

Abstract
We recently reported that an amide bond is unexpectedly formed by an acyl-CoA synthetase (which catalyzes the formation of a carbon-sulfur bond) when a suitable acid and l-cysteine are used as substrates. DltA, which is homologous to the adenylation domain of nonribosomal peptide synthetase, belongs to the same superfamily of adenylate-forming enzymes, which includes many kinds of enzymes, including the acyl-CoA synthetases. Here, we demonstrate that DltA synthesizes not only N-(d-alanyl)-l-cysteine (a dipeptide) but also various oligopeptides. We propose that this enzyme catalyzes peptide synthesis by the following unprecedented mechanism: (i) the formation of S-acyl-l-cysteine as an intermediate via its “enzymatic activity” and (ii) subsequent “chemical” S → N acyl transfer in the intermediate, resulting in peptide formation. Step ii is identical to the corresponding reaction in native chemical ligation, a method of chemical peptide synthesis, whereas step i is not. To the best of our knowledge, our discovery of this peptide synthesis mechanism involving an enzymatic reaction and a subsequent chemical reaction is the first such one to be reported. This new process yields peptides without the use of a thioesterified fragment, which is required in native chemical ligation. Together with these findings, the same mechanism-dependent formation of N-acyl compounds by other members of the above-mentioned superfamily demonstrated that all members most likely form peptide/amide compounds by using this novel mechanism. Each member enzyme acts on a specific substrate; thus, not only the corresponding peptides but also new types of amide compounds can be formed.
Keywords: amino acid, ATP, Bacillus, enzyme mechanism, peptide biosynthesis, peptides, protein domain, adenylate-forming superfamily, amide, native chemical ligation
Introduction
We previously reported that an acyl-CoA synthetase, AcsA, plays an essential role in acid utilization in the nitrile-degradative pathway of Pseudomonas chlororaphis B23 (1). The enzymes involved in the biological metabolism of nitriles have received much attention in both applied studies (2) and academic fields. Acyl-CoA synthetase, which is an acid-thiol ligase, catalyzes the ligation of an acid with CoA in the presence of ATP and Mg2+. The reaction mechanism and structure of acyl-CoA synthetase have been extensively investigated (3–8). During our studies of AcsA, we discovered a unique reaction. When l-cysteine was used as the substrate instead of CoA, N-isobutyryl-l-cysteine was surprisingly detected as the reaction product (9). This finding demonstrated that the enzyme formed a carbon-nitrogen bond involving the carboxyl group of the acid and the amino group of l-cysteine. Furthermore, this unexpected enzyme activity was also observed for acetyl-CoA synthetase (4, 7) and firefly luciferase (10, 11), both of which belong to the same superfamily of adenylate-forming enzymes. However, the mechanism underlying the carbon-nitrogen bond synthesis remains unknown.
The adenylation domain of nonribosomal peptide synthetase (NRPS)4 is also a member of this superfamily of adenylate-forming enzymes. This domain is responsible for the selective substrate recognition and formation of acyl-AMP as an intermediate during ATP consumption (12, 13). NRPSs are modular enzymes frequently used to synthesize small peptides such as antibiotics and antiviral agents (14, 15). These multienzyme complexes are organized into modules, each of which can be further subdivided into a series of domains that catalyze the individual steps of nonribosomal peptide synthesis. DltA,the d-alanine:d-alanyl carrier protein (DltC) ligase involved in the initial step of lipoteichoic acid d-alanylation in Gram-positive bacteria, is structurally homologous to the adenylation domain (16, 17) of NRPS. DltA not only activates d-alanine but also attaches the activated ester to the DltC-bound cofactor 4′-phosphopantetheine via a thioester bond (18, 19). The DltA features of ATP-dependent activation of the amino acid substrate and utilization of the carrier protein are also features of the adenylation domain of NRPSs.
Like the adenylation domain of NRPSs, DltA belongs to the superfamily of adenylate-forming enzymes, their three-dimensional structures being analogous to one another (20). Accordingly, we here examined the enzyme activity of DltA using l-cysteine instead of the physiological substrate DltC. We succeeded in using DltA to synthesize a dipeptide (N-(d-alanyl)-l-cysteine:d-Ala-l-Cys). Moreover, we revealed the mechanism underlying this previously unidentified peptide/amide bond formation reaction. In addition, we used DltA for this discovered reaction to obtain various oligopeptides.
Experimental Procedures
Materials
l-Cysteine, S-methyl-l-cysteine, and l-lysine were purchased from Sigma. d-Alanine, d-cysteine, d-serine, d-valine, d-asparagine, d-phenylalanine, l-alanine, l-serine, l-glutamic acid, l-glutamine, l-valine, l-histidine, l-isoleucine, l-phenylalanine, glycine, β-alanine, 3-mercaptopropionic acid, and thioglycolic acid were purchased from Nakalai Tesque (Kyoto, Japan). d-Lysine, d-methionine, d-isoleucine, d-tryptophan, and N-Boc-l-Ala were purchased from Peptide Institute, Inc. (Osaka, Japan). l-2-Aminobutyric acid, d-2-aminobutyric acid, and d-glutamine were purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). S-Aminoethyl-l-cysteine, S-acetamidomethyl-l-cysteine, and dipeptides were purchased from Bachem (Bubendorf, Switzerland). N-Boc-l-Cys was purchased from Merck Millipore. ATP and AMP were purchased from Kishida Chemical Co., Ltd. (Osaka, Japan). The pentapeptide (l-Cys-Gly-Gly-l-Arg-l-Glu), hexapeptide (d-Ala-l-Cys-Gly-Gly-l-Arg-l-Glu), decapeptide (l-Cys-Gly-Gly-l-Arg-l-Glu-l-Ser-Gly-l-Ser-Gly-l-Ser), and undecapeptide (d-Ala-l-Cys-Gly-Gly-l-Arg-l-Glu-l-Ser-Gly-l-Ser-Gly-l-Ser) wereprepared by Operon Biotechnology Inc. (Tokyo, Japan). Other reagents were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
Preparation of the Enzyme
The DNA fragment of the dltA gene was amplified by PCR using chromosomal DNA of Bacillus subtilis strain 168 as a template. The following two oligonucleotide primers were used: 5′-AGAATTCCATATGAAACTTTTACATGCTATTCAAACAC-3′ (NdeI recognition site is underlined) and 5′-TTCTCGAGTACAAGAACCTCTTCGCCAATG-3′ (XhoI recognition site is underlined). The PCR products were subcloned into vector pUC18 and checked by DNA sequencing. To construct C-terminally His6-tagged DltA, the insert DNA (dltA gene) was digested with NdeI and XhoI and then inserted into the corresponding sites of pET-24a(+) (Merck Millipore). The resultant plasmid was designated as pET-dltA.
Escherichia coli BL21-CodonPlus® (DE3)-RIL (Agilent Technologies, Inc., Palo Alto, CA) was transformed with pET-dltA. The transformants were grown in 2× YT medium containing kanamycin (50 μg/ml) and chloramphenicol (34 μg/ml) at 37 °C. When the cultures had grown to an optical density at 600 nm of 0.6, the incubation temperature was reduced to 24 °C, and protein expression was induced with 0.1 mm isopropyl β-d-thiogalactoside (IPTG). After 24 h of culture, the cells were harvested by centrifugation, washed twice, and then suspended in 20 mm sodium phosphate buffer (pH 7.4). The resuspended cells were sonicated, and the cell debris was removed by centrifugation (27,000 × g, 30 min). The resulting supernatant was applied to a nickel-chelating column of HisTrapTM HP (5 ml) (GE Healthcare, Ltd., UK), and washed with 20 mm sodium phosphate buffer (pH 7.4) containing 0.5 m NaCl and 0.01 m imidazole. DltA was eluted with a linear gradient (0.01–0.4 m) of imidazole. The fractions comprising the 57-kDa size peak were collected and dialyzed against 20 mm Tris-HCl buffer (pH 7.5). The homogeneity of the purified proteins was confirmed by SDS-PAGE. The N-terminal sequence of the purified DltA was analyzed with a model 490 Procise® protein sequencer (Applied Biosystems Inc., Foster City, CA). The purified fraction was used in all experiments in this study.
