

Abstract
Background
Inflammatory factors and β-cell dysfunction due to high-fat diets aggravate chronic diseases and their complications. However, omega-3 dietary fats have anti-inflammatory effects, and the involvement of autophagy in the etiology of diabetes has been reported. Therefore, we examined the protective effects of autophagy on diabetes using fat-1 transgenic mice with omega-3 self-synthesis capability.
Methods
Streptozotocin (STZ) administration induced β-cell dysfunction in mice; blood glucose levels and water consumption were subsequently measured. Using hematoxylin and eosin (H&E) and Masson's trichrome staining, we quantitatively assessed STZ-induced changes in the number, mass, and fibrosis of pancreatic islets in fat-1 and control mice. We identified the microtubule-associated protein 1A/1B light chain 3-immunoreactive puncta in β-cells and quantified p62 levels in the pancreas of fat-1 and control mice.
Results
STZ-induced diabetic phenotypes, including hyperglycemia and polydipsia, were attenuated in fat-1 mice. Histological determination using H&E and Masson's trichrome staining revealed the protective effects of the fat-1 expression on cell death and the scarring of pancreatic islets after STZ injection. In the β-cells of control mice, autophagy was abruptly activated after STZ treatment. Basal autophagy levels were elevated in fat-1 mice β-cells, and this persisted after STZ treatment. Together with autophagosome detection, these results revealed that n-3 polyunsaturated fatty acid (PUFA) enrichment might partly prevent the STZ-related pancreatic islet damage by upregulating the basal activity of autophagy and improving autophagic flux disturbance.
Conclusion
Fat-1 transgenic mice with a n-3 PUFA self-synthesis capability exert protective effects against STZ-induced β-cell death by activating autophagy in β-cells.
Keywords: Omega 3 fatty, Beta cell, Fat-1 transgenic mice
INTRODUCTION
Diabetes, a condition characterized by a gradual increase in blood glucose and chronic inflammation, is caused by constant deterioration of pancreatic function that results in microvascular and macrovascular complications as well as organ damage, thus leading to increased morbidity and mortality. Diabetes is caused by the irreversible destruction of β-cells, due to the persistent stress on β-cells caused by insulin resistance [1]. In addition, the incidence of diabetes-related risk factors for cardiovascular diseases, such as insulin resistance, metabolic syndrome, and abdominal obesity, is increasing.
The importance of a diabetic diet is its achievement of weight loss via an exclusion of high-calorie food and saturated fatty acids [2]. A healthy diabetic diet replaces saturated fatty acids with monounsaturated fatty acids or polyunsaturated fatty acids (PUFAs). It has been reported that n-3 PUFAs may reduce the risk of cardiovascular disease in patients with diabetes and reduce the risk of progression from prediabetes to diabetes [3]. n-3 PUFAs and n-6 PUFAs are essential nutrients in mammals, and n-6 PUFAs typically have pro-inflammatory effects, whereas n-3 PUFAs have anti-inflammatory effects [4].
Autophagy, which originates from the Greek words for 'self' (auto) and 'eat' (phagy), describes a process in which the materials inside the cell are removed by the cell itself. Waste from the cytoplasm, denatured or dysfunctional proteins and organelles, are isolated in a double membrane vesicle called an autophagosome. The vesicle then fuses with a lysosome in the same cell and is broken down by enzymes inside the lysosome. The degraded materials are used to create energy required for the survival of cells or to produce new organelles. In other words, autophagy can be understood as a recycling system occurring within the cells. In a recent study, the expression of Atg7, an essential gene required for the occurrence of autophagy, was suppressed in β-cells (Atg7Δβ-cell) using Cre-mediated gene recombination technology, and the corresponding knock-out mice showed an increase in blood glucose levels and a reduction in blood insulin concentrations [5,6].
The role of autophagy in diabetes has been investigated, but it has not yet been fully elucidated. Although some reports have suggested that autophagy may play an important role in the development and prevention of diabetes, no definite conclusions have been reached. Consequently, the present study examined the protective effects of autophagy on diabetes using fat-1 transgenic mice with n-3 PUFA self-synthesis capability [7].
