

Abstract
Notch receptors are determinants of cell fate and function and play a central role in skeletal development and bone remodeling. Hajdu Cheney syndrome, a disease characterized by osteoporosis and fractures, is associated with NOTCH2 mutations resulting in a truncated stable protein and gain-of-function. We created a mouse model reproducing the Hajdu Cheney syndrome by introducing a 6955C→T mutation in the Notch2 locus leading to a Q2319X change at the amino acid level. Notch2Q2319X heterozygous mutants were smaller and had shorter femurs than controls; and at 1 month of age they exhibited cancellous and cortical bone osteopenia. As the mice matured, cancellous bone volume was restored partially in male but not female mice, whereas cortical osteopenia persisted in both sexes. Cancellous bone histomorphometry revealed an increased number of osteoclasts and bone resorption, without a decrease in osteoblast number or bone formation. Osteoblast differentiation and function were not affected in Notch2Q2319X cells. The pre-osteoclast cell pool, osteoclast differentiation, and bone resorption in response to receptor activator of nuclear factor κB ligand in vitro were increased in Notch2Q2319X mutants. These effects were suppressed by the γ-secretase inhibitor LY450139. In conclusion, Notch2Q2319X mice exhibit cancellous and cortical bone osteopenia, enhanced osteoclastogenesis, and increased bone resorption.
Keywords: animal model, bone, Notch pathway, Notch protein, osteoblast, osteoclast, Hajdu Cheney syndrome
Introduction
Notch proteins are four single-pass transmembrane receptors that play a critical role in cell fate decisions (Fig. 1) (1–4). Notch regulates cell renewal and plays a role in skeletal development and homeostasis and in osteoblast and osteoclast differentiation (4–8). Jagged1 and -2 and DeltaLike1, -3, and -4 are the five classic Notch ligands (4). Notch-ligand interactions result in the proteolytic cleavage and release of the Notch intracellular domain (NICD),2 which translocates to the nucleus to form a complex with recombination signal-binding protein for immunoglobulin κJ region (Rbpjκ) and Mastermind-like to regulate transcription (9–12). This canonical signaling pathway leads to the transcription of Hairy Enhancer of Split (Hes)1, -5, and -7 and Hes related with YRPW motif (Hey)1, -2, and -L.
FIGURE 1.

Domains of Notch2 and engineering of the Hajdu Cheney Notch2Q2319X mutant allele. A, upper panel shows domains of the Notch2 receptor depicting the following: 1) extracellular domain containing multiple epidermal growth factor (EGF)-like tandem repeats upstream of Lin12-Notch repeats (LNR); 2) heterodimerization domain (HD) which, in association with the LNR, forms the negative regulatory region; 3) transmembrane domain (TMD); and 4) Notch intracellular domain (NICD) consisting of an Rbpjκ-association module (RAM) linked to ankyrin (ANK) repeats and a nuclear localization sequence (NLS), upstream from a PEST domain. Under the Notch2 protein domains, the genomic structure of mutant exon 34 aligned with the corresponding protein structure. Black bars represent exons (E) 30–33, black box exon 34 containing the 6955C→T mutation leading to a Notch2Q2319X truncated protein, and white box the 3′-untranslated region (UTR). Position of the loxP site remaining following the excision of the neo selection cassette is indicated by the white arrow. Lower panel A shows the region of the Notch2 locus surrounding the 6955C→T substitution; DNA coding for the nuclear localization sequence (green), the PEST domain (blue), and the Notch2 3′-UTR (red). B, identification of four targeted ES clones by long range PCR. Correct targeting of the 5′- and 3′- homology arms into the Notch2 locus was documented by the presence of a 4.4- (left) and 3.2-kb (right) PCR product in each of four clones. The middle lane represents molecular weight markers. Clone 2 was selected for ES cell aggregation. The lower panel in B shows the structure of the linearized targeting vector, consisting of a 4.6-kb 5′-homology arm containing exons 30–33 of Notch2, followed by a phosphoglycerate kinase promoter-driven neo selection cassette flanked by loxP sites ∼400 nucleotides upstream of exon 34. Vertical arrows on the bottom indicate the boundaries of the linearized targeting vector. Black boxes indicate coding sequences, and white box indicates the 3′-UTR. Horizontal arrows indicate binding sites for the primers used for the long range PCR. Primer pairs F1-R1 and F2-R2 were used for amplification of the 5′-homology arm, and primer pairs F3-R3 were used for amplification of the 3′-homology arm. C, genomic DNA from ear samples of F1 pups was used as a template for PCR, and products were sequenced by the Sanger method. The 1:1 signal ratio of C (blue) to T (red) demonstrates the presence of the 6955C→T substitution and heterozygosity of the mutation.
Skeletal cells express Notch1, Notch2, and low levels of Notch3 transcripts (13–15). Activation of Notch in undifferentiated and differentiated osteoblasts inhibits cell differentiation and function and causes osteopenia (16, 17). In contrast, activation of Notch1 in osteocytes causes a pronounced increase in bone mass due to a suppression of bone resorption (18). Results from the conditional inactivation of Notch1 and Notch2 in the developing skeleton confirmed the inhibitory role of Notch in osteoblastogenesis (6, 19). Whereas substantial work has characterized the consequences of Notch1 gain-of-function in the skeleton, there is limited knowledge on the function of Notch2 in the postnatal skeleton. This knowledge is particularly important because Notch1 and Notch2 do not have redundant functions, and Notch1 inhibits, whereas Notch 2 enhances, osteoclastogenesis (13, 20–24).
Hajdu Cheney syndrome is a devastating disease characterized by focal bone lysis of distal phalanges and by generalized osteoporosis (25, 26). Hajdu Cheney syndrome is transmitted as an autosomal dominant disease, although sporadic cases occur. Whole exome sequencing in families affected by Hajdu Cheney syndrome revealed the presence of point mutations or short deletions in exon 34 of NOTCH2 leading to the creation of a stop codon and the premature termination of the protein product upstream from the PEST (proline (P), glutamic acid (E), serine (S), and (T) threonine) domain (27–31). Because the PEST domain is necessary for the ubiquitinylation and degradation of Notch, the mutations lead to a stable NICD protein and persistence of NOTCH2 signaling because the sequences required for the formation of the Notch transcriptional complex are preserved (Fig. 1). Despite the pronounced skeletal abnormalities reported in Hajdu Cheney syndrome, little is known regarding the mechanisms underlying the bone loss or the effects of Notch2 gain-of-function in the skeleton. Information obtained from iliac crest bone biopsies has been inconclusive and reported in a small number of subjects with Hajdu Cheney syndrome (32–35).
The purpose of this work was to gain understanding on the Hajdu Cheney syndrome skeletal phenotype and the mechanisms involved. To this end, we created a mouse model reproducing the NOTCH2 mutation (6949C→T) found in a subject affected by the disease and presenting with pronounced osteoporosis and fractures (28, 30). To create a Hajdu Cheney syndrome mutant mouse, we introduced the human NOTCH2 mutation of 6949C→T into the corresponding base of the mouse Notch2 (6955C→T) gene, creating a stop codon in exon 34 and leading to a Q2319X change at the amino acid level with a truncated Notch2 protein of 2318 amino acids (Fig. 1). In this study, we define the skeletal phenotype of Notch2Q2319X mutant mice and the mechanisms responsible.
Experimental Procedures
Hajdu Cheney Mutant Mice
To create a mouse model of Hajdu Cheney syndrome, we reproduced the mutation reported in a subject with the disease (28, 30). In the individual, a C at nucleotide 6949 from the translational start of NOTCH2 mutated into a T (6949C→T) leading to the creation of a premature stop codon in exon 34. The mutation corresponding to the 6949C→T substitution (6955C→T) was introduced into the mouse Notch2 locus by homologous recombination. A targeting vector containing 4.6 kb of 5′-homology arm from exon 30 to exon 33 of Notch2, a phosphoglycerate kinase promoter-driven neomycin (neo) selection cassette flanked by loxP sites ∼400 nucleotides upstream of exon 34, a 6955C→T mutation, and a 3′-homology arm of 3.0 kb, was used (Fig. 1). Embryonic (ES) cells from 129Sv/C57BL/6j embryos were electroporated, and G418-resistant colonies were selected. Targeted clones were verified by long range polymerase chain reaction (PCR) of genomic DNA. To ensure proper integration of the 5′-homology arm, we used forward F1 5′-GGTTGACAGGTGATGCAGTGCCAG-3′ and reverse R1 5′-GGCTGGACGTAAACTCCTCTTCAG-3′ primers followed by nested forward F2 5′-GCACATACCACACGGTCAGCTGAG-3′ and reverse R25′-GATCGGAATTGGGCTGCAGGAATT-3′ primers. To ensure proper integration of the 3′-homology arm, forward F3 5′-GGCTTCTGAGGCGGAAAGAACCAG-3′ and reverse R3 5′-CAATGGGGAGCCGTCATCATCGG-3′ primers were used (Fig. 1B). The presence of the 6955C→T mutation in the selected clone was confirmed by DNA sequencing (GENEWIZ, South Plainfield, NJ) (Fig. 1). Targeted ES clones were used for aggregations to generate chimeric mice at the Gene Targeting and Transgenic Facility of University of Connecticut Health Center. Chimeric male mice were bred with female mice expressing Cre under the control of the hypoxanthine-guanine phosphoribosyltransferase (Hprt) promoter to remove the PGKneo cassette (36). The removal of the cassette was verified by PCR, and the Hprt-Cre transgene was segregated by crossing with C57BL/6j wild type mice. Genomic DNA was obtained from ear punches of F1 pups, and the Notch2 mutation was confirmed by DNA sequencing (GENEWIZ) as shown in Fig. 1C. Genotyping of Notch2Q2319X mice was conducted in tail DNA extracts by PCR using forward primer Nch2Lox gtF5′-CCCTTCTCTCTGTGCGGTAG-3′ and reverse primer Nch2Lox gtR 5′-CTCAGAGCCAAAGCCTCACTG-3′. In this study, we characterized 129Sv/C57BL/6j mutant mice and sex-matched littermate controls obtained by crossing heterozygous Notch2Q2319X Hajdu Cheney mutants with wild type mice. Studies were approved by the Institutional Animal and Care Use Committees of Saint Francis Hospital and Medical Center and University of Connecticut Health Center.
Microcomputed Tomography (μCT)
Bone microarchitecture of femurs from experimental and control mice was determined using a microcomputed tomography instrument (μCT 40; Scanco Medical AG, Bassersdorf, Switzerland), which was calibrated periodically using a phantom provided by the manufacturer (37, 38). Femurs were scanned in 70% ethanol at high resolution, energy level of 55 peak kV, intensity of 145 μA, and integration time of 200 ms. A total of 100 slices at midshaft and 160 slices at the distal metaphysis were acquired at an isotropic voxel size of 216 μm3 and a slice thickness of 6 μm and were chosen for analysis. Trabecular bone volume fraction and microarchitecture were evaluated starting ∼1.0 mm proximal from the femoral condyles. Contours were manually drawn every 10 slices a few voxels away from the endocortical boundary to define the region of interest for analysis. The remaining slice contours were iterated automatically. Trabecular regions were assessed for total volume, bone volume, bone volume fraction (bone volume/total volume), trabecular thickness, trabecular number, trabecular separation, connectivity density, and structure model index, using a Gaussian filter (σ = 0.8) and user-defined thresholds (37, 38). For analysis of femoral cortical bone, contours were iterated across 100 slices along the cortical shell of the femoral midshaft, excluding the marrow cavity. Analysis of bone volume/total volume, porosity, cortical thickness, total cross-sectional and cortical bone area, periosteal perimeter, endosteal perimeter, and material density was performed using a Gaussian filter (σ = 0.8, support = 1), and user defined thresholds.
