

Abstract
The present study was conducted to investigate a new pathway for the degradation of nicotinamide adenine dinucleotide (NAD) by Penicillium brevicompactum NRC 829 extracts. Enzymes involved in the hydrolysis of NAD, i.e. alkaline phosphatase, aminohydrolase and glycohydrolase were determined. Alkaline phosphatase was found to catalyse the sequential hydrolysis of two phosphate moieties of NAD molecule to nicotinamide riboside plus adenosine. Adenosine was then deaminated by aminohydrolase to inosine and ammonia. While glycohydrolase catalyzed the hydrolysis of the nicotinamide-ribosidic bond of NAD+ to produce nicotinamide and ADP-ribose in equimolar amounts, enzyme purification through a 3-step purification procedure revealed the existence of two peaks of alkaline phosphatases, and one peak contained deaminase and glycohydrolase activities. NAD deaminase was purified to homogeneity as estimated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis with an apparent molecular mass of 91 kDa. Characterization and determination of some of NAD aminohydrolase kinetic properties were conducted due to its biological role in the regulation of cellular NAD level. The results also revealed that NAD did not exert its feedback control on nicotinamide amidase produced by P. brevicompactum.
Keywords: NAD degradation, Penicillium brevicompactum NRC 829, Alkaline phosphatases, Deaminase, Glycohydrolase
Introduction
Increasing evidences have indicated that NAD and NADH play critical roles not only in energy metabolism, but also in several cellular functions including calcium homeostasis, gene expression, ageing, immunological mechanisms and cell death (Leonarda 2008). Nicotinamide adenine dinucleotide (NAD) and its reduced form NADH are essential cofactors for many redox biocatalysts. Because these cofactors are consumed in stoichiometric amounts, whole-cell biocatalysts have been routinely employed to reduce the costs. To further improve the efficacy of redox biocatalysts, it is essential to maintain the stability of nicotinamide cofactors, for which it is necessary to block degradation pathways for NAD (H). While the biosynthesis of NAD (H) has been well studied, it is less understood how NAD (H) are degraded (Wang et al. 2014).
Among studies on NAD degradation by non-pathogenic microorganisms, filamentous fungi are found to be particularly interesting because of their easy cultivation, high production of extracellular enzymes with large industrial potential. Besides, fungal enzymes are more resistant to harsh climatic conditions than other sources. These enzymes are applied in the industrialization of chemical and biomedical products (Guimarães et al. 2006). As newly discovered eukaryotic NAD precursors, NR and NaR have the potential to be important mammalian nutritional supplements and/or drugs (Ma et al. 2007; Bogan and Brenner 2008). Alkaline phosphatases involved in NAD degradation which cleave NAD at the phosphate linkage to produce ADP and nicotinamide riboside have been characterized earlier in our laboratory from A. niger (Elzainy and Ali 2000), A. terreus (Elzainy and Ali 2003, 2005). Subsequently, alkaline phosphatase splits ADP to AMP plus free inorganic phosphate. AMP was further cleaved by the same enzyme to produce adenosine plus Pi. Non-specific degradation of NAD to nicotinamide riboside was detected in Escherichia coli (Wang et al. 2014). The applications of alkaline phosphatase isolated from different sources are reviewed with special reference to the macromolecular structure and action mechanism of the enzyme in the reactions of phosphomonoester hydrolysis. The practicality of alkaline phosphatase as a helpful tool in conducting enzyme-linked immunoassays is demonstrated (Zueva et al. 1993).
N-glycosidic linkage of the released nicotinamide riboside was hydrolytically cleaved by glycohydrolase from the same organism producing nicotinamide (Nm) and free ribose. A glycohydrolase is highly specific for NMN. NAD, NADP, nicotinic acid-adenine dinucleotide, nicotinamide riboside catalyses (Imai 1979). On the other hand, studies on NAD+ biosynthesis originate from the involvement of NAD+ in skin cancer prevention mechanisms that include enhancing DNA repair and preventing photo-immune suppression. In addition, NAD+ serves as a cofactor for energy generation used in cornified epidermal barrier formation and release of agents with protective effects on skin (Leonarda 2008).