METHODS
Animals
Fat-1 transgenic mice carrying the fat-1 gene of Caenorhabditis elegans were kindly provided by Dr. Jing X. Kang (Department of Medicine, Massachusetts General Hospital and Harvard Medical School, USA) and backcrossed onto C57BL/6 background. WT C57BL/6 mice were purchased from a local animal facility (DBL, Eumseong, Korea). Male C57BL/6 mice between 6 and 10 weeks of age were used in our experiments (n=6/group). The concentrations of n-6 fatty acids in the tissues of the transgenic mice were significantly reduced, indicating that n-6 fatty acids were converted to n-3, causing the n-6 to n-3 ratio to decrease from 20 to 50 to almost 1. All mice were housed individually in cages under a standard 12:12 hours light:dark cycle. Water and food were available ad libitum until mice were transported to the laboratory, approximately 1 hour prior to the experiments. All experiments were performed with the approval of the Animal Care and Use Committee of the Konyang University and were consistent with the ethical guidelines of the National Institutes of Health and the International Association.
STZ administration
Diabetes was induced by streptozotocin (STZ), as described previously. Briefly, STZ (2-deoxy-2-3-[methyl-3-nitrosoureido]-Dglucopyranose, Sigma, St. Louis, MO, USA) was dissolved in 0.1 mol/L sodium citrate buffer (pH 4.5) and injected intraperitoneally at a dose of 45 mg/kg/day within 15 minutes of preparation for 5 consecutive days to produce a β-cell destruction model. Control wild-type (WT) and transgenic mice were injected with citrate buffer as vehicle. Blood glucose levels were measured in the venous blood of nonfasted animals using a glucometer (One Touch Vita, LifeScan, Issy les Moulineaux, France). Mice were evaluated every 2 days at 2:00 PM and were considered diabetic when blood glucose levels exceeded 250 mg/dL, usually 7 to 9 days after the final STZ injection (Fig. 1).
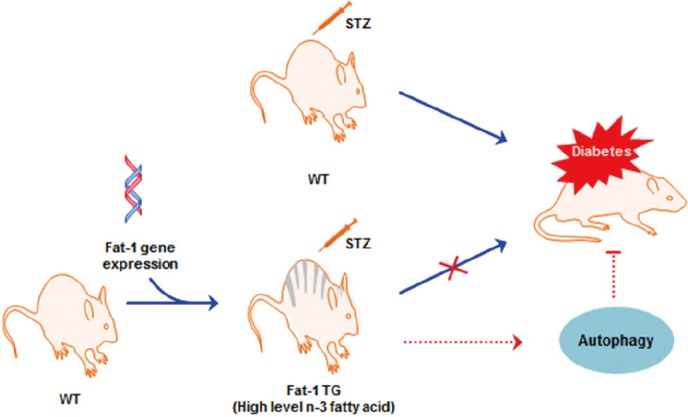
Fig. 1. Schematic Illustration of the hypothesis in this experiments. STZ, streptozotocin; WT, wild-type; TG, transgenic.
Blood glucose measurement
Blood obtained from the tail vein was used for measurements of glucose levels, which were performed using the OneTouch Blood Glucose Monitoring System (LifeScan, Milpitas, CA, USA). Mice were fasted for 4 hours before blood glucose measurement.
Confocal microscopy
Expression of microtubule-associated protein light chain 3 (LC3) was evaluated using confocal microscopy after immunofluorescent staining. After the STZ injection program was discontinued, tissue was isolated, fixed with 10% formalin. And deparaffinized tissue sections were incubated with primary antibody overnight at 4℃. Tissue sections were washed to remove excess primary antibody and incubated with the appropriate fluorescently labeled secondary antibodies for 1 hour at room temperature (RT). Nuclei were stained by incubation with 4',6-diamidino-2-phenylindole for 5 minutes. After mounting, fluorescence images were acquired using confocal laser-scanning microscopy (LSM 700, Zeiss, Oberkochen, Germany).
Histological analysis of the pancreas
Pancreases from WT and fat-1 mice were isolated. The tissue was fixed in 10% buffered formalin and processed for paraffin sectioning. Sections approximately 4-µm thick were stained with hematoxylin and eosin (H&E) and Masson trichrome for evaluation under a light microscope.
