

Abstract
Chromatin undergoes a rapid ATP-dependent, ATM and H2AX-independent decondensation when DNA damage is introduced by laser microirradiation. Although the detailed mechanism of this decondensation remains to be determined, the kinetics of decondensation are similar to the kinetics of poly(ADP-ribosyl)ation. We used laser microirradiation to introduce DNA strand breaks into living cells expressing a photoactivatable GFP-tagged histone H2B. We find that poly(ADP-ribosyl)ation mediated primarily by poly(ADP-ribose) polymerase 1 (PARP1) is responsible for the rapid decondensation of chromatin at sites of DNA damage. This decondensation of chromatin correlates temporally with the displacement of histones, which is sensitive to PARP inhibition and is transient in nature. Contrary to the predictions of the histone shuttle hypothesis, we did not find that histone H1 accumulated on poly(ADP-ribose) (PAR) in vivo. Rather, histone H1, and to a lessor extent, histones H2A and H2B were rapidly depleted from the sites of PAR accumulation. However, histone H1 returns to chromatin and the chromatin recondenses. Thus, the PARP-dependent relaxation of chromatin closely correlates with histone displacement.
Keywords: chromatin, chromatin remodeling, DNA damage, DNA damage response, histone modification, PARG, PARP-1, poly(ADP-ribosyl)ation, histone H1
Introduction
Chromatin is widely held to be a barrier to the execution of nuclear functions including DNA replication, DNA repair, and RNA transcription. Thus, it is important to define the mechanisms that regulate chromatin structure to orchestrate and execute these functions. The molecular delineation of cellular events occurring at the sites of DNA strand breaks has revealed an intricate network of sensing, signaling, and repair proteins that coordinate the accurate correction of DNA damage (1, 2). Accompanying this DNA strand break repair factor network is a remodeling of chromatin structure (3, 4). For example, modification of the histone H2A variant, H2AX, through phosphorylation of serine 139 is so characteristic of DNA double-strand break (DSB)4 signaling that it has been used as a quantitative marker to measure the number of DSBs in the cell (5).
In addition to biochemical modifications of chromatin proteins, morphological changes in chromatin structure have also been reported. In a landmark paper, Kruhlak and colleagues (6) used laser microirradiation to introduce DSBs at defined sites in the interphase nucleus while simultaneously photoactivating a GFP-tagged histone H2B. This enabled visualization of the changes in the organization of chromatin in response to the introduction of DSBs by virtue of the photoactivation of GFP-histone H2B solely in the region of the nucleus where DSBs were introduced into the chromatin. The large-scale decondensation of irradiated chromatin observed in this study was found to be extremely rapid, occurring within seconds of damage, and was ATM-independent but ATP-dependent. This chromatin relaxation was observed in both H2AX null and ATM null cells indicating that the phosphorylation of histone H2AX or any process dependent on either it or ATM kinase activity were not required for the observed decondensation. The mechanism(s) responsible for this chromatin decondensation and the contribution of any of the described biochemical changes in chromatin structure to this process is unknown.
Using live-cell imaging techniques, recent studies have identified the accumulation of PARP1 and PAR to be among the first events that take place at DNA double-stranded breaks, occurring within the first seconds after DNA injury (7, 8). PAR synthesis at sites of DNA damage is catalyzed by PARP1 and PARP2, whose activity is increased 10–500-fold by single and double-strand breaks in the DNA. PAR accumulation is transient and reversible due to its rapid catabolism by poly(ADP-ribose) glycohydrolase (PARG) (9). Although the accumulation kinetics of PARP2 are slower (8), PARP1 is recruited within 1 s of DNA damage (7). PARP1 synthesizes PAR, and contributes to the rapid activation of the ATM-dependent DNA damage signaling cascade, in part by recruiting MRE11 and NBS1 to DSBs (7) and to sites of stalled replication forks (10). ATM and MRE11 both have a functional PAR binding domain that is important for their function in the DNA damage response, which suggests that PAR deposition at DSBs is likely involved in the efficient recruitment of these repair mediators (7, 11, 12). Similarly, poly(ADP-ribosyl)ation has recently been reported to recruit the chromatin remodelers ALC1 and SMARCA5/SNF2H to sites of DNA damage as well as a wide range of transcriptional regulators (13–16). Based on the recruitment kinetics of PARP1 to the sites of DSBs (7) and its potential to decondense chromatin, we reasoned that PARP1 and/or PAR may function in the reported rapid decondensation of chromatin.
Several lines of evidence from in vivo studies strongly suggest that poly(ADP-ribose) (PAR) is critically involved in the local chromatin decondensation (17–19) necessary to give access to the DNA damage signaling and repair machineries (20). Poly(ADP-ribose) polymerase-1 (PARP1) synthesizes PAR on acceptor proteins, by adding the first ADP-ribose unit usually on the side chain of a glutamic acid residue and elongating the polymer by successive addition of ADP-ribose. Acceptor proteins are, for the most part, chromatin associated (21) and preferred substrates include PARP1 (automodification) and histones (20).
Histone H1 is of particular interest with respect to the impact of PARP activation. Upon maximal activation, such as what is observed at sites of DNA strand breaks, histone H1 is the preferred chromatin substrate (20). Histone H1 is also able to bind to PAR directly through non-covalent interactions between PAR and the lysine-rich C-terminal domain (22, 23). This has led to the hypothesis that PAR may both displace histone H1 from the chromatin while maintaining it in the vicinity of the strand break, serving as a “histone shuttle” (24). Upon degradation of the PAR polymer by poly(ADP-ribose) glycohydrolase activity, histone H1 is then able to re-bind the chromatin (24). Purified PARP1 leads to relaxation of the chromatin fiber through the interaction of PAR with histones H1, H2A, and H2B (25, 26). Upon treatment with PARG, the chromatin structure is restored to a condensed state, demonstrating that this decondensation is fully reversible (25). The local chromatin relaxation associated with transcription has also been attributed to the activity of PARP1 at specific genetic loci (17–19). PARP1 was also found to compete for promoter occupancy with histone H1 during transcriptional activation (27–29). This may reflect the ability of PARP1 to bind to nucleosomes and compete with H1 for binding to nucleosomes in the absence of PARP activity (30). The presence of PARP1 correlated with transcriptional activation and the absence of histone H1.