Identification of the Reaction Products of DltA
N-d-Alanyl-l-cysteine (d-Ala-l-Cys)
The reaction mixture, comprising 200 mm d-alanine, 200 mm l-cysteine, 5 mm ATP, 8 mm MgCl2, 100 mm (NH4)2SO4, and purified DltA (0.2 mg/ml) in 100 mm Tris-HCl buffer (pH 7.5), was incubated at 50 °C, and the reaction was stopped by adding an equal volume of a perchloric acid solution (pH 1.5). Then the mixture was analyzed by HPLC on a CROWNPAK CR(+) column (4.0 × 150 mm; Daicel Chemical Industries, Ltd., Osaka, Japan). The mobile phase was the perchloric acid solvent (pH 1.5), and chromatographic separation was performed at 25 °C at a flow rate of 1.0 ml/min. A new product peak (retention time, 3.0–3.7 min) distinct from that of AMP was detected on monitoring of the column effluent at 190 nm. The product peak was collected, concentrated, and then subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Ionelectrospray tandem mass spectrometric measurement of the product was carried out using a Shimadzu (Kyoto, Japan) LCMS-8030 triple quadrupole mass spectrometer equipped with a Shimadzu Nexera HPLC system and a TSK-gel ODS-100V column (4.6 × 150 mm; Tosoh, Tokyo, Japan). The mobile phase solvent was 0.1% (v/v) formic acid. The MS/MS analysis data were acquired under the following conditions: curved desolvation and heat block temperatures of 300 and 200 °C, respectively. Nitrogen was used as the nebulizer gas and drying gas at flow rates of 2 and 15 liters/min, respectively. The collision energy was varied from −35 to 35 V, and the ion source polarity was set in the positive or negative mode. 1H nuclear magnetic resonance (NMR), 13C NMR, and 1H-13C heteronuclear multiple-bond connectivity spectra of the product were measured with an Avance 600 spectrometer (Bruker, Ettlingen, Germany). Samples were prepared by dissolution in D2O, which was used as an internal standard.
Dipeptide (Xaa-l-Cys)
The composition of the reaction mixture, except for the omission of d-alanine, was the same as in the case of identification of d-Ala-l-Cys. β-Alanine, glycine, d-serine, l-alanine, or d-threonine was used instead of d-alanine. Each reaction mixture was analyzed by HPLC on a Develosil® RPAQUEOUS column (4.6 × 250 mm; Nomura Chemical Co., Ltd., Aichi, Japan). The mobile-phase solvent included 5% (v/v) acetonitrile containing 0.1% (v/v) trifluoroacetic acid (TFA) in water, and chromatographic separation was performed at 25 °C at a flow rate of 1.0 ml/min. Each product peak differing from that of AMP was fractionated, concentrated, and then subjected to LC-MS/MS analysis. The mixture was analyzed with a Shimadzu LCMS-8030 equipped with a Shimadzu Nexera HPLC system and a Develosil® RPAQUEOUS column (4.6 × 250 mm). The mobile-phase solvent was 5% (v/v) acetonitrile containing 0.1% (v/v) formic acid in water, and chromatographic separation was performed at 25 °C at a flow rate of 0.2 ml/min. The MS/MS analysis data were collected under the same conditions as for the d-Ala-l-Cys analysis except for the column and mobile-phase solvent.
Tripeptides (Xaa-l-Cys-Gly)
The composition of the reaction mixture, except for the omission of l-cysteine, was the same as in the case of identification of dipeptides (Xaa-l-Cys). l-Cysteinyl-glycine (l-Cys-Gly) was used instead of l-cysteine. The mixture was analyzed with a Shimadzu LCMS-8030 equipped with a Shimadzu Nexera HPLC system and a SeQuant® ZIC®-HILIC column (2.1 × 150 mm; Merck Millipore). The mobile phases for elution were as follows: mobile phase A, acetonitrile; and mobile phase B, 10 mm ammonium acetate. Samples were eluted at 0.2 ml/min using a linear gradient of 20–40% of mobile phase B over the course of 20 min. The final composition of 40% mobile phase B was held for 20 min from 20–40 min. Chromatographic separation was performed at 25 °C. The MS/MS analysis data were collected under the same conditions as for the d-Ala-l-Cys analysis except for the column and mobile-phase solvent.
Oligopeptides
The composition of the reaction mixture, except for the omission of l-cysteine, was the same as in the case of identification of d-Ala-l-Cys. The pentapeptide or decapeptide was used instead of l-cysteine. The mixture, including the pentapeptide, was analyzed with a Shimadzu LCMS-8030 and a SeQuant® ZIC®-HILIC column. The HPLC program for hexapeptide elution was the same as that for the dipeptides. The mixture, including the decapeptide, was analyzed with a Shimadzu LCMS-8030 and a SeQuant® ZIC®-HILIC column with acetonitrile, 10 mm ammonium acetate (6:4, v/v) as the mobile phase, and chromatographic separation was performed at 25 °C at a flow rate of 0.2 ml/min. The MS/MS analysis data were collected under the same conditions as for the d-Ala-l-Cys analysis except for the column and mobile-phase solvent.
S-(d-Alanyl)-3-mercaptopropionic Acid or S-(d-Alanyl)-thioglycolic acid
The reaction mixture, comprising 100 mm Tris-HCl buffer (pH 7.5), 200 mm d-alanine, 5 mm ATP, 8 mm MgCl2, 100 mm (NH4)2SO4, and 150 mm 3-mercaptopropionic acid or thioglycolic acid, was incubated with the purified DltA (0.2 mg/ml) at 50 °C, and the reaction was stopped by adding an equal volume of a perchloric acid solution (pH 1.5) to the reaction mixture. Then the mixture was analyzed by HPLC on a COSMOSIL® HILIC column (4.6 × 150 mm; Nakalai Tesque). The mobile phase solvent was 5 mm ammonium acetate in 50% acetonitrile, and chromatographic separation was performed at 25 °C at a flow rate of 0.5 ml/min. The product peak was fractionated, concentrated, and then subjected to LC-MS/MS analysis. The analysis was carried out using a Shimadzu LCMS-8030 equipped with a Shimadzu Nexera HPLC system and a Develosil® RPAQUEOUS column (2.0 × 150 mm). The mobile phase solvent was 10 mm ammonium acetate, and chromatographic separation was performed at 25 °C at a flow rate of 0.2 ml/min.
S-(d-Alanyl)-N-Boc-l-Cys and N-(d-alanyl)-N-Boc-l-Cys
The reaction mixture, comprising 100 mm Tris-HCl buffer (pH 7.5), 50 mm d-alanine, 10 mm ATP, 8 mm MgCl2, 100 mm (NH4)2SO4, and 50 mm N-Boc-l-Cys, was incubated with the purified DltA (1.0 mg/ml) at 50 °C. At several time points, the reaction mixture was subjected to LC-MS and LC-MS/MS analyses. These analyses were carried out using a Shimadzu LCMS-8030 equipped with a Shimadzu Nexera UPLC system and a Kinetex 1.7u C18 100A column (2.1 × 50 mm). The mobile phases for elution were as follows: mobile phase A, 0.1% formic acid; and mobile phase B, methanol. Samples were eluted at 0.4 ml/min using a linear gradient of 10–50% of mobile phase B over the course of 6 min. The final composition of 50% mobile phase B was held for 1 min from 6 to 7 min. Chromatographic separation was performed at 30 °C. The MS/MS analysis data were collected under the same conditions as for the d-Ala-l-Cys analysis except for the column and mobile-phase solvent.
Identification of the Reaction Products of Acetyl-CoA Synthetase or AcsA
S-Acetyl-3-mercaptopropionic Acid or S-Acetyl-thioglycolic Acid
The reaction mixture, comprising 200 mm Tris-HCl buffer (pH 7.5), 20 mm acetic acid, 5 mm ATP, 8 mm MgCl2, 100 mm (NH4)2SO4, and 100 mm 3-mercaptopropionic acid, was incubated with S-acetyl-CoA synthetase from bakers' yeast (Sigma, 0.5 mg/ml) at 28 °C. The mixture was analyzed with a Shimadzu LCMS-8030 and a Develosil® RPAQUEOUS column (2.0 × 150 mm) with 10 mm ammonium acetate as the mobile phase, and chromatographic separation was performed at 30 °C at a flow rate of 0.2 ml/min. When 100 mm thioglycolic acid was added as a substrate instead of 3-mercaptopropionic acid, a COSMOSIL® π-nap column (4.6 × 150 mm; Nakalai Tesque) was used with 0.05% (v/v) formic acid solvent as the mobile phase.
S-Isobutyryl-3-mercaptopropionic Acid or S-Isobutyrylthioglycolic Acid