Western blot analysis
Protein was collected by lysing the cells in 1 mL of ice-cold PRO-PRE (iNtRON, Seongnam, Korea) buffer. The protein concentration of the supernatant was evaluated using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, South Logan, UT, USA). Aliquots of protein (30 µg/lane) were separated by 10% to 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and then transferred onto polyvinylidene fluoride membranes. The membranes were blocked with blocking buffer containing 5% non-fat dry milk for 2 hours at RT and incubated with rabbit primary antibodies against p62 (1:500) at 4℃ overnight. The membranes were washed three times with Tris-buffered saline combined with 0.1% Tween 20 (TTBS) for 10 minutes each time and then incubated with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G secondary antibodies (1:1,000 each) for 2 hours at RT. After washing three times in TTBS, protein bands were visualized using a chemiluminescence detection kit (Thermo Scientific). The same membranes were subsequently used for β-actin immune detection, and equal protein loading was ensured.
Statistical analysis
Data are presented as the mean±standard deviation (SD). Statistical analyses of the data were performed using Student t test or a one-way analysis of variance. A P<0.05 was considered to indicate statistical significance.
RESULTS
Fat-1 mice protect against STZ-induced hyperglycemia
One week after completion of the STZ injection administration program, hyperglycemia was observed in the STZ-treated WT mice, which, as shown in Fig. 2A, persisted for the entire observation period (21 days). In contrast, blood glucose levels in the STZ-treated fat-1 mice remained at normal levels and were identical to the levels in WT and fat-1 citrate (vehicle)-treated mice. The mean blood glucose concentration was approximately 500 mg/dL (day 18) in the STZ-treated WT mice, whereas it was 150 mg/dL in the STZ-treated fat-1 mice and the control, vehicle-treated mice.
Fig. 2. Genetic enrichment of n-3 fatty acid confers resistance to diabetes-related physical status. (A) Changes of blood glucose concentrations of wild-type (WT) and fat-1 mice for 18 days after streptozotocin (STZ) treatment. (B) Changes of water intakes of WT and fat-1 mice for 18 days after STZ treatment. Values are expressed as mean±SD from three independent experiments (n=20 per group). aP<0.001 vs. WT+STZ.

To water consumption, water intake was monitored for 21 days. All groups showed no significant difference in water consumption for 9 days (Fig. 2B). However, the water consumption of STZ-treated WT mice increased after 9 days. STZ-treated WT mice consumed approximately 7 mL of water per day (calculated at the end of monitoring). Interestingly, STZ-treated fat-1 mice showed unchanged water intake over the same period.
Effects of n-3 fatty acid enrichment on STZ-induced pancreatic β-cell damage
To gain mechanistic insight into the failure to maintain normal conditions in STZ-induced hyperglycemia, we assessed β-cell mass. β-Cells are contained within Langerhans; thus, we evaluate whether changes in β-cell function were associated with changes in islet morphology. H&E staining of STZ-treated mice islets showed degenerative changes (Fig. 3). In STZ-treated fat-1 mice compared with STZ-treated WT mice, mild morphological change was shown in pancreatic islets. Islet shrinkage was observed in STZ-treated WT mice but not in vehicle-treated mice, whereas no histological changes were observed in STZ-treated fat-1 mice, even when compared with WT or fat-1 vehicle-treated mice. Therefore, fat-1 mice demonstrated protective effects, as shown by resistance to STZ-induced islet shrinkage and morphological changes.
Fig. 3. Genetic enrichment of n-3 fatty acid preserves pancreatic islets against streptozotocin (STZ)-induced damages. (A) Representative H&E stained pancreatic tissues of wild-type (WT) and fat-1 mice after STZ treatments. Arrowheads indicates the pancreatic islets (scale bar=100 µm). (B) Quantitative bar graphs for comparison of islets which were bigger than 10,000 µm2. Values are expressed as mean percentage±SD. aP<0.001 vs. WT+STZ.