Adenosine aminohydrolase (ADA) participates in the purine metabolism, whereas it degrades either adenosine or 2′-deoxyadenosine producing inosine or 2′-deoxyinosine, respectively (Kocic et al. 1991). The physiological function of ADA is critical in controlling the effects of these metabolites on immunological, neurological and vascular systems. ADA is also involved in the development of T. lymphocytes band as it is evident from the fact that ADA deficient animals suffer from B and T lymphopaenia (Ray and Sharma 2002; Ashok et al. 2008). Serum ADA levels increase in pancreatic disorders, especially in pancreatic cancer; therefore it may be used as a serum marker for the diagnosis of pancreatic cancer (Bi et al. 2007). The evidence of high ADA activity during rapid and stimulated growth of normal tissues is of importance in making a fully functional purine salvage pathway possible (Seiler 2004). Adenosine deaminase of Aspergillus oryzae (Ali et al. 2012, 2014) has been characterized in our laboratory. Both the adenosine and by-products of the NAD-consuming enzymes such as NMN and Nm can be recycled back to NAD (Sorcil et al. 2010; Gazzaniga et al. 2009). Therefore, the present research focused on studying in detail the degradation of NAD+ by Penicillium brevicompactum NRC 829 extracts. Besides, NAD aminohydrolase was purified to homogeneity and some of kinetic properties were characterized.
Materials and methods
Chemicals
Adenosine, adenine, inosine, cytosine, guanine, cytidine, adenosine 5′-monophosphate (AMP), guanosine 5′-monophosphate (GMP), NAD and cytidine 5′-monophosphate (CMP) were purchased from Sigma Chemical Company. Nicotinamide and nicotinic acid were purchased from Merck. DEAE-Sephadex A-25 and Sephadex G-100 were from Pharmacia Fine Chemical. All other reagents were prepared in Microbial Chemistry Department, National Research Centre.
Microorganism
Penicillium brevicompactum NRC 829 was obtained from culture collection of Microbial Chemistry Department, National Research Centre, Dokki, Egypt.
Medium
The selected fungal strain was grown and maintained on slants of solid modified Czapek Dox’s medium containing g l−1 distilled water: glucose, 30; NaNO3, 2.0; KH2PO4, 1.0; MgSO4·7H2O, 0.5; KCl, 0.5 and agar, 20.
Preparation of Penicillium brevicompactum extracts
The 4-day-old mats, grown on liquid modified potato dextrose medium, containing per litre 300 g of potato and 20 g dextrose at 28 °C, were harvested by filtration, washed thoroughly with distilled water and blotted to dry with absorbent paper. The mats were then ground with cold washed sand in a chilled mortar and extracted with cold distilled water. The slurry obtained was centrifuged at 1, 5229×g for 10 min and the supernatant was used as the crude enzyme preparation.
Enzyme assay
Alkaline phosphatase was determined according to the method described by Heninone and Lahti (1981), summarized as follows: the stock solutions consist of 10 mM (NH4)6Mo7O24.4H2O, 1 M citric acid and 5 N H2SO4, all in distilled water and stable at least for several weeks at 25 °C. Acetone–acid–molybdate (AAM) solution was prepared daily by mixing 1 vol of ammonium molybdate solution with 1 vol of 5 N H2SO4 and 2 vol of acetone. Inorganic phosphate determination: into a test tube containing 0.5 ml of sample, 4 ml of AAM solution was added. The contents were mixed carefully with a vortex mixer and 0.4 ml of 1 M citric acid was pipetted into each tube. After mixing, the yellow colour observed was measured at 390–420 nm. A sample with no added Pi was used as a blank. Adenosine deaminase activity was determined as ammonia according to the method described by Vogel (1961). Briefly, 0.5 ml of purified enzyme solution was mixed with 0.5 ml of Tris–acetate buffer (0.2 M, pH 5.0) containing 5 µM NAD. The mixture was incubated at 40 °C for 30 min and the reaction was terminated by the addition of 0.5 ml of Nessler’s reagent. The yellow colour formed was determined spectrophotometrically by measuring its absorbance at 450 nm. Nicotinamide riboside glycohydrolase was determined by the method described by Shin et al. (1999). Unit of enzyme activity was defined as the amount of enzyme required to produce 1 µM of product, i.e. NH3 or Pi or ribose, per min under the standard assay conditions.
Protein determination
Protein concentration was determined according to Lowry et al. (1951), using bovine serum albumin (BSA) as a standard. The protein content of the purified enzyme fractions was determined by the UV absorbance according to the method of Schleif and Wensink (1981).
Thin layer chromatography (TLC) analysis
Enzymatic reaction products were assayed by TLC Silica gel 60254 (Aluminium sheet) (20 × 20 cm) according to a procedure presented by Kemmer et al. (2001) and Lee et al. (2000). Reactions were done at 40 °C for 2 h in 80 µl of Tris–HCl (pH 7.5) that contained 5 µM NAD and 0.4 mg ml−1 enzyme of P. brevicompactum. Reactions were terminated by the addition of 5 µl of 2 M HCl and centrifuged at 12,000×g for 10 min. Samples were spotted onto TLC sheet, developed in a, n-butanol:acetone:acetic acid (glacial):ammonia (5 %):water (45:15:10:10:20) mixture (Smith and Seakins 1976) and visualized under UV at 254 nm.