The reaction mixture, comprising 200 mm Tris-HCl buffer (pH 7.5), 20 mm isobutyric acid, 5 mm ATP, 8 mm MgCl2, 100 mm (NH4)2SO4, and 100 mm 3-mercaptopropionic acid or thioglycolic acid, was incubated with the purified AcsA (0.5 mg/ml) at 28 °C. Then the mixture was subjected to LC-MS/MS analysis. The analysis was carried out using a Shimadzu LCMS-8030 equipped with a Shimadzu Nexera HPLC system and a Develosil® RPAQUEOUS column (2.0 × 150 mm). The mobile phases for elution were as follows: mobile phase A, 10 mm ammonium acetate; and mobile phase B, acetonitrile. Samples were eluted at 25 °C at a flow rate of 0.2 ml/min using a linear gradient of 0–20% of mobile phase B over the course of 15 min. The composition of 20% mobile phase B was held for 5 min from 15 to 20 min, and then the final composition of 0% mobile phase B was held for 5 min from 20 to 25 min.
S-Acetyl-N-Boc-l-Cys and N-acetyl-N-Boc-l-Cys
The reaction mixture, comprising 200 mm Tris-HCl buffer (pH 7.5), 10 mm acetic acid, 5 mm ATP, 8 mm MgCl2, 100 mm (NH4)2SO4, and 50 mm N-Boc-l-Cys, was incubated with S-acetyl-CoA synthetase (1.0 mg/ml) at 28 °C. At two time points (10 and 60 min), the reaction mixture was subjected to LC-MS and LC-MS/MS analyses. These analyses were carried out using a Shimadzu LCMS-8030 equipped with a COSMOSIL® π-nap column. The mobile phases for elution were as follows: mobile phase A, 0.05% formic acid; and mobile phase B, acetonitrile. Samples were eluted at 1 ml/min using a linear gradient of 2–100% of mobile phase B over the course of 8 min after 1 min from the start. The final composition of 100% mobile phase B was held for 2 min from 9 to 11 min. Chromatographic separation was performed at 40 °C.
Enzyme Assays of DltA
The standard assay A mixture (100 μl) included 5–200 mm d-alanine, 5–500 mm l-cysteine, 0.5–20 mm ATP, 8 mm MgCl2, 100 mm (NH4)2SO4, and purified DltA in 100 mm Tris-HCl (pH 7.5). The reaction was started by adding DltA (0.2 mg/ml), followed by incubation at 50 °C for 5 min. The reaction was stopped by adding 100 μl of a perchloric acid solution (pH 1.5). Then the mixture was analyzed by HPLC on a CROWNPAK CR(+) column. The mobile phase was the perchloric acid solvent (pH 1.5), and chromatographic separation was performed at 25 °C at a flow rate of 1.0 ml/min. The d-Ala-l-Cys product peak was quantitated by monitoring the column effluent at 210 nm with authentic d-Ala-l-Cys as the standard. One unit of d-Ala-l-Cys synthetic activity was defined as the amount of enzyme that catalyzed the formation of 1 μmol of d-Ala-l-Cys/min under the assay conditions used. Standard assay A was used to routinely measure d-Ala-l-Cys synthetic activity.
To determine the specificity of the enzyme for various amino acids, we assayed its activity using standard assay B, which measures acid-dependent AMP formation. Although the ATP-[32P]PPi exchange assay (21) or the colorimetric assay (22) has been used for measuring the specific activity of adenylating enzymes (involved in the reaction ATP → AMP + PPi), these assays are based upon indirect quantification of acyl-AMP or PPi, respectively. Therefore, in our study the modified method to measure directly the amount of AMP by HPLC (23) was used, because the HPLC method allows easier and more accurate quantification than the methods reported previously. The composition of the standard assay B mixture, except for d-alanine, was the same as that for standard assay A. AMP and ATP in each of the reaction series were separated on a Titan sphere TiO HPLC column (4.6 × 150 mm; GL Science Inc., Tokyo, Japan). The mobile phase was 50 mm potassium phosphate buffer (pH 7.0) containing 55% (v/v) acetonitrile, and chromatographic separation was performed at 50 °C at a flow rate of 1 ml/min, with monitoring at 254 nm. One unit was defined as the amount of enzyme that catalyzed the formation of 1 μmol of AMP/min under the assay conditions used. kcat values were calculated using a Mr of 56,874 for DltA. In the pH profile experiments, a three-component buffer system that maintained the ionic strength at a set value throughout each range was used (24).
Stoichiometry
To determine the stoichiometry, substrate consumption and product formation during the synthesis of N-d-alanyl-l-cysteine were examined in a reaction mixture comprising 5 mm ATP, 200 mm d-alanine, 200 mm l-cysteine, 8 mm MgCl2, 10 mm (NH4)2SO4, 100 mm Tris-HCl buffer (pH 7.5), and 0.6 mg of DltA, in a final volume of 1000 μl. The reaction was carried out at 50 °C. At several time points, the molar amounts of the formed products (d-Ala-l-Cys and AMP) and accompanying decreases in the substrate (ATP) were measured by means of standard assays.
Analytical Methods
Protein concentrations were measured with a Nakalai Tesque protein assay kit with bovine serum albumin as the standard. SDS-PAGE was performed in a 12.5% polyacrylamide slab gel. The gel was stained with Coomassie Brilliant Blue R-250. The relative molecular mass of the enzyme was calculated from the mobilities of the marker proteins, phosphorylase b (97 kDa), bovine serum albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (30 kDa), soybean trypsin inhibitor (20.1 kDa), and α-lactalbumin (14.4 kDa).
Results
Expression and Purification of DltA
DltA was simply purified as described under “Experimental Procedures.” The purity of the DltA was confirmed by migration of the protein as a single band corresponding to a molecular mass of 57 kDa on SDS-PAGE (Fig. 1). The purified DltA had the N-terminal sequence of MKLLHAIQTH, which is the same as that deduced from the DNA sequence.
FIGURE 1.
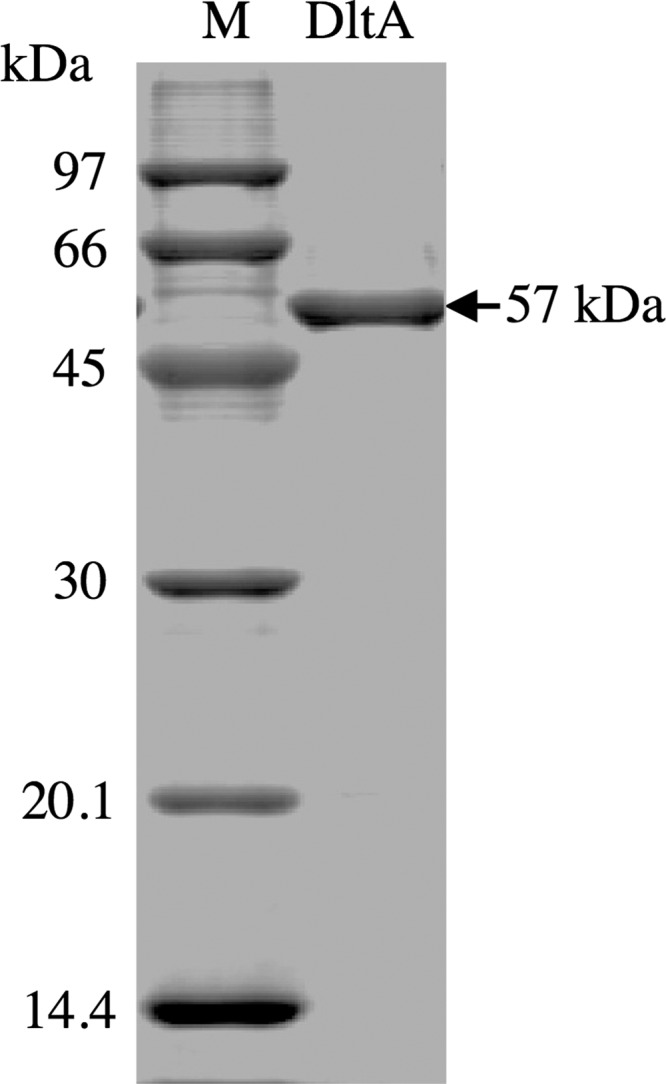
SDS-PAGE of the purified DltA protein. Protein bands were detected by staining with Coomassie Brilliant Blue. Lane M, marker proteins; lane DltA, purified DltA (1 μg).
Identification of the Novel Product of the Reaction Containing DltA
DltA of B. subtilis uses d-alanine (H3C-CH(NH2)-COOH) and 4′-phosphopantetheine-bound DltC as substrates and forms a thioester bond involving the carboxyl group of d-alanine and the thiol group of 4′-phosphopantetheine. Therefore, we examined whether or not DltA could catalyze the N-acyl-l-cysteine synthetic reaction when l-cysteine (HS-CH2-CH(NH2)-COOH) was used instead of DltC. When the reaction mixture, including the purified DltA, ATP, d-alanine, and l-cysteine, was analyzed by HPLC on a Titan sphere TiO column, a marked increase in AMP was observed (see under “Stoichiometry”). This increase in AMP with consumption of ATP indicates that the reaction proceeded. In addition, when the same reaction mixture was analyzed on a CROWNPAK CR(+) column, a new product peak (retention time, 3.0 min) distinct from that of AMP appeared. No other new peaks were detected. Without DltA, the peaks due to the new product and AMP were not detected. We therefore used LC-MS/MS and NMR to characterize the product. The molecular mass of the product, as determined by LC-ESI-MS, was 192 Da. Next, the structure of the product was determined by means of LC-MS/MS (Fig. 2). A fragment ion at m/z 113 (negative ion mode) derived from the precursor ion at m/z 191 was observed, suggesting that the new product was not S-(d-alanyl)-l-cysteine (H3C-CH(NH2)-CO-S-CH2-CH(NH2)-COOH) but N-(d-alanyl)-l-cysteine (H3C-CH(NH2)-CO-NH-CH(COOH)-CH2-SH) (namely d-Ala-l-Cys). This identification was supported by the NMR spectra of the following product: 1H NMR (600 MHz, D2O): δ 1.44 (d, J = 7.1 Hz, 3H, C4-Me), 2.85 (ddd, J = 14.2, 6.9 Hz, 1H, H-5), 2.88 (ddd, J = 14.2, 4.4 Hz, 1H, H-5), 4.04 (q, J = 7.1 Hz, 1H, H-4), 4.48 (dd, J = 6.9, 4.4 Hz, 1H, H-2); 13C NMR (150 MHz, D2O): δ 16.7 (C4-Me), 25.4 (C-5), 49.1 (C-4), 55.4 (C-2), 170.7 (C-3), 174.1 (C-1); and authentic d-Ala-l-Cys, 1H NMR (600 MHz, D2O): δ 1.44 (d, J = 7.1 Hz, 3H, C4-Me), 2.83 (ddd, J = 14.2, 6.8 Hz, 1H, H-5), 2.88 (ddd, J = 14.2, 4.3 Hz, 1H, H-5), 4.04 (q, J = 7.1 Hz, 1H, H-4), 4.43 (dd, J = 6.8, 4.4 Hz, 1H, H-2); 13C NMR (150 MHz, D2O): δ 16.7 (C4-Me), 25.5 (C-5), 49.1 (C-4), 55.7 (C-2), 170.7 (C-3), 174.5 (C-1). Additionally, some key heteronuclear multiple bond connectivity correlations were observed between H-2 and each of C-1, C-3, and C-5, indicating that the product had a peptide bond in its structure. These findings demonstrate that DltA produced a dipeptide from d-alanine and l-cysteine, even though it usually catalyzes thioester bond formation (Fig. 3).
FIGURE 2.

