

Abstract
Crystallographic evidence suggests that the pH-dependent affinity of IgG molecules for the neonatal Fc receptor (FcRn) receptor primarily arises from salt bridges involving IgG histidine residues, resulting in moderate affinity at mildly acidic conditions. However, this view does not explain the diversity in affinity found in IgG variants, such as the YTE mutant (M252Y,S254T,T256E), which increases affinity to FcRn by up to 10×. Here we compare hydrogen exchange measurements at pH 7.0 and pH 5.5 with and without FcRn bound with surface plasmon resonance estimates of dissociation constants and FcRn affinity chromatography. The combination of experimental results demonstrates that differences between an IgG and its cognate YTE mutant vary with their pH-sensitive dynamics prior to binding FcRn. The conformational dynamics of these two molecules are nearly indistinguishable upon binding FcRn. We present evidence that pH-induced destabilization in the CH2/3 domain interface of IgG increases binding affinity by breaking intramolecular H-bonds and increases side-chain adaptability in sites that form intermolecular contacts with FcRn. Our results provide new insights into the mechanism of pH-dependent affinity in IgG-FcRn interactions and exemplify the important and often ignored role of intrinsic conformational dynamics in a protein ligand, to dictate affinity for biologically important receptors.
Keywords: antibody, biophysics, hydrogen exchange mass spectrometry, immunoglobulin G (IgG), protein stability, structural biology, HDX-MS, neonatal Fc receptor (FcRn), affinity, conformational dynamics
Introduction
Since the era of structural biology began, researchers have sought to understand the determinants of binding affinity. Even for the most rigid interaction interfaces, a method to universally predict dissociation constants with high accuracy remains beyond reach. A possible reason relates to the unknown contribution(s) of conformational dynamics (1). Although not represented in static crystal structures, conformational dynamics have been shown to modulate dissociation rates (2–5), influence substrate transport (6), and even regulate protein activity (7, 8).
The interaction between IgG and neonatal Fc receptor (FcRn+β2-microglobulin, abbreviated here as FcRn)3 is of critical importance in the human immune system and also of considerable pharmaceutical interest (9). IgG molecules have moderate affinity for FcRn at the mildly acidic pH of recycling endosomes and lose all measureable affinity at or slightly above neutral pH. This interaction with FcRn is essential for the long serum half-lives of antibodies in circulation (10). Comparisons of available crystal structures of FcRn-bound Fc domain (11–15) with free antibody lead to Cα root mean square deviation values of less than 1 Å, implying a rigid interface stabilized by salt bridges that centrally involve IgG heavy chain His-310/435, whose ionization states define pH-dependent affinity.
In 2009, researchers seeking to improve serum half-life in a therapeutic antibody, motavizumab, found a triple mutant in the Fc4 domain, M252Y,S254T,T256E (YTE mutant), that led to improved serum half-life by 4× and improved binding affinity by 10× at pH 6.0 (16). The YTE mutation does not induce a structural change in the Fc domain (17); however, these mutations do appear to reduce the Fc domain stability and increase aggregation propensity in the mutant (18) drawn from HX measurements, showing that YTE is less stable than WT.
In the case of motavizumab-YTE, affinity increases have been attributed to salt bridge formation with Glu-256 (16, 17); however, in most other examples, inspection of crystal structures does not explain how mutations, external to the FcRn interaction interface, modulate affinity (19). Diversity in affinity between antibodies with nearly identical constant-domain sequences (20–22) cannot be rationalized by available crystallographic data.
Furthermore, recent work shows that the mechanism of pH dependence involving only the protonation of His-310 and His-435, needed for salt bridge formation with FcRn, is incomplete. First, many non-histidine mutations far from the interaction interface modify affinity to FcRn (19–22). Second, the highest resolution Fc domain in complex with FcRn presently available was solved in 2014 (17), and this showed that His-435 does not actually contact FcRn. Third, reduction in mobility of the loop containing His-435 reduces FcRn affinity for unknown reasons (9). Why does the mobility of this loop play any role in FcRn affinity? If His-435 does not actually interact with FcRn, why is this residue essential for binding (9, 11, 13, 19)?
Currently, there is no proposed mechanism by which a reduction in the stability of the unbound IgG with pH contributes to an increase in FcRn affinity. The idea that differences in sub-global Fc domain stability prior to binding FcRn might influence affinity has not been explored. In fact, the locations of structural regions in the Fc domain sensitive to pH are also unknown. Does pH reduction cause sub-global Fc domain destabilization in a manner that could influence FcRn affinity? If so, how do conformational dynamics prior to binding FcRn relate to binding affinity?
To address this, we have performed a comprehensive analysis of the role of protein dynamics on the pH-dependent affinity of the IgG-FcRn complex by measuring the HX rates of a wild-type IgG1 antibody (WT) and its cognate YTE mutant (YTE) at both pH 7 and pH 5.5±FcRn. We account for changes in hydroxide concentrations on HX rates, enabling a direct assessment of the change in local Fc domain stability as a function of pH. In doing so, pH dependence profiles for structural free energy are constructed between WT and YTE in the absence of FcRn along with the stability conferred to each molecule upon binding FcRn. Magnitudes of changes in structural free energy between all relevant states (pH 7, pH 5.5±FcRn) and between molecules, WT and YTE, are compared with SPR affinity measurements to explore whether pH-sensitive changes to conformational dynamics in the absence of FcRn play any causal role in pH-dependent FcRn affinity.