Separation and partial purification of NAD degrading enzymes
Acetone fractionation
Cold acetone (−20 °C) was added to the crude extract at various concentrations of 0–30, 30–60, 60–90 %, respectively. The suspension formed was centrifuged at 10,000 rpm for 15 min at 4 °C. The precipitate formed was collected and dissolved in a minimal volume of Tris–acetate buffer, pH 7.0 (0.02 M).
Dialysis
The sample obtained after acetone fractionation was dialysed for 3 h against cold distilled water at 7 °C using a dialysis bag (Size 3, 20/32–15.9 mm, Medicell International Ltd).
DEAE-Sephadex A-25 chromatography
The dialysed partially purified enzymes were loaded onto a DEAE-Sephadex A-25 column (1.0 × 45 cm) that was pre-equilibrated with the same buffer (0.1 M). Elution was carried at room temperature by batch-wise additions of 50 ml portions of increasing molarities (0.0–0.5 M) of sodium chloride solutions in 0.1 M Tris–acetate buffer pH 7, at flow rate 20 ml h−1. Fractions of 5.0 ml were collected and analysed for protein and the enzyme activities were determined as described previously. Fractions with high enzyme activities were pooled together, dialysed against the same buffer and finally concentrated by lyophilization (−50 °C) for further analysis.
Sephadex G-100 gel filtration
The lyophilized concentrated solution was further loaded onto Sephadex G-100 column (46 × 2.0 cm), which was equilibrated in 0.1 M Tris–acetate buffer pH 7.0. The enzyme was eluted from the column using the same buffer at a flow rate of 30 ml h−1, at room temperature (25 °C). The fractions were analysed for protein, Pi, NH3 and ribose determination.
Specific activity was expressed as µmol NH3 or Pi or ribose liberated from substrate (NAD) per mg protein per min. Appropriate control reaction mixtures, i.e. the enzyme source or the substrate was used as blanks throughout the work. All the experiments cited in this work were conducted in triplicate and the mean values were reported and all the results recorded were reproducible.
Molecular mass determination by SDS-PAGE
SDS-gel electrophoresis technique was used to detect the purity of enzyme and to determine molecular mass of the purified enzyme from P. brevicompactum NRC 829 according to the method described by Laemmli (1970), by using the following proteins which were used as molecular weight standards (Fermentas) (spectra TM multicolor broad range protein ladder): 250, 150, 100, 70, 50, 40, 30, 20 and 10 kDa.
Extent of NAD degradation by P. brevicompactum extracts
The experiment aimed to determine the extent of NAD degradation when P. brevicompactum extracts were incubated with NAD as a substrate, at optimum pH and temperature. The release Pi, NH3 and ribose were colourimetrically analysed at different time intervals over a period of 3 h.
Determination of optimal pH, temperature and stability
The optimal pH of the purified enzymes was determined by performing the enzyme assays in the appropriate buffers: KCl–HCl (pH 1.0–2.0); Tris–acetate (pH 3–6.0); Tris–HCl (pH 6.0–9.0) and carbonate–bicarbonate (pH 9.0–10.0). Temperatures of the enzyme were determined by performing the enzyme assays within the temperature range of 20–80 °C. The thermal stability of the purified enzymes was examined by measuring the residual activity after incubating the enzyme at each desired temperature for 30 min.
Influence of different compounds on the purified enzyme activity
The purified enzymes were pre-incubated with different compounds (10 mM) at 40 °C for 30 min in Tris–acetate buffer (50 mM, pH 6.0). The metal ions and some modulators were Fe2+, Fe3+, CO2+, Ca2+, Mg2+, Zn2+ and Mn2+. The enzyme activities was determined as described above using NAD as substrate. The enzyme activity without any agent was taken as 100 %.
Substrate specificity
Substrate specificity was investigated by replacing NAD in the assay mixture with an equal concentration of the representative phosphorylated, aminated riboside compounds.
Kinetics of P. brevicompactum NAD aminohydrolase
Kinetic parameters of P. brevicompactum aminohydrolase against NAD were determined in triplicates at 40◦C for 30 min. Various concentrations of NAD+ (1.5–20 μM) in 50 mM Tris-acetate buffer (pH 6.0) were used in the reaction mixture. Released ammonia was assayed discontinuously as previously described.
