

Background: The role of Gα13 in mediating invasion in three-dimensional collagen is not well known.
Results: Gα13 promotes invasion in three-dimensional collagen by disrupting cell-cell adhesion, whereas DDR1-Par3 signaling counteracts the effects of Gα13 by enhancing cell-cell adhesion.
Conclusion: Gα13 and DDR1-Par3 differentially regulate cell-cell junctions to mediate three-dimensional invasion.
Significance: Better understanding of how cells invade in three-dimensional collagen may identify potential therapeutic targets.
Keywords: cell polarity, collagen, G protein, invasion, metalloprotease
Abstract
Cancer cells can invade in three-dimensional collagen as single cells or as a cohesive group of cells that require coordination of cell-cell junctions and the actin cytoskeleton. To examine the role of Gα13, a G12 family heterotrimeric G protein, in regulating cellular invasion in three-dimensional collagen, we established a novel method to track cell invasion by membrane type 1 matrix metalloproteinase-expressing cancer cells. We show that knockdown of Gα13 decreased membrane type 1 matrix metalloproteinase-driven proteolytic invasion in three-dimensional collagen and enhanced E-cadherin-mediated cell-cell adhesion. E-cadherin knockdown reversed Gα13 siRNA-induced cell-cell adhesion but failed to reverse the effect of Gα13 siRNA on proteolytic invasion. Instead, concurrent knockdown of E-cadherin and Gα13 led to an increased number of single cells rather than groups of cells. Significantly, knockdown of discoidin domain receptor 1 (DDR1), a collagen-binding protein that also co-localizes to cell-cell junctions, reversed the effects of Gα13 knockdown on cell-cell adhesion and proteolytic invasion in three-dimensional collagen. Knockdown of the polarity protein Par3, which can function downstream of DDR1, also reversed the effects of Gα13 knockdown on cell-cell adhesion and proteolytic invasion in three-dimensional collagen. Overall, we show that Gα13 and DDR1-Par3 differentially regulate cell-cell junctions and the actin cytoskeleton to mediate invasion in three-dimensional collagen.
Introduction
Pancreatic ductal adenocarcinoma (PDAC)2 is a highly lethal malignancy, in part because of the presence of distant metastases at the time of diagnosis (1, 2). PDAC tumors in particular demonstrate a pronounced collagen-rich stromal reaction (3, 4) that, as we have shown previously, facilitates tumor invasion by inducing the expression of the key collagenase membrane type 1 matrix metalloproteinase (MT1-MMP) (5–7). Although cancer cells in high-grade tumors may invade as single cells, most low-grade tumors invade as a cohesive group of cells (“collective migration”), which can range from strands of only one or two cells in diameter to a cluster of cells (8). It has been postulated that collective migration requires the regulation of cell-cell junctions to transmit force between cells as well as the coordination of cytoskeletal reorganization within a group of cells to drive organized migration (9). Because collective migration requires cells to maintain intact cell-cell junctions, proteins such as E-cadherin maintain the integrity of cell-cell junctions. The collagen-binding protein discoidin domain receptor 1 (DDR1) has been shown to localize to cell-cell junctions and mediate cell-cell adhesion (10, 11). In addition to regulating E-cadherin stability at the cell surface (12, 13), DDR1, through its interaction with the Par3 polarity complex, can also regulate actin cytoskeleton dynamics (11).
Gα13, a member of the G12 family of heterotrimeric G proteins, activates Rho signaling downstream of G protein-coupled receptor activation (14–16) and has been implicated in cell migration downstream of both G protein-coupled receptors and receptor tyrosine kinases (17). For example, mouse embryonic fibroblasts from Gα13-null mice are deficient in lysophosphatidic acid-stimulated cell migration (18). Gα13, but not the closely related Gα12, also mediates EGF-driven migration (18). In addition to regulating cell migration, Gα13 can weaken adherens junctions by binding to E-cadherin and displacing β-catenin from adherens junctions (19, 20). Recently, Gα13 has also been shown to disrupt endothelial adherens junctions by binding to VE-cadherin and causing VE-cadherin internalization (21). Significantly, Gα13 is up-regulated in metastatic breast and prostate cancers (22, 23) and has been shown to play a role in pancreatic cancer cell invasion (24). However, the extent to which Gα13 contributes to pancreatic cancer cell invasion in three-dimensional collagen is not well known.
In this study, we show that Gα13 is up-regulated in human PDAC tumors and, using a quantitative method to track cell invasion in three-dimensional collagen gels, we demonstrate that knockdown of Gα13 decreased MT1-MMP-driven proteolytic invasion of PDAC cells in three-dimensional collagen and enhanced E-cadherin-mediated cell-cell adhesion. Although E-cadherin knockdown reversed the effect of Gα13 siRNA-induced cell-cell adhesion, E-cadherin knockdown failed to reverse the effect of Gα13 siRNA on proteolytic invasion. Instead, concurrent knockdown of E-cadherin and Gα13 led to an increased number of single cells rather than groups of cells. Significantly, knockdown of DDR1 reversed the effect of Gα13 knockdown on cell-cell adhesion and proteolytic invasion. Additionally, knockdown of the polarity protein Par3, which can function downstream of DDR1, reversed the effect of Gα13 knockdown on cell-cell adhesion and proteolytic invasion. Overall, we show that Gα13 and DDR1-Par3 signaling differentially regulate cell-cell junctions and the actin cytoskeleton to mediate invasion in three-dimensional collagen.
Experimental Procedures
Antibodies and Reagents
General tissue culture supplies were purchased from VWR (West Chester, PA). Antibodies against MT1-MMP (catalog no. ab38971) and Gα13 (catalog no. ab128900) were purchased from Abcam (Cambridge, MA), whereas Gα13 (catalog no. B860) antibody (25) was a gift from Tohru Kozasa. Antibodies against pERK1/2 Thr-202/Tyr-204 (catalog no. 4370P), ERK1/2 (catalog no. 4695S), pMLC2 Ser-19 (catalog no. 3671), and MLC2 (catalog no. 3672) were purchased from Cell Signaling Technology (Danvers, MA), whereas antibodies against E-cadherin for Western blotting and immunofluorescence (catalog no. 610181) and the E-cadherin function-blocking antibody HECD-1 were purchased from BD Biosciences and Invitrogen, respectively. Antibody against DDR1 (catalog no. AF2396) was purchased from R&D Systems (Minneapolis, MN), antibodies against GAPDH (catalog no. MAB374) and Par3 (catalog no. 07330) were purchased from Merck/Millipore (Billerica, MA), and antibody against α-tubulin (catalog no. sc-8035) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). HRP-conjugated secondary antibodies were purchased from Sigma. Alexa Fluor 546 phalloidin (catalog no. A22283) and Alexa Fluor 488-conjugated secondary antibody (catalog no. A11029) were purchased from Life Technologies. The MMP inhibitor GM6001 (catalog no. 561206), EGFR inhibitor (catalog no. 658458), the Rac1 inhibitor NSC23766 (catalog no. 553508), and the ROCK1/2 inhibitor Y27632 (catalog no. 688001) were purchased from Calbiochem (Ellisville, MO), and the MEK1/2 inhibitor U0126 (catalog no. 9903) was purchased from Cell Signaling Technology. EGF (catalog no. E9644) was purchased from Sigma, and doxycycline was purchased from Clontech. SMARTpool siGenome siRNAs against Gα13, E-cadherin, DDR1, and Par3 were purchased from GE Healthcare Dharmacon. Rat tail type I collagen (catalog no. 354249) was purchased from Corning. Collagenase (catalog no. 4196) was purchased from Worthington Biochemical Co.
Oncomine Dataset Analysis
Datasets analyzing mRNA from both normal and PDAC tumor samples were selected from the publicly available Oncomine database.
