infections, drug-induced forms of hepatic steatosis, and acute fatty liver of pregnancy. Regarding the pathophysiology of NAFLD, this work focuses on the close correlation between insulin resistance and hepatic triglyceride accumulation, highlighting the potential harmful effects of systemic insulin resistance on hepatic metabolism of fatty acids on the one side and the role of lipid intermediates on insulin signalling on the other side. Current studies on lipid droplet morphogenesis have identified novel candidate proteins and enzymes in NAFLD.
Keywords: Steatosis, Lipid accumulation, Fatty acids, Insulin resistance, Triglycerides
Core tip: Fatty liver disease is a highly prevalent condition in the Western World. This article summarizes the most frequent causes and states of hepatic steatosis, including non-alcoholic fatty liver disease (NAFLD), alcoholic fatty liver, drug-induced forms, hepatitis C virus infections, and acute fatty liver of pregnancy. Important pathophysiological and cellular aspects of various forms of fatty liver disease are reviewed as well as the clinically relevant close interaction between hepatic triglyceride accumulation and insulin resistance in NAFLD.
INTRODUCTION
Hepatic steatosis is defined by the accumulation of triglycerides resulting in more than 5% of hepatocytes containing visible lipid droplets in either a micro- or macrovesicular pattern. Most frequently, steatosis is histologically assessed by using a classification that was originally suggested for grading of non-alcoholic fatty liver disease (NAFLD) by Javor et al[1]: grade 0: < 5% hepatocytes involved; grade 1: 5%-33% hepatocytes involved; grade 2: > 33%-66% hepatocytes involved, grade 3: > 66% hepatocytes involved. At the cellular level, steatosis is an imbalance of hepatic fatty acid uptake, lipogenesis, β-oxidation, and triglyceride export in very low density lipoprotein (VLDL) particles.
Fatty liver might be caused by several conditions, diseases, or drugs; however, obesity, insulin resistance, and dyslipidemia as well as excess alcohol intake are the most frequent causes of NAFLD or alcoholic fatty liver disease (AFLD). Beyond metabolic or alimentary causes, fatty liver can also be found in patients with hepatitis C virus (HCV) infection and rarely in pregnancy. Other rare causes of fatty liver disease are summarized in Table 1[2]. This review will focus on the most frequent and important causes of fatty liver disease.
Table 1.
| Etiologies of fatty liver disease |
| Frequent causes: |
| Non-alcoholic fatty liver disease |
| Alcoholic fatty liver disease |
| Chronic hepatitis C virus infection |
| Drug-induced steatosis |
| Less common or rare causes: |
| Inborn errors of metabolism (Wilson disease, abetalipoproteinemia, tyrosinaemia, hypobetalipoproteinemia) |
| Lipodystrophy (congenital or acquired) |
| Total parenteral nutrition |
| Surgical procedures (e.g., biliopancreatic diversion extensive small bowel resection) |
The natural course of fatty liver is strongly dependent on etiology and comorbidities. For example, in NAFLD, 20%-30% of patients with steatosis will develop non-alcoholic steatohepatitis (NASH), which is associated with increased total and cardiovascular mortality[3]. NASH is complicated by liver cirrhosis in 2%-5% of patients[2]. Co-occurrence of steatosis and chronic HCV infection has been found to be associated with poorer outcome when compared to patients without steatosis[4].
NAFLD
NAFLD has become the most common cause of elevated liver enzymes in the Western World, affecting 30%-40% of men and 15%-20% of women in the general population[5] and up to 70% of type 2 diabetics[6]. It is now considered the hepatic manifestation of the metabolic syndrome, which comprises a spectrum of metabolic disorders, including overweight or obesity, insulin resistance, dyslipidemia, and hypertension[7,8]. According to estimates, NAFLD will become the most frequent indication for liver transplantation by 2030[9]. Beyond excess mortality rate from advanced liver disease, NAFLD is associated with significantly increased cardiovascular risk, mostly due to associated disturbances in glucose and lipid metabolism[9].
Intracellular lipid accumulation in NAFLD results from an imbalance between hepatic fatty acid uptake, lipid synthesis, lipid oxidation, and export via VLDL particles. Several studies have stressed the importance of adipose tissue lipolysis in the development of hepatic steatosis. Increased total fat mass in obesity is associated with elevated whole body lipolysis[10,11] and excess fatty acid uptake into the liver. Further highlighting the role of adipose tissue in triglyceride metabolism, Nye and colleagues[12] found that glycerol 3-phosphate, which is essential for triglyceride synthesis, primarily originates from glyceroneogenesis and only to a lesser extent from glycolysis. Accordingly, mice lacking fatty acid transporter protein 5 (FATP 5), which is the primary fatty acid transporter in the liver, are protected from diet-induced steatosis[13]. Underlining the importance of the adipose tissue in development of NAFLD, lipodystrophic patients who lack visceral and peripheral fat accumulation due to leptin deficiency display severe hepatic steatosis and insulin resistance[14,15]. Similarly, subjects with mutations of perilipin-1 - an inhibitor of adipose triglyceride lipase (ATGL) and stabilizer of lipid droplets in adipose tissue - have also profound hepatic steatosis and insulin resistance[16].
In addition to increased influx of fatty acids and their esterification, de novo lipogenesis (DNL), which from a quantitative point of view only plays a minor role in hepatic lipid accumulation, is also increased in NAFLD. In affected patients, DNL is significantly increased when compared to healthy controls[17-19]. Increased levels of ligand-activated transcription factor α (LXR α) and sterol regulating element binding protein 1c (SREBP-1c) have been described as major contributors to increased DNL in NAFLD[20,21]. Further highlighting the close connection between glucose and fatty acid metabolism, high glucose levels induce pyruvate production via stimulation of carbohydrate response element binding protein (ChREBP). Pyruvate is the principle source of acetyl-CoA in fatty acid synthesis[22]. Very recently, Perry and colleagues[23] found that white adipose tissue-derived hepatic acetyl-CoA is the critical determinant of insulin-induced suppression of hepatic glucose production. By decreasing oxidation of fatty acids on the one side and inducing DNL on the other side, fructose consumption is a well-known highly effective driver of hepatic steatosis[24,25].