Bone Histomorphometric Analysis
Static and dynamic cancellous bone histomorphometry was carried out on experimental and control mice after they were injected with calcein, 20 mg/kg, and demeclocycline, 50 mg/kg, at an interval of 2 days in 1-month-old or of 5 days in 3-month-old animals. Five micron longitudinal sections of undecalcified femurs embedded in methyl methacrylate were cut on a microtome (Microm, Richards-Allan Scientific, Kalamazoo, MI) and stained with 0.1% toluidine blue or von Kossa. Static parameters of bone formation and resorption were measured in a defined area between 360 and 2160 μm from the growth plate, using an OsteoMeasure morphometry system (Osteometrics, Atlanta, GA). For dynamic histomorphometry, mineralizing surface per bone surface and mineral apposition rate were measured on unstained sections under ultraviolet light, using a triple diamidino-2-phenylindole/fluorescein/Texas red set long pass filter, and bone formation rate was calculated.
For cortical histomorphometry, femurs from 1- and 3-month-old mice were embedded in methyl methacrylate and cut through the mid-diaphysis with an EXAKT Precision Saw. Slides were ground using an EXAKT 400 CS micro grinding system to a thickness of ∼15 μm and surface-polished. Cortical sections were processed either in-house or at Alizee Pathology (Baltimore, MD). Slides were left unstained for fluorescence microscopy or stained with hematoxylin/eosin to establish cellular parameters and analyzed at a magnification of ×400 using OsteoMeasureXP software. Stained sections were used to draw the cortical bone, marrow space, osteoid, and cell surfaces as well as to count osteocytes within the cortex and osteoblasts and osteoclasts along the endocortical surface. Data from 1-month-old mice were generated from an all-inclusive cortical section, whereas data from 3-month-old mice were obtained from approximately half of a cortical section. Osteocyte number was expressed as cells/bone area measured. The terminology and units used for cancellous and cortical bone are those recommended by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research (39, 40).
Biochemical Parameters of Bone Turnover
Serum levels of C-terminal collagen cross-links, procollagen type 1 N-terminal propeptide, and tartrate-resistant acid phosphatase form 5b (TRACP5b) were measured by enzyme-linked immunosorbent assay (ELISA) (Immunodiagnostic Systems, Bolton, England). Serum from Notch2Q2319X and control littermate mice was obtained following an overnight fast, and assays were conducted according to the manufacturer's instructions.
Osteoblast-enriched Cell Cultures
The parietal bones of 3–5-day-old control and Notch2Q2319X mutant mice were exposed to type II collagenase from Clostridium histolyticum (Worthington) pretreated with N-α-tosyl-l-lysyl-chloromethyl ketone hydrochloride at 17 μg/ml (Calbiochem) (41). Bones were digested for 20 min at 37 °C; cells were extracted in five consecutive reactions, and cells from the last three digestions were pooled and seeded at a density of 10,000 cells/cm2, as described (42). Osteoblast-enriched cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with non-essential amino acids (both from Gibco and Life Technologies, Inc.), 20 mm HEPES, 100 μg/ml ascorbic acid (both from Sigma), and 10% heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals, Norcross, GA) in a humidified 5% CO2 incubator at 37 °C. To promote maturation, confluent osteoblasts were exposed to DMEM supplemented with 10% heat-inactivated FBS, 100 μg/ml ascorbic acid, and 5 mm β-glycerophosphate (Sigma).
Transient Transfections
Osteoblast-enriched cells were transfected with a plasmid containing six dimeric CSL or Rbpjκ consensus sequences upstream of the β-globin basal promoter (12×CSL-Luc; L. J. Strobl, Munich, Germany) or with 2.9- and 2.0-kb fragments of the Hey1 (Hey1-Luc; M. M. Maier, Wuerzburg, Germany) or Hey2 (Hey2-Luc; T. Iso, Los Angeles, CA) promoter and cloned upstream of luciferase (43–45). Transfections were conducted in cells cultured to 70% confluence using X-tremeGENE 9 (1.5 μl of X-tremeGENE 9:1 μg of DNA), according to manufacturer's instructions (Roche Applied Science). A construct where the cytomegalovirus (CMV) promoter directs the expression of β-galactosidase (CMV/β-galactosidase; Clontech) was used to correct for transfection efficiency. Cells were exposed to the X-tremeGENE 9/DNA mix for 16 h, and the medium was replaced after 24 h. Subsequently, cells were harvested in reporter lysis buffer (Promega, Madison, WI) and lysed by freezing at −80 °C and thawing at 37 °C. Luciferase and β-galactosidase activities were determined, respectively, with the luciferase assay system kit (Promega) and galacton plus (Life Technologies, Inc.) in accordance with manufacturer's instructions on an Optocomp luminometer (MGM Instruments, Hamden, CT).
Osteocyte-enriched Cultures
Osteocyte-enriched cells were obtained following a modification of a previously described method (46). Femurs were removed aseptically from 1-month-old experimental and control mice; the surrounding tissues were dissected; the proximal epiphyseal end was excised, and the bone marrow was removed by centrifugation. The distal epiphysis was excised, and femurs were digested for 20 min at 37 °C with type II bacterial collagenase pretreated with N-α-tosyl-l-lysyl-chloromethyl ketone hydrochloride and subsequently exposed to 5 mm EDTA for 20 min at 37 °C. The resulting osteocyte-enriched cortical femurs were cultured individually in DMEM supplemented with nonessential amino acids (both from Life Technologies, Inc.), 100 μg/ml ascorbic acid, and 10% FBS for 3 days at 37 °C in a humidified 5% CO2 incubator. Cells were cultured in the absence or presence of the γ-secretase inhibitor LY450139 (Selleck Chemicals, Houston, TX), dissolved in dimethyl sulfoxide (DMSO), and tested at 1 μm (47). An equal amount of DMSO was added to control cultures.
Bone Marrow Cell Cultures and Osteoclast Formation
Bone marrow cells were isolated from the femurs by a modification of previously published methods (48, 49). Briefly, bone marrow cells were collected by centrifugation and washed twice with α-minimum essential medium (MEM) (Life Technologies, Inc.) and cultured overnight in α-MEM containing 10% FBS. Non-adherent cells were collected, and bone marrow mononuclear cells were isolated using Ficoll-Hypaque (GE Healthcare) density gradient centrifugation. The interface between Ficoll-Hypaque and medium was collected, and cells were seeded at a density of 1 × 106 cells/cm2 and cultured in α-MEM with 10% heat-inactivated FBS (GE Healthcare) in the presence of macrophage colony-stimulating factor (M-Csf) at 30 ng/ml and receptor activator of nuclear factor κB ligand (Rankl) at 1–30 ng/ml (both from R&D Systems, Minneapolis, MN) for 6 days. In a subsequent experiment, total bone marrow cells were isolated from femurs by centrifugation, cultured in α-MEM in the presence of M-Csf at 100 ng/ml for 6 days, and switched to α-MEM containing M-Csf at 30 ng/ml and Rankl at 10–30 ng/ml for 8 days. Cells were cultured in the absence or presence of the γ-secretase inhibitor LY450139 at 1 μm or DMSO (47). In both experiments, cultured medium was changed every 3 days and cells were fixed for 30 s at room temperature prior to tartrate-resistant acid phosphatase (TRAP) enzyme histochemistry using a commercial kit (Sigma), in accordance with the manufacturer's instructions. TRAP-positive cells that contained more than three nuclei were considered osteoclast-like cells.
In Vitro Bone Resorption Assay
Bone marrow cells were isolated from femurs by centrifugation, expanded in the presence of M-Csf at 100 ng/ml for 5 days, and seeded at a density of 30,000 cells/cm2 on bovine cortical bone slices in α-MEM with 10% FBS and M-Csf at 30 ng/ml and Rankl at 30 ng/ml each for 16 days. Culture medium was replaced every 4 days with fresh medium containing M-Csf and Rankl. To visualize resorption pits, bone slices were sonicated to remove osteoclasts and stained with 1% toluidine blue in 1% sodium borate. To evaluate the ability of osteoclasts to resorb bone, the number of individual pits and resorption area were measured on images acquired with an Olympus DP72 camera using CellSens Dimension software version 1.6 (Olympus Corp., Center Valley, PA) (50).
Flow Cytometry
Bone marrow cells were obtained by flushing tibiae and collecting cells in α-MEM. After washing in α-MEM, the red blood cells were lysed with ammonium chloride/potassium lysis buffer (Life Technologies, Inc.), and the cell preparation was filtered through a nylon mesh and counted. Dead cells were excluded by their ability to incorporate propidium iodide. Anti-mouse antibodies used for flow cytometric analysis were anti-CD45R (B220) for B-cell lineage cells, anti-CD3 for T-cell lineage cells, anti-CD11b (Mac-1) for macrophage lineage cells, anti-CD117 (c-Kit), a hematopoietic stem cell marker, and anti-CD115 (c-Fms, the M-Csf receptor) (all from BD Biosciences). Antibodies conjugated to fluorochromes, or biotinylated, and secondary step reagents were obtained commercially (eBioscience, San Diego, CA). Labeling of bone marrow cells for flow cytometric analysis was performed by standard staining procedures in Hanks' balanced salt solution (Life Technologies, Inc.) containing 0.01 m HEPES (pH 7.4), supplemented with 2% FBS. Flow cytometric analysis was carried out on a FACSCalibur instrument and data analysis performed using FlowJo software (Tree Star Inc., Ashland, OR) (51).
Quantitative Reverse Transcription (qRT)-PCR
Total RNA was extracted from cells or femurs, following the removal of the bone marrow by centrifugation, and mRNA levels determined by qRT-PCR (52, 53). For this purpose, equal amounts of RNA were reverse-transcribed using iScript RT-PCR kit (Bio-Rad), according to manufacturer's instructions, and amplified in the presence of specific primers (Table 1, all primers from Integrated DNA Technologies (IDT), Coralville, IA), and iQ SYBR Green Supermix (Bio-Rad), at 60 °C for 35 cycles. Transcript copy number was estimated by comparison with a serial dilution of cDNA for acid phosphatase 5, tartrate-resistant (Acp5, from Thermo Scientific, Pittsburgh, PA), alkaline phosphatase (Alpl, from American Type Tissue Culture Collection, ATCC, Manassas, VA), bone γ-carboxyglutamate protein (Bglap, encoding for osteocalcin, from J. Lian, Burlington, VT), caspase 3 (Casp3) (from GE Healthcare), Hes1 (from ATCC), Hey1 and Hey2 (both from T. Iso), HeyL (from D. Srivastava, Dallas, TX), nuclear factor of activated T cells 1 (Nfatc1, from A. Rao, La Jolla, CA), tumor necrosis factor receptor superfamily 11b (Tnfrsf11b, encoding for osteoprotegerin, from ATCC), or tumor necrosis factor, member 11 (Tnfsf11, encoding for Rankl, from Source BioScience, Nottingham, UK) (54–58).
TABLE 1.