Human PDAC Tissue Analysis
The human pancreatic tissue microarray (TMA) was generated by the Pathology Core Facility at Northwestern University using specimens obtained on an institutional review board-approved protocol. The TMA was composed of 2-mm cores from 17 PDAC specimens and 15 normal pancreatic tissue specimens. Additional TMA slides (TMA-1421) with 2-mm cores from three normal pancreatic tissue samples and 20 PDAC cases were purchased from Protein Biotechnologies (Ramona, CA). Slides with 4-μm sections of the TMA were deparaffinized and stained with H&E or trichrome-stained using a Masson kit by the Pathology Core Facility (Northwestern University). The extent of fibrosis was determined by the blue trichrome stain in the TMA specimens: <25% of the TMA core with blue stain (low) or >25% of the TMA core with blue stain (high).
For immunostaining, TMA slides were deparaffinized in xylene and rehydrated in a decreasing ethanol gradient, and then antigen retrieval was performed using a decloaking chamber at 110 °C for 10 min in BD Retrievagen A (pH 6.0) target retrieval solution, followed by blocking with Protein Block (Dako) and Peroxidase Block (Dako). The TMA slides were incubated with the primary rabbit monoclonal antibody against Gα13 (EPR5436, catalog no. ab128900, Abcam) overnight at 4 °C at 1:500 dilution in Dako antibody diluent. Following multiple washings with Dako wash buffer, the slides were incubated with anti-rabbit HRP-labeled polymer (Dako) at room temperature for 1 h and incubated with 3,3′-diaminobenzidine substrate (Dako) for 1 min, and then the slides were counterstained with hematoxylin. As specified on the Abcam web site for the ab128900 antibody, positive staining for Gα13 was indicated by the presence of a brown precipitate in the cytoplasm. The TMA cores were evaluated and scored as follows: 0, no staining or background staining; 1+, mild specific staining of malignant cells; 2+, moderate staining of malignant cells; 3+, the most intense staining of malignant cells.
The specificity of the rabbit monoclonal ab128900 antibody for Gα13 was validated by knocking down Gα13 in CD18-tet-MT cells, creating a cell block and then embedding the cell block in paraffin. Control and Gα13 knockdown CD18-tet-MT cell blocks were then sectioned and stained as above for the TMA specimens. In additional controls, human PDAC tumor sections were processed for immunostaining using a rabbit IgG monoclonal antibody (catalog no. ab172730, Abcam) as an isotype control for the anti-Gα13 ab128900 antibody.
Cell Culture
CD18, Panc1, and SCC25 cells were purchased from the ATCC, whereas UMSCC1 cells were provided by Dr. E. Lengyel. MT1-MMP-expressing cancer cells were generated and maintained as described previously (26).
Down-regulation of Gα13, E-cadherin, DDR1, and Par3
Cells were transfected with 25 nm siRNA using Lipofectamine RNAiMAX (Invitrogen). Transfected cells were allowed to recover for 48 h, trypsinized, and plated in collagen or suspended in hanging drops. For immunofluorescence, cells were plated on glass coverslips prior to transfection.
Three-dimensional Collagen Invasion Assay
MT1-MMP-expressing cancer cells were transfected with siRNA, allowed to recover for 48 h, grown in three-dimensional type I collagen gels (2.2 mg/ml) as described previously (7), and then treated with doxycycline and various inhibitors. Cells grown in collagen gels were examined using a Zeiss Axiovert 40 CFL microscope, and pictures were taken with a Nikon Coolpix 4500 camera. Collagen gels were then fixed in 10% formalin for 24 h and embedded in paraffin by the Pathology Core Facility at Northwestern University. Six 8-μm sections from each of the collagen-paraffin blocks were cut from the top of the block to the same depth, stained with H&E, and examined using a Zeiss Axioskop 40 microscope and camera. Areas of cleaved collagen (“collagenolytic tracks”) were measured from a minimum of 10 fields/condition at ×10 magnification using ImageJ.
Two-dimensional Collagen Migration
Migration of PDAC cells over collagen-coated surfaces was assessed as described previously (6). PDAC cells were plated onto thin-layer type I collagen overlaid with colloidal gold. Cells were allowed to migrate for 24 h, phagokinetic tracks were monitored by visual examination using a Zeiss Axiovert 40 CFL microscope, and pictures were taken with a Nikon Coolpix 4500 camera.
Extraction of PDAC Cells from Collagen Gels
PDAC cells were grown in collagen gels as indicated above. After 72 h, cells were extracted from the gels using collagenase and lysed in radioimmune precipitation assay buffer containing protease and phosphatase inhibitors for Western blotting (27, 28).
Cell Surface Biotinylation
CD18-tet-MT cells were labeled with cell-impermeable Sulfo-NHS-LC-Biotin (Pierce) in ice-cold PBS as described previously (29). Equal amounts of protein were then incubated with streptavidin beads (Pierce) to isolate biotinylated cell surface proteins. The biotin-labeled samples were analyzed for MT1-MMP by Western blotting using Transferrin receptor as a labeling control.
Immunoblotting
Immunoblotting was done as described previously (29, 30) and detected by enhanced chemiluminescence Western blotting reagents (Pierce).
Three-dimensional Collagen Contractility Assay
Collagen contractility assays were performed as described previously (31, 32). Briefly, PDAC cells were embedded in three-dimensional collagen (2.2 mg/ml) at 2 × 105 cells/plug (for inhibitor treatment) or 5 × 105 cells/plug (after transfection with siRNA) to form a cylinder-shaped plug. The plug was then inserted in an additional layer of collagen (2.2 mg/ml) to allow the formation of a core of cells surrounded by collagen. After contraction over a period of 5 days, pictures of the contracted cores were taken using an 8-megapixel iPhone 5S camera.
Hanging Drop Assay
PDAC cells were transfected with the indicated siRNAs and suspended in hanging drops as described previously (33). For cells treated with the E-cadherin function-blocking antibody HECD-1, cells were incubated with 2 μm IgG or HECD-1 per drop for 30 min at room temperature prior to suspension in drops. After 18 h, cells were imaged, and pictures were taken using a Zeiss Axiovert 40 CFL microscope and a Nikon Coolpix 4500 camera.
Immunofluorescence Imaging
PDAC cells were plated on glass coverslips and transfected with the indicated siRNAs for 72 h. Cells were fixed in 4% paraformaldehyde and permeabilized with 0.01% Triton X-100 and then stained for E-cadherin and phalloidin. Images were taken on a Zeiss LSM510 Meta confocal microscope at the Northwestern University Center for Advanced Microscopy (Chicago, IL). The relative intensity of E-cadherin at cell-cell junctions was determined using Image J.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 5 (San Diego, CA).
Results
Gα13 Is Up-regulated in Human PDAC Tumors
Cell invasion requires precise coordination of the actin cytoskeleton (31, 34). Because Gα13, the α subunit of the heterotrimeric G protein G13, is a major regulator of the actin cytoskeleton (35, 36), we examined the extent to which Gα13 levels are altered in human PDAC tumors using the publically available Oncomine database (37–40). As shown in Fig. 1A, there is an ∼2-fold increase in GNA13 mRNA levels in human PDAC tumors compared with normal tissue. Furthermore, immunohistochemical staining of human pancreatic TMAs showed that there is increased cytoplasmic staining of Gα13 protein in tumors compared with normal pancreas (Fig. 1, B and C). Because PDAC tumors are associated with a pronounced collagen-rich stromal reaction, as demonstrated by increased trichrome staining (Fig. 1B), we examined the relationship between fibrosis and Gα13 protein expression. We show that Gα13 protein levels are associated with increased fibrosis in human PDAC tumors (Fig. 1C).
FIGURE 1.