DNL is closely related to β-oxidation via SREBP-1c. In addition to induction of lipogenic enzymes, SREBP-1c activates acetyl-CoA carboxylase 2 (ACC2), resulting in increased levels of malonyl-CoA, which decreases β-oxidation via diminishing shuttling of fatty acids into the mitochondrion by inhibiting carnitine palmitoyl transferase 1 (CPT-1). AMP activated protein kinase (AMPK) is also a key regulator of intracellular lipid metabolism. By sensing cellular AMP levels, activation of AMPK stimulates fatty acid β-oxidation and inhibits ATP-consuming processes, such as DNL[26]. Mitochondrial dysfunction and resulting impaired fatty acid oxidation have been suggested to significantly contribute to the pathogenesis of NAFLD[27,28].
On a cellular level, triglycerides are stored in lipid droplets (LD) that supply fatty acids for oxidation and VLDL secretion. As described in detail in a very recent review by Mashek and colleagues[29], LDs are formed within the lipid bilayer of the endoplasmic reticulum (ER) and subsequently budded. The latter process requires action of certain fat storage-inducible transmembrane proteins as well as seipin[30]. Growth of LDs is dependent on triglyceride and phospholipid synthetic enzymes that are present on the LD surface. LDs are catabolized via three different mechanisms: (1) ATGL/patatin-like phospholipase domain-containing protein 2 (PNPLA2) catalyzes the first step of triglyceride hydrolysis and partitions hydrolyzed fatty acids to β-oxidation[31]. Activity of ATGL is strongly influenced by LD-associated proteins, such as perilipins 2 and 5[32]. Sirtuin 1 (SIRT1) has been shown to regulate ATGL activity, probably via peroxisome proliferator-activated receptor α (PPARα)[33]; (2) LDs are degraded by lysosomal lipases, a process that is determined by the small guanosine triphosphatase RAB7[34,35]; and (3) LD catabolism is mediated by carboxylesterase 3 and cell death inducing DFF45-like effector B (CIDEB), which are involved in repacking of cytosolic LDs in the ER leading to synthesis of VLDL particles[36,37]. Increased expression of LD associated proteins, such as 17 β hydroxysteroid dehydrogenase 13 and perilipins 1, 2, 3, and 5, have been reported in NAFLD[38-42]. PNPLA3 polymorphisms have been identified as the strongest genetic factor associated with NAFLD[43].
Apolipoprotein A5 is a protein with extra- and intracellular effects on lipid metabolism[44]. In obese subjects undergoing bariatric surgery, improvements of hepatic steatosis were associated with decreased hepatic apoA5 expression. Direct involvement of apolipoprotein A5 in hepatic triglyceride accumulation has been shown in a hepatic cell culture model[45]. In adipocytes, apolipoprotein A5 has been found to surround lipid droplets[46], suggesting a potential role of apolipoprotein A5 in hepatic lipid droplet metabolism.
Stressing the metabolic consequences of steatosis, hepatic triglyceride accumulation is associated with increased gluconeogenesis, decreased glycogen synthesis, and inhibition of insulin signalling[47,48] as well as alterations in hepatokine production[49]. Lipid intermediates of long-chain fatty acids (LCAF) have a crucial role in hepatic insulin resistance in NAFLD. In hepatocytes, LCAF are esterified with glycerol-3-phosphate to form monoacylglycerol, diacylglycerol (DAG), and triacylglycerol[9]. Hepatic DAG content has been positively correlated with inhibition of insulin signalling in mice and humans with NAFLD[48,50,51]. Mechanistically, DAG induces increased translocation of protein kinase C ε to the plasma membrane, where it binds and inhibits the activity of the intracellular kinase domain of the insulin receptor, resulting in diminished activation of the canonical insulin pathway[52,53]. As a consequence, activation of glycogen synthesis and inhibition of gluconeogenesis is reduced[48]. Unesterified lipid intermediates also exert proinflammatory properties by inducing ER stress, which leads to the activation of c-janus N-terminal kinases and nuclear factor kappa B, resulting in decreased phosphorylation of insulin receptor substrate-2 (IRS-2)[9,54,55]. Accumulation of ceramides in the plasma membrane have also been found to alleviate insulin signalling[56]. Pathophysiological aspects of NAFLD are summarized in Figure 1.
Figure 1.
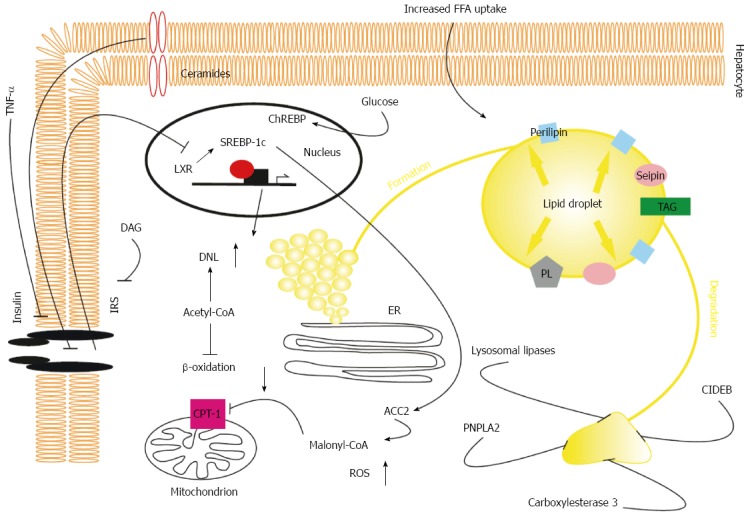
Pathophysiological aspects of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. Both increased uptake of fatty acids due to elevated whole body lipolysis in states of insulin resistance and enhanced fatty acid synthesis are key features of non-alcoholic fatty liver disease. Increased de novo lipogenesis results from enhanced activation of LXR, SREBP-1c, and ChREBP in insulin resistance. SREBP-induced activation of ACC2 leads to accumulation of malonyl-CoA, which in turn inhibits CPT-1 activity, resulting in reduced β-oxidation. In the liver, triglycerides are stored in LDs that are formed within the lipid bilayer of the ER. Stabilization and growth of LDs are dependent on transmembrane proteins, seipin, and triglyceride and phospholipid synthetic enzymes that are located on the LD surface. LDs are catabolized via ATGL-dependent hydrolysis and β-oxidation of fatty acids, lysosomal lipases, and carboxylesterase 3- and CIDEB- mediated repacking of cytosolic LDs in the ER, leading to synthesis of VLDL particle. Intermediates of long chain fatty acids (DAG) inhibit insulin signalling further, exacerbating hepatic insulin resistance by exerting proinflammatory effects and reducing activation of the insulin receptor. LXRα: Ligand-activated transcription factor α; SREBP1c: Sterol regulating element binding protein 1c; CPT-1: Carnitine palmitoyl transferase 1; LD: Lipid droplet; ATGL: Adipose triglyceride lipase; CIDEB: Cell death inducing DFFA like effector B; DAG: Diacylglycerol; IRS: Insulin receptor substrate; ER: Endoplasmic reticulum; ChREBP: Carbohydrate response element binding protein; LXR: Ligand-activated transcription factor; DNL: De novo lipogenesis; ACC2: Acetyl-CoA carboxylase 2; PNPLA2: Patatin-like phospholipase domain-containing protein 2; ROS: Reactive oxygen species.