Primers used for qRT-PCR determinations
GenBankTM accession numbers identify transcript recognized by primer pairs.
| Gene | Strand | Sequence 5′–3′ | GenBankTM accession no. |
|---|---|---|---|
| Acp5 | Forward | GACAAGAGGTTCCAGGAGAC | NM_001102404; NM_001102405; NM_007388 |
| Reverse | TTCCAGCCAGCACATACC | ||
| Alpl | Forward | TGGTATGGGCGTCTCCACAGTAACC | NM_007431; NM_001287172; NM_001287176 |
| Reverse | CTTGGAGAGGGCCACAAAGG | ||
| Bglap | Forward | GACTCCGGCGCTACCTTGGGTAAG | NM_001037939 |
| Reverse | CCCAGCACAACTCCTCCCTA | ||
| Casp3 | Forward | ATGGAGAACAACAAAACCTC | NM_009810; NM_001284409 |
| Reverse | CCATGTATGGTCTTTACTTCA | ||
| Gapdh | Forward | CCCCTCTGGAAAGCTGTGGCGT | NM_008084; NM_001289726 |
| Reverse | AGCTTCCCGTTCAGCTCTGG | ||
| Hes1 | Forward | ACCAAAGACGGCCTCTGAGCACAGAAAGT | NM_008235 |
| Reverse | ATTCTTGCCCTTCGCCTCTT | ||
| Hey1 | Forward | ATCTCAACAACTACGCATCCCAGC | NM_010423 |
| Reverse | GTGTGGGTGATGTCCGAAGG | ||
| Hey2 | Forward | AGCGAGAACAATTACCCTGGGCAC | NM_013904 |
| Reverse | GGTAGTTGTCGGTGAATTGGACCT | ||
| HeyL | Forward | CAGTAGCCTTTCTGAATTGCGAC | NM_013905 |
| Reverse | AGCTTGGAGGAGCCCTGTTTC | ||
| Nfatc1 | Forward | GCGCAAGTACAGTCTCAATGGCC | NM_198429; NM_001164110; NM_001164111; NM_001164112; NM_00116641091; NM_016791 |
| Reverse | GGATGGTGTGGGTGAGTGGT | ||
| Notch2 | Forward | CATCGTGACTTTCCA | NM_010928 |
| Reverse | GGATCTGGTACATAGAG | ||
| Rpl38 | Forward | AGAACAAGGATAATGTGAAGTTCAAGGTTC | NM_001048057; NM_001048058; NM_023372 |
| Reverse | CTGCTTCAGCTTCTCTGCCTTT | ||
| Tnfrsf11b | Forward | CAGAAAGGAAATGCAACACATGACAAC | NM_008764 |
| Reverse | GCCTCTTCACACAGGGTGACATC | ||
| Tnfsf11 | Forward | TATAGAATCCTGAGACTCCATGAAAAC | NM_011613 |
| Reverse | CCCTGAAAGGCTTGTTTCATCC |
To measure levels of the Notch26955C→T mutant transcript, total RNA was reverse-transcribed with Moloney murine leukemia virus reverse transcriptase, in accordance with the manufacturer's instructions (Life Technologies, Inc.), in the presence of reverse primers for Notch2 and of reverse primers for either ribosomal protein L38 (Rpl38) or glyceraldehyde-3-phosphate dehydrogenase (Gapdh) (Table 1). Notch2 cDNA was amplified by qPCR in the presence of specific primers (Table 1), a TET-labeled DNA probe of sequence 5′-CATTGCCTAGGCAGC-3′ covalently bound to a 3′-minor groove binder quencher (Life Technologies, Inc.), and SsoAdvanced Universal Probes Supermix (Bio-Rad) at 60 °C for 45 cycles (59). Rpl38 or Gapdh cDNA was amplified as described above. Notch26955C→T transcript copy number was estimated by comparison with a serial dilution of a synthetic DNA fragment (IDT) containing ∼200 bp surrounding the 6955C→T mutation in the Notch2 locus and cloned into pcDNA3.1(−) (Life Technologies, Inc.) by isothermal single reaction assembly using commercially available reagents (New England Biolabs, Ipswich, MA) (60).
Amplification reactions were conducted in a CFX96 qRT-PCR detection system (Bio-Rad), and fluorescence was monitored during every PCR cycle at the annealing step. Data are expressed as copy number corrected for Rpl38 or Gapdh copy number, estimated by comparison with a serial dilution of Rpl38 (from ATCC) or Gapdh (from R. Wu, Ithaca, NY), respectively (61, 62). To establish changes in gene expression in osteoblast-enriched cultures, data were obtained from three experiments and controls normalized to 1.
Statistics
Data are expressed as means ± S.D. Statistical differences were determined by unpaired Student's t test or analysis of variance with Holm-Sidak post hoc analysis for pairwise or multiple comparisons.
Results
Generation and General Appearance of Hajdu Cheney Notch2Q2319X Mutant Mice
To introduce a Hajdu Cheney syndrome mutation into the Notch2 locus, a targeting vector containing a 6955C→T substitution in exon 34 (Fig. 1) was introduced into ES cells derived from an F1 129Sv/C57LB/6j embryo by electroporation. A targeted clone was used to generate mutant mice, which were studied following the removal of the selection cassette. Breedings between heterozygous Hajdu Cheney Notch2Q2319X mutant mice resulted in perinatal lethality, whereas matings of heterozygous mutants with wild type mice resulted in no apparent lethality and a similar distribution of 56% wild type and 44% mutant mice in the offspring. Therefore, heterozygous Hajdu Cheney mutant mice were compared with wild type littermates following heterozygous crossings with wild type mice, all in a 129Sv/C57BL/6j genetic background. At 1 month of age, Notch2Q2319X heterozygous mice weighed ∼20% less than littermate controls, but their general appearance was not substantially different from controls. As the mice matured, there was less difference in the size of mutant and wild type mice, and mutant mice weighed ∼10% less than wild types at 3 months of age. Femoral length was 12% shorter in Notch2Q2319X heterozygous mice of both sexes than controls at 1 month of age, but at 3 months of age, femoral length in Notch2Q2319X mice was not different from controls in male mice and was only 5% shorter in female mice (Fig. 2).
FIGURE 2.

Weight and femoral length of male and female Hajdu Cheney Notch2Q2319X mutant mice (filled circles) and littermate wild type controls (open circles). Values are means ± S.D. The number of observations is as follows: male control mice n = 5 at 1 month and n = 9 at 3 months; female control mice n = 9 at 1 month and n = 4 at 3 months; male Notch2Q2319X n = 6 at 1 month and n = 10 at 3 months; and female Notch2Q2319X n = 8 at 1 month and n = 7 at 3 months of age. *, significantly different between Notch2Q2319X mutant mice and control, p < 0.05 by unpaired t test.
Femoral Microarchitecture and Histomorphometry of Hajdu Cheney Mutant Mice
μCT of the distal femur revealed that male Notch2Q2319X mutant mice had a 50% decrease in trabecular bone volume at 1 month of age, whereas female mice had a 20% decrease in trabecular bone volume (Table 2 and Fig. 3). The osteopenia was due to a reduction in the number of trabeculae and to a lesser extent to a decrease in trabecular thickness. The decreased cancellous bone volume in male mice was associated with decreased connectivity. The decrease in trabecular number was sustained and observed in 3-month-old Notch2Q2319X mutant mice resulting in a non-significant decrease in cancellous bone volume of 30% in male mice and a significant decrease of 50% in female mice (Table 2). There were pronounced changes in the cortical bone structure of Notch2Q2319X mutants of both sexes; cortical bone was thin and porous, and total area as well as bone area were reduced (Table 2 and Fig. 3). The decrease in cortical bone and overall bone size was more pronounced in Notch2Q2319X at 1 month of age, although cortical bone architecture remained affected at 3 months of age in Notch2Q2319X mutants of both sexes.
TABLE 2.
Femoral microarchitecture assessed by μCT of 1- and 3-month-old Notch2Q2319X mutant mice and littermate controls
μCT was performed in distal femurs for trabecular bone and midshaft for cortical bone from 1- and 3-month-old male and female Notch2Q2319X mutant mice and control littermates (Control). Values are means ± SD.
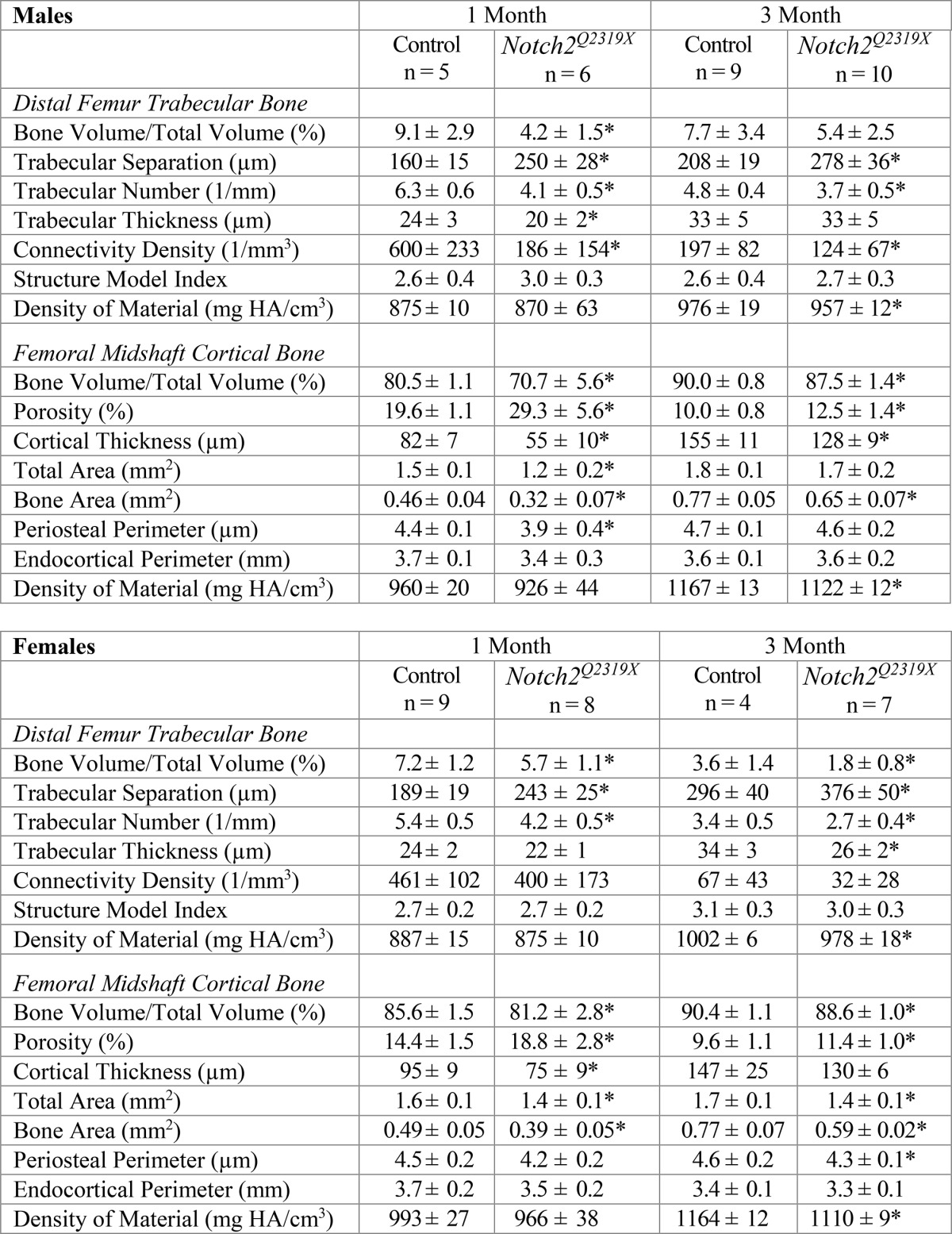
* Significantly different from controls, p < 0.05 by unpaired t-test.