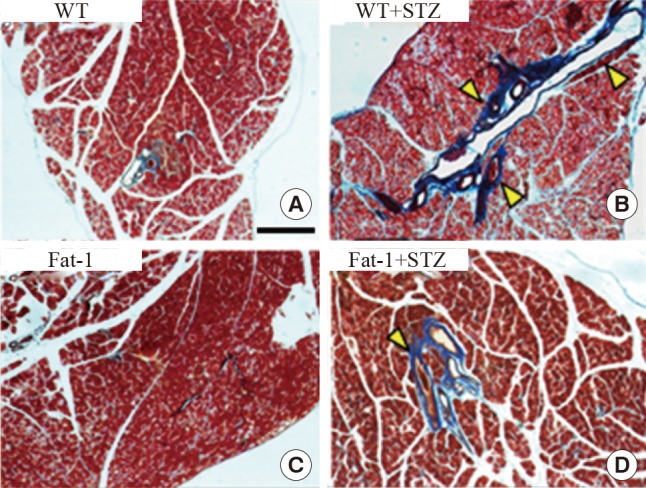
Fat-1 mice protect against STZ-induced fibrosis in the pancreas
The pancreases were evaluated using histological methods with Masson's trichrome staining, focusing on the fibrosis of the pancreas in the experimental groups. In STZ-treated WT mice, a significant amount of fibrosis was observed. However, STZ-treated fat-1 mice had fewer fibrotic lesions (Fig. 4). Collectively, these results demonstrated that fat-1 mice possess an essential factor involved in the protection against STZ-induced fibrosis.
Fig. 4. Genetic enrichment of n-3 fatty acid inhibits streptozotocin (STZ)-induced fibrosis in pancreas. Representative Masson trichrome-stained pancreatic tissues of wild-type (WT) and fat-1 mice after STZ treatments. (A, B) In STZ-treated WT mice, a significant amount of fibrosis was observed. (C, D) STZ-treated fat-1 mice had fewer fibrotic lesions. Arrowheads indicates fibrotic scars (scale bar=750 µm).
Fat-1 mice showed basally upregulated autophagic activity in islets and improved autophagic flux after STZ-induced toxicity
A beneficial role of autophagy in islet function and survival has been reported [6,8]. To investigate autophagy activity with and without STZ-induced islet toxicity in fat-1 mice, we performed immunofluorescence with anti-LC3. As shown in Fig. 5A, under normal conditions, fat-1 mice showed a significantly increased number and size of autophagosomes compared with WT mice. After STZ treatment, fat-1 and WT mice showed increased autophagosomes. Notably, a recent study reported that STZ-induced pancreatic islet injury was triggered by autophagic cell-death mechanisms characterized by abundant autophagosomes showing disturbed autophagic clearance [9]. Thus, we evaluated whether the genetic enrichment of n-3 PUFAs could ameliorate the autophagic cell death in β-cells using the immunologic detection method of p62, which is normally increased when cellular autophagic flux is disturbed. Interestingly, p62 levels in STZ-treated fat-1 mice were significantly lower than those in STZ-treated WT mice according to immunoblotting (Fig. 5B). These results suggest that pathological accumulation of autophagosomes induced by STZ treatment was inhibited in the β-cells of fat-1 mice. Together with the results from autophagosome detection, these results revealed that n-3 PUFA enrichment might prevent the diabetes-related pancreatic islet damage by upregulating, at least partly, the basal activity of autophagy and improving autophagic flux disturbance.
Fig. 5. Basal autophagy is upregulated and streptozotocin (STZ)-induced autophagic disturbance is attenuated in pancreatic islets of fat-1 mice. (A) Representative confocal microscopic images of light chain 3 (LC3)-immunostained pancreatic islet cells of wild-type (WT) and fat-1 mice with or without STZ treatments. Indicated rectangular areas magnified for clearer visualization of LC3 puncta. Arrowheads indicates the LC3-stained autophagosomes. 4',6-Diamidino-2-phenylindole was used for nuclear stains (scale bar=20 µm). (B) Representative immunoblot for quantification of p62 expression in pancreatic tissues of WT and fat-1 mice with or without STZ treatments. Actin was used for loading control.

DISCUSSION
As of 2010, the prevalence of diabetes was 9.7% or one in 10 adults in Korea, and the prevalence has continued to rapidly increase. This increase is closely associated with lifestyle; the use of walking as exercise has decreased dramatically compared with the past (from 75.5% in 2001 to 60.7% in 2005), the total calorie count for food intake has increased (from 1,985 and 1,976 kcal in 1998 and 2001, respectively, to 2,016 kcal in 2005), and the proportion of total fat intake has been increasing (from 41.5 and 41.6 g in 1998 and 2001, respectively, to 46.0 g in 2005) [10]. The rise in total fat intake has increased the risk of diabetes but, according to one report, the consumption of linoleic acid, an unsaturated fatty acid, was found to reduce the risk of diabetes [11]. In addition, a study showed that consuming more than 10 g of fish oil per day resulted in poor blood glucose control, whereas consuming less fish oil, between 1 and 2 g, reportedly had no adverse effects on blood glucose control. Therefore, the relationship between fish oil and glycometabolism is inconclusive [12,13].