MS/MS spectra of d-Ala-l-Cys formed by DltA in the positive (P) and negative (N) ion modes. The structural formula shows d-Ala-l-Cys. The theoretical m/z values of the fragments are provided with the structure. The fragment ion at m/z 113 (negative ion mode) suggests that the product is not S-(d-alanyl)-l-cysteine but d-Ala-l-Cys. CE, collision energy.
FIGURE 3.

Comparison of the dipeptide (d-Ala-l-Cys) synthetic reaction (a) with the original reaction (b) of DltA. In the original reaction, DltA produces d-alanyl-AMP, followed by the formation of S-(d-alanyl)-DltC with a covalent thioester bond between d-alanine and the phosphopantetheinyl prosthetic group of 4′-phosphopantetheine-bound DltC. The reaction product in the dotted box is d-Ala-l-Cys, which was identified in this study.
Stoichiometry
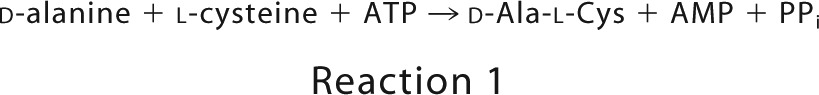
The stoichiometry of substrate consumption (ATP) and product formation (d-Ala-l-Cys and AMP) during the synthesis of d-Ala-l-Cys was examined. The results indicated that d-Ala-l-Cys and AMP were formed with the consumption of ATP in a stoichiometry of 1:1:1 (Fig. 4). DltA thus catalyzed Reaction 1,
 |
These findings also indicated that the amount of d-Ala-l-Cys produced is equivalent to that of AMP produced. In the following experiments, therefore, the amount of AMP produced was measured instead of the product, such as d-Ala-l-Cys, unless otherwise stated.
FIGURE 4.

Stoichiometry analysis of the d-Ala-l-Cys synthetic reaction of DltA. ATP (●); AMP (○); and d-Ala-l-Cys (♦). The methods used for analysis of the reactant and products are described under “Experimental Procedures.” All data points represent the mean values ± standard deviation for three experiments.
Characteristics of d-Ala-l-Cys Production by DltA
For d-Ala-l-Cys production by DltA, the Km and Vmax values for l-cysteine, d-alanine, and ATP were calculated from Michaelis-Menten kinetics. A Vmax value of 0.97 ± 0.02 units/mg and Km values of 0.97 ± 0.05 mm for ATP, 26 ± 2.0 mm for d-alanine, and 8.7 ± 0.8 mm for l-cysteine were obtained using d-Ala-l-Cys as the standard.
The effects of pH and temperature on d-Ala-l-Cys formation were examined. DltA exhibited maximum activity at pH 9.5, as shown in Fig. 5a. The optimal reaction temperature appeared to be 55 °C when the reaction was carried out for 5 min (Fig. 5b). Protein stability was investigated after incubation for 10 min at various temperatures. An aliquot of the enzyme solution was taken, and then the enzyme activity was assayed under the standard conditions. The enzyme exhibited the following activity: 0 °C, 91%; 10 °C, 90%; 20 °C, 95%; 30 °C, 100%; 40 °C, 100%; and 50 °C, 0%. Although the optimum temperature of DltA appeared to be 55 °C, DltA was completely inactivated on heating at 50 °C in the absence of a substrate. These results show that there may have been one or more compounds in the reaction mixture that increased the thermal stability of DltA. Thus, the stability of the enzyme was investigated in the presence of each of various compounds. Even after incubation for 10 min at 50 °C, DltA in the presence of ATP, d-alanine, or (NH4)2SO4 exhibited 100% of its initial activity, suggesting that the substrate or salt contributes to the thermostability of DltA.
FIGURE 5.

Effects of pH and temperature on the d-Ala-l-Cys synthetic activity of DltA. a, reactions were carried out for 5 min at 55 °C in the following buffers: buffer A consisting of 50 mm acetic acid, 50 mm MES, and 100 mm Tris (□) and buffer B consisting of 100 mm ACES, 52 mm Tris, and 52 mm ethanolamine (●). b, reactions were carried out for 5 min at various temperatures. Relative activity is expressed as a percentage of the maximum activity attained under the experimental conditions used.
Substrate Specificity for the Synthesis of Dipeptides (dl-Xaa-l-Cys) or Tripeptides (dl-Xaa-l-Cys-Gly)
Instead of d-alanine, another amino acid was added to the reaction mixture containing l-cysteine, and the ability of DltA to produce a peptide was measured as the amount of AMP produced. Among the tested amino acids listed under “Materials,” a significant increase in AMP was observed with glycine, d-serine, d-cysteine, d-threonine, or d-valine. These results suggest that the corresponding dipeptide would be produced. Also, when β-alanine or d-2-aminobutyric acid was used, the production of the corresponding amide compound was suggested. The structures of the products were also determined by LC-MS/MS. Each fragmentation pattern showed that the product was N-dl-Xaa-l-Cys rather than S-dl-Xaa-l-Cys, demonstrating that Gly-l-Cys, d-Ser-l-Cys, d-Cys-l-Cys, d-Thr-l-Cys, or d-Val-l-Cys was formed as the reaction product, respectively (Fig. 6, a–e). The Km and Vmax values for these substrates were determined (Table 1). According to the catalytic efficiencies (kcat/Km), the best substrate for DltA was d-alanine. When each of l-alanine, l-serine, and β-alanine was used, the corresponding products were identified as l-Ala-l-Cys, l-Ser-l-Cys, and β-Ala-l-Cys, respectively, after the long time reaction (5 h) (Fig. 6, f–h), although the Km and Vmax values could not be determined.
FIGURE 6.

MS/MS spectra of the dipeptides formed by DltA in the negative (N) ion mode. The structural formulae show the respective dipeptides: Gly-l-Cys (a); d-Ser-l-Cys (b); d-Cys-l-Cys (c); d-Thr-l-Cys (d); d-Val-l-Cys (e); l-Ala-l-Cys (f); l-Ser-l-Cys (g); and β-Ala-l-Cys (h). The theoretical m/z values of the fragments are given with the structures. CE, collision energy.
TABLE 1.
Substrate specificity (dl-Xaa-l-Cys and dl-Xaa-l-Cys-Gly)
The reactions were carried out in triplicate with measurement of amino acid-dependent AMP formation as described under “Experimental Procedures.” ND means not determined.
| d-Alanine side substrates | Km | Vmax | kcat/Km |
|---|---|---|---|
| mm | units/mg | s−1·mm−1 | |
| dl-Xaa-l-Cys | |||
| d-Alanine | 27 ± 2 | 0.97 ± 0.02 | 0.035 |
| Glycine | 110 ± 20 | 1.8 ± 0.1 | 0.016 |
| d-Serine | 81 ± 18 | 1.7 ± 0.1 | 0.019 |
| d-Cysteine | 89 ± 8 | 1.9 ± 0.1 | 0.021 |
| d-Threonine | 95 ± 24 | 0.94 ± 0.14 | 0.009 |
| d-Valine | 52 ± 30 | 0.60 ± 0.21 | 0.011 |
| l-Alanine | ND | ND | ND |
| l-Serine | ND | ND | ND |
| β-Alanine | ND | ND | ND |
| d-2-Aminobutyric acid | 56 ± 12 | 1.7 ± 0.1 | 0.029 |
| dl-Xaa-l-Cys-Gly | |||
| d-Alanine | 11 ± 4 | 0.70 ± 0.06 | 0.062 |
| Glycine | 97 ± 31 | 1.6 ± 0.2 | 0.016 |
| d-Serine | 71 ± 22 | 1.5 ± 0.2 | 0.019 |
| d-Cysteine | 190 ± 90 | 2.6 ± 0.7 | 0.013 |
| d-Threonine | ND | ND | ND |
| d-Valine | ND | ND | ND |
| l-Alanine | ND | ND | ND |
| l-Serine | ND | ND | ND |
| β-Alanine | 370 ± 160 | 2.6 ± 0.8 | 0.007 |
When the reaction mixture, comprising DltA, ATP, d-alanine, and l-cysteinyl-glycine (l-Cys-Gly), was analyzed, a considerable amount of AMP release was observed (Table 1). A reaction product other than AMP was detected on HPLC analysis and assessed by use of LC-MS/MS. The fragmentation detected on LC-MS/MS analysis indicated that d-Ala-l-Cys-Gly (tripeptide) was produced (Fig. 7a). Among the tested amino acids listed under “Materials,” we confirmed AMP release when glycine, d-serine, d-cysteine, or β-alanine was added to the reaction mixture containing l-Cys-Gly (Table 1), suggesting that the corresponding tripeptide was produced enzymatically. As the reaction product, each tripeptide, namely, Gly-l-Cys-Gly, d-Ser-l-Cys-Gly, d-Cys-l-Cys-Gly, or β-Ala-l-Cys-Gly, was identified on LC-MS/MS analysis (Fig. 7, b-d and i). For d-threonine, d-valine, l-alanine, and l-serine as the substrate, the corresponding products were identified as d-Thr-l-Cys-Gly, d-Val-l-Cys-Gly, l-Ala-l-Cys-Gly, and l-Ser-l-Cys-Gly, respectively, after the long time reaction (5 h) (Fig. 7, e–h).
FIGURE 7.