AFLD
Chronic alcohol exposure is another frequent cause of fatty liver disease. More than 90% of alcoholics have fatty liver disease that will potentially resolve with abstinence from alcohol[57-59]. Chronic consumption of more than 30 g of pure alcohol was demonstrated to significantly increase the risk of chronic liver disease[60]. Susceptibility factors included female sex, obesity, and cigarette smoking as well as coexistence of other hepatic disorders, such as hepatitis B or C virus infection, NAFLD, or hemochromatosis[61].
Alcohol is metabolized via two main pathways in the liver, the oxidative pathway, which is mediated by the alcohol dehydrogenase (ADH) and acetaldehyde dehydrogenase (ALDH), and the non-oxidative cytochrome P450 2E1 pathway[62-64].
Ethanol is converted by the cytosolic enzyme ADH to acetaldehyde and further metabolized to acetate by the mitochondrial enzyme ALDH. The reactions are both coupled to the reduction of nicotinamide adenine dinucleotide (NAD) to nicotinamide adenine dinucleotide-hydrogen (NADH)[65,66].
Excess NADH has deleterious effects on gluconeogenesis and fatty acid synthesis[67] Furthermore, acetaldehyde was found to induce lipogenesis by increasing SREBP-1c expression[68]. Increased fatty acid synthesis leads to the accumulation of fatty acid intermediates, such as malonyl-CoA, which suppresses fatty acid transport into the mitochondria and their oxidation by CPT-1[69]. Alcohol mainly via its metabolite acetaldehyde further inhibits PPARα, which is the main transcription factor for genes involved in oxidation, transport, and export of fatty acids[70-74]. In PPARα deficient mice, chronic ethanol feeding was associated with progressive intrahepatic triglyceride accumulation due to reduced β-oxidation and alterations in tricarboxylic acid cycle and the electron transfer chain[75]. In a mouse model, alcohol feeding resulted in dysfunction of ALDH2 and aldehyde accumulation. Pharmacological activation of ALDH2 reversed alcoholic steatosis in these mice, underlining the deleterious effects of accumulating metabolic products of alcohol[76]. Very recently, Guo et al[77] reported that the beneficial role of ALDH2 on hepatic steatosis and inflammation might be mediated through regulation of autophagy.
Ceramide-induced inhibition of AMPK and impaired hepatic adiponectin effects might further contribute to increased fatty acid synthesis and reduced fatty acid oxidation[78-80]. Besides β-oxidation and lipogenesis, VLDL export is also affected by alcohol: triglyceride export is impaired due to suppression of phosphatidylcholine, acetaldehyde-induced inhibition of microtubular assembly, and reduced apolipoprotein synthesis[81]. The role of perilipin 2, which is a major LD protein, was recently demonstrated in perilipin-deficient mice that are protected from ethanol-induced hepatic steatosis[82]. Several studies suggest that lipin-1, which is critically involved in triglyceride synthesis, might have a significant role in ALD. Liver-specific deficiency of lipin 1 was associated with increased hepatic triglyceride and cholesterol accumulation and inflammation in ethanol fed mice[83]. SIRT1, which is downregulated in mice models of ALD, was found to affect hepatic steatosis, inflammation, and fibrosis via lipin-1[84].
CHRONIC HCV INFECTION
Steatosis is also found in up to 80% of patients with chronic HCV infection[85]. Co-occurrence of steatosis and chronic HCV infection are associated with progression of liver disease and poor response in interferon-α based treatment[4,86-89]. Prevalence of steatosis in chronic HCV infection is double than that found in patients with hepatitis B virus infection, suggesting that co-occurrence of NAFLD and HCV does not fully explain the high prevalence of steatosis in patients with chronic HCV infection[85,90]. Additionally, probability of steatosis is significantly higher in genotype 3 than in non-3 genotype HCV infections, which argues in favor of viral induction of fatty liver in genotype 3 HCV infection[4]. Accordingly, fatty liver was found to be associated with HCV RNA levels in genotype 3a and body mass index in genotype 1 HCV infection[91]. Interestingly, infection of the immortalized hepatoma cell line HepG2 with HCV genotype 1b was associated with suppressor of cytokine signalling 3 mediated impairment of insulin signalling when compared to HCV genotype 2 infected cells[92]. All HCV genotypes, but genotype 3 to a higher extent, exert direct effects on hepatic lipid metabolism resulting in occurrence of large droplets in hepatocytes[85,93]. HCV makes use of VLDL assembly and secretion pathways in production of infectious virus. In virion morphogenesis, LDs are targeted by virus-encoded proteins. LDs have also been shown to influence the course of the infection. The role of LDs in HCV infection has recently been reviewed extensively by Filipe et al[93]. Increased intracellular triglyceride accumulation in HCV infection might be due to both direct viral effects and indirect effects resulting from accompanying insulin resistance[93-105]. Decreased β-oxidation, increased gluconeogenesis, and DNL are key features of steatosis in HCV patients. Recently, it was shown that HCV blocks phosphorylation of forkhead box protein O1 (FOXO1), resulting in enhanced gluconeogenesis and promotion of lipogenesis by increasing the levels of SREBP-1c and fatty acid synthase (FAS)[94,96,105]. In addition to reduced phosphorylation of FOXO1, increased expression of gluconeogenic enzymes phosphoenolpyruvate carboxykinase and glucose-6-phosphatase was found to be due to enhanced cyclic AMP responsive element-binding protein phosphorylation, linking HCV-induced ER stress with increased hepatic glucose output[106]. Viral proteins were also found to stimulate transcription of key enzymes of gluconeogenesis and diacylglycerol acyltransferase-1, which catalyses the final step in triglyceride synthesis and affects LD formation. Resulting insulin resistance might further contribute to increased lipogenesis. However, the exact mechanisms have not been fully elucidated in this setting. Furthermore infection with genotype 3 HCV strains has been found to be associated with reduced levels of phosphatase and tensin homolog, which is a negative regulator of insulin/phosphoinositide 3-kinase signalling. Remarkably, interaction of HCV with DDX3, a DEAD box protein, led to phosphorylation of inhibitor k B kinase α (IKKα) and, as a consequence, the induction of SREBP-induced lipogenesis and LD formation[102]. Beyond quantitative effects, HCV infection was recently shown to alter hepatic metabolic zonation by redistributing FAS from perilobular to mid-zone of the lobule[104]. Beta-oxidation is also reduced in virus-infected cells due to decreased expression of short-chain acyl-CoA dehydrogenase and medium-chain acyl-CoA dehydrogenase. In vivo data suggested that PPARα expression is also decreased as a consequence of increased suppression by microRNA 27 (miR27) in chronic HCV infection[99]. In contrast, Tanaka and colleagues[100] reported from a mouse model that permanent PPARα activation plays an essential role in development of hepatic steatosis in chronic HCV infection. Remarkably, HCV infection may also directly affect VLDL secretion, resulting in intracellular lipid accumulation. Triglyceride content of VLDL particles have been shown to be lower in HCV infected patients[101]. In the same study, hepatic lipogenesis was significantly higher and cholesterol synthesis lower in patients with chronic HCV infection when compared to healthy controls[101]. Low cholesterol levels have been associated with genotype 3 infection and, in contrast to non-responders, successful HCV treatment was accompanied by normalization of cholesterol levels[107].
