

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare, frequently under‐recognized condition associated with multi‐organ failure and very high mortality. A 44‐year‐old woman was admitted with a 4‐day history of fever, headache, delirium, and dyspnea. She progressed rapidly to type 1 respiratory failure and required intubation and mechanical ventilation. Laboratory tests showed pancytopenia, abnormal liver enzyme levels, elevated triglyceride level, and elevated ferritin level. Bone marrow biopsy showed features of HLH. Computed tomography scan showed bilateral consolidation. Bronchoalveolar lavage was positive for cytomegalovirus. She was treated with ganciclovir, methylprednisolone, broad spectrum antibiotics, and cytomegalovirus hyperimmunoglobulin without clinical response. Given the poor prognosis and reports of success in pediatric HLH, anakinra 100 µg subcutaneously daily was commenced. There was rapid defervescence, resolution of delirium, and improvement in gas exchange, leading to complete recovery. This case illustrates successful treatment of HLH associated with cytomegalovirus pneumonitis with the interleukin 1 inhibitor anakinra.
Keywords: Hemophagocytic lymphohistiocytosis, inflammation, pulmonary, virology
Introduction
Hemophagocytic syndrome, also known as hemophagocytic lymphohistiocytosis (HLH), is a rare and life‐threatening disease that is frequently under‐recognized in adults and associated with very high mortality despite treatment [1]. The syndrome is characterized by fever, altered mental status, hepatosplenomegaly, cytopenia, liver dysfunction, and hyperferritinemia. Hemophagocytosis is caused by dysregulation of natural killer cells and cytotoxic T cells, which may cause excessive activation of macrophages that ingest multiple hematopoietic elements in the bone marrow.
HLH may be primary (genetic etiology) or secondary to malignancies, autoimmune disease, or infections (most commonly viral). There have been multiple previous reports of cytomegalovirus (CMV) associated HLH developing in patients with ulcerative colitis receiving azathioprine [2]. We report the successful management of this syndrome with ganciclovir, corticosteroids and the new interleukin 1 inhibitor anakinra.
Case Report
A 44‐year‐old woman was admitted to a regional hospital because of a 4‐day history of fever, headache, delirium, and dyspnea. She progressed rapidly to type 1 respiratory failure and required intubation and mechanical ventilation. Past medical history was notable for ulcerative colitis that was well‐controlled with azathioprine (150 mg daily) and mesalazine (2.4 g, twice daily). Physical examination showed hepatomegaly. Laboratory tests showed pancytopenia (hemoglobin, 84 g/L; white blood cell count, 1.3 × 109/L; platelet count, 71 × 109/L), abnormal liver enzyme levels (alkaline phosphate, 644 U/L; gamma‐glutamyl transferase, 485 U/L; aspartate aminotransferase, 431 U/L; alanine aminotransferase, 231 U/L), elevated fasting triglyceride level (6.6 mmol/L), and elevated ferritin level (7000 μg/L). Chest radiography and computed tomography (CT) scan showed bilateral consolidation. Brain CT scan and lumbar puncture were normal. CMV DNA was detected in peripheral blood (titer, 6.8 × 105 copies/mL). CMV DNA was also isolated from the bronchoalveolar lavage, but no other pathogens were isolated. Bone marrow aspirate and biopsy confirmed hemophagocytosis (Fig. 1). CMV DNA was detected in the bone marrow. Treatment was started with intravenous (IV) ganciclovir (5 mg/kg), IV vancomycin, meropenem, oral doxycyline, oseltamivir, and CMV hyperimmunoglobulin on days 0, 7, and 15. The patient's gas exchange and clinical parameters deteriorated, and she was transferred to our center for consideration of veno‐venous extra‐corporeal membrane oxygenation (ECMO) support.
Figure 1.
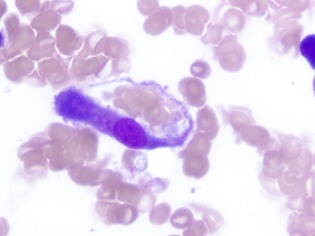
Histology of bone marrow aspirate: macrophage engulfing erythrocytes (May‐Grünwald‐Giemsa × 100).