Measurements show that in the presence of FcRn, the conformational dynamics of WT and YTE are nearly indistinguishable, suggesting that changes in affinity are the result of differences in stability prior to binding FcRn. Every structural element stabilized by binding FcRn is also destabilized by pH reduction in both molecules, suggesting that increased mobility, prior to binding FcRn, is indeed a determinant for affinity. The particular structural locations identified as sensitive to pH and FcRn binding allow us to propose that pH-driven destabilization increases affinity for FcRn by breaking specific intramolecular backbone H-bonds known to form intermolecular contacts with FcRn along with increasing side-chain adaptability in all residues that are known to reorient upon binding FcRn.
To our knowledge, this work is the first to suggest a mechanism by which pH-induced destabilization increases affinity for the FcRn receptor.
Experimental Procedures
Mass Analysis
Mass analysis was performed on WT and YTE for native and deglycosylated forms after reduction. Each antibody was deglycosylated by adding N-glycosidase F in sodium phosphate buffer, pH 7.1, to a substrate/enzyme ratio of 0.14 unit/μg of mAb and then incubated at 37 °C for 16 h. Both native and deglycosylated WT and YTE were reduced using 0.2 m tris(2-carboxyethyl)phosphine (TCEP) in 4 m guanidinium chloride at 37 °C for 30 min and desalted by size exclusion chromatography. Electrospray ionization mass spectra were acquired on a maXis Q-TOF (Bruker Daltonics, Bremen, Germany) equipped with a TriVersa NanoMate (Advion, Ithaca, NY). For data evaluation, software developed in-house was used. This verified that both WT and YTE had expected masses and the same glycan distributions, reported in Table 1.
TABLE 1.
Glycan composition of WT and YTE molecules in this study
The percentages of each glycoform (G0, G1, G2+fucose) present in solution are given in the table establishing that WT and YTE have nearly identical glycan profiles, with only a small amount of non-glycosylated molecules in the ensemble. The YTE mutations have negligible influence on post-translational glycosylation.
| Glycoform | WT | YTE |
|---|---|---|
| % | % | |
| No glycan | 6 | 9 |
| G0 + fucose | 59 | 52 |
| G1 + fucose | 29 | 32 |
| G2 + fucose | 7 | 7 |
Surface Plasmon Resonance
A Biacore T200 SPR spectrometer was utilized to determine the binding strength of WT and YTE antibodies at pH 5.5 and pH 7.0. Prior to the actual assay, a CM5 sensor chip was prepared by immobilizing about 50 response units of scFcRn5 (low density) onto flow cell two, about 400 response units of scFcRn onto flow cell three, and about 4000 response units of scFcRn onto flow cell four (high density), via amine coupling chemistry (BR-1000-50, GE Healthcare) at pH 4.5. Flow cell one was immobilized as a blank to be used as reference. A PBS-P+ (28-9950-84, GE Healthcare), pH 5.5, buffer was connected to the “buffer A” tubing, and a PBS-P+, pH 7.0, buffer was connected to the “buffer B” tubing of the Biacore instrument.
The assay consisted of two identical steps, of which one was carried out with buffer A and the other was carried out with buffer B. Each cycle consisted of a sample association (200 s, 50 μl/min), a sample dissociation (600 s, 50 μl/min), and two 30-s-long regeneration steps with PBS-P+, pH 8.8, at a flow speed of 30 μl/min. All samples were diluted independently for both steps in the corresponding running buffer. The sample set for each assay step included a 1:2 dilution series ranging from 1000 to 0.49 nm of the WT and YTE antibody variant.
FcRn Chromatography
The soluble extracellular domain of FcRn with His-Avi-Tag® associated with β-2-microglubulin was immobilized onto POROS® streptavidin beads that were packed into a 300-μl column (22). Approximately 30 μg of IgG was loaded in 20 mm MES, 150 mm NaCl, pH 6.0, where it was washed and eluted by pH gradient: 100% mobile phase A to 66% mobile phase B in 5 min; to 75% mobile phase B and 25% mobile phase C in 5 min; to 15% mobile phase B and 75% mobile phase C in 20 min; to 100% mobile phase C in 8 min; hold 100% mobile phase C for 9 min; to 100% mobile phase A in 2 min; hold 100% mobile phase A for 12 min. (Mobile phase A: 20 mm MES, 150 mm NaCl, pH 6.0; mobile phase B: 20 mm MES, 150 mm NaCl, pH 6.5; mobile phase C: 20 mm Tris/HCl, 150 mm NaCl, pH 8.8). The elution was monitored at UV 280 nm.
Hydrogen Exchange
Samples (30 μm IgG) were diluted 1:15 into a solution of D2O containing 20 mm histidine, 50 mm NaCl, at either pD 5.5 or pD 7.0, and allowed to exchange for seven time intervals from 0.5 to 1000 min. Time points taken in triplicate were randomized and collected by the LEAPv1 HX robotics platform (Leap Technologies, Carrboro, NC). For FcRn-bound experiments, IgG and FcRn were mixed at a 1:7 ratio and allowed to sit on the bench for 30 min before beginning labeling as described previously (23).