Gα13 is up-regulated in human PDAC tumors. A, the relative expression of GNA13 mRNA was evaluated in human PDAC tumors (T) relative to normal (N) pancreas using the publicly available Oncomine database. Only databases that contained at least 10 normal specimens were analyzed. Overall statistical analyses of Oncomine data were performed using Student's t test. ***, p < 0.001. B and C, human pancreatic TMAs generated by the Pathology Core Facility at Northwestern University or purchased from Protein Biotechnologies were H&E-stained, trichrome-stained (blue = fibrosis), or immunostained with anti-Gα13 antibody (B). The relative staining was graded as low (0 or 1+) or high (2+ or 3+) as detailed under “Experimental Procedures.” The relative expression of Gα13 in tumor tissue and adjacent normal was assessed by Fisher's exact test (C). The extent of fibrosis was determined by the blue trichrome stain in the TMA specimens: <25% of the TMA core with blue stain (low) or >25% of the TMA core with blue stain (high). The relationship between Gα13 expression in the tissue samples and the extent of fibrosis was assessed by Fisher's exact test (C). IHC, immunohistochemistry. Scale bars, 50 μm.
Establishment of a Quantitative Method to Track MT1-MMP-dependent Cell Invasion in Three-dimensional Collagen Gels
To examine the role of Gα13 in PDAC progression, we established a quantitative method to track MT1-MMP-dependent cell invasion in three-dimensional collagen gels using inducible expression of MT1-MMP in PDAC cells (hereafter referred to as PDAC-tet-MT cells; Fig. 2, A and B). Induction of MT1-MMP in CD18-tet-MT (Fig. 2C) and Panc1-tet-MT adenocarcinoma cells (data not shown) in three-dimensional type I collagen resulted in tracks generated by clearing of the pericellular collagen by invading cells, which was inhibited by the broad-spectrum MMP inhibitor GM6001. We also examined the extent to which our system can track MT1-MMP-dependent invasion of squamous cell cancer (SCC) cells in three-dimensional collagen. Induction of MT1-MMP in SCC cells (UMSCC1 and SCC25 cells) also resulted in tracks generated by clearing of the pericellular collagen by the invading cells that was also inhibited by GM6001 (data not shown). We additionally validated this quantitative method of tracking cells in three-dimensional collagen by examining the effect of EGF on cell invasion. EGF treatment enhanced the invasion of MT1-MMP-expressing CD18 (Fig. 2D) and Panc1 cells (data not shown). Significantly, there were morphological changes in PDAC-tet-MT cells upon induction of MT1-MMP expression. CD18 cells maintained their cohesive growth with a slight ellipsoid morphology (Fig. 2D) that morphed into a half moon-shaped group of cells following EGF treatment (Fig. 2D). Induction of MT1-MMP in established colonies also caused polarization of CD18-tet-MT cells, as demonstrated by increased numbers of half moon-shaped cell clusters, and increased invasion (Fig. 2E).
FIGURE 2.

Establishment of a quantitative method to track MT1-MMP-dependent cell invasion in three-dimensional collagen gels. A, to evaluate proteolytic invasion in three-dimensional collagen, collagen gels were fixed in formaldehyde (PFA) and embedded in paraffin, and then gel sections were stained with H&E. Proteolytic invasion was then quantified by measuring the cleared collagen using ImageJ. Dox, doxycycline. B, CD18 cells with inducible expression of MT1-MMP (CD18-tet-MT) were generated as detailed under “Experimental Procedures.” Treatment with doxycycline induced expression of both the catalytically active 55-kDa form of MT1-MMP and the inactive 43-kDa form of MT1-MMP. Concurrent treatment with the MMP inhibitor GM6001 (5 μm) blocked autodegradation of the 55-kDa form of MT1-MMP to the 43-kDa form of MT1-MMP. DMSO, dimethyl sulfoxide. C and D, CD18-tet-V and CD18-tet-MT cells were plated in three-dimensional (3D) type I collagen gels (2 mg/ml), treated with doxycycline, co-treated with GM6001 (5 μm) and/or EGF (20 ng/ml), and allowed to grow for 72 h. The effect on cell morphology was examined by phase microscopy, whereas three-dimensional collagen invasion was determined as detailed in A. E, CD18-tet-MT cells were allowed to grow in three-dimensional collagen in the absence of doxycycline for 72 h and then treated with doxycycline and/or EGF (20 ng/ml) for an additional 72 h. The effect on cell morphology and three-dimensional collagen invasion was examined and quantified. The number of cell-clusters demonstrating a half moon shape was also quantified. Statistics were performed using one-way ANOVA followed by Dunnett's post-test. V, vector; MT, MT1-MMP; AU, arbitrary units; ns, not significant; ***, p < 0.001. The results are representative of at least four independent experiments.
Knockdown of Gα13 Decreases MT1-MMP-driven Proteolytic Invasion of Cancer Cells in Three-dimensional Collagen
We next determined whether Gα13 was involved in MT1-MMP-driven invasion in three-dimensional collagen. Initially, we evaluated the effect of Gα13 knockdown on MT1-MMP expression and cell surface localization. Knockdown of Gα13 did not affect the levels of the catalytically active, 55-kDa form or the 43-kDa autodegradation form of MT1-MMP (Fig. 3A). Gα13 knockdown also did not affect cell surface expression of MT1-MMP in CD18-tet-MT cells (Fig. 3A). However, knockdown of Gα13 inhibited both basal and EGF-stimulated invasion of MT1-MMP-expressing CD18 (Fig. 3B) and Panc1 cells (Fig. 3D). Knockdown of Gα13 also inhibited invasion of MT1-MMP-expressing SCC cells (data not shown). In contrast, knockdown of Gα13 did not inhibit two-dimensional migration of PDAC cells over collagen-coated surfaces (Fig. 3C). Moreover, knockdown of Gα13 did not affect proliferation or apoptosis of PDAC cells in three-dimensional collagen (data not shown).
FIGURE 3.

Knockdown of Gα13 decreases MT1-MMP-driven proteolytic invasion of pancreatic cancer cells in three-dimensional collagen. A, CD18-tet-MT cells were transfected with control siRNA or Gα13 siRNA and treated with doxycycline to induce MT1-MMP for 72 h. Cell surface proteins were labeled with biotin as detailed under “Experimental Procedures,” and then biotin-labeled cell surface proteins were isolated using streptavidin beads. Total and cell surface expression of MT1-MMP was determined by Western blotting using Transferrin receptor as the labeling control for cell surface proteins. B, CD18-tet-MT cells transfected with Gα13 siRNA were grown in three-dimensional (3D) collagen for 72 h in the presence of doxycycline with or without exogenous EGF (20 ng/ml). Quantitation of invasion was performed as detailed in Fig. 2A. Dox, doxycycline. C, CD18-tet-MT cells transfected with Gα13 siRNA were plated onto thin-layer type I collagen matrix overlaid with colloidal gold and allowed to migrate in the presence of doxycycline for 24 h, and then the tracks were photographed and quantified. 2D, two-dimensional. D, Panc1 cells with inducible expression of MT1-MMP (Panc1-tet-MT) were generated as detailed under “Experimental Procedures.” Treatment with doxycycline induced expression of MT1-MMP, whereas co-treatment with the MMP inhibitor GM6001 inhibited autodegradation of the catalytically active 55-kDa form of MT1-MMP to the inactive 43-kDa form of MT1-MMP. Panc1-tet-MT cells transfected with siRNA against Gα13 were grown in three-dimensional collagen for 72 h in the presence of doxycycline ± EGF (20 ng/ml), and the effect on invasion was determined as detailed in Fig. 2A. Statistics were performed using one-way ANOVA followed by Dunnett's post-test. V, vector; MT, MT1-MMP; AU, arbitrary units; ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001. The results are representative of at least three independent experiments.