Results and discussion
Extent of NAD degradation
Penicillium brevicompactum NRC 829 extracts were incubated with NAD at different pH values. The results observed indicated that Pi and NH3 were optimally liberated from NAD at pH 8 and pH 6.0, respectively, while reducing sugar was optimally detected at pH 5.0. In addition, it was also found that the release of phosphate in the reaction adjusted at pH 8 was faster than the reactions at pH 6 or pH 5 (Fig. 1). Complete phosphate hydrolysis could be detected in the reaction mixtures after 3 h of incubation. The observed properties of phosphatases isolated from P. brevicompactum were similar to fungal phosphatases produced by A. niger (Elzainy and Ali 2000), A. terreus (Elzainy and Ali 2003) and A. oryzae (Ali et al. 2012). These enzymes are similar in being orthophosphate- non repressible enzymes and being not true phosphomonoesterases. This last property was based on their abilities to catalyse the hydrolysis of pyrophosphate linkage of ADP and the internal ester linkage between NR and ADP, in addition to the true phosphate ester linkages of NAD, ADP and AMP. At the end of the incubation period, almost all the phosphate links of the dinucleotide molecule were cleaved, while about 40 % either of the amido- or the amino-linkages were hydrolysed. However, this percentage may represent the sum of percentages of hydrolysed amido- and amino-bonds. Also the reducing power of 32 % may be formed as a result of the cleavage of N-glycosidic bond in adenosine or nicotinamide riboside.
Fig. 1.

Optimal pH of NAD degrading enzymes. The reaction mixture contained the following: substrate, NAD, 5 µmol; buffers of various pHs: 80 µmol (pH 3–6) citrate-buffer, (pH 6–9) Tris–acetate, (pH 9–10) carbonate–bicarbonate buffer; temp., 50 °C; protein extracts, 2.5 mg
NAD degradation pathway by the extracts of P. brevicompactum was summarized as shown in the following diagram.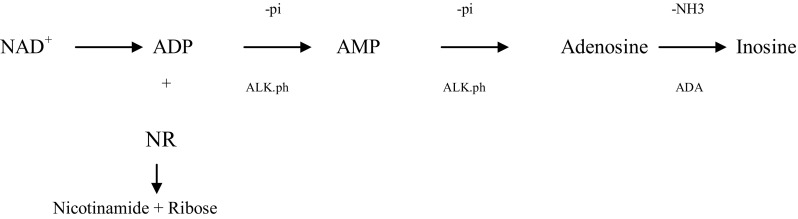
Identification of products and intermediates of NAD degradation
Analysis of the reaction mixture, in which inorganic phosphate, ribose and ammonia were formed, showed the extent of NAD degradation by P. brevicompactum enzymes. TLC clearly indicated that NAD was consumed, but there was apparently no accumulation of either ADP or AMP. Instead, the presence of NR, Nm, adenosine and inosine was evident for those lanes. These data showed that the end product NAD dephosphorylation was nicotinamide riboside (NR) and adenosine. Subsequently, adenosine was partially deaminated to inosine, while nicotinamide riboside was cleaved to nicotinamide and ribose. Adenosine, inosine, nicotinamide riboside and nicotinamide were located on TLC sheet as the final products (Fig. 2). These results are in agreement with that reported for NAD degradation in the extracts of A. niger (Elzainy and Ali 2000), A. terreus (Elzainy and Ali 2003) and A. oryzae (Ali et al. 2012); however, the present research differs regarding the formation of nicotinamide which formed due to the existence of NAD glycohydrolase, while, in case of other fungal strains, nicotinamide riboside remained without further degradation. The detection of inosine and adenosine in the chromatographic analysis demonstrated the partial deamination of adenosine. This result is in congruent with the result previously investigated in A. oryzae extracts (Ali et al. 2012).
Fig. 2.