Although the mechanistic details of the structural changes occurring at DNA strand breaks remain ill defined, based on the in vitro evidence, we propose that the rapid accumulation of PARP1 and PAR at DNA strand breaks is a key element inducing dynamic local chromatin structural changes necessary for efficient DNA repair. In the current study, we confirm that the rapid decondensation at sites of laser microirradiation-induced DNA damage is driven by PARP activity (15) and show that it results in the displacement of histone H1 and to a lesser extent core histones. Contrary to the prediction of the H1 shuttling model and prior in vitro work, histone H1 is displaced from both the damaged DNA and PAR-enriched sites associated with DNA damage. This is consistent with poly(ADP-ribosyl)ation playing a role in the displacement of histone H1 but would imply that, despite having a high affinity for PAR in vitro, histone H1 does not accumulate on PAR at sites of DNA damage. We use multiple approaches to modulate PAR content at sites of double-strand breaks and find that the regulation of PAR activity is highly correlated with chromatin decondensation and histone H1 displacement at sites of DNA damage in vivo.
Experimental Procedures
Cell Culture and Transfection
Mouse embryonic fibroblasts (MEFs) derived from PARP1-deficient mice (A1) and normal MEFs (C3H10T½) were maintained in minimal essential medium (α) containing 10% FBS and 2 mm glutamine. Human neuroblastoma (SK-N-SH) and human osteosarcoma (U2OS) cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). h-Tert immortalized human retina epithelium (RPE-1 H4-paGFP H2B-mRFP) (31) were grown in DMEM/F-12 medium supplemented with 10% FBS. All cell lines were maintained at 37 °C, under a controlled 5% CO2 atmosphere in a humidified incubator. The construct encoding photoactivatable histone H2B (H2B-paGFP) and the construct encoding H1.5-GFP were transiently transfected in both normal (C3H10T½) and PARP1-deficient (A1) cell lines as well as into SK-N-SH and U2OS cells using Effectene reagent (Qiagen) according to the manufacturer's protocol. Transient co-expression of FLAG-tagged PARP1 and H2B-paGFP in PARP1-deficient cells or FLAG-tagged PARG and H2B-paGFP in SK-N-SH cells was achieved under the same conditions. Constructs allowing the expression of tagged PARP1 and PARG have been described (7, 32). To establish conditions for co-transfection, the ratio between the experimental construct and the H2B-paGFP was varied until co-transfection was observed in 100% of the cells. This was determined by fixation and immunofluorescent staining of the cells for the transfected construct.
When indicated, cells were incubated with the PARP1 inhibitor AG14361, kindly provided by Pfizer/Aguron, or the newer and more potent inhibitor BMN673 (33) (Fig. 2D) 1 h prior to microirradiation in concentrations ranging between 1 nm and 5 μm. For each measurement, a minimum of 15 and a maximum of 30 cells were obtained from three separate experiments and used for the purpose of generating the mean ± S.D. of decondensation. In the original experiments, some cells show changes in nuclear shape or rotation that could not be accurately corrected. These cells were not included in the 15–30 cells used for measurements. There was no indication that decondensation differed visibly in these excluded cells.
FIGURE 2.

Poly(ADP-ribosyl)ation dependence of chromatin decondensation at sites of DNA damage. A, DNA damage was introduced by microirradiation in SK-N-SH cells treated with the PARP inhibitor AG14361. An SK-N-SH neuroblastoma cell nucleus is shown after treatment with 100 nm AG14361. The left and middle panels were collected immediately after and 2 min after microirradiation, respectively. The right panel is a composite of the immediate (red) and 2 min (green) panels. B, mouse A1 cells were microirradiated and examined by time lapse microscopy. The left panel shows an image collected immediately after the introduction of DNA damage. This is represented as red in the composite image in the right panel. The center panel shows an image collected 2 min after the introduction of DNA damage. This image is represented in green in the composite image. C, mouse A1 cells were transfected with FLAG-PARP1 to reconstitute PARP activity. The left and middle panels show images collected immediately and 2 min after the introduction of DNA damage, respectively. In the right panel, the first time point is represented in red, whereas the 2-min time point is represented in green. Scale bar is 3 μm. D, U2OS cells were laser microirradiated in the presence or absence of PARP inhibitors 100 nm AG14361 or 100 nm BMN673, fixed after 2 min, and then stained for PAR by immunofluorescence. The images show the distribution of PAR in cells. The image showing the cells treated with BMN673 contain arrows to facilitate locating the weak PAR signal. E, U2OS cells transfected with PARG-GFP were exposed to laser microirradiation, fixed after 2 min, and subsequently stained for PAR. F, quantitative measurements of the chromatin decondensation 3 min post-irradiation in U2OS cells in presence and absence of Hoechst 33342 and PARP inhibitors AG1436 and BMN673. G, a graphical representation of the dynamics of expansion following laser microirradiation in U2OS cells that have not been treated versus cells that have been sensitized with Hoechst in presence or absence of PARP inhibitor AG14361. H, quantification of the normalized decondensation in cells treated with PARP inhibitor or overexpressing PARG. The green bar is the control, the red bars are treatments with AG14361, and the blue bars are experiments with PARG overexpression. I, quantitative representation of the extent of decondensation observed in mouse 10T1/2 cells under control conditions or after treatment with increasing concentrations of AG14361. A1 cells and A1 cells transiently expressing PARP1 are also shown. All values were normalized to the control conditions for mouse 10T1/2 cells. The green bar is the control, the red bars are treatments with AG14361, and the blue bars are PARP1 null MEFs without or with PARP1 transfection.
Two-photon Microirradiation
We carried out laser microirradiation by modifying a previously described method. Cells were grown in MatTek 35-mm glass bottom dishes overnight to about 75% confluence.
The following day the medium was replaced with fresh media containing 0.5 μg/ml of Hoechst 33342 and incubated for 30 min. The media was then exchanged for 1 ml of new media. Cells were microirradiated and imaged on a Zeiss Axiovert 200M inverted microscope attached to a LSM510 NLO laser scanning system with a 25-milliwatt argon laser line. The system was connected to a Coherent Mira 900 2-photon laser. H2B-paGFP was photoactivated and DNA strand breaks were generated along a 2-μm wide region across the nucleus of a single living cell by excitation of the Hoechst 33342 dye using the 2-photon 750-nm titanium-sapphire laser line. The laser output was set to 5% (unless stated otherwise) and we used 10 iterations. The cells were visualized using a ×40 apochromatic 1.3 NA oil immersion objective lens and the 488-nm laser line (from 25 milliwatt argon laser).
405-nm Laser Microirradiation followed by Live-cell Observations at a Spinning Disc Microscope
Live-cell observations were carried out using a PerkinElmer Ultraview spinning-disk confocal microscope equipped with 405, 488, and 561 nm diode lasers. Cells were laser microirradiated using the FRAP module in the microscope acquisition software Volocity 6.3. A line of 165 nm (1 pixel, ×40 objective) was drawn across the midsection of the nuclei and microirradiation was carried out by scanning this region of interest 10 times using 10% of the laser power of the 405-nm laser. For the live-cell imaging and laser microirradiation the same Zeiss 1.3 NA ×40 objective lens was used. The spinning disk has the advantage of reduced phototoxicity in extended duration experiments but does photobleach GFP at the site of damage.