Knockdown of Gα13 Does Not Affect ERK1/2 Phosphorylation but Enhances MLC Phosphorylation and ROCK Activity in Three-dimensional Collagen
To understand the mechanism by which Gα13 regulated MT1-MMP-driven invasion in three-dimensional collagen, we examined the effect of Gα13 knockdown on ERK signaling. Notably, Gα13 knockdown has been shown previously to inhibit ERK1/2 phosphorylation in Jurkat T cells (41). Initially, we examined the extent to which EGFR-ERK1/2 signaling mediated invasion of MT1-MMP-expressing PDAC cells in three-dimensional collagen. Treatment of MT1-MMP-expressing CD18 cells with the EGFR kinase inhibitor AG1478 or the MEK inhibitor U0126 reduced invasion of MT1-MMP-expressing CD18 cells (Fig. 4A). Additionally, cells treated with AG1478 or U0126 blocked the EGF-induced morphological changes, indicating that EGFR-MEK-ERK signaling mediated the invasion of MT1-MMP-expressing CD18 cells. However, Gα13 knockdown in CD18-tet-MT cells did not affect ERK1/2 phosphorylation (Fig. 4B), suggesting that the inhibition of invasion by Gα13 knockdown was not due to its regulation of the ERK signaling pathway.
FIGURE 4.

Knockdown of Gα13 does not affect ERK1/2 phosphorylation but enhances MLC phosphorylation and ROCK activity in three-dimensional collagen. A, CD18-tet-MT cells were plated in collagen, treated with the EGFR inhibitor AG1478 (AG, 5 μm) or the MEK inhibitor U0126 (5 μm), allowed to grow for 72 h in the presence or absence of exogenous EGF (20 ng/ml), and the relative invasion was quantified. Dox, doxycycline; DMSO, dimethyl sulfoxide; 3D, three-dimensional. B, CD18-tet-MT cells were transfected with siRNA for 48 h on tissue culture plastic and then grown in three-dimensional collagen in the presence of doxycycline for 48 h. The effect on phospho-ERK1/2 (pERK1/2) and total ERK1/2 was determined by Western blotting. C, CD18-tet-MT cells were plated in three-dimensional collagen, treated with doxycycline and co-treated with Rac1 inhibitor NSC23766 (NSC, 20 μm) or ROCK1/2 inhibitor Y27632 (10 μm), and allowed to invade for 72 h in the presence or absence of exogenous EGF (20 ng/ml). The effect on collagen invasion was assessed as described in Fig. 2A. AU, arbitrary units. D, CD18-tet-MT cells were transfected with siRNA for 48 h and allowed to grow in three-dimensional collagen for 48 h in the presence of doxycycline, and then the effect on phospho-MLC (pMLC S19) was examined by Western blotting. E, CD18-tet-MT cells were plated in a plug of three-dimensional collagen and treated with doxycycline, the pan-MMP inhibitor GM6001 (5 μm), and/or EGF (20 ng/ml) as indicated. The ability of the cells to contract the collagen gels over 5 days was examined. The contracted gel area was measured using ImageJ. F, CD18-tet-MT cells were plated in a plug of three-dimensional collagen and treated with doxycycline and the Rac inhibitor NSC23766 (20 μm) or the ROCK1/2 inhibitor Y27632 (10 μm). The ability of the cells to contract the collagen gels over 5 days was examined. The contracted gel area was measured using ImageJ. G, CD18-tet-MT cells were transfected with siRNA against Gα13 for 48 h, embedded in a plug of three-dimensional collagen, and treated with doxycycline and the indicated reagents, and then the ability of the cells to contract the collagen gels over 3 days was examined. The contracted gel area was measured using ImageJ. Invasion quantitation statistics were performed using one-way ANOVA followed by Dunnett's post-test. Contractility assay statistics were performed using Student's t test. ns, not significant; ***, p < 0.001. The results are representative of three independent experiments.
Because a balance of Rho and Rac activity is required for the efficient invasion of cells (6, 42), we investigated the role of Rho and Rac signaling in regulating invasion in three-dimensional collagen using NSC23766, which inhibits the interaction between Rac1 and its guanine nucleotide exchange factor Tiam1 and the ROCK1/2 inhibitor Y27632 (43, 44). As shown in Fig. 4C, Y27632, but not NSC23766, blocked basal and EGF-driven invasion of MT1-MMP-expressing CD18 cells, indicating that ROCK1/2 signaling mediates the invasion of PDAC cells in three-dimensional collagen. However, Gα13 knockdown in three-dimensional collagen did not decrease ROCK1/2 activity, as measured by myosin light chain phosphorylation (phospho-MLC). Instead, Gα13 knockdown in three-dimensional collagen increased MLC phosphorylation (Fig. 4D). Because this was an unexpected result, we further evaluated the effect of Gα13 knockdown on ROCK1/2 activity using a collagen contraction assay. Because both MT1-MMP proteolytic activity and ROCK1/2 signaling mediate the contraction of collagen gels by PDAC cells (Fig. 4, E and F) (31), we examined the effect of Gα13 knockdown on collagen contraction by CD18-tet-MT cells. Gα13 knockdown enhanced collagen contraction, and this effect could be reversed by Y27632 (Fig. 4G), indicating that Gα13 knockdown in three-dimensional collagen increases ROCK1/2 activity. This suggests that the inhibitory effect of Gα13 knockdown on three-dimensional collagen invasion cannot be due to its effect on ROCK signaling.
Gα13 Knockdown Enhances E-cadherin-mediated Cell-Cell Adhesion, but E-cadherin Knockdown Fails to Reverse the Effect of Gα13 siRNA on Proteolytic Invasion
It has been shown previously that Gα13 can serve as a negative regulator of cadherin function by directly binding to the intracellular domain of E- and VE-cadherin (20, 21). Therefore, we examined the effect of Gα13 siRNA on cell-cell adhesion of MT1-MMP-expressing CD18 cells utilizing a hanging drop assay, which allows for analysis of cell-cell adhesion independent of cell matrix adhesion (33). As shown in Fig. 5A, MT1-MMP-expressing CD18 cells transfected with control siRNA formed spheroids in the hanging drop assay. Significantly, transfection with Gα13 siRNA caused more compact spheroids, indicating that Gα13 siRNA enhanced cell-cell adhesion. Although the spheroids formed by control siRNA-transfected cells became larger and less tightly packed after treatment with Y27632, treatment with Y27632 failed to loosen the compact spheroids formed by Gα13 siRNA-transfected cells, suggesting that the effect of knocking down Gα13 on cell-cell adhesion was independent of its ability to regulate ROCK signaling. In contrast, addition of the function-blocking E-cadherin antibody HECD1 in the hanging drop assay significantly reversed Gα13 siRNA-induced compaction of the MT1-MMP-expressing CD18 spheroids (Fig. 5B), suggesting that the increased cell-cell adhesion observed after knockdown of Gα13 was mediated through E-cadherin. Consistent with our findings in the hanging drop assay, E-cadherin localization was increased at cell-cell junctions in cells transfected with siRNA against Gα13 compared with control siRNA-transfected cells (Fig. 5C). Importantly, there was no significant change in either the E-cadherin mRNA (data not shown) or the E-cadherin protein levels (Fig. 5D) following knockdown of Gα13, indicating that the effects of Gα13 on E-cadherin-mediated cell-cell junctions occurs at the posttranslational level.
FIGURE 5.