The patient's clinical condition continued to deteriorate. She had a tracheotomy and was being ventilated with fraction of inspired oxygen (FiO2) 0.8. She was delirious and had ongoing fever and cytopenia. The ganciclovir plasma level was sub‐therapeutic (0.2 mg/L); the dose was increased from 5 to 10 mg/kg twice daily, but there was no clinical improvement (Fig. 2). Given the poor prognosis and reports of success in pediatric HLH, the IL‐1 inhibitor anakinra was commenced (100 µg subcutaneously daily) 17 days after initial presentation. Within 24 h the patient had defervesced and delirium had resolved (Fig. 2). The peripheral blood counts gradually improved. Seventy two hours later gas exchange had improved only marginally with persistent pulmonary infiltrates suggestive of organizing pneumonia. Intravenous methylprednisolone, 500 mg daily for 3 days, was commenced. The patient has extubated 7 days later and was discharged from hospital in excellent condition 18 days after anakinra was commenced.
Figure 2.
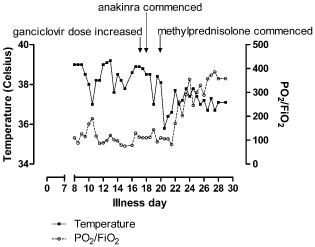
Treatment Response to Anakinra. FIO2, fraction of inspired oxygen; PO2, partial pressure of oxygen; Temp, temperature.
Discussion
HLH should be suspected in patients who present with fever, delirium, cytopenia, and liver dysfunction. Many cases of HLH are not diagnosed initially, likely contributing to poor outcomes. Since many features of HLH are nonspecific and are seen in common conditions such as sepsis and respiratory failure from multiple causes, it is important that respiratory physicians and intensive care specialists include the diagnosis in the list of differentials [1]. The present patient meets the 2004 Histiocyte Society diagnostic criteria: fever, cytopenia, hypertriglyceridemia, hemophagocytosis in bone marrow, and ferritin level > 500 μg/L [3].
Viral infection, which is often respiratory, is a common cause of secondary HLH and CMV‐associated HLH appear to be a particular risk in patients with ulcerative colitis treated with azathioprine [2]. In secondary HLH, it is important to treat the underlying cause [1]. The present case emphasizes the importance of therapeutic drug level monitoring with ganciclovir therapy.
HLH is characterized by markedly low natural killer and T‐cell‐mediated cytotoxicity. Deficient cytotoxicity may cause continuous immune activation with concomitant phagocytosis of bone marrow cells due to proliferation and activation of macrophages [1]. The cytokine storm that occurs during this process may cause a severe inflammatory reaction. This cytokine surge, not the original infection, causes the systemic manifestations of secondary HLH and eventually causes end‐organ damage and mortality. More than any other cytokine family, the IL‐1 family of ligands and receptors is associated with the inflammatory response. Recent studies have shown elevated IL‐1 in patients who have HLH [4]. Although the current treatment of choice is dexamethasone, etoposide, and cyclosporine [3], the use of these agents is often associated with opportunistic infections and treatment failure with secondary HLH [5]. Several reports of IL‐1 blockade by anakinra in pediatric HLH leading to rapid recovery [4, 5], alongside the poor prognosis in adult HLH using the HLH 2004, and recent knowledge of pathogenesis, led us to recommend anakinra for this adult with CMV related HLH.
In conclusion, physicians should be familiar with the clinical manifestations of HLH because it is often under‐recognized. IL‐1 blockade with anakinra may represent a lifesaving approach to the management of this often fatal condition.
Disclosure Statements
No conflict of interest declared.
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
Acknowledgments
Nina Raju: Haematologist, The Prince Charles Hospital, Queensland, Australia.
References
- 1. Wijsman CA, Roeters van Lennep JE, von dem Borne PA, et al. 2009. A diagnostic difficulty: two cases of haemophagocytic syndrome in adults. Neth. J. Med. 67(1):29–31. [PubMed] [Google Scholar]
- 2. Kullmann T, Kárász T, Gartner B, et al. 2013. Acute cytomegalovirus infection‐associated hemophagocytic syndrome in a patient treated with azathioprine [in Hungarian]. Orv. Hetil. 154(49):1959–1961. [DOI] [PubMed] [Google Scholar]
- 3. Henter JI, Horne A, Aricó M, et al. 2007. HLH‐2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 48(2):124–131. [DOI] [PubMed] [Google Scholar]
- 4. Rajasekaran S, Kruse K, Kovey K, et al. 2014. Therapeutic role of anakinra, an interleukin‐1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children. Pediatr. Crit. Care Med. 15(5):401–408. [DOI] [PubMed] [Google Scholar]
- 5. Miettunen PM, Narendran A, Jayanthan A, et al. 2011. Successful treatment of severe paediatric rheumatic disease‐associated macrophage activation syndrome with interleukin‐1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford) 50(2):417–419. [DOI] [PubMed] [Google Scholar]