Image Processing and Analysis of Photon Microirradiation
Line scan profiles were generated from H2B-paGFP time series images and the width of the photoactivated/microirradiated region was measured at 50% of the maximum intensity using MetaMorph (Molecular Devices) and ImageJ image processing and analysis software (34). For representing images in figures, the original 12-bit datasets were scaled to 8-bit images by subtracting background and then scaling the image such that the first image following laser microirradiation, which has the highest peak intensities, spanned the range of 0–255. In the color composites, it was sometimes necessary to further contract the range for both the red and green images to be maximally visible. The grayscale images serve as a reference for the initial relative intensities of the red and green images. Time-lapse series recorded at the spinning disc confocal microscope were exported from Volocity as OME-TIFFs and post processed in Imaris (Andor) or ImageJ (34).
Registration of two-dimensional time series was carried out by the ImageJ plug-in StackReg (35). The measurement at half-maximum intensity of the photoactivated H2Bpa-GFP was determined using an ImageJ macro that automatically used the best approximation for a Gaussian fit for the signal of the line scan for each time point of a time series. The H1.5-GFP intensity/time measurements were performed using another macro, which measured the relative change in intensity in a region of interest in the nucleus and normalized this intensity to the whole nuclear intensity. Plots were generated using Prism (Graph Pad) and MATLAB (MathWorks). For quantification of PAR content using the 10H antibody, the exposure time was set based on obtaining adequate exposure without saturation using the control cells. Using the same exposure setting, images were collected of both the 250 nm AG14361- and 2.5 μm AG14361-treated samples. The 12-bit images were imported into Adobe Photoshop and scaled using the “levels” function to represent the dynamic range of the PAR staining in the control cells on the 8-bit display. Identical scaling was used for the drug-treated samples and the composite image was converted to 8-bit grayscale. This allows direct comparison of the relative intensities of PAR staining across the samples.
Statistical Data Analysis
The effects on PARP inhibitor, PARP- and PARG-transfected cells in Fig. 2, F, H, and I, were calculated by comparing individual treatments with the control and using unpaired t tests. Significant differences were indicated when found. One star represents a significant difference between the experiment and the control group (5% level), three stars highly significant differences (0.01% level), and 4 stars very significant differences (0.001%). Sample sizes varied between 8 and 64 observations, depending on the experiment.
The H1.5 recovery slopes of the PARG-knocked down cells shown and the control cells in Fig. 5C were compared in the linear range of the curve (200–900 s) using the standard test for assessing the differences of slopes. This was done using the F-test (36) that is implemented in Prism®. The slopes of the curves were significantly different on a 0.01% level.
FIGURE 5.

Accumulation of poly(ADP-ribose) at sites of laser microirradiation-induced DNA damage. A, SK-N-SH cells were incubated with or without the PARP-inhibitor AG14361 at the concentrations indicated. DNA damage was introduced into SK-N-SH cells by laser microirradiation. The cells were fixed within 5 min of introducing damage and then stained with an antibody recognizing poly(ADP-ribose). The images were collected using identical imaging and data processing conditions to allow for visual inspection of quantitative differences. B, cells transfected with siRNA targeting PARG and stained for poly(ADP-ribose). C, the kinetics of histone H1.5 displacement and reassociation in cells transfected with siRNA targeting PARG.
Results
Chromatin Decondensation at Sites of Double-strand Breaks
We investigated chromatin decondensation during DNA strand break induction in several human and mouse cell lines using H2B-paGFP or H4-paGFP with H2B-mRFP, for chromatin decondensation. DNA strand breaks were introduced by microirradiation employing a 2-photon excitation of Hoechst 33342 as we have described previously (7). Under these conditions, the photoactivatable GFP associated with histone H2B or H4 is activated and fluoresces when excited using a 488-nm laser line. Fig. 1, A–D, shows examples of SK-N-SH, U2OS, 10T1/2, and RPE-1 cells following the introduction of DNA damage. The left panel shows the first image collected after the introduction of damage. This region is identified by the photoactivation of H2B-paGFP. The middle panel shows an image collected 2 min later. The composite image (right panel) superimposes the original chromatin organization in the damaged region (red) and the later time point (green). The composite images (and Fig. 1F, which is a line scan through the photoactivated area of a representative U2OS cell) show significant amounts of green (final time point) beyond the area occupied by the same chromatin in the first time point image, reflecting expansion of the photoactivated chromatin beyond the original dimensions. To make sure that the photoactivated chromatin represents the sites of damaged chromatin we have laser microirradiated cells and fixed them after 10 s and 2 min. After staining with γ-H2AX, the signals of the photoactivated and the damaged, γ-H2AX-stained chromatin were congruent (Fig. 1, E, G, and H), showing that the signal of the photoactivated H2A-GFP represents the damaged chromatin. The time course of decondensation recorded in U2OS cells at the spinning disc microscope (Fig. 2G) reveals that changes in chromatin structure are initiated within seconds after the introduction of DNA damage. We found no difference in the results between the 405-nm diode laser and the damage produced by the 750-nm 2-photon laser. Thus, our results confirm previous work showing that DNA damage is associated with a rapid decondensation of chromatin (6).
FIGURE 1.