FIGURE 3.
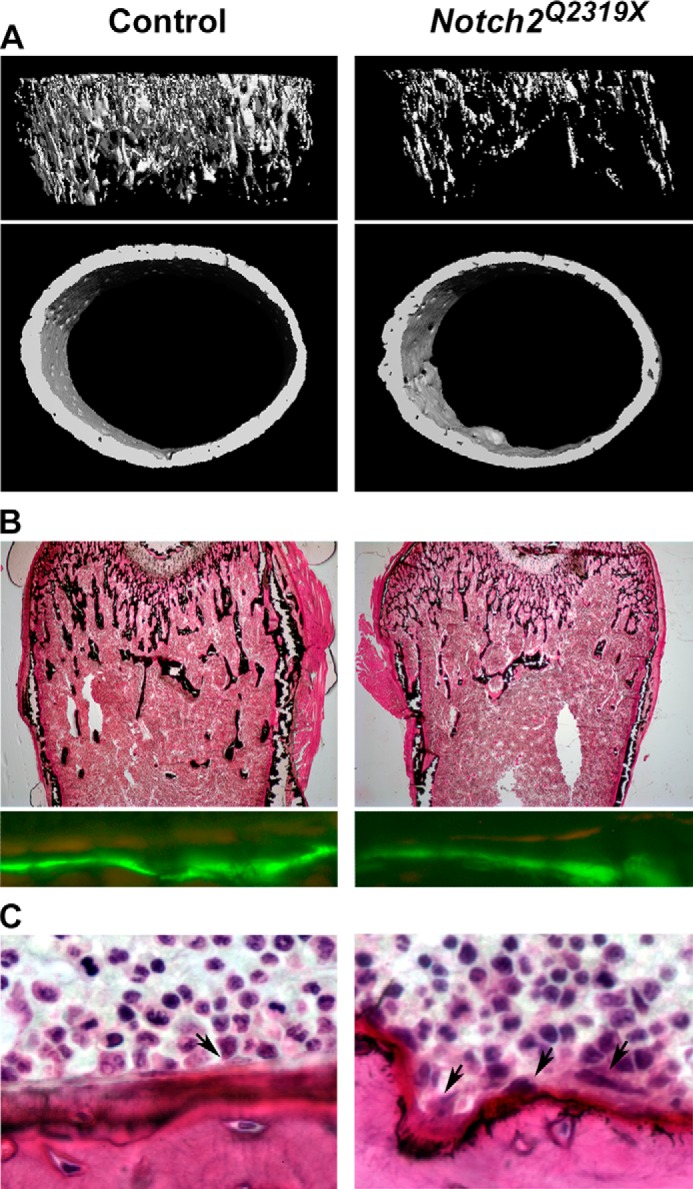
A, representative microcomputed tomography images of proximal trabecular bone and midshaft of femurs, showing cancellous bone osteopenia and decreased trabecular number and thinner and porous cortical bone in male Notch2Q2319X mutant mice. Complete data set is shown in Table 2. B, representative static cancellous bone histological sections stained with toluidine blue show decreased number of trabeculae in Hajdu Cheney Notch2Q2319X mice and calcein and demeclocycline labels show no differences in mineral apposition rate between control and Notch2Q2319X mice. Complete data set is in Table 3. C, cross-sectional cortical bone stained with hematoxylin and eosin. Arrows in C point to osteoclasts on the endocortical surface. Complete data set is in Table 4. All representative images are from femurs from 1-month-old male Hajdu Cheney Notch2Q2319X mutant and littermate wild type controls.
Cancellous bone histomorphometric analysis of femurs from Notch2Q2319X mutant mice at 1 month of age confirmed the microarchitectural findings and demonstrated decreased bone volume/tissue volume secondary to a decrease in trabecular number in male and female mutant mice (Table 3 and Fig. 3). At 1 month of age, there was an increase in osteoclast surface/bone surface and eroded surface in Notch2Q2319X mutant mice, and there was no change in osteoblast number/perimeter, osteoblast surface, bone formation rate, or osteocyte number/bone area. This indicates that enhanced bone resorption without a coupled bone-forming response was responsible for the skeletal phenotype. At 3 months of age, the cellular phenotype evolved. The numbers of osteoclasts and eroded surface were no longer significantly increased, and osteoblast surface and mineral apposition rates were increased in male mice, a possible reflection of increased bone remodeling or a compensatory bone-forming response to the enhanced bone resorption noted at 1 month of age. An increase in osteoblast number and mineralizing surface was not observed in female mice. Although trabecular number was decreased by ∼30% in 3-month-old male mice, bone volume/total volume was only modestly affected, possibly because, over time, bone formation compensated for the initial increase in bone resorption.
TABLE 3.
Cancellous bone histomorphometry of 1- and 3-month-old Notch2Q2319X mutant mice and littermate controls
Bone histomorphometry was performed in distal femurs from 1- and 3-month old male and female Notch2Q2319X mutant mice and control littermates (Control). Values are means ± SD.
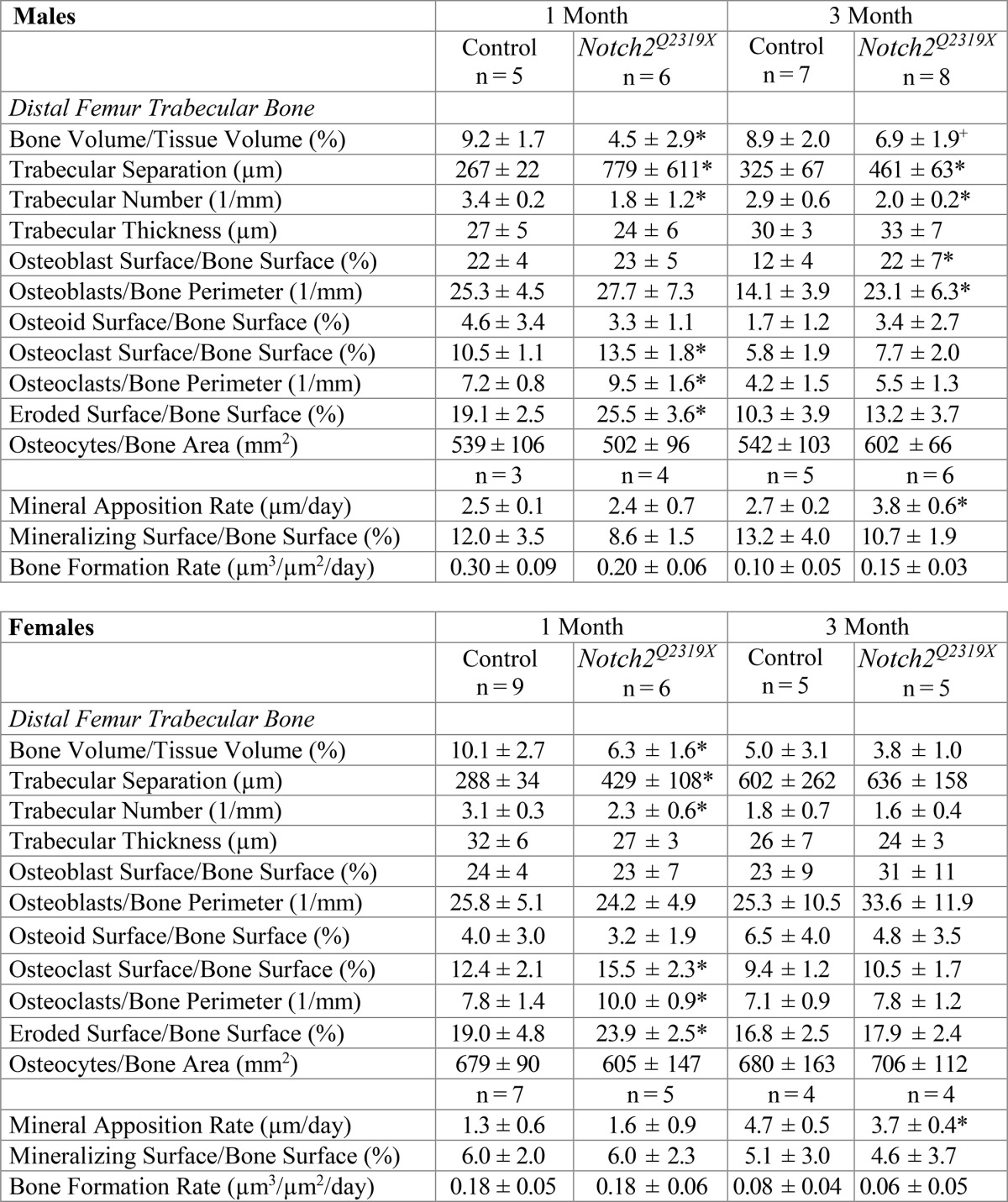
* Significantly different from controls, p < 0.05 by unpaired t-test.
+ p < 0.08.
Cortical bone histomorphometry confirmed a decrease in cortical thickness and revealed no difference in osteocyte number (Table 4). There was an increase in endocortical osteoclast number and eroded surface in 1-month-old Notch2Q2319X mutants, but the osteoblast number was not different from controls. At 3 months of age, osteoblast, osteoclast, and osteocyte number were not different between mutant and control mice. The identity of cells in the periosteal surface could not be determined with confidence. Mineral apposition rate was not determined in Notch2Q2319X mutants because limited areas contained well defined double labels. This may suggest that bone formation was impaired in cortical bone.
TABLE 4.
Cortical histomorphometry of 1- and 3-month-old Notch2Q2319X mutant male mice and littermate controls
Cortical bone histomorphometry was performed at mid-diaphysis in femurs from 1- and 3-month-old male Notch2Q2319X mutant mice and control littermates (control). Data from 1-month-old mice were generated from analysis of an all-inclusive cortical section, and data from 3-month-old mice were obtained from analysis of an approximate half-cortical section so that bone volume/total volume and total bone area were not calculated but are provided by μCT in Table 2. Osteocyte number was counted in either full or hemi-sections and expressed as cells/bone area measured. Values are means ± S.D.
| 1 Month |
3 Months |
|||
|---|---|---|---|---|
| Control n = 6 | Notch2Q2319X n = 4 | Control n = 4 | Notch2Q2319X n = 4 | |
| Cortical bone | ||||
| Bone volume/tissue volume (%) | 38.1 ± 7.5 | 29.7 ± 4.8 | ||
| Cortical thickness (μm) | 206 ± 30 | 168 ± 11* | 219 ± 70 | 157 ± 54 |
| Bone area (mm2) | 0.54 ± 0.11 | 0.42 ± 0.06 | ||
| Osteocyte no./bone area (mm2) | 1531 ± 385 | 1405 ± 263 | 796 ± 227 | 763 ± 140 |
| Endocortical surface | ||||
| Osteoblasts/bone perimeter (1/mm) | 26.0 ± 9.5 | 20.1 ± 7.4 | 10.4 ± 4.2 | 6.8 ± 2.3 |
| Osteoclasts/bone perimeter (1/mm) | 3.7 ± 0.6 | 6.9 ± 0.8* | 1.0 ± 0.1 | 1.0 ± 0.3 |
| Osteoclast surface/bone surface (%) | 5.5 ± 0.8 | 10.2 ± 1.4* | 1.7 ± 0.5 | 1.7 ± 0.4 |
| Eroded surface/bone surface (%) | 8.9 ± 2.6 | 15.5 ± 1.4* | 2.2 ± 0.6 | 2.2 ± 0.9 |
* Significantly different from controls, p < 0.05 by unpaired t test.