Gα13 knockdown enhances E-cadherin-mediated cell-cell adhesion, but E-cadherin knockdown fails to reverse the effect of Gα13 siRNA on proteolytic invasion. A, CD18-tet-MT cells were transfected with siRNA for 96 h and then trypsinized and resuspended (2.5 × 106 cells/ml of DMEM + 10% FBS +doxycycline) in the presence or absence of Y27632 (10 μm). A 10-μl drop of the cell suspension was hung on the undersurface of a tissue culture lid, incubated overnight at 37 °C, and photographed the next morning. The area covered by the cluster of cells was measured using ImageJ. B, CD18-tet-MT cells were transfected with siRNA for 96 h and then trypsinized and processed for the hanging drop assay in the presence of IgG or the function-blocking E-cadherin antibody HECD1. Hanging drops were incubated overnight at 37 °C and photographed the next morning. The area covered by the cluster of cells was measured using ImageJ. C, CD18-tet-MT cells were plated on glass coverslips, transfected with siRNA against Gα13, grown in the presence of doxycycline for 48 h, and fixed with 4% paraformaldehyde and permeabilized with 0.1% TritonX-100. Cells were stained for E-cadherin and phalloidin and imaged using a Zeiss Axiovert LSM510 Meta confocal microscope. The relative fluorescence intensity at cell-cell junctions was quantified using ImageJ. D and E, CD18-tet-MT cells were transfected with siRNA against Gα13, E-cadherin, or both Gα13 and E-cadherin. The effect on E-cadherin and Gα13 knockdown was determined by Western blotting. Prior to plating cells in three-dimensional (3D) collagen, cells were incubated with IgG or the E-cadherin (Ecad) function-blocking antibody HECD1. Cells were grown in three-dimensional collagen for 72 h with doxycycline in the presence of EGF (20 ng/ml). Gels were then processed to examine the effect on invasion as detailed in Fig. 2, and relative invasion was quantified (D). The number of single cells present in three-dimensional collagen per field in the invasion assay was quantified (E, left panel). CD18-tet-MT cells were plated on glass coverslips, transfected with siRNA against Gα13 and/or E-cadherin, grown in the presence of doxycycline for 48 h, fixed, permeabilized, and stained for E-cadherin and phalloidin (E, right panel). The statistics of the invasion assay were performed using one-way ANOVA followed by Dunnett's post-test. The statistics of the hanging drop assay were performed using Student's t test. ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001. The results are representative of four independent experiments. Ctrl, control.
We next examined the role of E-cadherin downstream of Gα13 in mediating PDAC cell invasion in three-dimensional collagen. We hypothesized that inhibiting E-cadherin-mediated cell-cell junctions in Gα13 siRNA-transfected cells would restore proteolytic invasion in three-dimensional collagen. However, concurrent knockdown of E-cadherin and Gα13 failed to increase proteolytic invasion of MT1-MMP-expressing CD18 cells in three-dimensional collagen (Fig. 5D). Significantly, concurrent knockdown of E-cadherin and Gα13 led to an increased number of single cells rather than groups of cells (Fig. 5E). Concurrent knockdown of Gα13 and E-cadherin led to dissociation of cell clusters grown on glass coverslips, which was associated with changes in phalloidin organization (Fig. 5E).
DDR1 Knockdown Reverses the Effect of Gα13 Knockdown on Cell-Cell Adhesion and Proteolytic Invasion
Because the collagen-binding protein DDR1 can regulate E-cadherin stability at cell-cell junctions and regulate actin cytoskeleton dynamics in skin cancer cells to regulate group invasion (11), we initially evaluated the extent to which DDR1 levels are altered in human PDAC tumors using the same Oncomine databases as described in Fig. 1A (37–40). As shown in Fig. 6A, there were increased DDR1 mRNA levels in human PDAC tumors compared with normal tissue. We next evaluated the extent to which DDR1 counteracts the effects of Gα13 in PDAC cells. As shown in Fig. 6B, cells co-transfected with siRNA against Gα13 and DDR1 formed looser clusters, suggesting that knockdown of DDR1 can reverse the effects of Gα13 siRNA on cell-cell adhesion. We next evaluated whether the effect of DDR1 on cell-cell adhesion was due to changes in E-cadherin expression or localization. Knockdown of DDR1 in MT1-MMP-expressing CD18 cells led to disruption of cell-cell junctions that was associated with diffuse localization of E-cadherin (Fig. 6C) but did not affect E-cadherin protein levels (Fig. 6C). Concurrent knockdown of Gα13 and DDR1 led to a further disruption of cell-cell junctions and changes in phalloidin staining at the cell periphery (Fig. 6C).
FIGURE 6.

DDR1 knockdown reverses the effect of Gα13 knockdown on cell-cell adhesion and proteolytic invasion. A, the relative expression of DDR1 mRNA was evaluated in human PDAC tumors relative to normal pancreas using the Oncomine database described in Fig. 1A. Overall statistics of Oncomine data were performed using Student's t test. **, p < 0.01. N, normal; T, tumor. B, CD-18-tet-MT cells were transfected with siRNA against Gα13, DDR1, or both Gα13 and DDR1. The effect on DDR1 and Gα13 knockdown was determined by Western blotting. The cells were also plated in suspension in a hanging drop for 18 h in the presence of doxycycline. Quantitation of the area of cell clusters was performed using ImageJ as detailed in Fig. 5. C, CD18-tet-MT cells were plated on glass coverslips, transfected with siRNA against Gα13 and/or DDR1, grown in the presence of doxycycline for 48 h, fixed, permeabilized, and stained for E-cadherin and phalloidin (left panel). CD18-tet-MT cells were transfected with siRNA against Gα13 and/or DDR1 for 48 h, plated in three-dimensional collagen in the presence of doxycycline, and allowed to grow for 48 h. Cell lysates were then analyzed for E-cadherin expression by Western blotting (right panel). D, CD18-tet-MT cells transfected with siRNA against Gα13 and/or DDR1 were grown in three-dimensional collagen in the presence of doxycycline and allowed to invade for 72 h. Quantitation of invasion was analyzed as detailed in Fig. 2A. The number of cell clusters demonstrating a half moon shape was also quantified. The statistics of the invasion assay and the number of half moon cell clusters were performed using one-way ANOVA followed by Dunnett's post-test. The statistics of the hanging drop assay were performed using Student's t test. ns, not significant; *, p < 0.05; ***, p < 0.001. The results are representative of at least three independent experiments. Ctrl, control.
We next examined the role of DDR1 downstream of Gα13 in mediating PDAC cell invasion in three-dimensional collagen. Although Gα13 knockdown decreased EGF-driven invasion of CD18-tet-MT cells, knockdown of DDR1 did not affect EGF-driven invasion of MT1-MMP-expressing CD18 cells. DDR1 knockdown also did not affect MT1-MMP expression or cell surface localization in CD18-tet-MT cells (data not shown). However, concurrent knockdown of Gα13 and DDR1 reversed the inhibitory effects of Gα13 knockdown on proteolytic invasion by MT1-MMP-expressing CD18 cells (Fig. 6D).
Par3 Knockdown Reverses the Effect of Gα13 siRNA on Cell-Cell Adhesion and Proteolytic Invasion
Our findings that CD18 cells invade in three-dimensional collagen in a “polarized” half moon shape (Fig. 2) suggest that the polarity complex may regulate the invasion of PDAC cells in three-dimensional collagen. Significantly, we show that Gα13 knockdown CD18-tet-MT cells fail to demonstrate any half moon cell clusters in three-dimensional collagen (Fig. 6D) and that concurrent knockdown of DDR1 can reverse the effects of Gα13 knockdown on the number of cell clusters with a half moon shape (Fig. 6D). Importantly, the polarity protein Par3 can function downstream of DDR1 to regulate the actin cytoskeleton in skin cancer cells (11). Therefore, we examined whether Par3 can regulate invasion of CD18-tet-MT cells in three-dimensional collagen. Initially, we examined the effect of DDR1 and Gα13 knockdown on Par3 expression and localization. Knockdown of DDR1 caused redistribution of Par3 without significantly affecting total Par3 levels (Fig. 7, A and B). In contrast to the effects of DDR1 knockdown, Gα13 knockdown caused increased localization of Par3 at cell-cell junctions (Fig. 7A).
FIGURE 7.