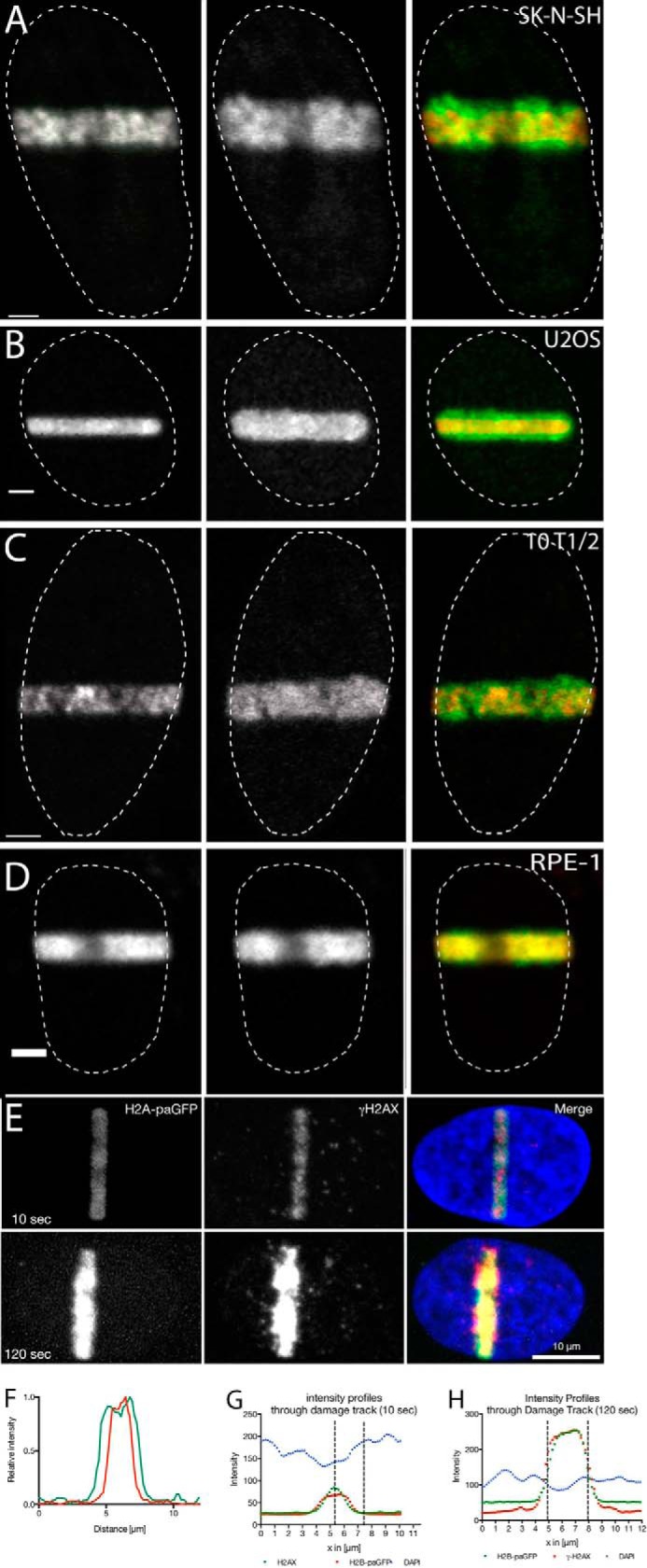
Rapid decondensation of chromatin at sites of laser micro-IR-induced DNA damage. Cells were transfected with photoactivatable histone H2B-GFP (or in case of the RPE-1 cells, stably expressing H4-paGFP) and DNA strand breaks were introduced by 2-photon (A and B) or 405-nm laser excitation (C and D) of Hoechst 33342 (see “Experimental Procedures”). Panels A and B show the nuclei of tumor cell lines (A, SK-N-SH neuroblastoma and B, U2OS osteosarcoma). Panels C and D show nuclei of non-tumor cell lines (C, 10T1/2 MEFs and D, RPE-1 immortalized retina pigment epithelium cells). Images were taken immediately after the introduction of DNA damage (left panel) and 2 min after the DNA damage induction (middle panel). Panel E, the photoactivated area coincides with the site of DNA damage. Cells were laser microirradiated and then fixed after 10 and 120 s, respectively. The width of the photoactivated chromatin was compared with the width of the γ-H2AX signal and the loss in DAPI intensity caused by the decondensation of the chromatin (also see G and H). Panel F, line scans showing the amount of chromatin decondensation (immediately (red line) and 2 min (green line) after DNA damage induction). Panels G and H, line scans through the damage tracks shown in E.
Inhibition of PARPs Prevents DNA Strand Break-dependent Chromatin Decondensation
We have recently shown that PARP1 is among the first proteins recruited to sites of laser microirradiation-induced DNA double-strand breaks (7). The PARP1-mediated accumulation of PAR at the site of DNA damage recruits DNA repair protein complexes, such as the MRN complex, thereby signaling the presence of DNA damage. Poly(ADP-ribosyl)ation has been shown to drive chromatin decondensation in vitro and in association with transcriptional activation (19, 25, 26). Thus, we tested the effect of the potent and highly specific PARP inhibitor, AG14361, on chromatin decondensation. Decondensation was strongly inhibited in SK-N-SH cells incubated in the presence of 100 nm AG14361 (Fig. 2A). At this concentration, AG14361 inhibited decondensation by over 85% (Fig. 2H). These results support the hypothesis that PARP activity is essential for chromatin decondensation and suggests that the rapid burst of PAR modification may be a crucial early component of the DNA repair pathway. Fig. 2, F and G (compiled from images acquired using a spinning disc microscope), show the difference between photoactivation of unsensitized U2OS cells, which do not result in DNA damage, the damage-related decondensation of Hoechst-sensitized U2OS cells and sensitized U2OS cells in the presence of 5 μm AG14361.
Further support for poly(ADP-ribosyl)ation driving chromatin decondensation upon induction of DNA damage was obtained by transfecting cells with PARG. Overexpression of PARG resulted in greater than 60% inhibition of the decondensation observed in untreated cells (Fig. 2H) by increasing the turnover kinetics of PAR (Fig. 2E).
The Role of PARP1 in DSB-associated Chromatin Relaxation
PARP1 is recruited rapidly to sites of DSBs. To determine whether PARP1 is required for the chromatin decondensation, we transfected PARP1 knock-out mouse embryonic fibroblasts (A1 cells) with photoactivatable H2B-GFP and introduced DNA strand breaks by laser microirradiation. Fig. 2B shows an example of an A1 nucleus immediately after (left panel, red in composite) and 2 min after (center panel, green in composite) microirradiation. Very little chromatin decondensation is observed in A1 cells. However, when A1 cells were transfected with a PARP1 expression vector to reconstitute PARP1 activity in the knock-out cells, the chromatin decondensation activity in these cells increased more than 2-fold (Fig. 2C). The influence of PARP1 on chromatin decondensation is summarized in Fig. 2I where the figure shows quantification of chromatin decondensation in mouse 10T1/2 cells (control) in the presence of increasing concentrations of AG14361 and in PARP1−/− mouse embryonic fibroblasts (A1 cells) in the presence or absence of transiently expressed PARP1. The A1 cells show an ∼45% reduction in decondensation.
Because neither PARP1 deletion nor AG14361 inhibition completely blocked decondensation, we wished to determine whether the residual decondensation was PARP-dependent or reflected a PARP-independent process. There are differences in the effectiveness of in vivo inhibition by PARP inhibitors. Consequently, we tested a more recently developed PARP inhibitor, BMN673 (33). When cells were treated with this inhibitor, there was no detectable decondensation (Fig. 2G). This suggests that the residual decondensation following AG14361 treatment and observed in PARP1 null MEFs is a consequence of PARP activity rather than a poly(ADP-ribosyl)ation-independent mechanism that occurs in parallel with the PARP-dependent response. However, it remains possible that a PARP-independent BMN673-sensitive molecule is involved.