Although cancellous and cortical bone histomorphometry revealed increased bone resorption in Notch2Q2319X mutants, serum levels of the biochemical markers of C-terminal collagen cross-links and procollagen type 1 N-terminal propeptide were not different between Notch2Q2319X mice and control littermates of both sexes at 1 and 3 months of age, and TRACP5b values were variable (data not shown). This may indicate that bone remodeling was not increased because a bone-forming response was delayed and observed by bone histomorphometry only in 3-month-old male mutant mice or may indicate limited sensitivity and variability of serum assays used to determine bone remodeling (63, 64).
Mechanisms Operational in Notch2Q2319X Mutant Mice
To explore mechanisms that may explain the phenotype of Notch2Q2319X mutant mice, RNA was extracted from femurs from mutant and control mice. qRT-PCR revealed expression of Notch26955C→T (Notch2Q2319X) and moderately increased mRNA levels of Notch target genes Hey1, Hey2, and HeyL, but not Hes1, in femurs from mutant mice confirming activation of Notch signaling in skeletal tissue (Fig. 4). In accordance with the resorptive phenotype observed in 1-month-old mice, there was an increased expression of Tnfsf11, encoding for Rankl, in femurs from Notch2Q2319X mutant mice, and no changes in Tnfrsf11b, encoding for osteoprotegerin.
FIGURE 4.

Notch6955C→T (Notch2Q2319X), Hey1, Hey2 and HeyL, Hes1, Tnfsf11 (Rankl), and Tnfrsf11b (osteoprotegerin) mRNA levels in femoral bones from 1-month-old Hajdu Cheney Notch2Q2319X mutant mice (black bars) and control littermate mice (white bars). Transcript levels are expressed as copy number corrected for Gapdh. Values are means ± S.D.; n = 8 for control; n = 9 for Notch2Q2319X for all transcripts. *, significantly different between Notch2Q2319X mutants and control, p < 0.05 by unpaired t test.
Osteoblast-enriched and Osteocyte-enriched Cell Cultures
To understand the consequences of the Hajdu Cheney Notch2Q2319X mutation in skeletal cells, osteoblast-enriched calvarial cells from mutant mice and littermate controls were cultured. Osteoblasts from Notch2Q2319X mice expressed Notch26955C→T transcripts, which were not detected in wild type littermate controls. The expression of the Notch target genes Hey1, Hey2, and HeyL and the activity of the transiently transfected 12×CSL-Luc and Hey2-Luc (both p < 0.05) and Hey1-Luc (p > 0.05) reporter constructs were increased in Notch2Q2319X osteoblasts confirming activation of Notch signaling (Fig. 5). In accordance with the histomorphometric findings revealing no changes in osteoblast number or function in 1-month-old Notch2Q2319X mice, Notch2Q2319X mutant cells expressed no significant changes in Bglap and Alpl mRNA levels (7, 17). In accordance with the increase in bone resorption observed in Notch2Q2319X mice, expression of Tnfsf11 was increased in osteoblast- (Fig. 5) and osteocyte-enriched preparations. This increase in osteocyte Tnfsf11 was dependent on activation of Notch signaling because it was prevented by the addition of the γ-secretase inhibitor LY450139. Tnfsf11/Rpl38 copy number was (means ± S.D.; n = 4 for all cultures) 1.2 ± 0.8 in control and 2.8 ± 0.7 (p < 0.05) in Notch2Q2319X osteocyte-enriched cultures. In the presence of the LY450139, the values were 1.3 ± 0.6 in control and 1.1 ± 0.6 in Notch2Q2319X cultures (not significant). In agreement with the lack of an effect of Notch2Q2319X mutant on osteocyte number in cancellous and cortical bone, Casp3 mRNA expression was not affected in osteocyte-rich cultures suggesting that osteocyte apoptosis was not enhanced in Notch2Q2319X mutants. Casp3/Rpl38 copy number was (means ± S.D.) 2.4 ± 1.6 (n = 7) in control and 2.4 ± 2.5 (n = 9) in Notch2Q2319X cultures.
FIGURE 5.

Calvarial osteoblast-enriched cells from Notch2Q2319X mutant (black bars) and wild type (white bars) littermate controls were isolated and cultured. A, 12× CSL-Luc, Hey1-Luc, and Hey2-Luc reporter constructs were transiently co-transfected with a CMV β-galactosidase construct; data are expressed as luciferase/β-galactosidase activity. Values are means ± S.D.; n = 6 for all data sets. B, total RNA was extracted, and gene expression was measured by qRT-PCR in the presence of specific primers and probes. Data are expressed as Notch26955C→T (Notch2Q2319X), Hey1, Hey2, HeyL, Alpl, Bglap, Tnfsf11 (Rankl), and Tnfrsf11b (osteoprotegerin) copy numbers and corrected for Rpl38. Values are means ± S.D.; number of observations for control, n = 12 at 0 days, n = 10 at 3 days, and n = 8 at 7 days; number of observations for Notch2Q2319X, n = 12 at 0 and at 3 days and n = 8 at 7 days for all transcripts. For mRNA expression, data were obtained from three experiments and controls normalized to 1. *, significantly different between Notch2Q2319X mutant and wild type control cells, p < 0.05 by unpaired t test.
In Vitro Osteoclast Formation and Flow Cytometry
To investigate the cause of the increase in osteoclast number and bone resorption, the number of osteoclast precursors was determined in bone marrow cells from Notch2Q2319X heterozygous and control littermates by flow cytometric analysis. There was a 25% increase in the fraction of B220− CD3− CD11b−/lo, CD115 (c-Fms)high CD117 (c-Kit)high cells in Notch2Q2319X mice compared with controls, and most of the early osteoclastogenic activity resides in these cells (50, 51). Further analysis of the monocyte/macrophage population present in the bone marrow B220− CD3− CD11b+ CD115+ fraction revealed no difference in additional osteoclast precursor cells between Notch2Q2319X and control mice. Somatic NOTCH2 gain-of-function mutations in exon 34 upstream from the PEST domain have been reported in B-cell lymphomas, and the B-cell fraction was analyzed by flow cytometry (65–67). There was no difference in the population of B-cells (B220+ fraction) in the bone marrow from Notch2Q2319X mice (means ± S.D.; n = 4), 34.8% ± 2.4, when compared with controls 31.9% ± 2.4.
To determine whether the Notch2Q2319X mutation resulted in enhanced osteoclastogenesis, non-adherent bone marrow mononuclear cells isolated by Ficoll-Hypaque gradient centrifugation were cultured in the presence of M-Csf at 30 ng/ml and Rankl at 1–30 ng/ml for 6 days and examined for the development of TRAP-positive multinucleated cells. There was a significant increase in the number of osteoclasts in bone marrow cell cultures from Notch2Q2319X mutants compared with controls (Fig. 6). In accordance with the reported induction of Nfatc1 by Notch2 in bone marrow macrophages, cells from Notch2Q2319X expressed higher levels of Nfatc1 mRNA than control cultures (20). Following treatment with Rankl at 30 ng/ml for 3 days, Nfatc1/Rpl38 copy number was (means ± S.D.; n = 4) 5.1 ± 1.6 in control and 8.0 ± 1.6 (p < 0.05) in Notch2Q2319X cultures. Hes1 mRNA was increased as control and Notch2Q2319X mutant cells matured, although the increase was more pronounced in Notch2Q2319X cells, which were the only cultures expressing Notch2 mutant transcripts (Fig. 6). Hey1, -2, and -L transcripts were not detected in either control or Notch2Q2319X bone marrow mononuclear cell cultures (data not shown). To verify these results and to determine whether Notch activation was required for the effects observed, in a subsequent experiment bone marrow mononuclear cells were isolated and expanded in the presence of M-Csf at 100 ng/ml for 6 days followed by the addition of Rankl at 30 ng/ml in the presence of M-Csf at 30 ng/ml. The cultures were conducted in the absence and presence of the γ-secretase inhibitor LY450139 at 1 μm, to prevent activation of Notch and release of the NICD. Osteoclasts formed 5 days following cellular exposure to Rankl, and the number of osteoclasts was greater in Notch2Q2319X cultures than in controls (Fig. 7). The γ-secretase inhibitor LY450139 decreased the formation of osteoclasts in the initial phases of osteoclast differentiation in control cultures, although not after 8 days of Rankl exposure. These findings confirm previous work suggesting that Notch activation is required for basal osteoclastogenesis (20). LY450139 precluded the effect of the Notch2Q2319X mutation on osteoclastogenesis demonstrating that Notch activation is required for this effect of Notch2. Notch2Q2319X osteoclasts expressed Notch2 mutant transcripts, whereas control cells did not. There was an increase in the levels of Hes1 mRNA, confirming activation of Notch signaling, and of Acp5 (encoding for TRAP) mRNA in Notch2Q2319X cells; LY450139 suppressed the expression of both genes in control and mutant cells (Fig. 7). Neither control nor Notch2Q2319X mutant cells expressed detectable levels of Hey1, -2, or -L transcripts (data not shown). These results demonstrate that, in the context of the Notch2Q2319X mutation, there is an increase in the pre-osteoclast cell pool as well as in its ability to differentiate into mature osteoclasts, and Notch activation is required for these effects.
FIGURE 6.