Following the exchange period, the reaction was quenched by reducing pH to 2.5 and the addition of 0.25 m tris(2-carboxyethyl)phosphine and 2 m guanidinium chloride. Chromatography mobile phases were prepared to maximize deuterium recovery as described previously (24). Quenched samples were injected into a cold online system (25) where they were digested at 0 °C by an immobilized pepsin column (2.1 × 30 mm, Applied Biosystems), desalted on a trap column (ACQUITY Vanguard C8), separated by reversed phase chromatography (ACQUITY UPLC BEH C18, 1.7-μm particle size, 1.0 × 50 mm), and introduced into the mass spectrometer (Thermo Orbitrap Elite) for measurement of carried deuterium at the resolving power of 120,000 Hz at m/z 400. Data were analyzed using the ExMS program (26) along with custom Python scripts that combine degenerate charge states and fit uptake traces with binomials to determine the amount of incorporated deuterium and assess modality as described in other work (24, 27, 28) with the addition of peptide slowing factor analysis described here.
This procedure gave 317 unique peptides with 83% sequence coverage in the absence of FcRn and 211 peptides (79% coverage) in FcRn-bound experiments. Nearly 100% of the constant domain was captured by many overlapping peptides in all experiments. Deuterium recoveries averaged 82% as measured by a fully deuterated control. We required that overlapping peptides corroborated information deemed significant by p value in isolation. Only one representative peptide will be discussed in this study for simplicity. Peptides are numbered according to the Kabat numbering scheme (29), and in the conserved IgG1 sequence, H represents heavy chain residues (for example, H241–H252) and L represents light chain residues (see Table 3 for examples). Where amino acid residues are meant, three-letter codes were used.
TABLE 3.
PFC1→C2
Shown is a quantitative description of the change in deuterium uptake rates between different experimental conditions, averaged over peptides measured in each experiment. When PFC1→C2 is less than 1, C1 exchanges more slowly than C2, and when it is greater than 1, C2 exchanges more slowly than C1, as described under the “Experimental Procedures.” In Fig. 5A, the bold entries in this table, indicating peptides that respond differentially to at least one of the experimental variables, are converted to changes in free energy between the indicated conditions (δδGC1→C2). H refers to heavy chain in constructions such as H5–H17, and L refers to light chain in constructions such as L136–L148.
| Peptide | PFYTE-WTa |
PFpH 5.5-7a |
PFpH 5.5 - pH5.5+FcRna |
||||||
|---|---|---|---|---|---|---|---|---|---|
| pH 7 | pH 5.5 | pH 5.5 + FcRn | WT | YTE | YTE/WT | WT | YTE | YTE/WT | |
| H5–H17 | 1 | 1 | 1 | 1.0–2.2 | 1.0–2.2 | 1 | 1 | 1 | 1 |
| H36–H47 | 1 | 1 | 1 | 1.5–1.8 | 1.5–1.8 | 1 | 1 | 1 | 1 |
| H97–H106 | 1 | 1 | 1 | 1–5 | 1–5 | 1 | 1 | 1 | 1 |
| H146–H155 | 1 | 1 | 1 | 1.5–1.8 | 1.5–1.8 | 1 | 1 | 1 | 1 |
| H241–H252 | 11 | 9.3 | 0.7–1.5b | 6.1 | 5.2 | 0.9 | 16 | 120 | 7.5 |
| H253–H262 | 0.4 | 0.5–1.5b | 0.3 | 4.5 | 13 | 2.9 | 31 | 110 | 3.5 |
| H263–H277 | 1 | 1 | 1 | 1.8–2.3 | 1.8–2.3 | 1 | 1 | 1 | 1 |
| H278–H293 | 0.5 | 0.85 | 1 | 1.8–2.2 | 1.8–2.2 | 1 | 1 | 1 | 1 |
| H306–H318 | 1 | 1.5 | 0.4–1.2b | 11 | 13 | 1.2 | 4.5 | 4.5 | 1 |
| H336–H348 | 5 | 2.9 | 1 | < 1.5 | 3.8 | 3.8 | 1 | 2 | 2.0 |
| H369–H379 | 1 | 0.4 | 0.2 | 2.4 | 3 | 1.3 | 1.6 | 3 | 1.9 |
| H379–H390 | 1 | 0.8 | 0.85 | 2.2 | 2.4 | 1.1 | 1 | 1 | 1 |
| H429–H440 | 2.3 | 6.3 | 0.2 | 21 | 24 | 1.1 | 25 | 90 | 3.6 |
| L136–L148 | 1 | 1 | 1 | 1.65 | 1.65 | 1 | 1 | 1 | 1 |
| L162–L172 | 1 | 1 | 1 | 2.1–2.3 | 2.1–2.3 | 1 | 1 | 1 | 1 |
| L196–L206 | 1 | 1 | 1 | 2–3 | 2–3 | 1 | 1 | 1 | 1 |
a PF = 1 if no significant differences were found by t test or if value was less than 1.5 threshold (see “Results”).
b Some sites are faster, others are slower.
Effective Labeling Time
To accurately compare HX measurements collected at different pH values, we referenced actual labeling times (tlabel) at pH 7 to corresponding effective labeling times (teffective) at a reference pH using teffective = tlabel 10pHlabel − pHreference (30, 31). This accounts for the first order dependence of HX reactions on hydroxide concentration (32, 33), enabling measurement of structural sensitivity to pH by HX.
Peptide Protection Factors
HX experiments fundamentally measure backbone H-bond energetics. H-bonds sequester protons from exchange with solvent, and thus observed deuterium uptake rates under native conditions are dictated by H-bond opening rates (kop) and reformation rates (kcl). The influences of changes in H-bond equilibria (kop/kcl) on HX uptake rates are well understood and have been discussed previously (34–38). We utilized these principles and developed a method to approximate changes in H-bond equilibria at the peptide level from HX MS data.