H1 Histone Is Rapidly Displaced from DNA Damage Sites via a PARP1-dependent Mechanism
The extent and rate of decondensation seen at laser micro-IR-induced DNA damage is remarkable. A previous study revealed that, although ATP-dependent, the chromatin decondensation occurring at these sites is independent of ATM, distinguishing this response from the previously characterized ATM-mediated regulation of the heterochromatin protein KAP1 (37, 38). To better understand the mechanism of chromatin decondensation, we tested several proteins thought to be involved in the regulation of chromatin structure including HP1, MeCP2, HMGA1, HMGA2, and histone H1. HMGA1, HMGA2, and MeCP2 all show increased density in heterochromatin domains, as expected, but none of these proteins show evidence of dissociation upon laser microirradiation. The HP1 proteins, in contrast, accumulate at the site of laser microirradiation, as reported previously (39–42). Although an initial rapid displacement of HP1 has been reported to occur prior to accumulation (39, 43), this was distinct from what was observed with histone H1 and, in our view, largely reflects a reduction in intensity as a result of decreasing the condensation of the heterochromatin as opposed to the displacement that we observe with histone H1. We found that only histone H1 responded in a manner consistent with a mechanistic role in the observed chromatin decondensation (see Fig. 3). Fig. 4A shows a time series collected from cells transfected with histone H1.5-GFP after the introduction of DNA damage by laser micro-IR in the presence or absence of the AG14361 PARP inhibitor. In untreated cells, the H1 histone is rapidly displaced (see Fig. 4B). Note that with the 2-photon induction of damage, there is no photobleaching taking place. This was not variant-specific as all histone H1 subtypes tested (H1.0, 1.1, 1.2, 1.3, 1.4, and 1.5) (Fig. 4C) showed displacement from the sites of DNA damage. The region of damage is evident as early as 10 s after the introduction of damage and rapid displacement continues until about 3 min post-damage. The timing of the displacement of histone H1 correlates closely with the extent of decondensation. In the presence of PARP inhibitor, the displacement of histone H1 is dramatically reduced, and may even be completely prevented with the higher concentration of PARP inhibitor. Notably, even in the presence of AG14361, some poly(ADP-ribosyl)ation remains detectable at the site of DNA damage (Fig. 5A).
FIGURE 3.

Lack of displacement of non-histone heterochromatin proteins at sites of DNA damage. MEFs were transfected with GFP-tagged versions of HMGA1, HMGA2, MeCP2, and the three isoforms of HP1. DNA damage was introduced by laser microirradiation and the dynamics of these proteins were followed by time lapse microscopy. The figure shows images collected immediately before and ∼2 min after the introduction of DNA damage. The lower panel shows the displacement of example histone H1 variants after laser microirradiation.
FIGURE 4.

Histone H1 is displaced from sites of microirradiation in a PARP-dependent manner. A, SK-N-SH cells were transfected with histone H1.5-GFP and DNA damage was introduced by laser microirradiation. A time series is shown of representative cells collected in the presence or absence of the indicated concentrations of AG14361. B, the relative intensity of histone H1, H2A, and histone H4 in the presence or absence of PARP inhibitors was plotted during the first 2 min after irradiation. C, histone H1 subtypes H1.0, H1.1, H1.2, H1.3, and H1.4 intensities were plotted relative to time following laser microirradiation in the presence or absence of PARP inhibition.
Core Histone Displacement Is Poly(ADP-ribosyl)ation Dependent
We also tested whether histone displacement occurred with core histones. When we examined histone H2A, to estimate histone H2A/H2B dimer displacement, and histone H4, to estimate H3/H4 tetramer displacement, we found damage-induced PARP-dependent reduction in fluorescence intensity of H2A and H4, although at lower levels than linker histones, in the laser microirradiated regions (see Fig. 4B). If we assume that the loss of histone H4 reflects decondensation rather than displacement, this provides a baseline to determine the minimum displacement of histone H2A. The results in Fig. 4B indicate that at least 5% of histone H2A is displaced within the damaged region.
PARP-dependent Chromatin Decondensation Is Transient
Poly(ADP-ribosyl)ation is a transient process in laser microirradiation experiments, where it is very abundant over the first few minutes post-DNA damage (8). Thus, we wished to determine whether or not this decondensation was transient, which might reflect a more direct role of poly(ADP-ribosyl)ation in chromatin decondensation, as suggested by the in vitro experiments (25, 26) or a more stable change, which might be expected following the action of PARP-dependent chromatin remodeling complexes. Thus, we followed the chromatin decondensation in U2OS cells for an extended period of time post-DNA damage induction by measuring the full width at half-maximum intensity of the photoactivated H2B-paGFP region. We observed that the chromatin returns to its initial degree of compaction after ∼15 min (Fig. 6A). The recondensation began 150 s after damage induction and proceeded at a slower rate than the decondensation.
FIGURE 6.

Chromatin decondensation is transient. A, the nucleus of a U2OS cell transfected with H2B paGFP was laser microirradiated and then monitored over the course of 15 min. A plot of the relative condensation state versus time is also shown. B, a U2OS cell expressing H1.5-GFP was laser microirradiated and the fluorescent intensity monitored over time. At 2.5 min a clear region of damage is found (arrow in B). Note that the width of the laser damage track is the same in both nuclei. The different appearance of the stripe thickness is due to the use of a large pinhole and thus an underrepresentation of the displaced chromatin in B coming from light emitted above and under the focal plane.
Rebinding of Histone H1 during Recovery from Damage
Because chromatin decondensation is transient, we next wanted to determine whether and when histone H1 returns to the chromatin. To test this, we extended the duration of the time lapse observation. We introduced the laser damage in U2OS cells and then examined the incorporation of histone H1.5 that was associated with chromatin outside of the damage site at the time of damage induction. We found that the recovery of fluorescence initiated around 4 min and continued through to 15 min (Fig. 6B). When this is compared with chromatin recondensation, very similar kinetics are observed. Knocking down PARG by siRNA and thus slowing down the PAR turnover at the sites of DNA damage leads to a slower recovery of H1.5-eGFP (Fig. 5, B and C), shown by significantly reduced recovery slopes.
U2OS Cells Hypercondense Chromatin after Damage
The U2OS human osteosarcoma cell line is one of the most commonly used cell lines in the study of the DNA damage response. We noted that U2OS cells behaved somewhat differently than the other cell lines that we tested. A large population of U2OS cells that were laser microirradiated using the 405-nm laser at the spinning disc microscope recondensed chromatin beyond the initial state of condensation. This hypercondensation of chromatin that could be observed by expression with FP-labeled histones (H2B, H1.5) (Fig. 7, A and E) leads to a local increase of 20% in fluorescence intensity (Fig. 7C). This increase persisted over several hours and was resolved before or during S-phase (Fig. 7E).