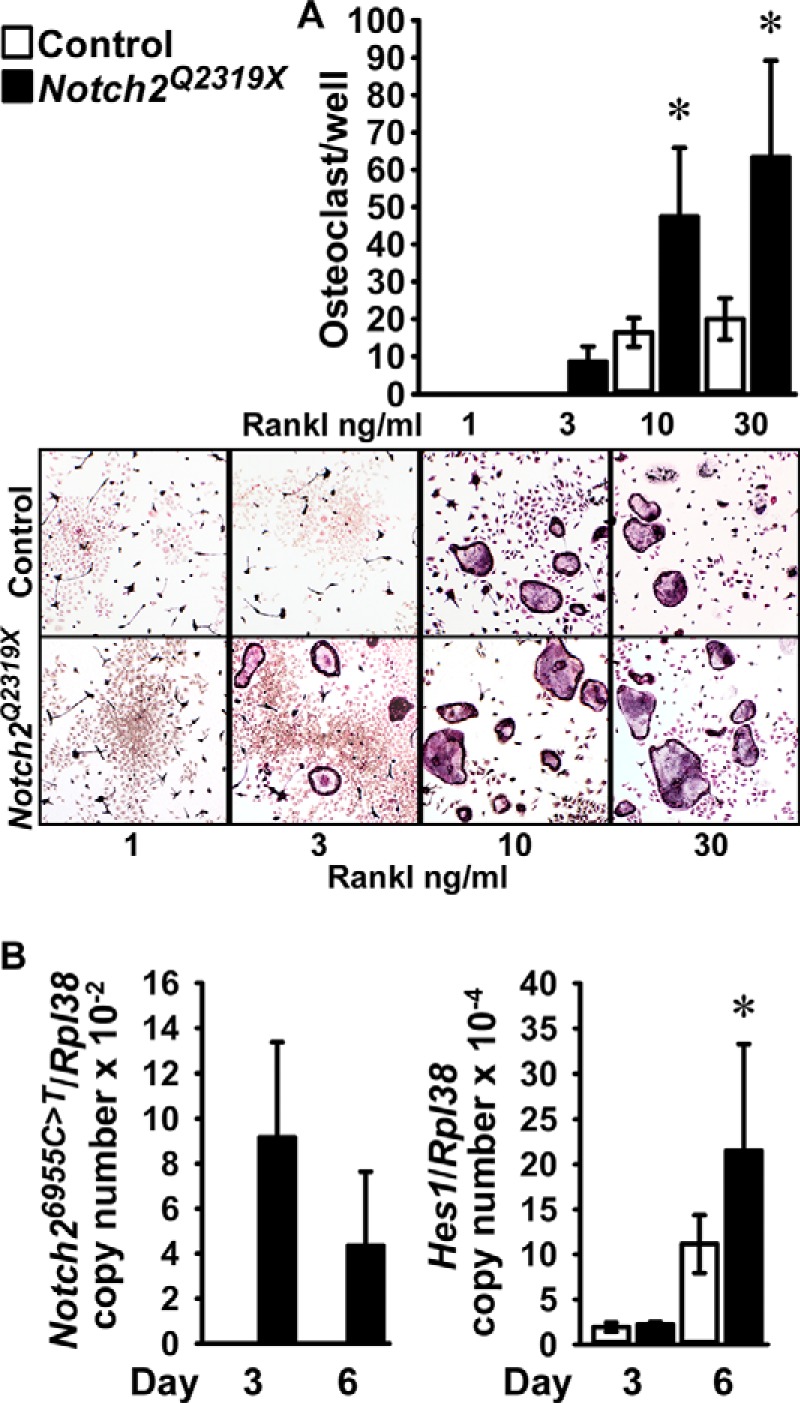
Bone marrow mononuclear cells, harvested from femurs of Notch2Q2319X mutant (black bars) and wild type littermate controls (white bars) were isolated by Ficoll-Hypaque gradient centrifugation and cultured for 6 days in the presence of M-Csf at 30 ng/ml and Rankl at 1–30 ng/ml. A, cultures were assessed for TRAP by enzyme histochemistry. Values are means ± S.D.; n = 6 for all data sets. The lower panel shows representative culture fields of TRAP-stained multinucleated cells. B, cultures were assessed for Notch26955C→T (Notch2Q2319X) and Hes1 mRNA levels expressed as copy number corrected for Rpl38. Values are means ± S.D. Number of observations: n = 4 for control and Notch2Q2319X at 3 and 6 days. *, significantly different between Notch2Q2319X mutant and control cells, p < 0.05 by two-way analysis of variance with Holm-Sidak post hoc analysis.
FIGURE 7.

Bone marrow mononuclear cells, harvested from femurs of Notch2Q2319X mutant (black bars) and wild type littermate controls (white bars) were cultured in the presence of M-Csf at 100 ng/ml for 6 days and switched to 30 ng/ml M-Csf- and 30 ng/ml Rankl-containing medium. Cultures were assessed in A for TRAP by enzyme histochemistry 5–8 days following exposure to Rankl. Values are means ± S.D.; n = 6 for all data sets except for Notch2Q2319X DMSO at day 5 where n = 5; the right upper panel shows representative culture fields of TRAP-stained multinucleated cells 8 days after the addition of Rankl. B, cultures were assessed for Notch26955C→T (Notch2Q2319X), Hes1, and Acp5 mRNA expression 8 days following exposure to Rankl. Values are means ± S.D.; n = 4 for all data sets. C, cultures were assessed for bone resorption as determined by resorption pit number and area 16 days following exposure to Rankl. Values are means ± S.D.; n = 6 for all data sets in control and in Notch2Q2319X in DMSO or LY450139 (LY). A representative culture showing resorption pits is presented in the right lower panel. Cultures were conducted in the presence of LY450139 at 1 μm or DMSO vehicle started at the time of Rankl addition and continued throughout the culture period. *, significantly different between Notch2Q2319X mutant and control cells, p < 0.05. #, significantly different between LY450139 and DMSO, p < 0.05, both two-way analysis of variance with Holm-Sidak post hoc analysis.
To determine the effect of the Notch2Q2319X mutation on bone resorption, bone marrow mononuclear cells from control and Notch2Q2319X mutants were expanded in the presence of M-Csf and seeded on bovine bone slices and cultured in the presence of M-Csf and Rankl. Sixteen days following the addition of Rankl, there was a greater number of pits and resorption area in the context of the Notch2Q2319X mutation (Fig. 7). The effect was partially dependent on the activation of Notch because the number of pits, but not the resorption area, was significantly lower in Notch2Q2319X cells in the presence of the γ-secretase inhibitor LY450139 (Fig. 7). An increase in bone resorption was observed in the presence of LY450139 alone. This may represent the inhibition of other signals targeted by γ-secretase because the inhibitor is not specific for Notch2 activation. It is possible that LY450139 inhibited Notch1 activation which, in contrast to Notch2, has been shown to decrease bone resorption (13).
Discussion
Our findings indicate that a global Notch2Q2319X gain-of-function mutation causes osteopenia affecting both cancellous and cortical bone. The phenotype appeared as early as 1 month of age and was accompanied by a shortening of the femoral length suggesting a possible effect on endochondral bone formation and an influence by the Notch2Q2319X mutation on skeletal development. The phenotype of 1-month-old mice harboring the Notch2Q2319X mutation could be attributed to an increase in osteoclast number and bone resorption. It is of interest that there was a subsequent increase in osteoblast number and mineral apposition rate observed in 3-month-old male, but not female, mutant mice possibly representing a delayed compensatory response or increased bone remodeling. This response may explain a partial recovery in cancellous and cortical bone architecture. Although 1-month-old female mice also exhibited a cancellous bone osteopenic phenotype, it was less pronounced than in male mice, but as female mice matured the osteopenia became more evident. Biochemical markers of bone turnover were not affected in Notch2Q2319X mutant mice, but this can be explained by limited sensitivity of the assays and variability of results (63, 64).
The phenotype of the Notch2Q2319X mutant mouse recapitulates aspects of Hajdu Cheney syndrome, a progressive disorder characterized by a high degree of phenotypical pleiotropy. Some of the facial features of the syndrome appear in the first few months of life, but the clinical manifestations of the syndrome, including osteoporosis and acral osteolysis, are progressive and more evident during adolescence and adulthood (68, 69). It is of interest that Notch2Q2319X mutant Hajdu Cheney mice did not exhibit detectable acral osteolysis or obvious neurological manifestations reported in humans affected by the disease. However, our work is limited to the study of young mice and additional phenotypic manifestations may appear in aging mice.
The phenotype observed in Hajdu Cheney Notch2Q2319X mutant mice is distinct from the one reported following the activation of Notch1 in osteoblasts (7, 16). Notch1 activation in immature and mature osteoblasts leads to pronounced osteopenia due to an arrest of osteoblast maturation and function. Because the Notch2Q2319X global mutation affects all cell lineages, an osteopenic phenotype secondary to a decrease in osteoblast number or function was conceivable. However, there was no evidence that Hajdu Cheney mutants had decreased osteoblast number or function in cancellous bone, and in vitro experiments revealed no changes in the expression of osteoblast gene markers in cultures of calvarial osteoblasts from Notch2Q2319X mutants. Importantly, the phenotype of 1-month-old mice revealed a lack of a bone-forming response to enhanced bone resorption, possibly invoking a direct or indirect inhibition of this process under conditions of Notch2 activation. In this context, we have found that Notch2 induces Nfatc2 in skeletal cells, and Nfatc2 suppresses osteoblast differentiation and function and may contribute to the uncoupling of a bone-forming response to the increase in bone resorption (70, 71).3 Histomorphometric analysis of cortical bone confirmed an increase in osteoclast number and bone resorption in the endocortical surface of 1-month-old Notch2Q2319X mutants.
The osteopenic phenotype of the Hajdu Cheney Notch2Q2319X mutant mouse can be explained by an increase in bone resorption and osteoclastogenesis, an effect that has been reported selectively for Notch2 but not for Notch1 (13, 20). Notch2 enhances osteoclastogenesis by interacting with NF-κB on Nfatc1 regulatory elements in cells of the osteoclast lineage (20). A recent study conducted in Rbpjκ null mice demonstrated that Rbpjκ inhibits osteoclastogenesis, an effect similar to that reported for Notch1 (72). This would suggest that canonical Notch signaling is responsible for the inhibitory effect of Notch1 on osteoclastogenesis because Rbpjκ is required for the activation of this signaling pathway. It may also suggest that the induction of osteoclastogenesis by Notch2 operates by non-canonical signaling pathways and that the direct interactions between the Notch2 intracellular domain and NF-κB on the Nfatc1 promoter do not require activation of Notch canonical signaling. It is also possible that interactions between the Notch2 intracellular domain and Rbpjκ are distinct from those of Notch1, and they may result in different downstream events than those reported for Notch1 (73). It is of interest that Notch2, like Notch1, induced the expression of the canonical target genes Hes1, Hey1, Hey2, and HeyL in skeletal cells indicating a capacity to activate Notch canonical signaling. However, induction of Notch target genes may simply represent activation of Notch signaling and not necessarily imply that the canonical target genes are responsible for the effects observed. Although there was a non-preferential increase in canonical Notch target genes, Hey1, Hey2, and HeyL in Notch2Q2319X mutant osteoblasts, this was not the case in cells of the osteoclast lineage that expressed Hes1 but not Hey1, Hey2, and HeyL. Confirming previous work, the induction of Hes1 in osteoclasts was dependent not only on Notch2 activation but also on the degree of osteoclast maturation (20). Moreover, the phenotype observed in Notch2Q2319X male mice is in accordance with the known effects of Hes1 on osteoclastogenesis and bone resorption (74). This may suggest that Hes1 is responsible for selected actions of Notch2 or of the Notch2Q2319X mutants in the skeleton.
Although these studies demonstrate induction of Notch target gene mRNA and transactivation, both representing enhanced Notch activation by the Notch2Q2319X mutation, we were not able to determine the mechanism responsible for the gain-of-function. Technical difficulties prevented us from determining Notch2 protein levels and demonstrating a more stable Notch2 protein product in Notch2Q2319X mutant cells. This is because antibodies to detect cleaved Notch2 are not available (75). We suggest that stabilization of the truncated Notch2 protein is the cause of enhanced Notch2 signaling in mice. The mutation in the mouse, like in humans, was created upstream from the PEST domain, which is required for protein degradation (76). Moreover, mutations in the same region of exon 34 of either Notch1 or Notch2 are associated with Notch gain-of-function and signal activation (66, 78, 79). Another limitation of the studies presented is the use of a global knock-in Notch2Q2319X mutation. Although the intent was to reproduce the human syndrome, we cannot exclude systemic effects of the mutant Notch2Q2319X on the skeleton.