To quantify approximate changes in H-bond equilibria between any two experimental conditions, C1 and C2, from changes in deuterium uptake traces at the peptide level, we computed peptide protection factors (PFC1→C2) by comparing the amount of time required for equivalent deuterium incorporation between experimental conditions. In thermodynamic terms, taking Kcl = kcl/kop to represent the association constant for H-bonds, PFC1→C2, scales changes from one condition to the other by Kcl,C1 × PFC1→C2 ≅ Kcl,C1 in the same way as an increase in affinity, represented by Ka,C1→C2, scales association constants between two conditions by Ka,C1 × Ka,C1→C2 = Ka,C2. Peptide protection factors are directly related to changes in free energy by ΔΔGC1→C2 = −RTln(PFC1→C2).
An extended mathematical synthesis to establish the validity of our approach and a visual diagram that delineates each step of the PFC1→C2 computation for peptide level HX are given in the supplemental text. This discussion reaches far beyond the scope of this report; HX practitioners may find this information to be particularly enriching and necessary to appreciate and understand the idiosyncrasies of our approach.
Results
pH Dependence and Affinity for FcRn
We investigated the affinity of both IgG WT and YTE to FcRn using SPR. Here FcRn was conjugated to an SPR chip at three different ligation densities, which were simultaneously monitored as solutions containing known concentrations of WT and YTE flowed over the chip and bound FcRn at the indicated pH for 200 s followed by a solvent-only wash to estimate dissociation kinetics (Fig. 1, A–F). Conventional determination of affinity for FcRn using instrument software was obtained by kinetic and steady-state analysis of SPR data over a range of protein concentrations, and these results demonstrated an increase in affinity for YTE over WT at pH 5.5 that ranged from 4- to 12-fold depending on FcRn coupling density (Table 2)
FIGURE 1.
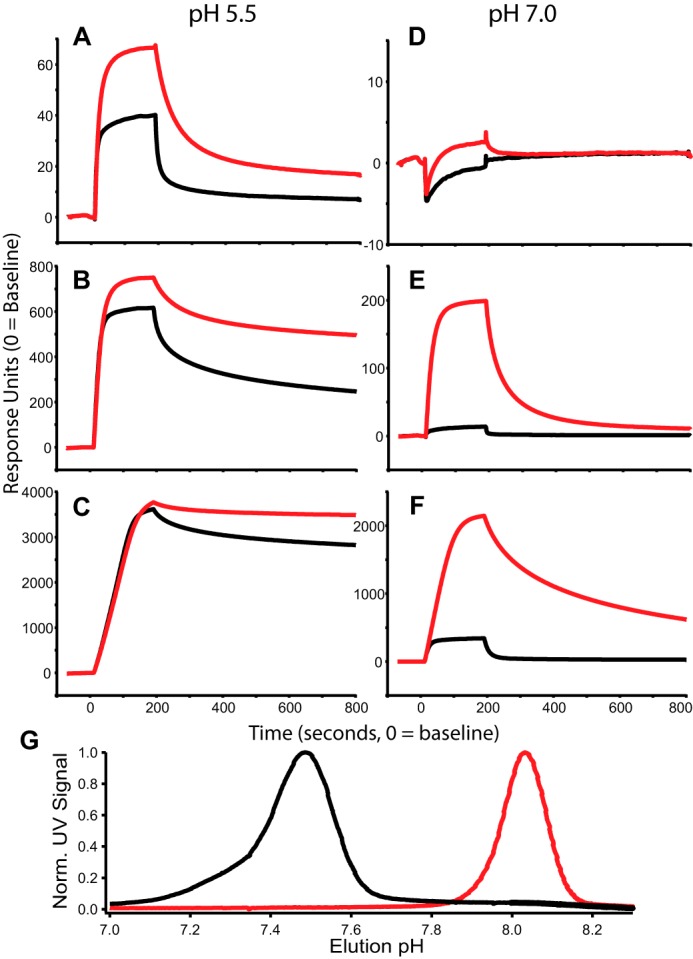
pH-dependent FcRn binding. A–F, SPR sensorgrams using 60 nm WT (black) and YTE (red) at pH 5.5 (A–C) and pH 7.0 (D–F). FcRn ligation densities were varied as discussed under “Experimental Procedures” and shown as low (A and D), medium (B and E), and high (C and F) density. G, FcRn chromatography of WT (black) and YTE (red). YTE elutes with a pH of 8.1 and WT elutes with a pH of 7.5; pH was measured online with UV absorbance. Norm. UV Signal, normalized UV signal.
TABLE 2.