Most studies regarding omega-3 (n-3 PUFA) have been limited to lipids. The combination of statins and omega-3 fatty acids in diabetic patients with hyperlipidemia reduces triglycerides and has a positive effect on low-density lipoprotein particle size [14] however, studies on the effects on pancreatic cells are limited. In general, the ratio of omega-3 to omega-6 (n-6 PUFA) is approximately 1:6 to 10 in the organs because omega-6 fatty acids are more widely distributed. This ratio should remain constant in organs. If this ratio is changed, an increase in omega-6 fatty acids can induce pro-inflammatory cytokine production during fatty acid metabolism and promote an inflammatory response [15], or the anti-proliferative effects of n-3 PUFA, eicosapentaenoic acid, and docosahexaenoic acid (DHA) may decrease and cause cancer [16]. The fat-1 transgenic mice used in this experiment were rich in desaturase enzymes converting omega-6 to omega-3 and therefore maintained the omega-3 to omega-6 ratio close to 1:1, showing an ability to reduce pro-inflammatory cytokine levels and increase anti-inflammatory cytokine levels, resulting in reduced occurrence of inflammation after β-cell damage. Because the mechanism underpinning this effect is not clearly understood, the authors of the current study observed the role of autophagy in β-cells and investigated the association between autophagy and these anti-inflammatory mechanisms. The role of autophagy in diabetes has been recently investigated but remains unclear. However, several recent reports suggest that autophagy may play an important role in the development and prevention of diabetes.
Cell death is largely classified into apoptosis and necrosis, but a new type of cell death, in the form of autophagy, has recently been reported. This autophagy is called type 2 programmed cell death, whereas apoptosis is regarded as type 1 programmed cell death [17]. Although autophagy may be a mechanism of cell death, it probably plays a more important role in the survival of cells, as the cell is supplied with energy and metabolites. Morphological changes in mitochondria, vesicles, and the Golgi apparatus as well as reduced insulin granules and accumulated ubiquitin were observed in autophagy-deficient β-cells [6]. As the accumulation of ubiquitin, the abnormality of mitochondria, and an increase in vesicle stress are known to cause β-cell death and deteriorated function, these factors probably mediate the morphological and functional changes in autophagy-deficient β-cells. In the present study, the basal autophagosome in fat-1 transgenic mice was increased, leading to basal autophagy activation when compared with WT mice. In addition, after STZ injection, p62 was reduced in fat-1 mice compared with STZ-treated WT mice, leading to increased autophagy clearance and subsequent protection of β-cells. A study conducted by Jung et al. [6] reported a rise in blood glucose levels and a reduction in insulin concentration after suppressing Atg7, a gene essential for the occurrence of autophagy and for reducing the quantity of β-cells in a morphological analysis of the pancreas. Additionally, increased β-cell death and suppressed proliferation were observed, both of which contributed to the reduction in the quantity of β-cells. A study conducted by Shin et al. [18] regarding the mechanism underpinning the autophagy activation of omega-3 found that ROS-regulated apoptosis and autophagy occurred via Akt-mTOR signaling when mutant p53 prostate cancer cells were treated with DHA.
To date, autophagy has been recognized as important for maintaining the function, structure, and quantity of β-cells, but its role in the development of diabetes remains unclear. The present study is the first to investigate the autophagy activity of omega-3 fatty acids. We found that the basal autophagy in transgenic mice was activated and that the autophagy activation exerted protective effects on β-cells through autophagy clearance. Future studies on the β-cell protection of omega-3 are necessary and may advance the development of diabetes drugs.
ACKNOWLEDGMENTS
This study was supported by research funds from the 2012 Konyang University Myunggok Research Fund and the Daejeon and Chungcheong Branch of the Korean Endocrine Society. We thank Dr. J.X. Kang at the Harvard Medical School for providing the fat-1 transgenic mice.