TLC analysis of products from NAD degradation activity of P. brevicompactum aminohydrolase. Right lane was the sample of reaction mixture containing 5 µM NAD being treated at 50 °C for 2 h by aminohydrolase at an initial concentration of 20 µg. Concentrations of chemical standards were 2 mM. Abbreviations of authentic: NAD, nicotinamide adenine dinucleotide; AMP, adenosine 5′-monophosphate; ADP, adenosine 5′-diphosphate; Ado, adenosine; Ade, adenine; Ino, inosine; Nm, nicotinamide; NR, nicotinamide riboside
Separation of NAD deaminating and NAD glycohydrolase activities from NAD dephosphorylating activity
The data presented in Table 1 showed the purification summary of the three enzymes responsible for NAD degradation. During all purification steps, the activity of alkaline phosphatase was detected in protein fractions with two peaks; this result indicates the existence of two molecular forms of alkaline isozymes. In addition, the activities of NAD glycohydrolase and NAD deaminating were recorded in one peak. The separation of NAD glycohydrolase and NAD deaminase was illustrated by Sephadex G-100 column chromatography described under ‘‘Materials and Methods’’ section. The enzyme was purified to homogeneity. SDS-PAGE showed that it had a molecular mass of 91 kDa (Fig. 3). In this respect, the molecular masses of 94 and 85 kDa were reported for the enzymes produced by A. oryzae and A. fumigatus 4 as investigated by Ali et al. (2014) and Yoshimune et al. (2005), respectively. On the other hand, the purified enzyme from A. oryzae was found to exhibit a smaller molecular mass of 14.5 kDa as reported by Rosinova et al. (1978).
Table 1.
Purification of the NAD degrading enzymes from P. brevicompactum NRC 829
| Purification steps | Total activity (units) | Protein (mg) | Sp. activity | Recovery (%) | Purification fold |
|---|---|---|---|---|---|
| Alkaline phosphatase | |||||
| Crude extracts | 600 | 390 | 1.5 | 100 | 1 |
| Acetone fraction | 562 | 150 | 3.7 | 93 | 2.5 |
| DEAE-Sephadex A25 | |||||
| ALK1 | 230 | 3.0 | 60.6 | 38 | 51.1 |
| ALK2 | 200 | 2.8 | 71.4 | 33 | 47.6 |
| NAD aminohydrolase | |||||
| Crude extracts | 450 | 390 | 1.1 | 100 | 1 |
| Acetone fraction | 400 | 150 | 2.6 | 88 | 2.2 |
| DEAE-Sephadex A25 | 150 | 5 | 30 | 30 | 27.3 |
| Sephadex G-100 | 100 | 2 | 50 | 22 | 41 |
| NAD glycohydrolase | |||||
| Crude extracts | 290 | 390 | 0.75 | 100 | 1 |
| Acetone-fraction | 245 | 150 | 1.6 | 84 | 2.1 |
| DEAE-Sephadex A 25 | 130 | 5 | 27 | 44 | 36 |
| Sephadex G-100 | 76 | 1.9 | 40 | 26 | 50 |
Fig. 3.

Electrophoretic analysis of Penicillium brevicompactum NRC 829 NAD aminohydrolase. From left to right: lane 1 molecular mass markers, lane 2 fractional precipitation by chilled acetone, lane 3 partial purified NAD aminohydrolase on DEAE-Sephadex A-25, lane 4 purified NAD aminohydrolase on Sephadex G-100
Characterization of purified NAD deaminase
Optimal pH and temperature
The effect of pH on deaminase activity was examined over a wide pH range of 3.0–10.0. The results illustrated in Fig. 4 demonstrated that the deaminase displayed optimal activity at pH 6.0. The enzyme was stable when pre incubated in the pH range of 6.0 to 8.0, while 50 and 55 % of its original activity were observed at pHs 4.0 and 9.0, respectively (Fig. 5). This is similar to the optimal pH (5–7) of adenosine-phosphate deaminase of A. fumigatus (Yoshimune et al. 2005), NAD deaminase produced by A. oryzae (Ali et al. 2014). At pH 8.0, the deaminase loses 40 % of its activity as A. oryzae NAD deaminase, which had its optimum activity at pH 5 and abroad pH stability. The optimal temperature for the deaminase activity was in the range of 50–70 °C (Fig. 6). This is about 10–20 °C higher than optimum temperature of the adenosine-phosphate deaminase of A. fumigatus (Yoshimune et al. 2005) and NAD deaminase A. oryzae (Ali et al. 2014), respectively. The enzyme was found to thermostable up to 70 °C; the results also showed that 80 and 88 % of its original activity still remained at 50 and 60 °C, respectively, after 30 min of incubation (Data not shown). The enzyme purified from P. brevicompactum was considerably more thermostable with higher specific activity than deaminase of A. fumigatus and A. oryzae.
Fig. 4.

pH dependence of the purified deaminase activity
Fig. 5.

pH stability of the NAD aminohydrolase. The enzyme was stored in buffers of various pHs (100 mM) at 50 °C for 30 min, and the residual activities were measured
Fig. 6.