Par3 knockdown reverses the effect of Gα13 siRNA on cell-cell adhesion and proteolytic invasion. A, CD18-tet-MT cells were plated on glass coverslips, transfected with siRNA against DDR1, Gα13, or Par3, and grown in the presence of doxycycline for 48 h. Cells on coverslips were fixed, permeabilized, and stained for E-cadherin and Par3. B, CD18-tet-MT cells transfected with siRNA against Par3, Gα13, or DDR1 were grown in three-dimensional collagen for 48 h. Collagenase-extracted cell lysates were then analyzed for E-cadherin, Par3, Gα13, and DDR1 expression by Western blotting. C, CD-18-tet-MT cells were transfected with siRNA against Gα13, Par3, or both Gα13 and Par3. The cells were plated in suspension in a hanging drop for 18 h in the presence of doxycycline. Quantitation of the area of cell clusters was performed using ImageJ. D, CD18-tet-MT cells transfected with siRNA against Gα13 and/or Par3 were grown in three-dimensional (3D) collagen in the presence of doxycycline and EGF (20 ng/ml) and allowed to invade for 72 h. Quantitation of invasion was analyzed as detailed in Fig. 2. The number of cell clusters demonstrating a half moon shape was also quantified. The statistics of the invasion assay and the number of half moon cell clusters were performed using one-way ANOVA followed by Dunnett's post-test. The statistics of the hanging drop assay were performed using Student's t test. ns, not significant; **, p < 0.01; ***, p < 0.001. The results are representative of three independent experiments.
Finally, we analyzed the effect of co-transfecting Par3 siRNA with Gα13 siRNA in MT1-MMP-expressing CD18 cells on cell-cell adhesion. Similar to the DDR1 siRNA (Fig. 6B), Par3 siRNA reversed the effects of Gα13 siRNA on cell-cell adhesion in the hanging drop assay (Fig. 7C). Moreover, although Par3 siRNA did not significantly increase three-dimensional collagen invasion (Fig. 7D) or affect MT1-MMP levels (data not shown), concurrent knockdown of Par3 and Gα13 reversed the inhibitory effects of Gα13 siRNA on collagen invasion by MT1-MMP-expressing CD18 cells (Fig. 7D). Concurrent knockdown of Par3 also reversed the effects of Gα13 knockdown on the number of cell clusters with a half moon shape (Fig. 7D).
Discussion
Patients with pancreatic cancer, the fourth leading cause of cancer-related death, often present with metastatic disease or locally advanced disease with invasion into surrounding structures (1, 2). Therefore, there is increasing interest in identifying mechanisms that regulate the invasion of pancreatic cancer cells. Previously, we have shown that the collagenase MT1-MMP mediated invasion of pancreatic cancer cells in three-dimensional collagen (5–7). In this report, we show that Gα13 is overexpressed in human PDAC tumors, and, by utilizing a quantitative method to track proteolytic invasion in three-dimensional collagen gels, we show that Gα13 functions downstream of MT1-MMP to mediate the invasion of pancreatic cancer cells in three-dimensional collagen.
Gα13 is known to regulate the actin cytoskeleton, and therefore cell migration and invasion, through activation of the Rho/ROCK/myosin pathway (14–16). Previously, it has been shown that overexpression of constitutively active Gα13 increases invasion by breast and prostate cancer cells through Matrigel-coated transwell filters (22, 23), whereas inhibition of Gα13 using shRNA decreases lysophosphatidic acid-mediated invasion of pancreatic cancer cells through collagen-coated transwell filters (24). The increased invasion by Gα13-overexpressing breast and prostate cancer cells was due to increased activation of Rho signaling (23). Even though we show that ROCK signaling mediates MT1-MMP-driven collagen invasion, Gα13 knockdown did not significantly attenuate Rho/ROCK signaling in pancreatic cancer cells. In fact, Gα13 knockdown in three-dimensional collagen increased ROCK activity, as demonstrated by increased myosin light chain phosphorylation and increased collagen contraction. This suggests that the inhibitory effect of Gα13 knockdown on three-dimensional collagen invasion occurs independently of its effect on ROCK signaling.
Additionally, we demonstrate that Gα13 siRNA enhances cell-cell adhesion, suggesting that Gα13 may regulate pancreatic cancer cell invasion in three-dimensional collagen by disrupting cell-cell junctions. Gα13 overexpression has been shown previously to disrupt E-cadherin-mediated cell-cell adhesion in MDA-MB-435 cells (20). Consistent with our findings that the ROCK1/2 inhibitor Y27632 fails to block Gα13-induced pancreatic cancer cell-cell adhesion, the effect of Gα13 on cadherin function in MDA-MB-435 cells was also independent of its effect on Rho signaling (20). Gα13 siRNA enhances E-cadherin-mediated cell-cell adhesion in our study. However, E-cadherin down-regulation does not increase group proteolytic invasion but, rather, increases the number of single cells in three-dimensional collagen. In contrast, knockdown of DDR1, which also localizes to cell-cell junctions (10, 11), reverses the inhibitory effect of Gα13 siRNA on group proteolytic invasion in three-dimensional collagen. Significantly, even though concurrent knockdown of Gα13 and DDR1 leads to single cells when grown on two-dimensional surfaces, knockdown cells lacking both Gα13 and DDR1 are able to undergo group migration in three-dimensional collagen. As DDR1 knockdown cells continue to express E-cadherin protein, it is very likely that DDR1 knockdown cells, in contrast to E-cadherin knockdown cells, can re-establish cell-cell junctions in three-dimensional collagen for group migration.
DDR1 has been shown to either attenuate or promote invasion depending on the type of cancer. DDR1 overexpression increased invasion of lung cancer cells and is associated with increased lymph node metastasis in human lung cancer tumor samples (45). Recently, it has been shown that DDR1 expression was increased in human PDAC tumor samples compared with normal adjacent tissue and that high DDR1 expression in PDAC tumors is associated with a poor prognosis (46). In contrast, DDR1 expression in breast cancer cells was associated with decreased invasion and was linked with increased breast cancer cell differentiation (47, 48). Moreover, DDR1 expression in breast cancer cells was primarily restricted to cell lines that demonstrate minimal to no evidence of epithelial-mesenchymal transition (13), suggesting that DDR1 in breast tumors functions to maintain cells in a more differentiated state. It has also been shown that knockdown of DDR1 enhanced cell-cell dissociation by decreasing the membrane stability of E-cadherin (12). We also show that DDR1 knockdown in pancreatic cancer cells decreases cell-cell adhesion. Additionally, we show that DDR1 knockdown decreases cortical actin structures and increases stress fiber formation, suggesting that DDR1 regulates cellular invasion through modulation of the actin cytoskeleton. Although we do not exclude the possibility that DDR1 may additionally be acting as a collagen-binding protein in our system, the effect of DDR1 siRNA in the absence of extracellular matrix on the hanging drop assay strongly suggests that its effect on invasion is not through collagen binding. Additionally, a previous report has demonstrated that the DDR1 regulation of collective cancer cell invasion was through its effect on cell-cell junctions (11). Moreover, the collagen-binding defective mutant of DDR1 had a similar effect on F-actin organization at cell-cell junctions as the wild-type DDR1 (11), suggesting that the DDR1 regulation of collective cancer cell invasion of squamous cancer cells may be independent of its collagen-binding function.
In this report, we also show that the knocking down the polarity protein Par3, which has been shown previously to function downstream of DDR1 in skin cancer cells (11), counteracts the inhibitory effects of Gα13 siRNA on pancreatic cancer cell invasion. Significantly, Par3 has been shown recently to regulate tumor progression. For example, Par3 loss increased Ras-induced skin keratoacanthomas and also cooperated with oncogenes to induce breast cancer invasion and metastasis in vivo (49–51). Loss of Par3 cooperated with the ErbB2 oncogene to induce invasion of mammary epithelial cells (51). Importantly, Par3 protein levels are either reduced significantly or localized abnormally in a large majority of breast tumors compared with normal tissue (51). Moreover, Par3, like DDR1, also regulates E-cadherin junction stability in breast cancer cells. Loss of Par3 in breast cancer cells compromises E-cadherin junctions and decreases cohesion between cancer cells (51). We also show that Par3 knockdown decreases cell-cell cohesion in pancreatic cancer cells. Significantly, we found that Gα13 knockdown, in contrast to DDR1 knockdown, caused increased localization of Par3 at cell-cell junctions, suggesting that Gα13 and DDR1 differentially regulate Par3 function in pancreatic cancer cells.