MS/MS spectra of the tripeptides formed by DltA in the positive (P) and negative (N) ion modes. The structural formulae show the respective tripeptides: d-Ala-l-Cys-Gly (a); Gly-l-Cys-Gly (b); d-Ser-l-Cys-Gly (c); d-Cys-l-Cys-Gly (d); d-Thr-l-Cys-Gly (e); d-Val-l-Cys-Gly (f); l-Ala-l-Cys-Gly (g); l-Ser-l-Cys-Gly (h); and β-Ala-l-Cys-Gly (i). The theoretical m/z values of the fragments are given with these structures. CE, collision energy.
Substrate Specificity for the Synthesis of Oligopeptides, d-Ala-dl-Cys-(dl-Xaa)n
DltA synthesized not only d-Ala-d-Cys (dipeptide) from d-alanine and d-cysteine but also d-Ala-d-Cys-d-Cys (tripeptide) from d-alanine and d-Cys-d-Cys (Table 2). We also observed the formation of these products in the reaction mixture containing DltA, ATP, d-alanine, and l-Cys-Gly-Gly-l-Arg-l-Glu or l-Cys-Gly-Gly-l-Arg-l-Glu-l-Ser-Gly-l-Ser-Gly-l-Ser. LC-MS/MS analysis showed that the products were N-d-Ala-l-Cys-(Xaa)n but S-d-Ala-l-Cys-(Xaa)n. Namely, a hexapeptide (d-Ala-l-Cys-Gly-Gly-l-Arg-l-Glu) or an undecapeptide (d-Ala-l-Cys-Gly-Gly-l-Arg-l-Glu-l-Ser-Gly-l-Ser-Gly-l-Ser), respectively, was identified as the corresponding reaction product (Fig. 8). When the concentration of each of the substrates was 25 mm, the hexapeptide synthetic activity of 0.36 units/mg and the undecapeptide synthetic activity of 0.45 units/mg were also similar to the d-Ala-l-Cys synthetic activity of 0.50 units/mg.
TABLE 2.
Substrate specificity (d-Ala-oligopeptide synthesis)
The reactions were carried out in triplicate with measurement of acid-dependent AMP formation as described under “Experimental Procedures.” The reactions were carried out as described except that various amino acids were used as substrates in place of l-cysteine. NT, not tested, because of the insolubility and expensiveness of the substrates.
| Cysteine side oligopeptides | Km | Vmax | kcat/Km |
|---|---|---|---|
| mm | units/mg | s−1·mm−1 | |
| l-Cysteine | 8.7 ± 0.8 | 0.90 ± 0.01 | 0.098 |
| d-Cysteine | 35 ± 10 | 0.75 ± 0.08 | 0.021 |
| l-Cys-Gly | 15 ± 1 | 0.35 ± 0.09 | 0.023 |
| d-Cys-d-Cys | NT | NT | NT |
| l-Cys-Gly-Gly-l-Arg-l-Glu | NT | NT | NT |
| l-Cys-Gly-Gly-l-Arg-l-Glu-l-Ser-l-Gly-l-Ser-l-Gly-l-Ser | NT | NT | NT |
FIGURE 8.

MS/MS spectra of oligopeptides formed by DltA in the positive ion mode. The structural formulae show the hexapeptide (a) and undecapeptide (b). The fragmentation of the reaction products was consistent with that of authentic d-Ala-l-Cys-Gly-Gly-l-Arg-l-Glu (a) or d-Ala-l-Cys-Gly-Gly-l-Arg-l-Glu-l-Ser-Gly-l-Ser-Gly-l-Ser (b), respectively. CE, collision energy.
Substrate Specificity for Cysteine Derivatives
The cysteine derivatives used in the above experiments have both an amino group and a thiol group. We next measured the enzyme activity in reaction mixtures that contained various cysteine derivatives as substrates, focusing on these functional groups of cysteine.
(i) We used two cysteine derivatives lacking an amino group (3-mercaptopropionic acid (HS-CH2-CH2-COOH) and thioglycolic acid (HS-CH2-COOH)). When 3-mercaptopropionic acid was used as the substrate, the obtained Vmax value of 0.63 units/mg was close to that (0.90 units/mg) of l-cysteine (Table 3). On HPLC analysis of this reaction mixture on a DevelosilTM RPAQUEOUS column, a new product peak (retention time, 9.5 min), distinct from that of AMP, appeared. We determined the structure of the reaction product by LC-MS/MS and found its molecular mass to be 177 Da. Moreover, the fragment ion at m/z 88 or 104 (Fig. 9a) demonstrated that the corresponding product was S-(d-alanyl)-3-mercaptopropionic acid (H3C-CH(NH2)-CO-S-CH2-CH2-COOH), derived through ligation of 3-mercaptopropionic acid and d-alanine. When another cysteine derivative lacking an amino group, thioglycolic acid, was used as the substrate, an increase in AMP was also detected, indicating that the reaction proceeded. The fragment ion at m/z 88 (Fig. 9b) observed on LC-MS/MS analysis demonstrated that the thioester compound S-(d-alanyl)-thioglycolic acid (H3C-CH(NH2)-CO-S-CH2-COOH) was synthesized as a product.
TABLE 3.
Substrate specificity (d-Ala-cysteine derivative synthesis)
The reactions were carried out in triplicate with measurement of acid-dependent AMP formation as described under “Experimental Procedures.” The reactions were carried out as described except that various amino acids were used as substrates in place of l-cysteine.
| Cysteine side derivatives | Km | Vmax | kcat/Km |
|---|---|---|---|
| mm | units/mg | s−1·mm−1 | |
| l-Cysteine | 8.7 ± 0.8 | 0.90 ± 0.01 | 0.098 |
| 3-Mercaptopropionic acid | 280 ± 130 | 0.63 ± 0.16 | 0.002 |
| Thioglycolic acid | 27 ± 4 | 0.44 ± 0.02 | 0.015 |
| N-Boc-l-Cys | 20 ± 2 | 0.29 ± 0.01 | 0.014 |
FIGURE 9.

MS/MS spectra of S-(d-alanyl)-3-mercaptopropionic acid (a) and S-(d-alanyl)-thioglycolic acid (b) in the positive (P) and negative (N) ion modes. The structural formulae show S-(d-alanyl)-3-mercaptopropionic acid (a) and S-(d-alanyl)-thioglycolic acid (b). The theoretical m/z values of the fragments are given with the structures. The fragment ion at m/z 88 or m/z 104 (negative ion mode) suggests that each product is an S-aminoacyl compound, respectively. CE, collision energy.
(ii) When cysteine derivatives lacking a thiol group (l-2-aminobutyric acid (H3C-CH2-CH(NH2)-COOH) or l-alanine (H3C-CH(NH2)-COOH)) or had a protected thiol group (S-methyl-l-cysteine (H3C-S-CH2-CH(NH2)-COOH), S-aminoethyl-l-cysteine (H2N-CH2-CH2-S-CH2-CH(NH2)-COOH) or S-acetamidomethyl-l-cysteine (H3C-CO-NH-CH2-S-CH2-CH(NH2)-COOH)) was used as substrates, the reaction did not proceed. In addition, when l-serine (HO-CH2-CH(NH2)-COOH) was used as the substrate instead of l-cysteine, an increase in AMP was not detected.
(iii) For cysteine derivatives with a protected amino group, N-(tert-butoxycarbonyl)- l-cysteine (N-Boc-l-Cys) {HS-CH2-CH(NH-COOC(CH3)3)-COOH}, an increase in AMP was detected, indicating that N-Boc-l-Cys acted as a substrate. However, when an N-Boc-l-Cys derivative lacking a thiol group, N-(tert-butoxycarbonyl)-l-alanine (N-Boc-l-Ala) {H3C-CH(NH-COOC(CH3)3)-COOH}, was used as the substrate, no AMP release was detected. LC-MS analysis of the reaction mixture containing N-Boc-l-Cys on a Kinetex® 1.7-μm C18 100 Å column revealed two product peaks (retention times, 1.5 and 3.2 min) in the LC-MS chromatogram at m/z 293 (positive ion mode). The structures of the reaction products were determined by LC-MS/MS. The fragment ion at m/z 104 (Fig. 10a) of the peak at 1.5 min demonstrated that the corresponding product was S-(d-alanyl)-N-Boc-l-Cys {H3C-CH(NH2)-CO-S-CH2-CH(NH-COOC(CH3)3)-COOH}. The fragment ion at m/z 113 (Fig. 10b) of the other peak (at 3.2 min) demonstrated that the corresponding product was N-(d-alanyl)-N-Boc-l-Cys {H3C-CH(NH2)-CO-N(COOC(CH3)3)-CH(COOH)-CH2-SH}. When the amount of each compound in the reaction mixture was monitored at several time points by LC-MS, we found an increase in the amount of N-(d-alanyl)-N-Boc-l-Cys with a decrease in that of S-(d-alanyl)-N-Boc-l-Cys as the reaction progressed (Fig. 10c).
FIGURE 10.