FIGURE 7.

U2OS cells hypercondense chromatin following laser microirradiation. A, U2OS cell expressing H1.5-GFP was monitored for 60 min after damage induction by a 405-nm laser. A montage of the same nucleus at various times post-irradiation is shown. The arrows indicate a region of hypercondensation of the chromatin occurring along the irradiation track. B, changes in H1.5 intensity over time represented by a heat map. Each vertical line represents the line profile averaged over 15 pixels across the damaged area in A. Blue and green colors indicate low intensity (low levels of chromatin compaction), yellow orange and red indicate high intensity (high levels of chromatin compaction). C, pooled data from 10 different cells showing consistent timing of recondensation and hypercondensation. D, a laser microirradiated cell to simultaneously damage and photoactivate histone H2B, highlighting the microirradiated region, and then stained with DAPI to compare the density of chromatin (DAPI) with the sites of damage. E, time lapse experiment showing the resolution of hypercondensed chromatin along the damaged area at the onset of S-phase.
To determine that the effect reflects in fact condensation of chromatin and not a recruitment of H1.5 we have examined U2OS cells stably expressing H2B-mRFP (see Fig. 7E) and U2OS cells fixed 1 h after laser irradiation and stained them with Hoechst subsequently. In all cases we could observe an increase of fluorescence along the laser track (Fig. 7D).
Discussion
The hypercondensation we and colleagues could observe in U2OS cells is an interesting effect that seems to be important for DNA damage response signaling (44), and proper recruitment of some repair proteins (45). Of all the cell types we tested, we only observed this in U2OS cells. Interestingly, compared with other cell lines, the nuclei of U2OS cells show very decondensed chromatin, even without DNA breaks. This might be the reason why the hypercondensation could not be detected with fluorescence microscopes in other cell lines. Although dramatic in U2OS, it is less clear how much the hypercondensation results from U2OS cells can be generalized to other normal and cancer cell types.
The recondensation of chromatin following the initial PARP-dependent chromatin opening triggered from the introduction of DNA breaks seems to be important for efficient upstream DNA damage response signaling (47) as well as the establishment of chromatin modifications that enhance the proper recruitment of some repair proteins (41). Thus, a waning of the initial PARP-dependent chromatin opening and recondensation of the chromatin appears to be important for the progression of the DDR response. The re-establishment of histone H1 may be an important component in this progression. Curiously, the change in chromatin structure after the introduction of DNA breaks from initial chromatin expansion to then recondensation is most dramatic in U2OS cells compared with other cell types and ultimately becomes hypercondensed. It will be important to determine whether the cycle of chromatin remodeling after the introduction of DNA breaks has an equally significant influence on the DDR in all cell types. Interestingly, the hypercondensed chromatin state persists until S phase. It would be interesting to determine whether some of the chromatin marks that are associated with the chromatin during the DDR are also cleared during this transition.
A long standing unresolved question in fully understanding the cellular response to DNA damage is “what are the first sensing events triggered by DNA strand breaks?” We have previously demonstrated that PARP1 is recruited in less than 300 ms to a microirradiated track, preceding the recruitment of MRE11, NBS1, and ATM (7). Furthermore, we have shown that the lack of PARP1 substantially impedes the recruitment of MRE11. MRE11 has a PAR-binding domain, suggesting that its recruitment to DNA strand breaks may depend on an interaction with PAR.
Collectively, these findings suggested a very early key role for PARP1 in the sensing of DNA strand breaks (7). Poly(ADP-ribosyl)ation may function as a matrix to locally concentrate proteins that function at sites of DNA damage. The work presented here reveals that the rapid decondensation of chromatin at sites of DNA damage is also driven by poly(ADP-ribosyl)ation. Thus, whereas PAR is concentrating DNA damage response proteins near the site of damage, it is simultaneously opening up the chromatin to increase accessibility of the DNA to these same DNA damage factors. Thus, poly(ADP-ribosyl)ation may establish a nuclear microenvironment that is highly conducive to the rapid processing of DNA damage.
Our results provide a mechanistic explanation for the decondensation of chromatin at sites of micro-IR-induced DNA damage reported by Kruhlak and colleagues (6, 15). The modification of histones would be expected to drive decondensation through the accumulation of negative charge. This would be consistent with the reversibility of PARP1-mediated chromatin decondensation in vitro (25). The displacement of histone H1, which may not occur in vitro (26), could occur either through the direct modification of histone H1 and its repulsion from chromatin or through the disruption of histone H1 binding sites on chromatin that could result from modification of nucleosomal histones. A third possibility, that histone H1 is displaced from chromatin through a higher affinity binding to PAR than chromatin (22), is not consistent with our results. Our results, therefore, do not support the histone shuttle hypothesis (24) of PAR function at sites of DNA damage. Indeed, based on the higher affinity that histone H1 shows for PAR relative to DNA in vitro and the relatively high mobility of histone H1 in vivo, it is surprising that unmodified histone H1 displaced from regions outside of the DNA damage sites does not accumulate at sites of PAR accumulation (even after PARG knockdown).
Similarly, there is recent evidence for a PAR-dependent phase separation and the accumulation of intrinsically disordered positively charged proteins at sites of PAR accumulation following laser microirradiation (46). Our results are also not consistent with a recent study that reports that intrinsically disordered proteins are accumulating around sites of PAR deposition and lead to a membrane-less compartment by phase separation. The CTD of histone H1 is an intrinsically disordered positively charged domain (47) but is displaced from rather than accumulates in the PAR-rich domain following laser microirradation.
There is also a modest loss of histone H2B from the damaged chromatin. This is unlikely to be due to decondensation because there is about 5% more H2A lost than histone H4. Because there is decondensation and we cannot uncouple decondensation from histone displacement, we cannot be certain that there is no loss of histone H4. Thus, we conclude that there is at minimum ∼5% of the H2A that is displaced from the damage site. Using a different surrogate measure, the size of the rapidly diffusing pool of histone H2B, Morisaki and McNally (48) concluded that there was no H2A-H2B displacement. Because we have the reference behavior of histone H4, our results indicate that there is indeed a small amount of displacement of histone H2A-H2B dimers. Interestingly, macroH2A1.1 is assembled onto chromatin in a PARP-dependent manner (45, 49). It may be that displacement of H2A-H2B dimers enables the incorporation of macroH2A1.1 into nucleosomes at sites of damage. The participation of histone variants, such as macroH2A, in the repertoire may influence the rate of chromatin remodeling/recondensation and the targeting of posttranslational modifications which in turn may influence which DNA repair pathway is selected to repair the damage.