Effect of temperature on purified deaminase
Effect of various agents on the purified enzyme activity
In the present study, results in Table 2 clearly showed that the enzyme was enhanced about 30 % by Zn2+ (1 mM), while no significant enhancement of the enzyme activity was reported by Ca2+, Co2+, Fe2+, Mg2+, Mn2+ and Fe3+. In addition, the present data revealed that the enzyme activity was not inhibited by iodoacetate, used at 10 mM final concentration, which clearly indicates the absence of evidence for the involvement of an SH group(s) in the catalytic action of enzyme. The effect of EDTA, known as a metal chelating agent, on enzyme activity was tested to find out whether this enzyme is a metalloenzyme or not. It was found that the addition of EDTA (10 mM) to the reaction mixture did not inhibit enzyme activity indicating that NAD deaminase is not a metalloenzyme. This property closely resembles that of a nonspecific NAD aminohydrolase produced from A. oryzae (Ali et al. 2014); whereas the enzyme was slightly activated by addition of Na+ and K+, while inhibited by addition of Mn2+, Ag+, Hg2+, and Cu2+. In this concern, Yoshimune et al. (2005) reported that the adenosine-phosphate deaminase produced by A. fumigatus was inhibited by 38 % in the presence of Cu2+ (1 mM), while no significant enhancement of the enzyme activity was reported by Ba2+, Ca2+, Co2+, Fe2+, Mg2+, Mn2+, Ni2+, Sn2+, Zn2+ and Fe3+. Yoshino and Murakami (1980) reported that the baker’s yeast AMP deaminase was activated in the presence of various divalent cations.
Table 2.
Effect of metal ions and various chemical reagents on the enzyme activity
| Reagents (1 mM) | Relative activity (%) |
|---|---|
| None | 100 |
| Zn2+ | 132 |
| Ca2+ | 92 |
| Co2+ | 94 |
| Fe2+ | 95 |
| Mg2+ | 90 |
| Mn2 | 91 |
| Fe3+ | 89 |
| EDTA | 92 |
| Iodoacetate | 93 |
| Mercaptoethanol | 96 |
Substrate specificity
The substrate specificity of purified NAD deaminase was examined and the results were tabulated in Table 3, from which it was found that the enzyme was active toward adenine and various adenine derivatives (adenosine, AMP, ADP, NAD, ATP and NAD) while no activity was detected towards nicotinamide or nicotinamide riboside as a nonspecific NAD aminohydrolase from A. oryzae (Ali et al. 2014). In contrast, A. terreus NAD deamidase catalysed amide group of the intact NAD molecule and could also cleave the amide group from NMN, NR, Nm, glutamine and asparagine (Elzainy and Ali 2005). The microbial AMP-deaminating activity was catalysed by adenosine-phosphate deaminase (EC 3.5.4.17) and adenylic acid deaminase (EC 3.5.4.6) (Yoshimune et al. 2005). The enzyme from Takadiastase (A. oryzae) was identified as a nonspecific adenosine deaminase (EC 3.5.4.4, adenosine aminohydrolase) by Minato et al. (1965); this enzyme deaminated various adenosine derivatives (3′-AMP, 5′-AMP, and NAD), but not adenine or NADP. The enzymes from A. oryzae and A. fumigatus 4 were different in substrate specificity, as described above. The enzyme from S. aureofaciens catalyses the deamination of adenosine, 5′-AMP, ADP and ATP, but not that of adenine (Rosinova et al. 1978). The enzyme from Streptomyces sp. is active towards adenosine, 2′-deoxyadenosine, 3′-AMP, 5′-AMP and cAMP (Jun et al. 1991), but the enzyme activities towards 3′-AMP, 5′-AMP and cAMP were 4– 8 % of those of adenosine, indicating that Streptomyces sp. enzyme is classified as adenosine deaminase. As described above, the substrate specificities of microbial adenosine-phosphate deaminases are different depending on the origin of the enzymes.
Table 3.
Kinetic parameters of P. brevicompactum aminohydrolase
| Substrate | K m (µM) | K cat (s−1) | K cat/K m (M−1 s−1) |
|---|---|---|---|
| NAD | 5.2 | 10.08 | 1.93 |
| AMP | 8.33 | 8.12 | 0.98 |
| ADP | 6.25 | 0.93 | 0.207 |
| Adenosine | 4.5 | 11.2 | 1.79 |
| Nicotinamide riboside | ND | ND | ND |
ND not detected
Kinetic parameters of NAD aminohydrolase
Kinetic parameters of P. brevicompactum aminohydrolase catalysis were determined using NAD, ADP, AMP and adenosine, as substrates (Table 3). While K m values were close for all of these substrates, K cat values for AMP and ADP were orders of magnitude higher than those for NAD and adenosine, indicating that aminohydrolase had substantially higher catalytic efficiency for ammonia hydrolysis. It should be pointed out that the P. brevicompactum had the highest NAD and adenosine aminohydrolase activity compared to the enzyme produced by A. oryzae (Ali et al. 2014). The K m values for NAD (5.2 µM) and adenosine (4.5 µM) of P. brevicompactum were substantially lower; indicating aminohydrolase has much stronger binding affinity towards NAD and adenosine compared to other enzymes for NAD degradation.