MS/MS spectra of the intermediate, S-(d-alanyl)-N-Boc-l-Cys (a), or the final product, N-(d-alanyl)-N-Boc-l-Cys (b), in the positive (P) and negative (N) ion modes, and LC-MS chromatogram of m/z 293 (positive ion mode) (c). The structural formulae show S-(d-alanyl)-N-Boc-l-Cys (a) (retention time, 1.5 min) and N-(d-alanyl)-N-Boc-l-Cys (b) (retention time, 3.2 min). The theoretical m/z values of the fragments are given with the structures. The fragment ions at m/z 104 (a, negative ion mode) and 113 (b, negative ion mode) suggest that the products are an S-aminoacyl compound and an N-aminoacyl compound, respectively. CE, collision energy. c, LC-MS chromatograms of S-(d-alanyl)-N-Boc-l-Cys and N-(d-alanyl)-N-Boc-l-Cys (m/z 293, positive ion mode). The peak with the retention time of 1.5 min corresponds to S-(d-alanyl)-N-Boc-l-Cys; the other peak at 3.2 min is N-(d-alanyl)-N-Boc-l-Cys.
Generality of the Novel Catalytic Mechanism
We investigated the generality of the novel catalytic mechanism for other enzymes. Previously, we reported that AcsA and Saccharomyces cerevisiae acetyl-CoA synthetase produce N-acyl-l-cysteine in a reaction mixture containing a suitable acid and l-cysteine as the substrates (9). Like DltA, both enzymes were confirmed here to form an S-acyl product as the reaction product, respectively, when a cysteine derivative lacking an amino group, 3-mercaptopropionic acid or thioglycolic acid was used as the substrate (Fig. 11). The fragment ion at m/z 75 (Fig. 11a) or m/z 103 (Fig. 11b) observed on LC-MS/MS analysis demonstrated that the thioester compounds S-acetyl-3-mercaptopropionic acid and S-acetyl-thioglycolic acid or S-isobutyryl-3-mercaptopropionic acid and S-isobutyryl-thioglycolic acid were synthesized by acetyl-CoA synthetase or AcsA, respectively. When a cysteine derivative lacking a thiol group (l-2-aminobutyric acid) or having a protected thiol group (S-acetamidomethyl-l-cysteine) was used as the substrate, an increase in AMP was not detected. In addition, when N-Boc-l-Cys was used as the substrate, both S-acetyl-N-Boc-l-Cys and N-acetyl-N-Boc-l-Cys were detected as reaction products with acetyl-CoA synthetase. Although we could only detect one product peak (retention time, 7.2 min) in the LC-MS chromatogram of m/z 262 (negative ion mode), fragment ions at m/z 75 and m/z 33 were detected in the MS/MS spectrum of the precursor ion m/z 262. Because the fragment ion at m/z 75 or m/z 33 is derived from S-acetyl-N-Boc-l-Cys or N-acetyl-N-Boc-l-Cys, respectively, the peak in the LC-MS chromatogram of m/z 262 was demonstrated to contain both S-acetyl-N-Boc-l-Cys and N-acetyl-N-Boc-l-Cys. On monitoring of the LC-MS/MS chromatogram of m/z 75 or m/z 33, we found that the increase in the amount of N-acetyl-N-Boc-l-Cys formed (m/z 33) was significantly delayed compared with that of S-acetyl-N-Boc-l-Cys formed (m/z 75) (Fig. 12). In the case of AcsA, the predicted product (S-isobutyryl-N-Boc-l-Cys or N-isobutyryl-N-Boc-l-Cys) was not separated on LC-MS/MS. When N-Boc-l-Ala was used as the substrate for acetyl-CoA synthetase or AcsA, an increase in AMP was not detected, respectively. These findings were the same as those obtained for DltA, indicating that AcsA and acetyl-CoA synthetase synthesize N-acyl products by the same mechanism as that used by DltA.
FIGURE 11.

MS/MS spectra of S-acetyl-3-mercaptopropionic acid and S-acetyl-thioglycolic acid formed by acetyl-CoA synthetase (a), S-isobutyryl-3-mercaptopropionic acid, and S-isobutyryl-thioglycolic acid formed by AcsA (b) in the negative (N) ion mode. The theoretical m/z values of the fragments are given with the structures. The fragment ion at m/z 75 or m/z 103 suggests that each product is an S-acyl compound, respectively. CE, collision energy.
FIGURE 12.

LC-MS chromatogram of the precursor ion m/z 262 (negative ion mode) (S-acetyl-N-Boc-l-Cys and N-acetyl-N-Boc-l-Cys) and LC-MS/MS chromatogram of the fragment ions m/z 75 (negative ion mode) derived from the intermediate, S-acetyl-N-Boc-l-Cys, and m/z 33 (negative ion mode) derived from the final product, N-acetyl-N-Boc-l-Cys. The structural formulae show S-acetyl-N-Boc-l-Cys and N-acetyl-N-Boc-l-Cys. The theoretical m/z values of the fragments are given with the structures. CE, collision energy.
Discussion
We previously reported that acyl- and acetyl-CoA synthetases produce N-acyl-l-cysteine when l-cysteine is used as the substrate instead of CoA (9). In general, acyl-CoA synthetases catalyze the ligation of an acid with CoA through a two-step reaction (7). The initial reaction involves the formation of an acyl-AMP intermediate with the release of inorganic pyrophosphate (step 1, acid + ATP → acyl-AMP + PPi). Next, the AMP of the acyl-AMP intermediate is displaced rapidly by CoA to yield acyl-CoA with a thioester bond (step 2, acyl-AMP + CoA → acyl-CoA + AMP). The discovery of the N-acyl-l-cysteine synthetic activity of acyl-CoA synthetases, which belong to the superfamily of adenylate-forming enzymes, demonstrates that these enzymes can produce products with an amide bond.
Here, we found that in addition to DltC (which is a d-alanyl carrier protein), l-cysteine can also serve as a substrate for DltA. Thus, both an amino acid (l-cysteine) and a protein containing 4′-phosphopantetheine can be a substrate for DltA. In addition, the product of the novel reaction was a dipeptide (d-Ala-l-Cys), demonstrating for the first time that a member of the superfamily of adenylate-forming enzymes, DltA, catalyzes peptide bond formation (Fig. 3), although we previously reported amide bond formation by acyl-CoA synthetases and luciferase. For d-Ala-l-Cys synthesis by DltA, we obtained a Vmax value of 0.97 units/mg and a Km value of 8.7 mm for l-cysteine, whereas for N-isobutyryl-l-cysteine synthesis by AcsA, a Vmax value of 0.20 units/mg and a Km value of 71 mm for l-cysteine were previously reported (9). The Km value (8.7 mm) of DltA for l-cysteine was about one-eighth that of AcsA (71 mm). This finding suggests that DltA binds with higher affinity to l-cysteine than AcsA does, although l-cysteine is not a physiological substrate for DltA in vivo. The Vmax value of DltA for d-Ala-l-Cys synthetic activity (0.97 units/mg) is 5-fold greater than that of AcsA for N-isobutyryl-l-cysteine synthetic activity (0.20 units/mg). These characteristics of a higher Vmax value and a lower Km value of DltA compared with those of AcsA are favorable for the enzymatic synthesis of peptides containing l-cysteine.
We further discovered that some amino acids, namely glycine, d-serine, d-cysteine, d-threonine, d-valine, l-alanine, l-serine, or β-alanine (as well as d-alanine), and l-cysteine, or the dipeptide l-Cys-Gly were also active as substrates for DltA, various peptides being produced (Figs. 6 and 7). The results of our analysis of the substrate specificity of DltA for different amino acids (Table 1) suggest that DltA would likely activate not only d-alanine, its natural substrate, but also some of its analogs.
Instead of DltC, l-cysteine was found to be an active substrate for DltA to yield various peptides. In contrast, amines that lack a thiol group (l-2-aminobutyric acid, l-alanine, or l-serine) or have a protected cysteine thiol group (S-methyl-l-cysteine, S-aminoethyl-l-cysteine, or S-acetamidomethyl-l-cysteine) were inert as substrates (Fig. 13a). The finding that a peptide bond was not formed between the amino group of these amines and the carboxyl group of d-alanine in the above reactions suggests that these two groups are not directly linked by DltA. When 3-mercaptopropionic acid or thioglycolic acid (both lacking an amino group) was used as the substrate instead of l-cysteine, a thioester compound (S-(d-alanyl)-3-mercaptopropionic acid or S-(d-alanyl)-thioglycolic acid) was synthesized as the reaction product (Fig. 13a and Table 3). In these cases, the thiol group of 3-mercaptopropionic acid or thioglycolic acid was ligated with the carboxyl group of d-alanine by DltA to form a thioester bond. Given that DltA is an acid-thiol ligase (EC 6.2.1), we speculated that the thioester compound (S-d-alanyl-l-cysteine) was first synthesized as a reaction intermediate when l-cysteine and d-alanine were used as the substrates. Subsequently, the resulting thioester intermediate would rapidly undergo spontaneous and irreversible S- to N-acyl transfer to yield a peptide bond (Fig. 14a). To confirm our hypothesis regarding this potential reaction mechanism, we attempted to detect the thioester intermediate during the peptide/amide bond synthetic reaction. When N-Boc-l-Cys was used instead of l-cysteine, we detected AMP release. Through LC-MS/MS, we identified the two detected compounds as S-(d-alanyl)-N-Boc-l-Cys (with a thioester bond) and N-(d-alanyl)-N-Boc-l-Cys (with an amide bond). However, when N-Boc-l-Ala, which lacks the thiol group of N-Boc-l-Cys, was used instead of N-Boc-l-Cys, N-(d-alanyl)-N-Boc-l-Ala (with an amide bond) was not produced (Fig. 13b). This finding indicates that direct d-alanylation of the Boc-protected amino group of N-Boc-l-Cys does not occur in the enzyme reaction catalyzed by DltA and that the thiol group of the substrate is essential for the formation of the N-(d-alanyl)-N-Boc compound. Considering these findings together with the fact that DltA produces a compound with a thioester bond as a product (i.e. DltA acts as an acid-thiol ligase), it is reasonable to presume that S-(d-alanyl)-N-Boc-l-Cys is a reaction intermediate synthesized by DltA and that N-(d-alanyl)-N-Boc-l-Cys is the final reaction product produced through spontaneous and irreversible S- to N-(d-alanyl) transfer. We monitored the amount of each of the two compounds in the reaction mixture containing N-Boc-l-Cys at several time points. Two minutes after the initiation of the reaction, the S-(d-alanyl)-N-Boc-l-Cys peak was detected in the LC-MS chromatogram of m/z 293 (positive ion mode), yet the N-(d-alanyl)-N-Boc-l-Cys peak was hardly detectable. As the reaction progressed, an increase in the amount of N-(d-alanyl)-N-Boc-l-Cys was observed with a concomitant decrease in that of S-(d-alanyl)-N-Boc-l-Cys (Fig. 10c). These findings strongly suggest that S-(d-alanyl)-N-Boc-l-Cys was converted to N-(d-alanyl)-N-Boc-l-Cys. We therefore propose that the mechanism for the peptide/amide bond synthetic reaction by DltA consists of the formation of the S-(d-alanyl)-intermediate by an enzymatic reaction and the subsequent synthesis of the N-(d-alanyl)-final product via spontaneous chemical intramolecular S- to N-acyl transfer. To our knowledge, there have been no previous reports of a peptide synthesis mechanism involving an enzymatic reaction and a subsequent chemical reaction. Both acetyl-CoA synthetase and AcsA, which belong to the superfamily of adenylate-forming enzymes, synthesized thioester compounds (S-acyl-3-mercaptopropionic acid and S-acyl-thioglycolic acid) when 3-mercaptopropionic acid or thioglycolic acid was used as the substrate instead of l-cysteine. In contrast, both enzymes could not synthesize amide compounds when a cysteine derivative lacking a thiol group (l-2-aminobutyric acid) or having a protected thiol group (S-acetamidomethyl-l-cysteine) was used as the substrate instead of l-cysteine. Furthermore, S-acetyl-N-Boc-l-Cys intermediates and N-acetyl-N-Boc-l-Cys final products were detected when acyl-CoA synthetase was used instead of DltA in the reaction mixtures containing a suitable acid and N-Boc-l-Cys as the substrates. These findings demonstrate that both enzymes form N-acyl-compounds via spontaneous chemical intramolecular S- to N-acyl transfer. Our findings demonstrate that all enzymes belonging to the superfamily of adenylate-forming enzymes would synthesize amides through this mechanism.
FIGURE 13.