SPR affinity
FcRn dissociation constants and affinity increases in the YTE mutant were estimated from SPR binding data. Steady-state analysis (SS) considers only the magnitude of SPR response versus concentration of analyte at 300 seconds, whereas kinetic analysis determines affinity by consideration of the on- and off- rates versus concentration. ND, not determined.
| Condition | Low density FcRn |
High density FcRn |
||||
|---|---|---|---|---|---|---|
| WT | YTE | WT/YTE | WT | YTE | WT/YTE | |
| nm | nm | nm | nm | |||
| SS — pH 5.5 | 96 | 20 | 4.8 | 26 | 28 | 1 |
| Kinetics 5.5 | 8 | 2 | 4 | 5 | 0.4 | 12.5 |
| SS — pH 7.4 | 1561 | 701 | 2.2 | 407 | 30 | 13.5 |
| Kinetics 7.4 | ND | 318 | ND | 528 | 9 | 58 |
To complement the SPR analyses, we performed FcRn affinity chromatography on samples of IgG WT and YTE. FcRn affinity chromatography is a useful tool for characterizing the pH dependence of FcRn affinity (22). Briefly, FcRn was conjugated to the solid phase; antibodies were loaded onto the column and bound under isocratic flow at pH 5.5 before being separated by a pH gradient. The WT protein eluted at a pH of 7.5, whereas YTE remained bound until ∼pH 8.0, as shown in Fig. 1G.
IgG Conformational Dynamics at pH 7 and pH 5.5 in the Presence and Absence of FcRn
HX MS experiments were performed on IgG WT and YTE in the presence and absence of FcRn at pH 5.5, and at pH 7 in the absence of FcRn. HX experiments as performed here assess the frequency of backbone H-bond opening and reclosing rates by measuring the rate at which backbone protons exchange with solvent deuterium. H-bonds involved in stable structure exchange more slowly than H-bonds involved in less stable structure; the rate of exchange is related directly to structural stability as discussed under “Experimental Procedures.”
HX measurements address the magnitudes of changes in structural stabilities upon binding FcRn, between pH 5.5 and 7.0 in the absence of FcRn, and between WT and YTE for each condition. These data were quantitatively assessed by computing peptide protection factors (PFC1→C2, see “Experimental Procedures”), and all peptides with differences in deuterium uptake rate between any of these conditions are listed in Table 3. In Table 3, non-bold entries show peptides that were mildly sensitive to pH and insensitive to binding FcRn; these peptides are not discussed but are listed in Table 3 for full disclosure. Bold entries in Table 3 are discussed below.
All peptides whose uptake rates change when bound to FcRn were localized to the CH2/3 domain interface and are shown in Fig. 2. Five structural regions marked by peptides H241–H252 (Fig. 2A), H253–H262 (Fig. 2B), H306–H318 (Fig. 2C), H336–H348 (Fig. 2D), H369–H379 (Fig. 2E), and H429–H440 (Fig. 2F) were observed to have significantly slower exchange profiles upon binding FcRn, similar to the Fc domain regions identified by Jensen et al. (23) to be sensitive to binding FcRn. Peptide H369–H379 is also shown because this was the only peptide where WT and YTE exhibited differences in stability at pH 5.5 without being affected by FcRn binding (Fig. 2E). The locations of these peptides in the Fc domain are illustrated in Fig. 2G.
FIGURE 2.

IgG conformational stabilization upon binding FcRn at pH 5.5. A–F, representative uptake traces at pH 5.5 (solid) and pH 5.5+FcRn (dashed) for WT (black) and YTE (red) for all regions whose deuterium uptake profiles change in response to binding FcRn. Dash-dot traces in black (and red where applicable for YTE mutations) represent the theoretical uptake rate, scaled to account for back exchange, for the same amino acid sequence devoid of H-bonds. G, structural locations of peptides are indicated on the zoomed view. Locations of the YTE mutations are shown in yellow; histidines 310, 433, and 435 are shown in green. Peptides stabilized by binding FcRn (orange) are shown in blue along with those regions sensitive to pH and not responding to FcRn in red. Error bars indicate ± S.E. for n = 3.
All peptides stabilized by binding FcRn at pH 5.5 (Fig. 2, A–D and F) are also destabilized by pH reduction of the mAb in the absence of FcRn binding as shown in Fig. 3; the only additional peptide sensitive to pH in YTE was H336–H348 in panel E; binding FcRn did not change the exchange rate for this peptide in WT (Fig. 2D). The largest responses to pH depression are shown in peptides H241–H252 (Fig. 3A), H306–H318 (Fig. 3D), and H429–H440 (Fig. 3F).
FIGURE 3.

pH reduction destabilizes IgG. A–F, representative HX uptake curves for YTE (red) and WT (black) at pH 5.5 (solid) and pH 7.0 (dashed). Theoretical traces are shown here, as in Fig. 2, in dot-dashed traces. Error bars indicate ± S.E. for n = 3.
To test the generality of our results, we measured four additional IgG1 molecules at pH 7 and 5.5. The same structural regions most sensitive to pH in WT and YTE were also most sensitive to pH in these antibodies: H241–H242, H306–H318, and H429–H440 (Fig. 4, A–C).
FIGURE 4.

General IgG pH sensitivity observed in other IgG molecules. A–C, HX traces are shown for four additional IgG molecules. pH 5.5 data are shown in solid traces, and pH 7 data are shown in dashed traces; the color of each antibody is maintained for all panels. Error bars indicate ± S.E. for n = 3.
Discussion
FcRn Affinity Differences between YTE and WT
In direct comparison by SPR, YTE always displays faster on-rates and slower off-rates than WT, resulting in the observed higher FcRn affinity (Fig. 1, A–F). SPR models suggest a 4–12-fold difference in affinity (KD,WT/KD,YTE) at pH 5.5 with much larger differences, up to 111-fold at pH 7.4.