DRUG-INDUCED HEPATIC STEATOSIS
Several drugs have been identified that may induce reversible intrahepatic triglyceride accumulation. These include antimicrobials, such as tetracycline, glucocorticoids, tamoxifen, chemotherapeutic agents, methotrexate, anti-arrhythmics such as amiodarone as well as antiepileptic valproic acid, several antiviral agents, lipid lowering mipomersen and lomitapide, and non-steroidal anti-inflammatory drugs such as acetylsalicylic acid[108]. Mechanistically, drug-induced (e.g., valproic acid) diminished mitochondrial β-oxidation of fatty acids leads to excessive reactive oxygen species generation and depletion of ATP, commonly resulting in development of microvesicular steatosis. Other pathophysiological mechanisms include enhanced cellular uptake of fatty acid due to increased fatty acid translocase expression or increased DNL resulting from elevated expression levels of SREBP1c or decreased AMPK levels. In contrast to the microvesicular steatotic pattern, macrovesicular steatosis is usually considered benign and is often reversible. Detailed mechanisms and outcome have recently been reviewed by Amacher et al[108] (Table 2).
Table 2.
Overview on drug-induced micro- or macrovesicular forms of fatty liver disease (adapted from[108,123])
| Causes of drug-induced fatty liver disease |
| Predominantly microvesicular pattern: |
| Tetracyclin |
| Valproic acid |
| Amiodarone |
| Glucocorticoids |
| 5-FU |
| Margosa oil |
| Predominantly macrovesicular pattern: |
| Acetylsalicylic acid (intoxication) |
| Tamoxifen |
| Mixed pattern: |
| Antiretroviral agents (e.g., didanosine, stavudine) |
| Methotrexate |
ACUTE FATTY LIVER OF PREGNANCY
Acute fatty liver of pregnancy is a rare life-threatening disease in late pregnancy. It is characterized by microvesicular pattern of hepatic steatosis[109]. Incidence was estimated as 5/100000 pregnancies in the United Kingdom[110]. Mechanistically, fatty liver is due to a defect in mitochondrial β-oxidation. In late pregnancy, when the woman is increasingly dependent on fat as an energy source, the hitherto compensated defect in fatty acid metabolism results in mitochondrial dysfunction[111,112]. Remarkably, risk of acute fatty liver of pregnancy (AFLP) is highly increased when the fetus is homozygous or compound heterozygous for a defect in fatty acid oxidation (FAO)[113,114]. Urgent termination of pregnancy usually leads to significant improvements or normalization of AFLP[115], suggesting a pathophysiological role of the placenta in this disease. Mitochondrial dysfunction due to a defect in placental FAO leads to oxidative stress and, in parallel, accumulation of circulating toxic fatty acid intermediates, such as arachidonic acid. The latter has been shown to induce damage, apoptosis, and lipid accumulation in hepatocytes[116].
FUTURE PERSPECTIVES
Although knowledge on pathophysiology of fatty liver disease has significantly improved in the past years, treatment options, especially of NAFLD, are still very limited. Several studies have suggested the beneficial effects of weight loss on the course of NAFLD. Pharmacologically, thiazolidinedione (glitazones) and antioxidative vitamin E are the most promising therapies today. Novel expectant concepts include activation of SIRT-1 (e.g., reservatrol), which was found to be beneficial in murine models of NAFLD by exerting insulin-sensitizing, anti-inflammatory, and antioxidative effects[117,118]. Very recently, a liver specific LXR inverse agonist significantly reduced hepatic steatosis by reducing de novo lipogenesis[119]. The effect of simtuzumab, which is a humanized antifibrotic monoclonal antibody against lysyl oxidase like molecule 2, is currently under investigation in patients with advanced NAFLC[120]. FXR agonists, such as obeticholic acid, are probably the most promising therapeutic option under investigation in NAFLD.
ACKNOWLEDGMENTS
We would like to thank Anna Kirschner, MSc for proofreading and assistance in manuscript preparation.
Footnotes
Supported by Austrian Federal Ministry of Science, Research and Economy and the National Foundation for Research, Technology and Development.