Conclusion
The present work demonstrated the occurrence of two alkaline phosphatases, aminohydrolase and glycohydrolase in P. brevicompactum NRC 829. These enzymes are involved in NAD degradation. Purification and separation showed high aminohydrolase activity with the catalytic efficiencies for hydrolysis of NAD and adenosine at 1.9 and 1.8 μM−1 s−1, respectively. These results significantly enriched our understanding on NAD metabolism and should facilitate many applications including designing redox biocatalysts.
Acknowledgments
This work was supported by National Research Centre of Egypt.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interest.
References
- Ali TH, Haroun BM, Tantawy AE. Optimization of culture conditions for the production of NAD-degrading enzymes by Aspergillus oryzae NRRL 447. Adv Food Sci. 2012;34(4):195–201. [Google Scholar]
- Ali TH, Ali NH, Haroun BM, Tantawy AE. Purification and properties of Aspergillus oryzae NRRL 447 aminohydrolase. World J Microbiol Biotechnol. 2014;30:819–825. doi: 10.1007/s11274-013-1483-1. [DOI] [PubMed] [Google Scholar]
- Ashok KJ, Pinto GJ, Kavitha AK, Palathra MJ. The diagnostic and prognostic value of serum adenosine deaminase levels in head and neck cancer. J Clin Diagn Res. 2008;3:833–837. [Google Scholar]
- Bi M, Koklu S, Meric Y, Basar O, Yilmaz G, Yuksel O, Yildirim E, Ozturk Z. Serum adenosine deaminase levels in pancreatic diseases. Pancreat. 2007;7:5–6. doi: 10.1159/000108970. [DOI] [PubMed] [Google Scholar]
- Bogan KL, Brenner C. Nicotinic acid, nicotinamide and nicotinamide riboside: a molecular evaluation of NAD precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- Elzainy TA, Ali TH. Pathways of NADP and NAD degradation by Aspergillus niger extracts. Ann Microbiol. 2000;50:65–75. [Google Scholar]
- Elzainy TA, Ali TH. Nature and mode of action of NAD and ATP dephosphorylating enzyme from Aspergillus terreus DSM 826. Antonie van Leeuwenhoek J Microbiol. 2003;84:255–260. doi: 10.1023/A:1026028311575. [DOI] [PubMed] [Google Scholar]
- Elzainy TA, Ali TH. NAD deamidation, a non-previously recognized reaction, by an enzyme from Aspergillus terreus DSM 826. Antonie van Leeuwenhoek J Microbiol. 2005;87:119–129. doi: 10.1007/s10482-004-0136-7. [DOI] [PubMed] [Google Scholar]
- Gazzaniga F, Stebbins R, Chang SZ, McPeek MA, Brenner C. Microbial NAD metabolism. Microbiol Mol Biol Rev. 2009;73(3):529. doi: 10.1128/MMBR.00042-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães LHS, Peixoto-Nogueira SC, Michele Michelin M, Rizzatti ACS, Sandrim VC, Zanoelo FF, Aquino ACMM, Junior AB, Maria de Lourdes TM, Polizeli MLTM. Screening of filamentous fungi for production of enzymes of biotechnological interest. Braz J Microbiol. 2006;37:474–480. doi: 10.1590/S1517-83822006000400014. [DOI] [Google Scholar]
- Heninone JK, Lahti RJ. A new and convenient colorimetric determination of inorganic orthophosphate and its application to the assay of inorganic pyrophosphatase. J Biochem. 1981;113:313–317. doi: 10.1016/0003-2697(81)90082-8. [DOI] [PubMed] [Google Scholar]
- Imai T. Isolation and properties of a glycohydrolase specific for nicotinamide mononucleotide from Azotobacter vinelandii. J Biochem. 1979;85(4):887–899. doi: 10.1093/oxfordjournals.jbchem.a132420. [DOI] [PubMed] [Google Scholar]
- Jun HK, Kim TS, Sakai T. Purification and characterization of extracellular adenosine deaminase from a Streptomyces sp. J Ferment Bioeng. 1991;71:6–11. doi: 10.1016/0922-338X(91)90295-R. [DOI] [Google Scholar]