SPR measurements confirm that YTE binds with higher affinity but are substantially influenced by avidity effects, as discussed previously (39). Still, the range reported is likely to be a fairly good estimate of affinity differences at these fixed conditions. SPR measurements show that affinity differences are much larger at pH 7.4 (Table 2), as expected given that this pH is close to the elution midpoint for WT (Fig. 1G).
FcRn chromatography data demonstrate that the pH dependence of FcRn affinity is augmented by the YTE mutation. As expected, the binding rate was undeterminable by SPR kinetics at pH 7.4, the place where the molecule elutes. This confirms that the mutation affects association and dissociation kinetics differentially, as was suggested by SPR measurements (Table 2).
Conformational Dynamics in the Absence of FcRn Determine the Difference in FcRn Affinity between WT and YTE
We originally hypothesized that the YTE mutant would have slower deuterium uptake than WT while bound to FcRn, reflective of its higher affinity. To test this hypothesis, we compared the exchange rates of WT and YTE, free in solution at pH 5.5, with their exchange profiles while bound to FcRn at the same pH.
Unexpectedly, there were only subtle differences in uptake rates between WT and YTE while bound to FcRn, suggesting that the conformational stabilities (hydrogen bonding of backbone amides) of both systems are equivalent in this state. Exchange profiles between YTE and WT differed to a much greater extent in the absence of FcRn at pH 5.5. Together, these two observations unambiguously demonstrate that affinity differences between WT and YTE for FcRn must reside in the unbound state.
Changes in equilibrium constants assessed by orthogonal techniques quantitatively agree and further support this view. We measured a 7.5-fold increase in YTE PFpH 5.5→pH 5.5+FcRn as compared with WT in H241–H252 (Table 3), within the 4–12.5-fold range of affinity increase measured by SPR (Table 2).
pH Dependence of Local Structural Stability Implies a Relationship to FcRn Affinity
Given the importance of local loop mobility recently identified to influence FcRn affinity (9), we next measured the exchange profiles of WT and YTE in the absence of FcRn at pH 7 and pH 5.5 to determine whether structural pH sensitivity coincides with regions that are important for interacting with FcRn.
The destabilizing influence of pH reduction of specific structural locations in both WT and YTE is shown in Fig. 3. These HX measurements interpreted as changes in free energy are shown in Fig. 5A, and are shaded onto structural models in panels B and C. Although there were areas with mild pH sensitivity that are unaffected by FcRn interaction, all peptides reporting stabilization upon binding FcRn are destabilized by pH reduction.
FIGURE 5.

pH-dependent FcRn affinity is determined by the stability of the unbound state of the IgG. A, change in structural free energy for WT (black bars) and YTE (gray bars) in response to pH (top) and FcRn binding (bottom) for all peptides with significant differences in deuterium uptake identified in the data set (see “Results” and Table 3); the values shown are computed using ΔΔGC1→c2 = −RTln(PFC1→C2) = −RTln(PFC2→C1−1). B, regions that destabilize in response to pH reduction are shown in red. C, regions that stabilize when bound to FcRn (FcRn is in orange) at pH 5.5 are shown in blue. In both panels (B and C), histidine residues potentially important in the process are colored green, sites of the YTE mutations are shown in yellow, and portions of the sequence with no coverage in the HX data are colored in black. D, relative free energy diagram of the IgG molecule based on HX measurements of different IgG states.
The same three structural regions most affected by pH are also most affected by binding FcRn, as shown in peptides H241–H252, H253–H262, H306–H318, and H429–H440 (Table 3, Fig. 5A). These measurements, combined with what has been previously established about these three regions, clearly show how destabilization of canonical structure converts the mAb from low to high FcRn affinity.
Fc side chains Ile-253, Arg-311, and His-435 assume different rotamers in complex with FcRn than when unbound; these are the largest observed conformational differences between free and FcRn-bound IgG crystal structures (11). These side chains reside in the three structural regions most destabilized by pH (Fig. 3, A, B, E, and F). We confirmed similar behavior in four additional IgG1 molecules (Fig. 4). This observation is a general feature of antibodies. Thus, pH reduction causes the greatest local structural destabilization in those regions of the molecule containing these important residues.
Local structural destabilization means that these atoms sample greater conformational space at pH 5.5 than at pH 7. Reduction of pH and subsequent destabilization for this system appear to unlock Fc side chains Ile-253, Arg-311, and His-435 from their native orientations, reminiscent of folding upon binding mechanisms, which explains the preponderance of intrinsically disordered proteins in biology.
Destabilization also directly increases affinity by breaking internal H-bonds that must dissociate before forming H-bonds with FcRn. FcRn residue Glu-135 makes backbone hydrogen bonds with Fc residues 253 and 254 (11). Energy is required to break intramolecular hydrogen bonds in order for these residues to interact with FcRn. Fig. 2, A and B, shows that peptides reporting on H-bonds in this region experience much faster deuterium uptake at pH 5.5 as compared with pH 7.0. Disruption of these H-bonds is prerequisite for H-bond formation to FcRn Glu-135. Peptides containing H253 and H254 residues are among the most stabilized regions upon binding FcRn (Figs. 2, A and B, and 4A), highlighting the importance of these contacts.
Destabilization, induced by pH reduction, effectively lowers the barrier for intermolecular H-bond formation between FcRn Glu-135 and Fc residues 253 and 254. Because destabilization of the unbound state has no influence on the dissociation energy, it selectively increases the binding rate without influencing dissociation rate. In this way, pH-driven local destabilization directly enhances affinity.