Footnotes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
References
- 1.Kahn CR, Weir GC, King GL, Moses AC, Smith RJ, Jacobson AM. Joslin's diabetes mellitus. 14th ed. Philadelphia: Lippincott Williams & Wilkins; 2005. Chapter 24, Insulin resistance and its role in the pathogenesis of type 2 diabetes; pp. 425–448. [Google Scholar]
- 2.Franz MJ, Bantle JP, Beebe CA, Brunzell JD, Chiasson JL, Garg A, et al. Evidence-based nutrition principles and recommendations for the treatment and prevention of diabetes and related complications. Diabetes Care. 2003;26(Suppl 1):S51–S61. doi: 10.2337/diacare.26.2007.s51. [DOI] [PubMed] [Google Scholar]
- 3.Kesavulu MM, Kameswararao B, Apparao C, Kumar EG, Harinarayan CV. Effect of omega-3 fatty acids on lipid peroxidation and antioxidant enzyme status in type 2 diabetic patients. Diabetes Metab. 2002;28:20–26. [PubMed] [Google Scholar]
- 4.Calder PC. n-3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr. 2006;83(6 Suppl):1505S–1519S. doi: 10.1093/ajcn/83.6.1505S. [DOI] [PubMed] [Google Scholar]
- 5.Marchetti P, Masini M. Autophagy and the pancreatic beta-cell in human type 2 diabetes. Autophagy. 2009;5:1055–1056. doi: 10.4161/auto.5.7.9511. [DOI] [PubMed] [Google Scholar]
- 6.Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427:504. doi: 10.1038/427504a. [DOI] [PubMed] [Google Scholar]
- 8.Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez CD, Lee MS, Marchetti P, Pietropaolo M, Towns R, Vaccaro MI, et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7:2–11. doi: 10.4161/auto.7.1.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim S, Shin H, Song JH, Kwak SH, Kang SM, Won Yoon J, et al. Increasing prevalence of metabolic syndrome in Korea: the Korean National Health and Nutrition Examination Survey for 1998-2007. Diabetes Care. 2011;34:1323–1328. doi: 10.2337/dc10-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Dam RM, Willett WC, Rimm EB, Stampfer MJ, Hu FB. Dietary fat and meat intake in relation to risk of type 2 diabetes in men. Diabetes Care. 2002;25:417–424. doi: 10.2337/diacare.25.3.417. [DOI] [PubMed] [Google Scholar]
- 12.Luo J, Rizkalla SW, Vidal H, Oppert JM, Colas C, Boussairi A, et al. Moderate intake of n-3 fatty acids for 2 months has no detrimental effect on glucose metabolism and could ameliorate the lipid profile in type 2 diabetic men. Results of a controlled study. Diabetes Care. 1998;21:717–724. doi: 10.2337/diacare.21.5.717. [DOI] [PubMed] [Google Scholar]
- 13.Sirtori CR, Crepaldi G, Manzato E, Mancini M, Rivellese A, Paoletti R, et al. One-year treatment with ethyl esters of n-3 fatty acids in patients with hypertriglyceridemia and glucose intolerance: reduced triglyceridemia, total cholesterol and increased HDL-C without glycemic alterations. Atherosclerosis. 1998;137:419–427. doi: 10.1016/s0021-9150(97)00298-0. [DOI] [PubMed] [Google Scholar]
- 14.Lee MW, Park JK, Hong JW, Kim KJ, Shin DY, Ahn CW, et al. Beneficial effects of omega-3 fatty acids on low density lipoprotein particle size in patients with type 2 diabetes already under statin therapy. Diabetes Metab J. 2013;37:207–211. doi: 10.4093/dmj.2013.37.3.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaikkonen JE, Kresanov P, Ahotupa M, Jula A, Mikkila V, Viikari JS, et al. High serum n6 fatty acid proportion is associated with lowered LDL oxidation and inflammation: the Cardiovascular Risk in Young Finns Study. Free Radic Res. 2014;48:420–426. doi: 10.3109/10715762.2014.883071. [DOI] [PubMed] [Google Scholar]
- 16.Williams CD, Whitley BM, Hoyo C, Grant DJ, Iraggi JD, Newman KA, et al. A high ratio of dietary n-6/n-3 polyunsaturated fatty acids is associated with increased risk of prostate cancer. Nutr Res. 2011;31:1–8. doi: 10.1016/j.nutres.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shin S, Jing K, Jeong S, Kim N, Song KS, Heo JY, et al. The omega-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p53. Biomed Res Int. 2013;2013:568671. doi: 10.1155/2013/568671. [DOI] [PMC free article] [PubMed] [Google Scholar]