Overall, we show that Gα13 regulates the invasion of pancreatic cancer cells by disrupting cell-cell adhesion (Fig. 8). We also show that DDR1 blocks three-dimensional collagen invasion by enhancing cell-cell adhesion. Finally, we show that Par3, which is regulated differentially by DDR1 and Gα13, can also attenuate the effects of Gα13 on three-dimensional collagen invasion by enhancing cell-cell adhesion.
FIGURE 8.
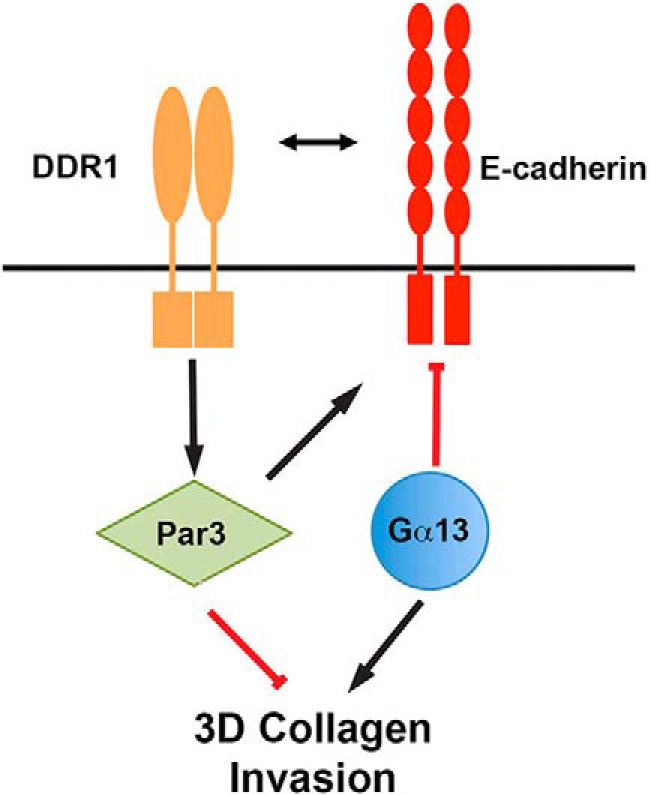
Gα13 and DDR1-Par3 differentially regulate cancer cell invasion in three-dimensional collagen. Gα13 enhances the invasion of pancreatic cancer cells in three-dimensional collagen by disrupting cell-cell adhesion. In contrast, DDR1 blocks three-dimensional collagen invasion by enhancing cell-cell adhesion. Par3, which is differentially regulated by DDR1 and Gα13, can also attenuate the effects of Gα13 on three-dimensional collagen invasion by enhancing cell-cell adhesion.
Author Contributions
C. R. C., K. E., L. M. K., and K. K. performed the experiments and data analysis. D. J. B. provided human tumor samples. C. R. C. and H. G. M. designed the research and wrote the manuscript.
This work was supported NCI/National Institutes of Health Grants R01CA126888 and R01CA186885 (to H. G. M.) and Department of Veterans Affairs Merit Award I01BX001363 (to H. G. M.). This work was also supported by NCI/National Institutes of Health Training Grant T32CA070085 (to C. R. C.). The authors declare that they have no conflicts of interest with contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PDAC
- pancreatic ductal adenocarcinoma
- MMP
- matrix metalloproteinase
- MT1
- membrane type 1
- TMA
- tissue microarray
- SCC
- squamous cell cancer
- MLC
- myosin light chain
- ANOVA
- analysis of variance
- ROCK
- Rho-associated protein kinase.
References
- 1. Hidalgo M. (2010) Pancreatic cancer. N. Engl. J. Med. 362, 1605–1617 [DOI] [PubMed] [Google Scholar]
- 2. Vincent A., Herman J., Schulick R., Hruban R. H., and Goggins M. (2011) Pancreatic cancer. Lancet 378, 607–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chu G. C., Kimmelman A. C., Hezel A. F., and DePinho R. A. (2007) Stromal biology of pancreatic cancer. J. Cell Biochem. 101, 887–907 [DOI] [PubMed] [Google Scholar]
- 4. Shields M. A., Dangi-Garimella S., Redig A. J., and Munshi H. G. (2012) Biochemical role of the collagen-rich tumor microenvironment in pancreatic cancer progression. Biochem. J. 441, 541–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ottaviano A. J., Sun L., Ananthanarayanan V., and Munshi H. G. (2006) Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-β1. Cancer Res. 66, 7032–7040 [DOI] [PubMed] [Google Scholar]
- 6. Shields M. A., Krantz S. B., Bentrem D. J., Dangi-Garimella S., and Munshi H. G. (2012) Interplay between β1-integrin and Rho signaling regulates differential scattering and motility of pancreatic cancer cells by snail and Slug proteins. J. Biol. Chem. 287, 6218–6229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shields M. A., Dangi-Garimella S., Krantz S. B., Bentrem D. J., and Munshi H. G. (2011) Pancreatic cancer cells respond to type I collagen by inducing Snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion. J. Biol. Chem. 286, 10495–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Christiansen J. J., and Rajasekaran A. K. (2006) Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 66, 8319–8326 [DOI] [PubMed] [Google Scholar]
- 9. Friedl P., Locker J., Sahai E., and Segall J. E. (2012) Classifying collective cancer cell invasion. Nat. Cell Biol. 14, 777–783 [DOI] [PubMed] [Google Scholar]
- 10. Wang C.-Z., Yeh Y.-C., and Tang M.-J. (2009) DDR1/E-cadherin complex regulates the activation of DDR1 and cell spreading. Am. J. Physiol. Cell Physiol. 297, C419–C429 [DOI] [PubMed] [Google Scholar]
- 11. Hidalgo-Carcedo C., Hooper S., Chaudhry S. I., Williamson P., Harrington K., Leitinger B., and Sahai E. (2011) Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat. Cell Biol. 13, 49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eswaramoorthy R., Wang C.-K., Chen W.-C., Tang M.-J., Ho M.-L., Hwang C.-C., Wang H.-M., and Wang C.-Z. (2010) DDR1 regulates the stabilization of cell surface E-cadherin and E-cadherin-mediated cell aggregation. J. Cell Physiol. 224, 387–397 [DOI] [PubMed] [Google Scholar]
- 13. Yeh Y. C., Wu C. C., Wang Y. K., and Tang M. J. (2011) DDR1 triggers epithelial cell differentiation by promoting cell adhesion through stabilization of E-cadherin. Mol. Biol. Cell 22, 940–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kozasa T., Hajicek N., Chow C. R., and Suzuki N. (2011) Signalling mechanisms of RhoGTPase regulation by the heterotrimeric G proteins G12 and G13. J. Biochem. 150, 357–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kurose H. (2003) Gα12 and Gα13 as key regulatory mediator in signal transduction. Life Sci. 74, 155–161 [DOI] [PubMed] [Google Scholar]
- 16. Kelly P., Casey P. J., and Meigs T. E. (2007) Biologic functions of the G12 subfamily of heterotrimeric G proteins: growth, migration, and metastasis. Biochemistry 46, 6677–6687 [DOI] [PubMed] [Google Scholar]
- 17. Dhanasekaran D. N. (2006) Transducing the signals: a G protein takes a new identity. Science's STKE 2006, pe31. [DOI] [PubMed] [Google Scholar]
- 18. Shan D., Chen L., Wang D., Tan Y. C., Gu J. L., and Huang X. Y. (2006) The G protein Gα13 is required for growth factor-induced cell migration. Dev. Cell 10, 707–718 [DOI] [PubMed] [Google Scholar]
- 19. Meigs T. E., Fields T. A., McKee D. D., and Casey P. J. (2001) Interaction of Gα12 and Gα13 with the cytoplasmic domain of cadherin provides a mechanism for β-catenin release. Proc. Natl. Acad. Sci. U.S.A. 98, 519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meigs T. E., Fedor-Chaiken M., Kaplan D. D., Brackenbury R., and Casey P. J. (2002) Gα12 and Gα13 Negatively regulate the adhesive functions of cadherin. J. Biol. Chem. 277, 24594–24600 [DOI] [PubMed] [Google Scholar]
- 21. Gong H., Gao X., Feng S., Siddiqui M. R., Garcia A., Bonini M. G., Komarova Y., Vogel S. M., Mehta D., and Malik A. B. (2014) Evidence of a common mechanism of disassembly of adherens junctions through Gα13 targeting of VE-cadherin. J. Exp. Med. 211, 579–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kelly P., Stemmle L. N., Madden J. F., Fields T. A., Daaka Y., and Casey P. J. (2006) A Role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J. Biol. Chem. 281, 26483–26490 [DOI] [PubMed] [Google Scholar]