It is important to note that Notch operates by distinct mechanisms in different cellular compartments. In bone Notch2Q2319X mutants expressed increased levels of Rankl without changes in osteoprotegerin expression so that the Rankl/osteoprotegerin ratio was increased at the mRNA level, possibly contributing to the resorptive phenotype observed. It is of interest that Rankl expression was increased in both osteoblasts and osteocytes of mutant mice, and this increase was dependent on Notch activation because it was not observed in osteocyte-rich cultures treated with a γ-secretase inhibitor. The increased number of osteoclasts and bone resorption may be secondary to a diversity of mechanisms in addition to the enhanced Rankl expression. The osteoclast cell precursor pool was increased in Notch2Q2319X mutants by ∼25%. Moreover, their capacity to differentiate into mature osteoclasts, capable of resorbing bone, in response to Rankl was enhanced, and these mechanisms serve to explain the increased osteoclast number and bone resorption in Notch2Q2319X mutants. These results are consistent with the reported stimulatory effects of Notch2 on osteoclastogenesis and are congruent with a mutation causing a Notch2 gain-of-function.
There was no change in the number of osteocytes in either cancellous or cortical bone of Notch2Q2319X mutants, and caspase 3 expression in osteocytes was not increased, suggesting that osteocyte apoptosis was not affected and probably not responsible for the increased cancellous and cortical bone resorption. Although osteocytes are a rich source of Rankl and osteocyte apoptosis precedes osteoclast recruitment during unloading, inhibition of osteocyte apoptosis prevents the increase in Rankl but does not stop bone resorption suggesting that additional mechanisms play a role in the regulation of bone resorption by these cells (77, 80–82). The cortical porosity observed is likely secondary to the increased osteoclastogenesis and bone resorption in Notch2Q2319X mutants.
Although one needs to be cautious with the extrapolation of these results to human disease, an increase in bone resorption could explain the pronounced osteoporosis suffered by subjects with Hajdu Cheney syndrome. The osteolytic lesions observed in these patients reflect a localized resorptive event as well as an inflammatory process (68). If enhanced bone resorption is responsible for the disease, anti-resorptive therapy could prove beneficial to patients with Hajdu Cheney syndrome; however, clinical data on its effectiveness are sparse. Although enhanced bone resorption was observed, additional undiscovered mechanisms may contribute to the bone loss.
In conclusion, Notch2Q2319X mutant mice replicating the mutation found in subjects with Hajdu Cheney syndrome exhibit marked osteopenia; enhanced osteoclastogenesis and bone resorption are in part responsible for the phenotype observed.
Author Contributions
E. C. designed the research studies, analyzed the data, and wrote the manuscript. L. S. conducted the experiments. S. P. Y, designed and created the mouse model. K. L. conducted the experiments. S. Z. acquired and analyzed the data, conducted the experiments, and edited the manuscript.
Acknowledgments
We thank J. Lian for Bglap cDNA, D. Srivastava for HeyL cDNA; A. Rao for Nfatc1 cDNA; R. Wu for Gapdh cDNA; L. J. Strobl for 12×CSL-Luc reporter construct; M. M. Maier for Hey1-Luc promoter construct; T. Iso for Hey2-Luc promoter construct and Hey1 and Hey2 cDNAs; Dr. Lorenzo for helpful advice and for providing bovine bone slices for resorption assays; Allison Kent and David Bridgewater for technical assistance; and Mary Yurczak for secretarial support.
This work was supported by grant DK045227 from the National Institute of Diabetes and Digestive and Kidney Diseases. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.
S. Zanotti and E. Canalis, unpublished observations.
- NICD
- Notch intracellular domain
- Alpl
- alkaline phosphatase
- αMEM
- α-minimum essential medium
- μCT
- microcomputed tomography
- neo
- neomycin
- NF
- nuclear factor
- qRT-PCR
- quantitative reverse transcription-PCR
- Rankl
- receptor activator of NF-κB ligand
- Rbpjκ
- recombination signal binding protein for immunoglobulin κJ region
- TRAP
- tartrate resistant acid phosphatase.
References
- 1. Fortini M. E. (2009) Notch signaling: the core pathway and its posttranslational regulation. Dev. Cell 16, 633–647 [DOI] [PubMed] [Google Scholar]
- 2. Mumm J. S., and Kopan R. (2000) Notch signaling: from the outside in. Dev. Biol. 228, 151–165 [DOI] [PubMed] [Google Scholar]
- 3. Sahlgren C., and Lendahl U. (2006) Notch signaling and its integration with other signaling mechanisms. Regen. Med. 1, 195–205 [DOI] [PubMed] [Google Scholar]
- 4. Zanotti S., and Canalis E. (2010) Notch and the skeleton. Mol. Cell. Biol. 30, 886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Engin F., Yao Z., Yang T., Zhou G., Bertin T., Jiang M. M., Chen Y., Wang L., Zheng H., Sutton R. E., Boyce B. F., and Lee B. (2008) Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 14, 299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hilton M. J., Tu X., Wu X., Bai S., Zhao H., Kobayashi T., Kronenberg H. M., Teitelbaum S. L., Ross F. P., Kopan R., and Long F. (2008) Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 14, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zanotti S., Smerdel-Ramoya A., Stadmeyer L., Durant D., Radtke F., and Canalis E. (2008) Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology 149, 3890–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zanotti S., and Canalis E. (2012) Notch regulation of bone development and remodeling and related skeletal disorders. Calcif. Tissue Int. 90, 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kovall R. A. (2008) More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene 27, 5099–5109 [DOI] [PubMed] [Google Scholar]
- 10. Nam Y., Sliz P., Song L., Aster J. C., and Blacklow S. C. (2006) Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 124, 973–983 [DOI] [PubMed] [Google Scholar]
- 11. Schroeter E. H., Kisslinger J. A., and Kopan R. (1998) Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 [DOI] [PubMed] [Google Scholar]
- 12. Wilson J. J., and Kovall R. A. (2006) Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell 124, 985–996 [DOI] [PubMed] [Google Scholar]
- 13. Bai S., Kopan R., Zou W., Hilton M. J., Ong C. T., Long F., Ross F. P., and Teitelbaum S. L. (2008) NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J. Biol. Chem. 283, 6509–6518 [DOI] [PubMed] [Google Scholar]
- 14. Dallas D. J., Genever P. G., Patton A. J., Millichip M. I., McKie N., and Skerry T. M. (1999) Localization of ADAM10 and Notch receptors in bone. Bone 25, 9–15 [DOI] [PubMed] [Google Scholar]
- 15. Pereira R. M., Delany A. M., Durant D., and Canalis E. (2002) Cortisol regulates the expression of Notch in osteoblasts. J. Cell. Biochem. 85, 252–258 [DOI] [PubMed] [Google Scholar]
- 16. Canalis E., Parker K., Feng J. Q., and Zanotti S. (2013) Osteoblast lineage-specific effects of Notch activation in the skeleton. Endocrinology 154, 623–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zanotti S., Smerdel-Ramoya A., and Canalis E. (2011) Reciprocal regulation of notch and nuclear factor of activated T-cells (NFAT)c1 transactivation in osteoblasts. J. Biol. Chem. 286, 4576–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Canalis E., Adams D. J., Boskey A., Parker K., Kranz L., and Zanotti S. (2013) Notch signaling in osteocytes differentially regulates cancellous and cortical bone remodeling. J. Biol. Chem. 288, 25614–25625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tu X., Chen J., Lim J., Karner C. M., Lee S. Y., Heisig J., Wiese C., Surendran K., Kopan R., Gessler M., and Long F. (2012) Physiological notch signaling maintains bone homeostasis via RBPjk and Hey upstream of NFATc1. PLoS Genet. 8, e1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fukushima H., Nakao A., Okamoto F., Shin M., Kajiya H., Sakano S., Bigas A., Jimi E., and Okabe K. (2008) The association of Notch2 and NF-κB accelerates RANKL-induced osteoclastogenesis. Mol. Cell. Biol. 28, 6402–6412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conlon R. A., Reaume A. G., and Rossant J. (1995) Notch1 is required for the coordinate segmentation of somites. Development 121, 1533–1545 [DOI] [PubMed] [Google Scholar]
- 22. Swiatek P. J., Lindsell C. E., del Amo F. F., Weinmaster G., and Gridley T. (1994) Notch1 is essential for postimplantation development in mice. Genes Dev. 8, 707–719 [DOI] [PubMed] [Google Scholar]
- 23. Weinmaster G., Roberts V. J., and Lemke G. (1992) Notch2: a second mammalian Notch gene. Development 116, 931–941 [DOI] [PubMed] [Google Scholar]
- 24. Bigas A., Martin D. I., and Milner L. A. (1998) Notch1 and Notch2 inhibit myeloid differentiation in response to different cytokines. Mol. Cell. Biol. 18, 2324–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheney W. D. (1965) Acro-Osteolysis. Am. J. Roentgenol. Radium. Ther. Nucl. Med. 94, 595–607 [PubMed] [Google Scholar]
- 26. Hajdu N., and Kauntze R. (1948) Cranio-skeletal dysplasia. Br. J. Radiol. 21, 42–48 [DOI] [PubMed] [Google Scholar]
- 27. Gray M. J., Kim C. A., Bertola D. R., Arantes P. R., Stewart H., Simpson M. A., Irving M. D., and Robertson S. P. (2012) Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome. Eur. J. Hum. Genet. 20, 122–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Isidor B., Lindenbaum P., Pichon O., Bézieau S., Dina C., Jacquemont S., Martin-Coignard D., Thauvin-Robinet C., Le Merrer M., Mandel J. L., David A., Faivre L., Cormier-Daire V., Redon R., and Le Caignec C. (2011) Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat. Genet. 43, 306–308 [DOI] [PubMed] [Google Scholar]
- 29. Majewski J., Schwartzentruber J. A., Caqueret A., Patry L., Marcadier J., Fryns J. P., Boycott K. M., Ste-Marie L. G., McKiernan F. E., Marik I., Van Esch H., FORGE Canada Consortium, Michaud J. L., and Samuels M. E. (2011) Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 32, 1114–1117 [DOI] [PubMed] [Google Scholar]
- 30. Simpson M. A., Irving M. D., Asilmaz E., Gray M. J., Dafou D., Elmslie F. V., Mansour S., Holder S. E., Brain C. E., Burton B. K., Kim K. H., Pauli R. M., Aftimos S., Stewart H., Kim C. A., et al. (2011) Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 43, 303–305 [DOI] [PubMed] [Google Scholar]
- 31. Zhao W., Petit E., Gafni R. I., Collins M. T., Robey P. G., Seton M., Miller K. K., and Mannstadt M. (2013) Mutations in NOTCH2 in patients with Hajdu-Cheney syndrome. Osteoporos. Int. 24, 2275–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Udell J., Schumacher H. R. Jr., Kaplan F., and Fallon M. D. (1986) Idiopathic familial acroosteolysis: histomorphometric study of bone and literature review of the Hajdu-Cheney syndrome. Arthritis Rheum. 29, 1032–1038 [DOI] [PubMed] [Google Scholar]
- 33. Blumenauer B. T., Cranney A. B., and Goldstein R. (2002) Acro-osteolysis and osteoporosis as manifestations of the Hajdu-Cheney syndrome. Clin. Exp. Rheumatol. 20, 574–575 [PubMed] [Google Scholar]