A New Role for Fc Residue His-435
If pH-induced destabilization is relevant as we propose, revisiting previously unexplained observations in the literature with this new concept of pH-induced destabilization influencing FcRn affinity should provide productive insight into the phenomenon.
Ionization of heavy chain His-435 is centrally important to FcRn binding (9, 11, 13, 19); substitution with other amino acids either reduces FcRn binding rates at lower pH (for neutral amino acid substitutions) or reduces dissociation rates at neutral pH (positively charged residue substitutions), both of which are equally necessary for optimal functioning of the IgG recycling system. The first crystal structures solved for WT bound to FcRn (13, 14) led to the suggestion that His-435 contacts FcRn; however, higher resolution structures of Fc-YTE and Fc-WT (17) revealed that the orientation and distance of His-435 from FcRn residues are inconsistent with any contact forming between FcRn and this residue. What role does His-435 play in FcRn affinity? If it does not contact FcRn, why does substitution with other amino acids hinder pH-dependent affinity for FcRn?
Recent evidence highlights the importance of mobility in the His-435 loop. Rigidifying this loop by introduction of non-native disulfide bonds led to a reduction in FcRn affinity (9). Our HX measurements show that peptides spanning this loop are the most affected by pH reduction with a 25× increase in deuterium uptake rate in ∼2–4 exchangeable sites (Table 3, Figs. 3F and 4C). This is direct evidence that pH reduction drives increased loop mobility. We propose that His-435 serves the role of transmitting this pH change into local structural destabilization through columbic repulsion with ionized His-433 and His-429, among others, upon adopting a positive charge at acidic pH.
Mechanism for Destabilization as a Determinant of FcRn Affinity
We propose that destabilization of the FcRn binding region in the IgG Fc domain by pH reduction increases affinity. This mechanism operates in two ways: first, by disruption of intramolecular H-bonds in structural regions that form backbone intermolecular H-bonds with FcRn, and second, by non-specifically increasing the occupancy of binding-competent side-chain rotamers in residues that are known to reorient upon binding FcRn. Diagrammed in Fig. 5D as discovered here, destabilization and salt-bridge formation between charged histidine residues at acidic pH synergistically explain why IgG molecules have high affinity for FcRn under acidic solution conditions.
This mechanism provides new insight into how the pH-dependent affinity switch operates. With this view, we are able to rationalize the importance of His-435 and explain why reduced His-435 loop mobility decreases FcRn affinity. It will be interesting to explore the influence on the structural stability of many mutants identified by Monnet et al. (19) that influence FcRn affinity but are not in the FcRn epitope.
Concluding Remarks
As observed for the IgG-FcRn complex, similar mechanisms for regulating protein-protein interactions that originate in localized changes in conformational dynamics may be in effect in the formation of a wider range of biologically important protein complexes. Furthermore, for pharmaceutical applications, tuning the FcRn affinity of the IgG molecule by mutation presents an attractive strategy for the development of novel therapeutic IgGs with long serum lifetimes. Understanding the presently observed dynamic determinants of affinity is a prerequisite for rational protein engineering.
Author Contributions
B. T. W. conceived and coordinated the study. B. T. W., P. F. J., K. D. R., and J. Z. wrote the paper. V. L. designed, performed, and analyzed experiments shown in Table 1. T. S. and K. L. designed, performed, and analyzed experiments shown in Figure 1 and Table 2. B. T. W., K. D. R., P. F. J., and T. P. designed experiments; B. T. W. and P. F. J. performed and analyzed experiments shown in Figures 2–4 and Table 3. B. T. W. and T. P. developed and performed peptide protection factor analysis. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Ben Moore, and Alec Ricciuti for reviewing the manuscript; Kathleen Abadie and Adrian Zwick for assistance in the lab; Alex Bujotzek for helpful discussions; and Hans Koll, John Stults, and Yung-Hsiang Kao for supporting this research.
This work was supported by Genentech, Inc. and Roche, Inc. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental text.
Fc stands for fragment crystallizable or constant domain. This is roughly heavy chain residues 220 through the C terminus and includes the CH2 and CH3 IgG domains.
scFcRn indicates single-chain FcRn, which is FcRn and β2-microglobulin fused together as a single polypeptide chain. This prevents dissociation of the FcRn complex.
- FcRn
- neonatal Fc receptor
- HX
- amide hydrogen-deuterium exchange
- PF
- protection factor.