- 23. Kelly P., Moeller B. J., Juneja J., Booden M. A., Der C. J., Daaka Y., Dewhirst M. W., Fields T. A., and Casey P. J. (2006) The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. U.S.A. 103, 8173–8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gardner J. A., Ha J. H., Jayaraman M., and Dhanasekaran D. N. (2013) The gep proto-oncogene Gα13 mediates lysophosphatidic acid-mediated migration of pancreatic cancer cells. Pancreas 42, 819–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Singer W. D., Miller R. T., and Sternweis P. C. (1994) Purification and characterization of the α subunit of G13. J. Biol. Chem. 269, 19796–19802 [PubMed] [Google Scholar]
- 26. Dangi-Garimella S., Redig A. J., Shields M. A., Siddiqui M. A., and Munshi H. G. (2010) Rho-ROCK-myosin signaling mediates membrane type 1 matrix metalloproteinase-induced cellular aggregation of keratinocytes. J. Biol. Chem. 285, 28363–28372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dangi-Garimella S., Krantz S. B., Barron M. R., Shields M. A., Heiferman M. J., Grippo P. J., Bentrem D. J., and Munshi H. G. (2011) Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 71, 1019–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dangi-Garimella S., Strouch M. J., Grippo P. J., Bentrem D. J., and Munshi H. G. (2011) Collagen regulation of let-7 in pancreatic cancer involves TGF-β1-mediated membrane type 1-matrix metalloproteinase expression. Oncogene 30, 1002–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Munshi H. G., Wu Y. I., Ariztia E. V., and Stack M. S. (2002) Calcium regulation of matrix metalloproteinase-mediated migration in oral squamous cell carcinoma cells. J. Biol. Chem. 277, 41480–41488 [DOI] [PubMed] [Google Scholar]
- 30. Munshi H. G., Wu Y. I., Mukhopadhyay S., Ottaviano A. J., Sassano A., Koblinski J. E., Platanias L. C., and Stack M. S. (2004) Differential regulation of membrane type 1-matrix metalloproteinase activity by ERK 1/2- and p38 MAPK-modulated tissue inhibitor of metalloproteinases 2 expression controls transforming growth factor-β1-induced pericellular collagenolysis. J. Biol. Chem. 279, 39042–39050 [DOI] [PubMed] [Google Scholar]
- 31. Wolf K., Te Lindert M., Krause M., Alexander S., Te Riet J., Willis A. L., Hoffman R. M., Figdor C. G., Weiss S. J., and Friedl P. (2013) Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 201, 1069–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sassano A., Mavrommatis E., Arslan A. D., Kroczynska B., Beauchamp E. M., Khuon S., Chew T. L., Green K. J., Munshi H. G., Verma A. K., and Platanias L. C. (2015) Human Schlafen 5 (SLFN5) is a regulator of motility and invasiveness of renal cell carcinoma cells. Mol. Cell Biol. 35, 2684–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Foty R. (2011) A simple hanging drop cell culture protocol for generation of 3D spheroids. J. Vis. Exp. 10.3791/2720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sahai E., and Marshall C. J. (2003) Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5, 711–719 [DOI] [PubMed] [Google Scholar]
- 35. Gohla A., Offermanns S., Wilkie T. M., and Schultz G. (1999) Differential involvement of Gα12 and Gα13 in receptor-mediated stress fiber formation. J. Biol. Chem. 274, 17901–17907 [DOI] [PubMed] [Google Scholar]
- 36. Buhl A. M., Johnson N. L., Dhanasekaran N., and Johnson G. L. (1995) Gα12 and Gα13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 270, 24631–24634 [DOI] [PubMed] [Google Scholar]
- 37. Badea L. (2008) Extracting gene expression profiles common to colon and pancreatic adenocarcinoma using simultaneous nonnegative matrix factorization. Pac. Symp. Biocomput. 13, 279–290 [PubMed] [Google Scholar]
- 38. Grützmann R., Pilarsky C., Ammerpohl O., Lüttges J., Böhme A., Sipos B., Foerder M., Alldinger I., Jahnke B., Schackert H. K., Kalthoff H., Kremer B., Klöppel G., and Saeger H. D. (2004) Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia 6, 611–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ishikawa M., Yoshida K., Yamashita Y., Ota J., Takada S., Kisanuki H., Koinuma K., Choi Y. L., Kaneda R., Iwao T., Tamada K., Sugano K., and Mano H. (2005) Experimental trial for diagnosis of pancreatic ductal carcinoma based on gene expression profiles of pancreatic ductal cells. Cancer Sci. 96, 387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pei H., Li L., Fridley B. L., Jenkins G. D., Kalari K. R., Lingle W., Petersen G., Lou Z., and Wang L. (2009) FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16, 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tan W., Martin D., and Gutkind J. S. (2006) The Gα13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J. Biol. Chem. 281, 39542–39549 [DOI] [PubMed] [Google Scholar]
- 42. Vial E., Sahai E., and Marshall C. J. (2003) ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 4, 67–79 [DOI] [PubMed] [Google Scholar]
- 43. Gao Y., Dickerson J. B., Guo F., Zheng J., and Zheng Y. (2004) Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. U.S.A. 101, 7618–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ishizaki T., Uehata M., Tamechika I., Keel J., Nonomura K., Maekawa M., and Narumiya S. (2000) Pharmacological properties of Y-27632, a specific inhibitor of Rho-associated kinases. Mol. Pharmacol. 57, 976–983 [PubMed] [Google Scholar]
- 45. Yang S. H., Baek H. A., Lee H. J., Park H. S., Jang K. Y., Kang M. J., Lee D. G., Lee Y. C., Moon W. S., and Chung M. J. (2010) Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung carcinomas. Oncol. Rep. 24, 311–319 [DOI] [PubMed] [Google Scholar]
- 46. Huo Y., Yang M., Liu W., Yang J., Fu X., Liu D., Li J., Zhang J., Hua R., and Sun Y. (2015) High expression of DDR1 is associated with the poor prognosis in Chinese patients with pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 34, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Turashvili G., Bouchal J., Baumforth K., Wei W., Dziechciarkova M., Ehrmann J., Klein J., Fridman E., Skarda J., Srovnal J., Hajduch M., Murray P., and Kolar Z. (2007) Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hansen C., Greengard P., Nairn A. C., Andersson T., and Vogel W. F. (2006) Phosphorylation of DARPP-32 regulates breast cancer cell migration downstream of the receptor tyrosine kinase DDR1. Exp. Cell Res. 312, 4011–4018 [DOI] [PubMed] [Google Scholar]
- 49. Iden S., van Riel W. E., Schäfer R., Song J. Y., Hirose T., Ohno S., and Collard J. G. (2012) Tumor type-dependent function of the par3 polarity protein in skin tumorigenesis. Cancer Cell 22, 389–403 [DOI] [PubMed] [Google Scholar]
- 50. McCaffrey L. M., Montalbano J., Mihai C., and Macara I. G. (2012) Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 22, 601–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xue B., Krishnamurthy K., Allred D. C., and Muthuswamy S. K. (2013) Loss of Par3 promotes breast cancer metastasis by compromising cell-cell cohesion. Nat. Cell Biol. 15, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]