Conflict-of-interest statement: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 27, 2015
First decision: July 20, 2015
Article in press: September 30, 2015
P- Reviewer: La Mura V, Marin JJG S- Editor: Ma YJ L- Editor: Filipodia E- Editor: Ma S
References
- 1.Javor ED, Ghany MG, Cochran EK, Oral EA, DePaoli AM, Premkumar A, Kleiner DE, Gorden P. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005;41:753–760. doi: 10.1002/hep.20672. [DOI] [PubMed] [Google Scholar]
- 2.Hübscher SG. Histological assessment of non-alcoholic fatty liver disease. Histopathology. 2006;49:450–465. doi: 10.1111/j.1365-2559.2006.02416.x. [DOI] [PubMed] [Google Scholar]
- 3.Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 4.Leandro G, Mangia A, Hui J, Fabris P, Rubbia-Brandt L, Colloredo G, Adinolfi LE, Asselah T, Jonsson JR, Smedile A, et al. Relationship between steatosis, inflammation, and fibrosis in chronic hepatitis C: a meta-analysis of individual patient data. Gastroenterology. 2006;130:1636–1642. doi: 10.1053/j.gastro.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 5.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 6.Leleu H, Blachier M, Rosa I. Cost-effectiveness of sofosbuvir in the treatment of patients with hepatitis C. J Viral Hepat. 2015;22:376–383. doi: 10.1111/jvh.12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Alwis NM, Day CP. Non-alcoholic fatty liver disease: the mist gradually clears. J Hepatol. 2008;48 Suppl 1:S104–S112. doi: 10.1016/j.jhep.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Kaser S, Ebenbichler CF, Tilg H. Pharmacological and non-pharmacological treatment of non-alcoholic fatty liver disease. Int J Clin Pract. 2010;64:968–983. doi: 10.1111/j.1742-1241.2009.02327.x. [DOI] [PubMed] [Google Scholar]
- 9.Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62:S47–S64. doi: 10.1016/j.jhep.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Mittendorfer B, Magkos F, Fabbrini E, Mohammed BS, Klein S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity (Silver Spring) 2009;17:1872–1877. doi: 10.1038/oby.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pardina E, Baena-Fustegueras JA, Llamas R, Catalán R, Galard R, Lecube A, Fort JM, Llobera M, Allende H, Vargas V, et al. Lipoprotein lipase expression in livers of morbidly obese patients could be responsible for liver steatosis. Obes Surg. 2009;19:608–616. doi: 10.1007/s11695-009-9827-5. [DOI] [PubMed] [Google Scholar]
- 12.Nye CK, Hanson RW, Kalhan SC. Glyceroneogenesis is the dominant pathway for triglyceride glycerol synthesis in vivo in the rat. J Biol Chem. 2008;283:27565–27574. doi: 10.1074/jbc.M804393200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doege H, Grimm D, Falcon A, Tsang B, Storm TA, Xu H, Ortegon AM, Kazantzis M, Kay MA, Stahl A. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J Biol Chem. 2008;283:22186–22192. doi: 10.1074/jbc.M803510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savage DB, Murgatroyd PR, Chatterjee VK, O’Rahilly S. Energy expenditure and adaptive responses to an acute hypercaloric fat load in humans with lipodystrophy. J Clin Endocrinol Metab. 2005;90:1446–1452. doi: 10.1210/jc.2004-1494. [DOI] [PubMed] [Google Scholar]
- 15.Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA. 2007;104:12587–12594. doi: 10.1073/pnas.0705408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petersen KF, Dufour S, Morino K, Yoo PS, Cline GW, Shulman GI. Reversal of muscle insulin resistance by weight reduction in young, lean, insulin-resistant offspring of parents with type 2 diabetes. Proc Natl Acad Sci USA. 2012;109:8236–8240. doi: 10.1073/pnas.1205675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flannery C, Dufour S, Rabøl R, Shulman GI, Petersen KF. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes. 2012;61:2711–2717. doi: 10.2337/db12-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higuchi N, Kato M, Shundo Y, Tajiri H, Tanaka M, Yamashita N, Kohjima M, Kotoh K, Nakamuta M, Takayanagi R, et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol Res. 2008;38:1122–1129. doi: 10.1111/j.1872-034X.2008.00382.x. [DOI] [PubMed] [Google Scholar]
- 21.Lima-Cabello E, García-Mediavilla MV, Miquilena-Colina ME, Vargas-Castrillón J, Lozano-Rodríguez T, Fernández-Bermejo M, Olcoz JL, González-Gallego J, García-Monzón C, Sánchez-Campos S. Enhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis C. Clin Sci (Lond) 2011;120:239–250. doi: 10.1042/CS20100387. [DOI] [PubMed] [Google Scholar]
- 22.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delarue J, Normand S, Couet C, Pachiaudi C, Urbain C, Lamisse F, Riou JP. Effects of free fatty acids on the metabolic response to oral fructose in lean healthy humans. Int J Obes Relat Metab Disord. 1996;20:130–136. [PubMed] [Google Scholar]
- 25.Deer J, Koska J, Ozias M, Reaven P. Dietary models of insulin resistance. Metabolism. 2015;64:163–171. doi: 10.1016/j.metabol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 27.Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:8713–8742. doi: 10.3390/ijms15058713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP. Hepatic lipid droplet biology: Getting to the root of fatty liver. Hepatology. 2015;62:964–967. doi: 10.1002/hep.27839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan JS, Seow CJ, Goh VJ, Silver DL. Recent advances in understanding proteins involved in lipid droplet formation, growth and fusion. J Genet Genomics. 2014;41:251–259. doi: 10.1016/j.jgg.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology. 2011;53:116–126. doi: 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coleman RA, Mashek DG. Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem Rev. 2011;111:6359–6386. doi: 10.1021/cr100404w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan SA, Sathyanarayan A, Mashek MT, Ong KT, Wollaston-Hayden EE, Mashek DG. ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1α/PPAR-α signaling. Diabetes. 2015;64:418–426. doi: 10.2337/db14-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 2015;61:1896–1907. doi: 10.1002/hep.27667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lian J, Wei E, Wang SP, Quiroga AD, Li L, Di Pardo A, van der Veen J, Sipione S, Mitchell GA, Lehner R. Liver specific inactivation of carboxylesterase 3/triacylglycerol hydrolase decreases blood lipids without causing severe steatosis in mice. Hepatology. 