- Kemmer G, Reilly T, Schmidt-Brauns J, Zlotnik GW, Green BA, Fiske MJ, Herbert M, Kraiβ A, Schlör S, Smith A, Reidl J. NadN and e (P4) are essential for the utilization of NAD and nicotinamide mononucleotide but not nicotinamide riboside in Haemophilus influenzae. J Bacteriol. 2001;183:3974–3981. doi: 10.1128/JB.183.13.3974-3981.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocic G, Stanojevic G, Nagorni A, Brankvic B, Pavlovic D, Jevtovic T. Diagnostic importance of adenosine deaminase activity for progression and invasion of human colon tumors. Med Biol. 1991;10(2):76–78. [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee KS, Song SB, Kim KE, Kim YH, Kim SK, Kho BH, Ko DK, Choi YK, Lee K, Kim CK, Kim YC, Lim JY, Kim Y, Min KH, Wanner BL. Cloning and characterization of the UDP-sugar hydrolase gene (ushA) of Enterobacter aerogenes IFO 12010. Biochem Biophys Res Commun. 2000;269:526–531. doi: 10.1006/bbrc.2000.2328. [DOI] [PubMed] [Google Scholar]
- Leonarda S (2008) NAD Metabolism: aspects of biosynthesis and degradation. PhD thesis, School of Advanced Studies—Doctorate Course in Biology
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Ma B, Pan SJ, Zupancic ML, Cormack BP. Assimilation of NAD precursors in Candida glabrata. Mol Microbiol. 2007;66:14–25. doi: 10.1111/j.1365-2958.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- Minato S, Tagawa T, Nakanishi K. Studies on nonspecific adenosine deaminase from Takadiastase. I. Purification and properties. J Biochem. 1965;58:519–525. doi: 10.1093/oxfordjournals.jbchem.a128236. [DOI] [PubMed] [Google Scholar]
- Ray I, Sharma R. Dietary regulation of adenosine deaminase activity in stomach, small intestine and spleen of mice. Indian J Biochem Biophys. 2002;39:419–421. [PubMed] [Google Scholar]
- Rosinova M, Zeliinkova E, Zelinka J. Adenosine aminohydrolase from Streptomyces aureofaciens. Collect Czechoslov Chem Commun. 1978;43:2324–2329. doi: 10.1135/cccc19782324. [DOI] [Google Scholar]
- Schleif RF, Wensink PC. Practical methods in molecular biology. New York: Springer; 1981. p. 74. [Google Scholar]
- Seiler N. Catabolism of polyamines. Amino Acids. 2004;26:217–233. doi: 10.1007/s00726-004-0070-z. [DOI] [PubMed] [Google Scholar]
- Shin PP, Park YC, Chung CS. Biological control of Fusarium by chitinolytic bacteria. Phytopathology. 1999;89:92–99. doi: 10.1094/PHYTO.1999.89.1.92. [DOI] [PubMed] [Google Scholar]
- Smith I, Seakins JWT. Chromatographic and electrophoretic techniques. London: William Heinemann Medical Books Ltd; 1976. pp. 158–159. [Google Scholar]
- Sorcil LK, Rodionov DA, Osterman AL (2010) Comprehensive natural products: II. Chemistry and biology (Mander L, Lui H-W, editors). Elsevier, Oxford, pp 213–251
- Vogel AI. Quantitative inorganic analysis, theory and practice. 3. London: Longmann, Green and Co.; 1961. p. 783. [Google Scholar]
- Wang L, Zhou YJ, Debin JI, Lin X, Liu Y, Zhang Y, Liu W, Zhao ZK. Identification of UshA as a major enzyme for NAD degradation in Escherichia coli. Enzyme Microb Technol. 2014;58–59:75–79. doi: 10.1016/j.enzmictec.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Yoshimune K, Kugimiya K, Moriguchi M. Purification and characterization of a flavour-enhancing enzyme, thermostable adenosine- phosphate deaminase, from thermophilic Aspergillus fumigatus no. 4. Ann Microbiol. 2005;55(4):267–272. [Google Scholar]
- Yoshino M, Murakami K. Role of cations in the regulation of baker’s yeast AMP deaminase. Biochim Biophys Acta. 1980;616:82–88. doi: 10.1016/0005-2744(80)90265-X. [DOI] [PubMed] [Google Scholar]
- Zueva NN, Dalev PG, Lazarova DL. Properties, production, and practical uses of alkaline phosphatase. Biokhimiia. 1993;58(7):1009–1023. [PubMed] [Google Scholar]