- 34. Brown D. M., Bradford D. S., Gorlin R. J., Desnick R. J., Langer L. O., Jowsey J., and Sauk J. J. (1976) The acro-osteolysis syndrome: morphologic and biochemical studies. J. Pediatr. 88, 573–580 [DOI] [PubMed] [Google Scholar]
- 35. Avela K., Valanne L., Helenius I., and Makitie O. (2011) Hajdu-Cheney syndrome with severe dural ectasia. Am. J. Med. Genet. 155, 595–598 [DOI] [PubMed] [Google Scholar]
- 36. Tang S. H., Silva F. J., Tsark W. M., and Mann J. R. (2002) A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis 32, 199–202 [DOI] [PubMed] [Google Scholar]
- 37. Bouxsein M. L., Boyd S. K., Christiansen B. A., Guldberg R. E., Jepsen K. J., and Müller R. (2010) Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 25, 1468–1486 [DOI] [PubMed] [Google Scholar]
- 38. Glatt V., Canalis E., Stadmeyer L., and Bouxsein M. L. (2007) Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J. Bone Miner. Res. 22, 1197–1207 [DOI] [PubMed] [Google Scholar]
- 39. Dempster D. W., Compston J. E., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., Recker R. R., and Parfitt A. M. (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parfitt A. M., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., and Recker R. R. (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2, 595–610 [DOI] [PubMed] [Google Scholar]
- 41. Hefley T., Cushing J., and Brand J. S. (1981) Enzymatic isolation of cells from bone: cytotoxic enzymes of bacterial collagenase. Am. J. Physiol. 240, C234–C238 [DOI] [PubMed] [Google Scholar]
- 42. Zanotti S., Kalajzic I., Aguila H. L., and Canalis E. (2014) Sex and genetic factors determine osteoblastic differentiation potential of murine bone marrow stromal cells. PLoS ONE 9, e86757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strobl L. J., Höfelmayr H., Stein C., Marschall G., Brielmeier M., Laux G., Bornkamm G. W., and Zimber-Strobl U. (1997) Both Epstein-Barr viral nuclear antigen 2 (EBNA2) and activated Notch1 transactivate genes by interacting with the cellular protein RBP-Jκ. Immunobiology 198, 299–306 [DOI] [PubMed] [Google Scholar]
- 44. Maier M. M., and Gessler M. (2000) Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem. Biophys. Res. Commun. 275, 652–660 [DOI] [PubMed] [Google Scholar]
- 45. Iso T., Chung G., Hamamori Y., and Kedes L. (2002) HERP1 is a cell type-specific primary target of Notch. J. Biol. Chem. 277, 6598–6607 [DOI] [PubMed] [Google Scholar]
- 46. Halleux C., Kramer I., Allard C., and Kneissel M. (2012) Isolation of mouse osteocytes using cell fractionation for gene expression analysis. Methods Mol. Biol. 816, 55–66 [DOI] [PubMed] [Google Scholar]
- 47. Borgegard T., Juréus A., Olsson F., Rosqvist S., Sabirsh A., Rotticci D., Paulsen K., Klintenberg R., Yan H., Waldman M., Stromberg K., Nord J., Johansson J., Regner A., Parpal S., et al. (2012) First and second generation γ-secretase modulators (GSMs) modulate amyloid-β (Aβ) peptide production through different mechanisms. J. Biol. Chem. 287, 11810–11819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mun S. H., Oh D., and Lee S. K. (2014) Macrophage migration inhibitory factor down-regulates the RANKL-RANK signaling pathway by activating Lyn tyrosine kinase in mouse models. Arthritis Rheumatol. 66, 2482–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mun S. H., Won H. Y., Hernandez P., Aguila H. L., and Lee S. K. (2013) Deletion of CD74, a putative MIF receptor, in mice enhances osteoclastogenesis and decreases bone mass. J. Bone Miner. Res. 28, 948–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jacome-Galarza C. E., Lee S. K., Lorenzo J. A., and Aguila H. L. (2011) Parathyroid hormone regulates the distribution and osteoclastogenic potential of hematopoietic progenitors in the bone marrow. J. Bone Miner. Res. 26, 1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aguila H. L., Mun S. H., Kalinowski J., Adams D. J., Lorenzo J. A., and Lee S. K. (2012) Osteoblast-specific overexpression of human interleukin-7 rescues the bone mass phenotype of interleukin-7-deficient female mice. J. Bone Miner. Res. 27, 1030–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nazarenko I., Pires R., Lowe B., Obaidy M., and Rashtchian A. (2002) Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes. Nucleic Acids Res. 30, 2089–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nazarenko I., Lowe B., Darfler M., Ikonomi P., Schuster D., and Rashtchian A. (2002) Multiplex quantitative PCR using self-quenched primers labeled with a single fluorophore. Nucleic Acids Res. 30, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lian J., Stewart C., Puchacz E., Mackowiak S., Shalhoub V., Collart D., Zambetti G., and Stein G. (1989) Structure of the rat osteocalcin gene and regulation of vitamin D-dependent expression. Proc. Natl. Acad. Sci. U.S.A. 86, 1143–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Iso T., Sartorelli V., Chung G., Shichinohe T., Kedes L., and Hamamori Y. (2001) HERP, a new primary target of Notch regulated by ligand binding. Mol. Cell. Biol. 21, 6071–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nakagawa O., Nakagawa M., Richardson J. A., Olson E. N., and Srivastava D. (1999) HRT1, HRT2, and HRT3: a new subclass of bHLH transcription factors marking specific cardiac, somitic, and pharyngeal arch segments. Dev. Biol. 216, 72–84 [DOI] [PubMed] [Google Scholar]
- 57. Glinka A., Wu W., Delius H., Monaghan A. P., Blumenstock C., and Niehrs C. (1998) Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 391, 357–362 [DOI] [PubMed] [Google Scholar]
- 58. Monticelli S., and Rao A. (2002) NFAT1 and NFAT2 are positive regulators of IL-4 gene transcription. Eur. J. Immunol. 32, 2971–2978 [DOI] [PubMed] [Google Scholar]
- 59. Kutyavin I. V., Afonina I. A., Mills A., Gorn V. V., Lukhtanov E. A., Belousov E. S., Singer M. J., Walburger D. K., Lokhov S. G., Gall A. A., Dempcy R., Reed M. W., Meyer R. B., and Hedgpeth J. (2000) 3′-minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 28, 655–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 61. Kouadjo K. E., Nishida Y., Cadrin-Girard J. F., Yoshioka M., and St-Amand J. (2007) Housekeeping and tissue-specific genes in mouse tissues. BMC. Genomics 8, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tso J. Y., Sun X. H., Kao T. H., Reece K. S., and Wu R. (1985) Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res. 13, 2485–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Herrmann M. (2011) in Osteoporosis Research- Animal Models (Duque G., Watanabe K., ed) pp. 57–82, Springer, New York [Google Scholar]
- 64. Seibel M. J. (2005) Biochemical markers of bone turnover: part I: biochemistry and variability. Clin. Biochem. Rev. 26, 97–122 [PMC free article] [PubMed] [Google Scholar]
- 65. Kiel M. J., Velusamy T., Betz B. L., Zhao L., Weigelin H. G., Chiang M. Y., Huebner-Chan D. R., Bailey N. G., Yang D. T., Bhagat G., Miranda R. N., Bahler D. W., Medeiros L. J., Lim M. S., and Elenitoba-Johnson K. S. (2012) Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J. Exp. Med. 209, 1553–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rossi D., Trifonov V., Fangazio M., Bruscaggin A., Rasi S., Spina V., Monti S., Vaisitti T., Arruga F., Famà R., Ciardullo C., Greco M., Cresta S., Piranda D., Holmes A., et al. (2012) The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J. Exp. Med. 209, 1537–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang X., Shi Y., Weng Y., Lai Q., Luo T., Zhao J., Ren G., Li W., Pan H., Ke Y., Zhang W., He Q., Wang Q., and Zhou R. (2014) The truncate mutation of Notch2 enhances cell proliferation through activating the NF-κB signal pathway in the diffuse large B-cell lymphomas. PLoS ONE 9, e108747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Canalis E., and Zanotti S. (2014) Hajdu-Cheney syndrome: a review. Orphanet J. Rare Dis. 9, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Descartes M., Rojnueangnit K., Cole L., Sutton A., Morgan S. L., Patry L., and Samuels M. E. (2014) Hajdu-Cheney syndrome: phenotypical progression with de novo NOTCH2 mutation. Clin. Dysmorphol. 23, 88–94 [DOI] [PubMed] [Google Scholar]
- 70. Zanotti S., and Canalis E. (2015) Activation of Nfatc2 in osteoblasts causes osteopenia. J. Cell. Physiol. 230, 1689–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zanotti S., Smerdel-Ramoya A., and Canalis E. (2013) Nuclear factor of activated T-cells (Nfat)c2 inhibits Notch signaling in osteoblasts. J. Biol. Chem. 288, 624–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li S., Miller C. H., Giannopoulou E., Hu X., Ivashkiv L. B., and Zhao B. (2014) RBP-J imposes a requirement for ITAM-mediated costimulation of osteoclastogenesis. J. Clin. Invest. 124, 5057–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yuan Z., Friedmann D. R., VanderWielen B. D., Collins K. J., and Kovall R. A. (2012) Characterization of CSL (CBF-1, Su(H), Lag-1) mutants reveals differences in signaling mediated by Notch1 and Notch2. J. Biol. Chem. 287, 34904–34916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zanotti S., Smerdel-Ramoya A., and Canalis E. (2011) Hairy and enhancer of split (HES)1 is a determinant of bone mass. J. Biol. Chem. 286, 2648–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Habets R. A., Groot A. J., Yahyanejad S., Tiyanont K., Blacklow S. C., and Vooijs M. (2015) Human NOTCH2 is resistant to ligand-independent activation by metalloprotease Adam17. J. Biol. Chem. 290, 14705–14716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rogers S., Wells R., and Rechsteiner M. (1986) Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234, 364–368 [DOI] [PubMed] [Google Scholar]
- 77. Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., and O'Brien C. A. (2011) Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Weng A. P., Ferrando A. A., Lee W., Morris J. P. 4th, Silverman L. B., Sanchez-Irizarry C., Blacklow S. C., Look A. T., and Aster J. C. (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 [DOI] [PubMed] [Google Scholar]
- 79. Breit S., Stanulla M., Flohr T., Schrappe M., Ludwig W. D., Tolle G., Happich M., Muckenthaler M. U., and Kulozik A. E. (2006) Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood 108, 1151–1157 [DOI] [PubMed] [Google Scholar]
- 80. Aguirre J. I., Plotkin L. I., Stewart S. A., Weinstein R. S., Parfitt A. M., Manolagas S. C., and Bellido T. (2006) Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner. Res. 21, 605–615 [DOI] [PubMed] [Google Scholar]
- 81. Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F., Penninger J. M., and Takayanagi H. (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 [DOI] [PubMed] [Google Scholar]
- 82. Plotkin L. I., Gortazar A. R., Davis H. M., Condon K. W., Gabilondo H., Maycas M., Allen M. R., and Bellido T. (2015) Inhibition of osteocyte apoptosis prevents the increase in osteocytic receptor activator of nuclear factor-κB ligand (RANKL) but does not stop bone resorption or the loss of bone induced by unloading. J. Biol. Chem. 290, 18934–18942 [DOI] [PMC free article] [PubMed] [Google Scholar]