2012;56:2154–2162. doi: 10.1002/hep.25881. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Ye J, Zhou L, Gu W, Fisher EA, Li P. Opposing roles of cell death-inducing DFF45-like effector B and perilipin 2 in controlling hepatic VLDL lipidation. J Lipid Res. 2012;53:1877–1889. doi: 10.1194/jlr.M026591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su W, Wang Y, Jia X, Wu W, Li L, Tian X, Li S, Wang C, Xu H, Cao J, et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2014;111:11437–11442. doi: 10.1073/pnas.1410741111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imai Y, Varela GM, Jackson MB, Graham MJ, Crooke RM, Ahima RS. Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology. 2007;132:1947–1954. doi: 10.1053/j.gastro.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 40.Carr RM, Patel RT, Rao V, Dhir R, Graham MJ, Crooke RM, Ahima RS. Reduction of TIP47 improves hepatic steatosis and glucose homeostasis in mice. Am J Physiol Regul Integr Comp Physiol. 2012;302:R996–1003. doi: 10.1152/ajpregu.00177.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C, Zhao Y, Gao X, Li L, Yuan Y, Liu F, Zhang L, Wu J, Hu P, Zhang X, et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology. 2015;61:870–882. doi: 10.1002/hep.27409. [DOI] [PubMed] [Google Scholar]
- 42.Szendroedi J, Roden M. Perilipin 5: from fatty liver to hepatic lipodystrophy? Hepatology. 2015;61:751–753. doi: 10.1002/hep.27618. [DOI] [PubMed] [Google Scholar]
- 43.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forte TM, Ryan RO. Apolipoprotein A5: Extracellular and Intracellular Roles in Triglyceride Metabolism. Curr Drug Targets. 2015;16:1274–1280. doi: 10.2174/1389450116666150531161138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ress C, Moschen AR, Sausgruber N, Tschoner A, Graziadei I, Weiss H, Schgoer W, Ebenbichler CF, Konrad RJ, Patsch JR, et al. The role of apolipoprotein A5 in non-alcoholic fatty liver disease. Gut. 2011;60:985–991. doi: 10.1136/gut.2010.222224. [DOI] [PubMed] [Google Scholar]
- 46.Zheng XY, Zhao SP, Yu BL, Wu CL, Liu L. Apolipoprotein A5 internalized by human adipocytes modulates cellular triglyceride content. Biol Chem. 2012;393:161–167. doi: 10.1515/hsz-2011-0259. [DOI] [PubMed] [Google Scholar]
- 47.Gao Z, Zhang J, Kheterpal I, Kennedy N, Davis RJ, Ye J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J Biol Chem. 2011;286:22227–22234. doi: 10.1074/jbc.M111.228874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 49.Kantartzis K, Machann J, Schick F, Fritsche A, Häring HU, Stefan N. The impact of liver fat vs visceral fat in determining categories of prediabetes. Diabetologia. 2010;53:882–889. doi: 10.1007/s00125-010-1663-6. [DOI] [PubMed] [Google Scholar]
- 50.Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, Still CD, Gerhard GS, Han X, Dziura J, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2011;108:16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, Varela JE, Brunt EM, Patterson BW, Klein S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology. 2012;142:1444–1446.e2. doi: 10.1053/j.gastro.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282:826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 54.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tilg H, Moschen AR. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol Metab. 2008;19:371–379. doi: 10.1016/j.tem.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Lipina C, Hundal HS. Sphingolipids: agents provocateurs in the pathogenesis of insulin resistance. Diabetologia. 2011;54:1596–1607. doi: 10.1007/s00125-011-2127-3. [DOI] [PubMed] [Google Scholar]
- 57.Liu JD, Leung KW, Wang CK, Liao LY, Wang CS, Chen PH, Chen CC, Yeh EK. Alcohol-related problems in Taiwan with particular emphasis on alcoholic liver diseases. Alcohol Clin Exp Res. 1998;22:164S–169S. doi: 10.1111/acer.1998.22.s3_part1.164s. [DOI] [PubMed] [Google Scholar]
- 58.Stickel F, Hoehn B, Schuppan D, Seitz HK. Review article: Nutritional therapy in alcoholic liver disease. Aliment Pharmacol Ther. 2003;18:357–373. doi: 10.1046/j.1365-2036.2003.01660.x. [DOI] [PubMed] [Google Scholar]
- 59.Diehl AM. Alcoholic liver disease: natural history. Liver Transpl Surg. 1997;3:206–211. [PubMed] [Google Scholar]
- 60.Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, Saveria Crocè L, Sasso F, Pozzato G, Cristianini G, et al. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut. 1997;41:845–850. doi: 10.1136/gut.41.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis. 2004;24:217–232. doi: 10.1055/s-2004-832936. [DOI] [PubMed] [Google Scholar]
- 62.Seth D, Haber PS, Syn WK, Diehl AM, Day CP. Pathogenesis of alcohol-induced liver disease: classical concepts and recent advances. J Gastroenterol Hepatol. 2011;26:1089–1105. doi: 10.1111/j.1440-1746.2011.06756.x. [DOI] [PubMed] [Google Scholar]
- 63.Lieber CS. Role of oxidative stress and antioxidant therapy in alcoholic and nonalcoholic liver diseases. Adv Pharmacol. 1997;38:601–628. doi: 10.1016/s1054-3589(08)61001-7. [DOI] [PubMed] [Google Scholar]
- 64.Rocco A, Compare D, Angrisani D, Sanduzzi Zamparelli M, Nardone G. Alcoholic disease: liver and beyond. World J Gastroenterol. 2014;20:14652–14659. doi: 10.3748/wjg.v20.i40.14652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grant BF, Dufour MC, Harford TC. Epidemiology of alcoholic liver disease. Semin Liver Dis. 1988;8:12–25. doi: 10.1055/s-2008-1040525. [DOI] [PubMed] [Google Scholar]
- 66.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 67.Berg JM, Tymoczko JL, Stryer L. Lipids and cell membranes. In: Biochemistry , fifth edition, editors. New York: W. H. Freeman; 2002. p. 1050. [Google Scholar]
- 68.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP) J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 69.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 70.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 71.Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med. 2003;3:561–572. doi: 10.2174/1566524033479537. [DOI] [PubMed] [Google Scholar]