Our results, together with our previous results, reveal that the poly(ADP-ribosyl)ation of PARPs and histone proteins at sites of DNA damage play two important and complementary roles. They sequester molecules that are involved in the processing of double-strand breaks (50–52) and drive the local decondensation of higher-order chromatin packaging, which can increase the accessibility of these proteins, chromatin remodeling complexes, and transcription factors (16) to the damaged DNA. Thus, it is easy to see how these two complementary functions could act synergistically to stimulate the repair of DNA double-strand breaks.
Author Contributions
M. J. H. conceived and designed the study, coordinated its execution, and wrote the manuscript. H. S. contributed to the design of the experiments, developed analytical approaches for the analysis of the data, and conceived, designed, and executed the experiment in Fig. 7, as well as wrote parts of the manuscript. D. M. performed experiments and contributed to study design and the writing of the manuscript. J-F. H., M. R., J. P. H. T., and G. P. contributed to the design of the study and the writing of the manuscript. M. J. K. contributed to experimental design and the writing of the manuscript. T. I. and G. N. C. designed and characterized reagents used in Fig. 3. J. A. and D. A. U. contributed to the design of the experiments presented in Fig. 3 and contributed to the writing of the manuscript.
Acknowledgments
We thank Dr. Thomas Cremer for the H4-paGFP H2B-mRFP cell-line and Dr. Xuejun Sun for help in acquiring 2-photon laser power measurement and Sanita Ghosh for statistical advice. We thank Randall Gieni for critically reading the manuscript.
This work was supported in part by grants from the Canadian Institutes of Health Research, the Canadian Cancer Society, and the Alberta Cancer Research Institute. The authors declare that they have no conflicts of interest with the contents of this article.
- DSB
- double-strand break
- MEF
- mouse embryonic fibroblast
- PARG
- poly(ADP-ribose) glycohydrolase
- PARP
- poly(ADP-ribose) polymerase
- IR
- irradiation.
References
- 1. Lavin M. F. (2007) ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 26, 7749–7758 [DOI] [PubMed] [Google Scholar]
- 2. Williams R. S., Williams J. S., and Tainer J. A. (2007) Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 85, 509–520 [DOI] [PubMed] [Google Scholar]
- 3. Altaf M., Saksouk N., and Côté J. (2007) Histone modifications in response to DNA damage. Mutat. Res. 618, 81–90 [DOI] [PubMed] [Google Scholar]
- 4. Bao Y., and Shen X. (2007) Chromatin remodeling in DNA double-strand break repair. Curr. Opin. Genet. Dev. 17, 126–131 [DOI] [PubMed] [Google Scholar]
- 5. Ismail I. H., and Hendzel M. J. (2008) The γ-H2A.X: is it just a surrogate marker of double-strand breaks or much more? Environ. Mol. Mutagen. 49, 73–82 [DOI] [PubMed] [Google Scholar]
- 6. Kruhlak M. J., Celeste A., Dellaire G., Fernandez-Capetillo O., Müller W. G., McNally J. G., Bazett-Jones D. P., and Nussenzweig A. (2006) Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 172, 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haince J. F., McDonald D., Rodrigue A., Déry U., Masson J. Y., Hendzel M. J., and Poirier G. G. (2008) PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 283, 1197–1208 [DOI] [PubMed] [Google Scholar]
- 8. Mortusewicz O., Amé J. C., Schreiber V., and Leonhardt H. (2007) Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 35, 7665–7675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brochu G., Duchaine C., Thibeault L., Lagueux J., Shah G. M., and Poirier G. G. (1994) Mode of action of poly(ADP-ribose) glycohydrolase. Biochim. Biophys. Acta 1219, 342–350 [DOI] [PubMed] [Google Scholar]
- 10. Bryant H. E., Petermann E., Schultz N., Jemth A.-S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P., and Helleday T. (2009) PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haince J. F., Kozlov S., Dawson V. L., Dawson T. M., Hendzel M. J., Lavin M. F., and Poirier G. G. (2007) Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 282, 16441–16453 [DOI] [PubMed] [Google Scholar]
- 12. Aguilar-Quesada R., Muñoz-Gámez J. A., Martín-Oliva D., Peralta A., Valenzuela M. T., Matínez-Romero R., Quiles-Pérez R., Menissier-de Murcia J., de Murcia G., Ruiz de Almodóvar M., and Oliver F. J. (2007) Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol. Biol. 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahel D., Horejsí Z., Wiechens N., Polo S. E., Garcia-Wilson E., Ahel I., Flynn H., Skehel M., West S. C., Jackson S. P., Owen-Hughes T., and Boulton S. J. (2009) Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325, 1240–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gottschalk A. J., Timinszky G., Kong S. E., Jin J., Cai Y., Swanson S. K., Washburn M. P., Florens L., Ladurner A. G., Conaway J. W., and Conaway R. C. (2009) Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U.S.A. 106, 13770–13774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smeenk G., Wiegant W. W., Marteijn J. A., Luijsterburg M. S., Sroczynski N., Costelloe T., Romeijn R. J., Pastink A., Mailand N., Vermeulen W., and van Attikum H. (2013) Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J. Cell Sci. 126, 889–903 [DOI] [PubMed] [Google Scholar]
- 16. Izhar L., Adamson B., Ciccia A., Lewis J., Pontano-Vaites L., Leng Y., Liang A. C., Westbrook T. F., Harper J. W., and Elledge S. J. (2015) A systematic analysis of factors localized to damaged chromatin reveals PARP-dependent recruitment of transcription factors. Cell Rep. 11, 1486–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thomas C. J., Kotova E., Andrake M., Adolf-Bryfogle J., Glaser R., Regnard C., and Tulin A. V. (2014) Kinase-mediated changes in nucleosome conformation trigger chromatin decondensation via poly(ADP-ribosyl)ation. Mol. Cell 53, 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kolesnikova T. D., Semeshin V. F., Andreyeva E. N., Zykov I. A., Kokoza E. B., Kalashnikova D. A., Belyaeva E. S., and Zhimulev I. F. (2011) Induced decondensation of heterochromatin in Drosophila melanogaster polytene chromosomes under condition of ectopic expression of the Supressor of underreplication gene. Fly 5, 181–190 [DOI] [PubMed] [Google Scholar]
- 19. Tulin A., and Spradling A. (2003) Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 299, 560–562 [DOI] [PubMed] [Google Scholar]
- 20. Rouleau M., Aubin R. A., and Poirier G. G. (2004) Poly(ADP-ribosyl)ated chromatin domains: access granted. J. Cell Sci. 117, 815–825 [DOI] [PubMed] [Google Scholar]