Substrate specificity for cysteine derivatives and their reaction products. Cysteine derivatives used in this study and their reaction products. *, **, and ***; see Figs. 2, 9, and 10, respectively. N.D., not detected.
FIGURE 14.

Comparison of reaction mechanisms for peptide bond synthesis. Mechanisms of oligopeptide synthesis by DltA (a) and peptide synthesis by NCL (b). Both reactions proceed via the formation of a thioester intermediate and subsequent spontaneous intramolecular S → N acyl transfer.
Because of the potential usefulness of oligopeptides, many kinds of procedures have been developed for their production by means of enzymatic and chemical methods. To date, various peptidases (25, 26), an Empedobacter peptide-forming enzyme utilizing an amino acid methyl ester as a substrate (27) and Bacillus l-amino acid α-ligase (Fig. 15, d–f) (28, 29) have been utilized for peptide synthesis. NRPSs are also involved in peptide synthesis. In the general NRPS system of bacteria, peptides are produced by multienzyme complexes organized into modules, each of which can be further subdivided into a series of domains (14, 15, 30). Naturally produced peptide-based iron chelators such as bacillibactin (31) and erythrochelin (32) are synthesized by such multienzyme complexes. In these NRPS systems, a module consisting of at least three domains (an adenylation domain (Fig. 15b1), a thiolation domain, and a condensation domain (Fig. 15b2) is needed to form a peptide bond. Furthermore, very recently, Hamano and co-workers (33) reported amide bond formation by ORF19 of Streptomyces rochei NBRC12908, i.e. the stand-alone adenylation domain involved in streptothricin biosynthesis. Hamano and co-workers (33) showed that ORF19 catalyzes amide bond formation only between l-β-lysine and an l-β-lysine oligopeptide (Fig. 15c); it does not utilize cysteine as a substrate, namely intramolecular S- to N-acyl transfer does not occur in the enzyme reaction of ORF19. Therefore, the mechanism of peptide bond formation by DltA (Fig. 15a) that we discovered and present here is distinctly different from those of the above-mentioned enzymes.
FIGURE 15.