- 72.Lee GY, Kim NH, Zhao ZS, Cha BS, Kim YS. Peroxisomal-proliferator-activated receptor alpha activates transcription of the rat hepatic malonyl-CoA decarboxylase gene: a key regulation of malonyl-CoA level. Biochem J. 2004;378:983–990. doi: 10.1042/BJ20031565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nanji AA, Dannenberg AJ, Jokelainen K, Bass NM. Alcoholic liver injury in the rat is associated with reduced expression of peroxisome proliferator-alpha (PPARalpha)-regulated genes and is ameliorated by PPARalpha activation. J Pharmacol Exp Ther. 2004;310:417–424. doi: 10.1124/jpet.103.064717. [DOI] [PubMed] [Google Scholar]
- 74.Galli A, Pinaire J, Fischer M, Dorris R, Crabb DW. The transcriptional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J Biol Chem. 2001;276:68–75. doi: 10.1074/jbc.M008791200. [DOI] [PubMed] [Google Scholar]
- 75.Li HH, Tyburski JB, Wang YW, Strawn S, Moon BH, Kallakury BV, Gonzalez FJ, Fornace AJ. Modulation of fatty acid and bile acid metabolism by peroxisome proliferator-activated receptor α protects against alcoholic liver disease. Alcohol Clin Exp Res. 2014;38:1520–1531. doi: 10.1111/acer.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhong W, Zhang W, Li Q, Xie G, Sun Q, Sun X, Tan X, Sun X, Jia W, Zhou Z. Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol-induced hepatic steatosis and cell death in mice. J Hepatol. 2015;62:1375–1381. doi: 10.1016/j.jhep.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo R, Xu X, Babcock SA, Zhang Y, Ren J. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. J Hepatol. 2015;62:647–656. doi: 10.1016/j.jhep.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 78.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liangpunsakul S, Wou SE, Zeng Y, Ross RA, Jayaram HN, Crabb DW. Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1173–G1181. doi: 10.1152/ajpgi.90349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.García-Ruiz C, Colell A, Marí M, Morales A, Calvo M, Enrich C, Fernández-Checa JC. Defective TNF-alpha-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J Clin Invest. 2003;111:197–208. doi: 10.1172/JCI16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wehr H, Rodo M, Lieber CS, Baraona E. Acetaldehyde adducts and autoantibodies against VLDL and LDL in alcoholics. J Lipid Res. 1993;34:1237–1244. [PubMed] [Google Scholar]
- 82.Carr RM, Peralta G, Yin X, Ahima RS. Absence of perilipin 2 prevents hepatic steatosis, glucose intolerance and ceramide accumulation in alcohol-fed mice. PLoS One. 2014;9:e97118. doi: 10.1371/journal.pone.0097118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu M, Wang F, Li X, Rogers CQ, Liang X, Finck BN, Mitra MS, Zhang R, Mitchell DA, You M. Regulation of hepatic lipin-1 by ethanol: role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology. 2012;55:437–446. doi: 10.1002/hep.24708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, Odena G, Stevens SM, You M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146:801–811. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goossens N, Negro F. Insulin resistance, non-alcoholic fatty liver disease and hepatitis C virus infection. Rev Recent Clin Trials. 2014;9:204–209. doi: 10.2174/1574887109666141216101939. [DOI] [PubMed] [Google Scholar]
- 86.Bochud PY, Cai T, Overbeck K, Bochud M, Dufour JF, Müllhaupt B, Borovicka J, Heim M, Moradpour D, Cerny A, et al. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol. 2009;51:655–666. doi: 10.1016/j.jhep.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 87.Bugianesi E, Marchesini G, Gentilcore E, Cua IH, Vanni E, Rizzetto M, George J. Fibrosis in genotype 3 chronic hepatitis C and nonalcoholic fatty liver disease: Role of insulin resistance and hepatic steatosis. Hepatology. 2006;44:1648–1655. doi: 10.1002/hep.21429. [DOI] [PubMed] [Google Scholar]
- 88.Fartoux L, Poujol-Robert A, Guéchot J, Wendum D, Poupon R, Serfaty L. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut. 2005;54:1003–1008. doi: 10.1136/gut.2004.050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bedossa P, Moucari R, Chelbi E, Asselah T, Paradis V, Vidaud M, Cazals-Hatem D, Boyer N, Valla D, Marcellin P. Evidence for a role of nonalcoholic steatohepatitis in hepatitis C: a prospective study. Hepatology. 2007;46:380–387. doi: 10.1002/hep.21711. [DOI] [PubMed] [Google Scholar]
- 90.Czaja AJ, Carpenter HA, Santrach PJ, Moore SB. Host- and disease-specific factors affecting steatosis in chronic hepatitis C. J Hepatol. 1998;29:198–206. doi: 10.1016/s0168-8278(98)80004-4. [DOI] [PubMed] [Google Scholar]
- 91.Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology. 2001;33:1358–1364. doi: 10.1053/jhep.2001.24432. [DOI] [PubMed] [Google Scholar]
- 92.Persico M, Russo R, Persico E, Svelto M, Spano D, Andolfo I, La Mura V, Capasso M, Tiribelli C, Torella R, et al. SOCS3 and IRS-1 gene expression differs between genotype 1 and genotype 2 hepatitis C virus-infected HepG2 cells. Clin Chem Lab Med. 2009;47:1217–1225. doi: 10.1515/CCLM.2009.280. [DOI] [PubMed] [Google Scholar]
- 93.Filipe A, McLauchlan J. Hepatitis C virus and lipid droplets: finding a niche. Trends Mol Med. 2015;21:34–42. doi: 10.1016/j.molmed.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 94.Bernsmeier C, Calabrese D, Heim MH, Duong HT. Hepatitis C virus dysregulates glucose homeostasis by a dual mechanism involving induction of PGC1α and dephosphorylation of FoxO1. J Viral Hepat. 2014;21:9–18. doi: 10.1111/jvh.12208. [DOI] [PubMed] [Google Scholar]
- 95.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci USA. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Banerjee A, Meyer K, Mazumdar B, Ray RB, Ray R. Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin-induced metabolic gene expression. J Virol. 2010;84:5936–5946. doi: 10.1128/JVI.02344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Parvaiz F, Manzoor S, Iqbal J, McRae S, Javed F, Ahmed QL, Waris G. Hepatitis C virus nonstructural protein 5A favors upregulation of gluconeogenic and lipogenic gene expression leading towards insulin resistance: a metabolic syndrome. Arch Virol. 2014;159:1017–1025. doi: 10.1007/s00705-013-1892-3. [DOI] [PubMed] [Google Scholar]