- 21. Jungmichel S., Rosenthal F., Altmeyer M., Lukas J., Hottiger M. O., and Nielsen M. L. (2013) Proteome-wide identification of poly(ADP-ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 52, 272–285 [DOI] [PubMed] [Google Scholar]
- 22. Malanga M., Atorino L., Tramontano F., Farina B., and Quesada P. (1998) Poly(ADP-ribose) binding properties of histone H1 variants. Biochim. Biophys. Acta 1399, 154–160 [DOI] [PubMed] [Google Scholar]
- 23. Panzeter P. L., Zweifel B., Malanga M., Waser S. H., Richard M., and Althaus F. R. (1993) Targeting of histone tails by poly(ADP-ribose). J. Biol. Chem. 268, 17662–17664 [PubMed] [Google Scholar]
- 24. Althaus F. R. (1992) Poly ADP-ribosylation: a histone shuttle mechanism in DNA excision repair. J. Cell Sci. 102, 663–670 [DOI] [PubMed] [Google Scholar]
- 25. de Murcia G., Huletsky A., Lamarre D., Gaudreau A., Pouyet J., Daune M., and Poirier G. G. (1986) Modulation of chromatin superstructure induced by poly(ADP-ribose) synthesis and degradation. J. Biol. Chem. 261, 7011–7017 [PubMed] [Google Scholar]
- 26. Poirier G. G., de Murcia G., Jongstra-Bilen J., Niedergang C., and Mandel P. (1982) Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. U.S.A. 79, 3423–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krishnakumar R., Gamble M. J., Frizzell K. M., Berrocal J. G., Kininis M., and Kraus W. L. (2008) Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 319, 819–821 [DOI] [PubMed] [Google Scholar]
- 28. Krishnakumar R., and Kraus W. L. (2010) PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell 39, 736–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Messner S., and Hottiger M. O. (2011) Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 21, 534–542 [DOI] [PubMed] [Google Scholar]
- 30. Kim M. Y., Mauro S., Gévry N., Lis J. T., and Kraus W. L. (2004) NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119, 803–814 [DOI] [PubMed] [Google Scholar]
- 31. Strickfaden H., Zunhammer A., van Koningsbruggen S., Köhler D., and Cremer T. (2010) 4D chromatin dynamics in cycling cells: Theodor Boveri's hypotheses revisited. Nucleus 1, 284–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gagné J. P., Bonicalzi M. E., Gagné P., Ouellet M. E., Hendzel M. J., and Poirier G. G. (2005) Poly(ADP-ribose) glycohydrolase is a component of the FMRP-associated messenger ribonucleoparticles. Biochem. J. 392, 499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen Y., Rehman F. L., Feng Y., Boshuizen J., Bajrami I., Elliott R., Wang B., Lord C. J., Post L. E., and Ashworth A. (2013) BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 19, 5003–5015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thévenaz P., Ruttimann U. E., and Unser M. (1998) A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 [DOI] [PubMed] [Google Scholar]
- 36. Kleinbaum D., Kupper L., Nizam A., and Rosenberg E. (2013) Applied Regression Analysis and Other Multivariable Methods, Cengage Learning [Google Scholar]
- 37. Goodarzi A. A., Noon A. T., Deckbar D., Ziv Y., Shiloh Y., Löbrich M., and Jeggo P. A. (2008) ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 31, 167–177 [DOI] [PubMed] [Google Scholar]
- 38. Ziv Y., Bielopolski D., Galanty Y., Lukas C., Taya Y., Schultz D. C., Lukas J., Bekker-Jensen S., Bartek J., and Shiloh Y. (2006) Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 8, 870–876 [DOI] [PubMed] [Google Scholar]
- 39. Ayoub N., Jeyasekharan A. D., and Venkitaraman A. R. (2009) Mobilization and recruitment of HP1: a bimodal response to DNA breakage. Cell Cycle 8, 2945–2950 [PubMed] [Google Scholar]
- 40. Baldeyron C., Soria G., Roche D., Cook A. J., and Almouzni G. (2011) HP1α recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J. Cell Biol. 193, 81–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luijsterburg M. S., Dinant C., Lans H., Stap J., Wiernasz E., Lagerwerf S., Warmerdam D. O., Lindh M., Brink M. C., Dobrucki J. W., Aten J. A., Fousteri M. I., Jansen G., Dantuma N. P., Vermeulen W., Mullenders L. H., Houtsmuller A. B., Verschure P. J., and van Driel R. (2009) Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 185, 577–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sehnalová P., Legartová S., Cmarko D., Kozubek S., and Bártová E. (2014) Recruitment of HP1β to UVA-induced DNA lesions is independent of radiation-induced changes in A-type lamins. Biol. Cell 106, 151–165 [DOI] [PubMed] [Google Scholar]
- 43. Ayoub N., Jeyasekharan A. D., Bernal J. A., and Venkitaraman A. R. (2008) HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453, 682–686 [DOI] [PubMed] [Google Scholar]
- 44. Burgess R. C., Burman B., Kruhlak M. J., and Misteli T. (2014) Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 9, 1703–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khurana S., Kruhlak M. J., Kim J., Tran A. D., Liu J., Nyswaner K., Shi L., Jailwala P., Sung M. H., Hakim O., and Oberdoerffer P. (2014) A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell reports 8, 1049–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Altmeyer M., Neelsen K. J., Teloni F., Pozdnyakova I., Pellegrino S., Grøfte M., Rask M. B., Streicher W., Jungmichel S., Nielsen M. L., and Lukas J. (2015) Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 6, 8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hansen J. C., Lu X., Ross E. D., and Woody R. W. (2006) Intrinsic protein disorder, amino acid composition, and histone terminal domains. J. Biol. Chem. 281, 1853–1856 [DOI] [PubMed] [Google Scholar]
- 48. Morisaki T., and McNally J. G. (2014) Photoswitching-free FRAP analysis with a genetically encoded fluorescent tag. PloS One 9, e107730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Timinszky G., Till S., Hassa P. O., Hothorn M., Kustatscher G., Nijmeijer B., Colombelli J., Altmeyer M., Stelzer E. H., Scheffzek K., Hottiger M. O., and Ladurner A. G. (2009) A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16, 923–929 [DOI] [PubMed] [Google Scholar]
- 50. Fernandez-Capetillo O., Celeste A., and Nussenzweig A. (2003) Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle 2, 426–427 [PubMed] [Google Scholar]
- 51. Maser R. S., Monsen K. J., Nelms B. E., and Petrini J. H. (1997) hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol. 17, 6087–6096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haaf T., Golub E. I., Reddy G., Radding C. M., and Ward D. C. (1995) Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. U.S.A. 92, 2298–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]