Comparison of enzyme reactions involved in peptide/amide bond synthesis. a, oligopeptide synthesis by DltA. b1 and b2, peptide synthesis by the general NRPS system. The adenylation domain produces an S-aminoacyl-thiolation domain intermediate (b1). The reaction for peptide bond formation between two aminoacyl substrates bound to the thiolation domain proceeds under the catalytic control of the condensation domain (b2). T, 4′-Phosphopantetheine-bound thiolation domain. c, amide synthesis by ORF19 of S. rochei NBRC12908 (the stand-alone adenylation domain). ORF19 catalyzes amide bond formation only between l-β-lysine and an l-β-lysine oligopeptide. d, peptide synthesis through the reverse reaction of various peptidases. e, dipeptide synthesis by Empedobacter peptide-forming enzyme. This enzyme uses amino acid methyl esters as substrates. f, dipeptide synthesis by Bacillus l-amino acid α-ligase. This enzyme has been reported to belong to the ATP-dependent carboxylate-amine/thiol ligase superfamily.
In addition to enzymatic methods, chemical methods have also been developed for the production of oligopeptides. Native chemical ligation (NCL) (34–37) is a very well known technique used for the chemical synthesis of peptides and proteins. NCL involves the chemoselective coupling of two peptides, one containing a C-terminal thioester (-CO-S-R) and the other containing an N-terminal cysteine residue. The reaction, which gives rise to a peptide bond at the point of ligation, includes the following two chemical steps: (i) intermediate synthesis through trans-thioesterification (i.e. thiol-thioester exchange) between the C-terminal thioester in one peptide and the N-terminal cysteine residue in the other peptide, and (ii) rapid intramolecular S- to N-acyl transfer in the resulting intermediate (Fig. 14b). To the best of our knowledge, no such enzyme that catalyzes the NCL reaction has yet been found. The intramolecular S- to N-acyl transfer step in the reaction mechanism for the synthesis of a peptide/amide bond by DltA is identical to the corresponding step of NCL. A reaction mechanism involving an enzymatic reaction yielding a thioester intermediate and the subsequent chemical reaction yielding the final product with a peptide/amide bond has not been reported previously.
For NCL, thioesterification of the C-terminal residue of the substrate is required, and therefore, a large number of studies have examined the synthesis of thioesterified peptides (38–40). In our new reaction involving DltA, however, thioesterified peptides are not needed as substrates; peptides that have a free C terminus can be used.
In recent years, there has been an explosion of interest in protein-assembly technologies (41–43). In particular, NCL is currently the most powerful ligation method for chemically synthesizing moderately sized proteins (including modified proteins). The strength of NCL is that it allows two peptides to be coupled together. Although in our previous study we used amino acid analogs (namely d-cysteine, dl-homocysteine, and l-cysteine methyl ester) instead of l-cysteine, we here used peptides containing an N-terminal cysteine residue. Our substrate specificity studies showed that DltA can also synthesize tripeptides from d-alanine and l-Cys-Gly or d-Cys-d-Cys (Table 3). These tripeptide synthetic activities were almost the same as that for d-Ala-l-Cys. Thus, DltA can use any peptide with an N-terminal cysteine residue. Indeed, DltA also synthesized oligopeptides from d-alanine and l-Cys-Gly-Gly-l-Arg-l-Glu or l-Cys-Gly-Gly-l-Arg-l-Glu-l-Ser-Gly-l-Ser-Gly-l-Ser. Itshexapeptide and undecapeptide synthetic activities (0.36 and 0.45 units/mg) were also similar to that for d-Ala-l-Cys (0.50 units/mg), suggesting that synthesis of oligopeptides by DltA is independent of the length of the peptide containing the N-terminal cysteine residue, with almost the same yield as that for dipeptide (d-Ala-l-Cys) synthesis. In other words, DltA can synthesize oligopeptides longer than an undecapeptide. Glycine, d-serine, d-cysteine, d-threonine, d-valine, l-alanine, l-serine, β-alanine, and d-2-aminobutyric acid could all be used instead of d-alanine by DltA, but dipeptides such as d-Ala-d-Ala could not, indicating that DltA activates amino acids (to yield aminoacyl-AMP) but not peptides. Therefore, the N-terminal portion of the final product is limited to a single amino acid, whereas even a relatively long peptide can be used as the C-terminal portion. The unique reaction mechanism we have uncovered suggests that all enzymes belonging to the superfamily of adenylate-forming enzymes should be capable of synthesizing amides as well as peptides. Any enzyme in this superfamily that can utilize the respective peptide instead of a single amino acid as a substrate can be used to synthesize peptides or proteins in future studies. Moreover, we could produce various kinds of amides, through the enzymatic coupling of acids (including luciferin) to various thiols with an amino group such as cysteine, its derivatives, and peptides with an N-terminal cysteine, by using acyl-CoA synthetase, acetyl-CoA synthetase, or firefly luciferase (9). Given the vast array of enzymes that belong to this superfamily and could activate the respective substrates (44), these enzymes would form not only peptides but also new types of amide compounds, such as N-acyl-peptides and N-luciferyl-l-cysteine.
Author Contributions
M. K. is responsible for the project planning. T. A., Y. H., and M. K. designed the study and wrote the paper. T. A., Y. H., Y. Z., Y. G., and T. K. performed the experiments. T. A., Y. H., Y. Z., Y. G., T. K., and M. K. analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Shinji Kuroda, Dr. Shun-Ichi Suzuki, and Sachise Eto (Ajinomoto Co., Inc.); Prof. Masao Yamana and Kenta Kobayashi (Tokyo Denki University); and Dr. Toshihiko Sakurai (Tottori University) for the useful discussions and valuable advice, and Prof. Akira Nakamura (University of Tsukuba) for providing the chromosomal DNA of B. subtilis strain 168.
This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan. The authors declare that they have no conflicts of interest with the contents of this article.
- NRPS
- nonribosomal peptide synthetase
- NCL
- native chemical ligation
- d-Ala-l-Cys
- N-d-alanyl-l-cysteine
- N-Boc
- N-t-butoxycarbonyl
- ACES
- 2-[(2-amino-2-oxoethyl)amino]ethanesulfonic acid.
References
- 1. Hashimoto Y., Hosaka H., Oinuma K., Goda M., Higashibata H., and Kobayashi M. (2005) Nitrile pathway involving acyl-CoA synthetase: overall metabolic gene organization and purification and characterization of the enzyme. J. Biol. Chem. 280, 8660–8667 [DOI] [PubMed] [Google Scholar]
- 2. Kobayashi M., and Shimizu S. (1998) Metalloenzyme nitrile hydratase: structure, regulation, and application to biotechnology. Nat. Biotechnol. 16, 733–736 [DOI] [PubMed] [Google Scholar]
- 3. Faergeman N. J., and Knudsen J. (1997) Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochem. J. 323, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Starai V. J., Celic I., Cole R. N., Boeke J. D., and Escalante-Semerena J. C. (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 [DOI] [PubMed] [Google Scholar]
- 5. Schneider K., Hövel K., Witzel K., Hamberger B., Schomburg D., Kombrink E., and Stuible H. P. (2003) The substrate specificity-determining amino acid code of 4-coumarate:CoA ligase. Proc. Natl. Acad. Sci. U.S.A. 100, 8601–8606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gulick A. M., Starai V. J., Horswill A. R., Homick K. M., and Escalante-Semerena J. C. (2003) The 1.75 Å crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 42, 2866–2873 [DOI] [PubMed] [Google Scholar]
- 7. Starai V. J., and Escalante-Semerena J. C. (2004) Acetyl-coenzyme A synthetase (AMP forming). Cell. Mol. Life Sci. 61, 2020–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morgan-Kiss R. M., and Cronan J. E. (2004) The Escherichia coli fadK (ydiD) gene encodes an anaerobically regulated short chain acyl-CoA synthetase. J. Biol. Chem. 279, 37324–37333 [DOI] [PubMed] [Google Scholar]
- 9. Abe T., Hashimoto Y., Hosaka H., Tomita-Yokotani K., and Kobayashi M. (2008) Discovery of amide (peptide) bond synthetic activity in acyl-CoA synthetase. J. Biol. Chem. 283, 11312–11321 [DOI] [PubMed] [Google Scholar]
- 10. Nakatsu T., Ichiyama S., Hiratake J., Saldanha A., Kobashi N., Sakata K., and Kato H. (2006) Structural basis for the spectral difference in luciferase bioluminescence. Nature 440, 372–376 [DOI] [PubMed] [Google Scholar]
- 11. Conti E., Franks N. P., and Brick P. (1996) Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure 4, 287–298 [DOI] [PubMed] [Google Scholar]
- 12. Conti E., Stachelhaus T., Marahiel M. A., and Brick P. (1997) Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. May J. J., Kessler N., Marahiel M. A., and Stubbs M. T. (2002) Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U.S.A. 99, 12120–12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marahiel M. A., Stachelhaus T., and Mootz H. D. (1997) Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 97, 2651–2674 [DOI] [PubMed] [Google Scholar]
- 15. Finking R., and Marahiel M. A. (2004) Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 58, 453–488 [DOI] [PubMed] [Google Scholar]
- 16. Yonus H., Neumann P., Zimmermann S., May J. J., Marahiel M. A., and Stubbs M. T. (2008) Crystal structure of DltA: implications for the reaction mechanism of non-ribosomal peptide synthetase adenylation domains. J. Biol. Chem. 283, 32484–32491 [DOI] [PubMed] [Google Scholar]
- 17. Osman K. T., Du L., He Y., and Luo Y. (2009) Crystal structure of Bacillus cereus d-alanyl carrier protein ligase (DltA) in complex with ATP. J. Mol. Biol. 388, 345–355 [DOI] [PubMed] [Google Scholar]
- 18. Kiriukhin M. Y., and Neuhaus F. C. (2001) d-Alanylation of lipoteichoic acid: role of the d-alanyl carrier protein in acylation. J. Bacteriol. 183, 2051–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neuhaus F. C., and Baddiley J. (2003) A continuum of anionic charge: structures and functions of d-alanyl-teichoic acids in Gram-positive bacteria. J. Microbiol. Mol. Biol. Rev. 67, 686–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gulick A. M. (2009) Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4, 811–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee S. G., and Lipmann F. (1975) Tyrocidine synthetase system. Methods Enzymol. 43, 585–602 [DOI] [PubMed] [Google Scholar]
- 22. Katano H., Tanaka R., Maruyama C., and Hamano Y. (2012) Assay of enzymes forming AMP + PPi by the pyrophosphate determination based on the formation of 18-molybdopyrophosphate. Anal. Biochem. 421, 308–312 [DOI] [PubMed] [Google Scholar]
- 23. Kimura Y., Shibasaki S., Morisato K., Ishizuka N., Minakuchi H., Nakanishi K., Matsuo M., Amachi T., Ueda M., and Ueda K. (2004) Microanalysis for MDR1 ATPase by high-performance liquid chromatography with a titanium dioxide column. Anal. Biochem. 326, 262–266 [DOI] [PubMed] [Google Scholar]
- 24. Ellis K. J., and Morrison J. F. (1982) Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 87, 405–426 [DOI] [PubMed] [Google Scholar]
- 25. Morihara K. (1987) Using proteases in peptide synthesis. Trends Biotechnol. 5, 164–170 [Google Scholar]
- 26. Usuki H., Yamamoto Y., Arima J., Iwabuchi M., Miyoshi S., Nitoda T., and Hatanaka T. (2011) Peptide bond formation by aminolysin-A catalysis: a simple approach to enzymatic synthesis of diverse short oligopeptides and biologically active puromycins. Org. Biomol. Chem. 9, 2327–2335 [DOI] [PubMed] [Google Scholar]
- 27. Yokozeki K., and Hara S. (2005) A novel and efficient enzymatic method for the production of peptides from unprotected starting materials. J. Biotechnol. 115, 211–220 [DOI] [PubMed] [Google Scholar]
- 28. Tabata K., Ikeda H., and Hashimoto S. (2005) ywfE in Bacillus subtilis codes for a novel enzyme, l-amino acid ligase. J. Bacteriol. 187, 5195–5202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tabata K., and Hashimoto S. (2007) Fermentative production of l-alanyl-l-glutamine by a metabolically engineered Escherichia coli strain expressing l-amino acid α-ligase. Appl. Environ. Microbiol. 73, 6378–6385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lautru S., Deeth R. J., Bailey L. M., and Challis G. L. (2005) Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat. Chem. Biol. 1, 265–269 [DOI] [PubMed] [Google Scholar]
- 31. May J. J., Wendrich T. M., and Marahiel M. A. (2001) The dhb operon of Bacillus subtilis encodes the biosynthetic template for the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threonine trimeric ester bacillibactin. J. Biol. Chem. 276, 7209–7217 [DOI] [PubMed] [Google Scholar]
- 32. Lazos O., Tosin M., Slusarczyk A. L., Boakes S., Cortés J., Sidebottom P. J., and Leadlay P. F. (2010) Biosynthesis of the putative siderophore erythrochelin requires unprecedented crosstalk between separate nonribosomal peptide gene clusters. Chem. Biol. 17, 160–173 [DOI] [PubMed] [Google Scholar]
- 33. Maruyama C., Toyoda J., Kato Y., Izumikawa M., Takagi M., Shin-ya K., Katano H., Utagawa T., and Hamano Y. (2012) A stand-alone adenylation domain forms amide bonds in streptothricin biosynthesis. Nat. Chem. Biol. 8, 791–797 [DOI] [PubMed] [Google Scholar]
- 34. Dawson P. E., Muir T. W., Clark-Lewis I., and Kent S. B. (1994) Synthesis of proteins by native chemical ligation. Science 266, 776–779 [DOI] [PubMed] [Google Scholar]
- 35. Bennett C. S., and Wong C. H. (2007) Chemoenzymatic approaches to glycoprotein synthesis. Chem. Soc. Rev. 36, 1227–1238 [DOI] [PubMed] [Google Scholar]
- 36. Crich D., and Banerjee A. (2007) Native chemical ligation at phenylalanine. J. Am. Chem. Soc. 129, 10064–10065 [DOI] [PubMed] [Google Scholar]
- 37. Shang S., Tan Z., and Danishefsky S. J. (2011) Application of the logic of cysteine-free native chemical ligation to the synthesis of human parathyroid hormone (hPTH). Proc. Natl. Acad. Sci. U.S.A. 108, 5986–5989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mende F., and Seitz O. (2011) 9-Fluorenylmethoxycarbonyl-based solid-phase synthesis of peptide α-thioesters. Angew. Chem. Int. Ed. Engl. 50, 1232–1240 [DOI] [PubMed] [Google Scholar]
- 39. Sewing A., and Hilvert D. (2001) Fmoc-compatible solid-phase peptide synthesis of long C-terminal peptide thioesters. Angew. Chem. Int. Ed. Engl. 40, 3395–3396 [DOI] [PubMed] [Google Scholar]
- 40. Ingenito R., Bianchi E., Fattori D., and Pessi A. (1999) Solid phase synthesis of peptide C-terminal thioesters by Fmoc/t-Bu chemistry. J. Am. Chem. Soc. 121, 11369–11374 [Google Scholar]
- 41. Dawson P. E., and Kent S. B. (2000) Synthesis of native proteins by chemical ligation. Annu. Rev. Biochem. 69, 923–960 [DOI] [PubMed] [Google Scholar]
- 42. Hackenberger C. P., and Schwarzer D. (2008) Chemoselective ligation and modification strategies for peptides and proteins. Angew. Chem. Int. Ed. Engl. 47, 10030–10074 [DOI] [PubMed] [Google Scholar]
- 43. Kent S. B. (2009) Total chemical synthesis of proteins. Chem. Soc. Rev. 38, 338–351 [DOI] [PubMed] [Google Scholar]
- 44. Omura S., Ikeda H., Ishikawa J., Hanamoto A., Takahashi C., Shinose M., Takahashi Y., Horikawa H., Nakazawa H., Osonoe T., Kikuchi H., Shiba T., Sakaki Y., and Hattori M. (2001) Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. U.S.A. 98, 12215–12220 [DOI] [PMC free article] [PubMed] [Google Scholar]