- 98.Dharancy S, Malapel M, Perlemuter G, Roskams T, Cheng Y, Dubuquoy L, Podevin P, Conti F, Canva V, Philippe D, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology. 2005;128:334–342. doi: 10.1053/j.gastro.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 99.Singaravelu R, Chen R, Lyn RK, Jones DM, O’Hara S, Rouleau Y, Cheng J, Srinivasan P, Nasheri N, Russell RS, et al. Hepatitis C virus induced up-regulation of microRNA-27: a novel mechanism for hepatic steatosis. Hepatology. 2014;59:98–108. doi: 10.1002/hep.26634. [DOI] [PubMed] [Google Scholar]
- 100.Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Invest. 2008;118:683–694. doi: 10.1172/JCI33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lambert JE, Bain VG, Ryan EA, Thomson AB, Clandinin MT. Elevated lipogenesis and diminished cholesterol synthesis in patients with hepatitis C viral infection compared to healthy humans. Hepatology. 2013;57:1697–1704. doi: 10.1002/hep.25990. [DOI] [PubMed] [Google Scholar]
- 102.Li Q, Pène V, Krishnamurthy S, Cha H, Liang TJ. Hepatitis C virus infection activates an innate pathway involving IKK-α in lipogenesis and viral assembly. Nat Med. 2013;19:722–729. doi: 10.1038/nm.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oshiumi H, Ikeda M, Matsumoto M, Watanabe A, Takeuchi O, Akira S, Kato N, Shimotohno K, Seya T. Hepatitis C virus core protein abrogates the DDX3 function that enhances IPS-1-mediated IFN-beta induction. PLoS One. 2010;5:e14258. doi: 10.1371/journal.pone.0014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moreau M, Rivière B, Vegna S, Aoun M, Gard C, Ramos J, Assenat E, Hibner U. Hepatitis C viral proteins perturb metabolic liver zonation. J Hepatol. 2015;62:278–285. doi: 10.1016/j.jhep.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 105.Bose SK, Kim H, Meyer K, Wolins N, Davidson NO, Ray R. Forkhead box transcription factor regulation and lipid accumulation by hepatitis C virus. J Virol. 2014;88:4195–4203. doi: 10.1128/JVI.03327-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yao W, Cai H, Li X, Li T, Hu L, Peng T. Endoplasmic reticulum stress links hepatitis C virus RNA replication to wild-type PGC-1α/liver-specific PGC-1α upregulation. J Virol. 2014;88:8361–8374. doi: 10.1128/JVI.01202-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Corey KE, Kane E, Munroe C, Barlow LL, Zheng H, Chung RT. Hepatitis C virus infection and its clearance alter circulating lipids: implications for long-term follow-up. Hepatology. 2009;50:1030–1037. doi: 10.1002/hep.23219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Amacher DE, Chalasani N. Drug-induced hepatic steatosis. Semin Liver Dis. 2014;34:205–214. doi: 10.1055/s-0034-1375960. [DOI] [PubMed] [Google Scholar]
- 109.Eapen CE, Ramakrishna B, Jose R, Loganathan G, Chandy G. Liver failure during pregnancy. Gut. 2008;57:83. doi: 10.1136/gut.2007.143305. [DOI] [PubMed] [Google Scholar]
- 110.Knight M, Nelson-Piercy C, Kurinczuk JJ, Spark P, Brocklehurst P. A prospective national study of acute fatty liver of pregnancy in the UK. Gut. 2008;57:951–956. doi: 10.1136/gut.2008.148676. [DOI] [PubMed] [Google Scholar]
- 111.Goel A, Jamwal KD, Ramachandran A, Balasubramanian KA, Eapen CE. Pregnancy-related liver disorders. J Clin Exp Hepatol. 2014;4:151–162. doi: 10.1016/j.jceh.2013.03.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kamimura K, Abe H, Kawai H, Kamimura H, Kobayashi Y, Nomoto M, Aoyagi Y, Terai S. Advances in understanding and treating liver diseases during pregnancy: A review. World J Gastroenterol. 2015;21:5183–5190. doi: 10.3748/wjg.v21.i17.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ibdah JA, Bennett MJ, Rinaldo P, Zhao Y, Gibson B, Sims HF, Strauss AW. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med. 1999;340:1723–1731. doi: 10.1056/NEJM199906033402204. [DOI] [PubMed] [Google Scholar]
- 114.Yang Z, Yamada J, Zhao Y, Strauss AW, Ibdah JA. Prospective screening for pediatric mitochondrial trifunctional protein defects in pregnancies complicated by liver disease. JAMA. 2002;288:2163–2166. doi: 10.1001/jama.288.17.2163. [DOI] [PubMed] [Google Scholar]
- 115.Xiong HF, Liu JY, Guo LM, Li XW. Acute fatty liver of pregnancy: over six months follow-up study of twenty-five patients. World J Gastroenterol. 2015;21:1927–1931. doi: 10.3748/wjg.v21.i6.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Natarajan SK, Thangaraj KR, Eapen CE, Ramachandran A, Mukhopadhya A, Mathai M, Seshadri L, Peedikayil A, Ramakrishna B, Balasubramanian KA. Liver injury in acute fatty liver of pregnancy: possible link to placental mitochondrial dysfunction and oxidative stress. Hepatology. 2010;51:191–200. doi: 10.1002/hep.23245. [DOI] [PubMed] [Google Scholar]
- 117.Colak Y, Yesil A, Mutlu HH, Caklili OT, Ulasoglu C, Senates E, Takir M, Kostek O, Yilmaz Y, Yilmaz Enc F, et al. A potential treatment of non-alcoholic fatty liver disease with SIRT1 activators. J Gastrointestin Liver Dis. 2014;23:311–319. doi: 10.15403/jgld.2014.1121.233.yck. [DOI] [PubMed] [Google Scholar]
- 118.Xu F, Li Z, Zheng X, Liu H, Liang H, Xu H, Chen Z, Zeng K, Weng J. SIRT1 mediates the effect of GLP-1 receptor agonist exenatide on ameliorating hepatic steatosis. Diabetes. 2014;63:3637–3646. doi: 10.2337/db14-0263. [DOI] [PubMed] [Google Scholar]
- 119.Griffett K, Welch RD, Flaveny CA, Kolar GR, Neuschwander-Tetri BA, Burris TP. The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol Metab. 2015;4:353–357. doi: 10.1016/j.molmet.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Talal A, Feron-Rigodon M, Madere J, Subramanian G, Bornstein J. 1319 Simutuzumab, an antifibrotic monoclonal antibody against Lysyl Oxidase-Like 2(LOXL2) enzyme, appears safe and well tolerated in patients with liver disease of diverse etiology. J Hepatol. 2013;58 Suppl 1:S532. [Google Scholar]
- 121.Reid AE. Nonalcoholic steatohepatitis. Gastroenterology. 2001;121:710–723. doi: 10.1053/gast.2001.27126. [DOI] [PubMed] [Google Scholar]
- 122.Ramesh S, Sanyal AJ. Evaluation and management of non-alcoholic steatohepatitis. J Hepatol. 2005;42 Suppl:S2–S12. doi: 10.1016/j.jhep.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 123.Fromenty B. Drug-induced liver injury in obesity. J Hepatol. 2013;58:824–826. doi: 10.1016/j.jhep.2012.12.018. [DOI] [PubMed] [Google Scholar]