References
- 1. Kastritis P. L., and Bonvin A. M. (2013) Molecular origins of binding affinity: seeking the Archimedean point. Curr. Opin. Struct. Biol. 23, 868–877 [DOI] [PubMed] [Google Scholar]
- 2. Mercier E., Girodat D., and Wieden H. J. (2015) A conserved P-loop anchor limits the structural dynamics that mediate nucleotide dissociation in EF-Tu. Sci. Rep. 5, 7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seo M. H., Park J., Kim E., Hohng S., and Kim H. S. (2014) Protein conformational dynamics dictate the binding affinity for a ligand. Nat. Commun. 5, 3724. [DOI] [PubMed] [Google Scholar]
- 4. Boehr D. D., Nussinov R., and Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carroll M. J., Mauldin R. V., Gromova A. V., Singleton S. F., Collins E. J., and Lee A. L. (2012) Evidence for dynamics in proteins as a mechanism for ligand dissociation. Nat. Chem. Biol. 8, 246–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gouridis G., Schuurman-Wolters G. K., Ploetz E., Husada F., Vietrov R., de Boer M., Cordes T., and Poolman B. (2015) Conformational dynamics in substrate-binding domains influences transport in the ABC importer GlnPQ. Nat. Struct. Mol. Biol. 22, 57–64 [DOI] [PubMed] [Google Scholar]
- 7. Henzler-Wildman K., and Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972 [DOI] [PubMed] [Google Scholar]
- 8. Tzeng S. R., and Kalodimos C. G. (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240 [DOI] [PubMed] [Google Scholar]
- 9. Borrok M. J., Wu Y., Beyaz N., Yu X.-Q., Oganesyan V., Dall'Acqua W. F., and Tsui P. (2015) pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J. Biol. Chem. 290, 4282–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roopenian D. C., and Akilesh S. (2007) FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. 7, 715–725 [DOI] [PubMed] [Google Scholar]
- 11. Martin W. L., West A. P. Jr., Gan L., and Bjorkman P. J. (2001) Crystal structure at 2.8 Å of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol. Cell 7, 867–877 [DOI] [PubMed] [Google Scholar]
- 12. Burmeister W. P., Gastinel L. N., Simister N. E., Blum M. L., and Bjorkman P. J. (1994) Crystal structure at 2.2 Å resolution of the MHC-related neonatal Fc receptor. Nature 372, 336–343 [DOI] [PubMed] [Google Scholar]
- 13. Vaughn D. E., and Bjorkman P. J. (1998) Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure 6, 63–73 [DOI] [PubMed] [Google Scholar]
- 14. Burmeister W. P., Huber A. H., and Bjorkman P. J. (1994) Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature 372, 379–383 [DOI] [PubMed] [Google Scholar]
- 15. Raghavan M., Bonagura V. R., Morrison S. L., and Bjorkman P. J. (1995) Analysis of the pH dependence of the neonatal Fc receptor immunoglobulin-G interaction using antibody and receptor variant. Biochemistry 34, 14649–14657 [DOI] [PubMed] [Google Scholar]
- 16. Oganesyan V., Damschroder M. M., Woods R. M., Cook K. E., Wu H., and Dall'acqua W. F. (2009) Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol. Immunol. 46, 1750–1755 [DOI] [PubMed] [Google Scholar]
- 17. Oganesyan V., Damschroder M. M., Cook K. E., Li Q., Gao C., Wu H., and Dall'Acqua W. F. (2014) Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 289, 7812–7824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Majumdar R., Esfandiary R., Bishop S. M., Samra H. S., Middaugh C. R., Volkin D. B., and Weis D. D. (2015) Correlations between changes in conformational dynamics and physical stability in a mutant IgG1 mAb engineered for extended serum half-life. MAbs 7, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Monnet C., Jorieux S., Urbain R., Fournier N., Bouayadi K., De Romeuf C., Behrens C. K., Fontayne A., and Mondon P. (2015) Selection of IgG variants with increased FcRn binding using random and directed mutagenesis: impact on effector functions. Front. Immunol. 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang W., Lu P., Fang Y., Hamuro L., Pittman T., Carr B., Hochman J., and Prueksaritanont T. (2011) Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab. Dispos. 39, 1469–1477 [DOI] [PubMed] [Google Scholar]
- 21. Li B., Tesar D., Boswell C. A., Cahaya H. S., Wong A., Zhang J., Meng Y. G., Eigenbrot C., Pantua H., Diao J., Kapadia S. B., Deng R., and Kelley R. F. (2014) Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. MAbs 6, 1255–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlothauer T., Rueger P., Stracke J. O., Hertenberger H., Fingas F., Kling L., Emrich T., Drabner G., Seeber S., Auer J., Koch S., and Papadimitriou A. (2013) Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. MAbs 5, 576–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jensen P. F., Larraillet V., Schlothauer T., Kettenberger H., Hilger M., and Rand K. D. (2015) Investigating the interaction between the neonatal Fc receptor and monoclonal antibody variants by hydrogen/deuterium exchange mass spectrometry. Mol. Cell Proteomics 14, 148–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walters B. T., Ricciuti A., Mayne L., and Englander S. W. (2012) Minimizing back exchange in the hydrogen exchange-mass spectrometry experiment. J. Am. Soc. Mass. Spectrom. 23, 2132–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mayne L., Kan Z.-Y., Chetty P. S., Ricciuti A., Walters B. T., and Englander S. W. (2011) Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J. Am. Soc. Mass. Spectrom. 22, 1898–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kan Z. Y., Mayne L., Chetty P. S., and Englander S. W. (2011) ExMS: data analysis for HX-MS experiments. J. Am. Soc. Mass. Spectrom. 22, 1906–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walters B. T., Mayne L., Hinshaw J. R., Sosnick T. R., and Englander S. W. (2013) Folding of a large protein at high structural resolution. Proc. Natl. Acad. Sci. U.S.A. 110, 18898–18903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu W., Walters B. T., Kan Z.-Y., Mayne L., Rosen L. E., Marqusee S., and Englander S. W. (2013) Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 110, 7684–7689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elvin A. K. (1991) Sequences of Proteins of Immunological Interest, 5th Ed., U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, Bethesda, MD [Google Scholar]
- 30. Walters B. T., Patapoff T., and Zhang J. (2014) The combination of molecular dynamics, amide hydrogen exchange, and mass spectrometry to understand mAb conformational dynamics. in 62nd Annual ASMS Conference on Mass Spectrometry and Allied Topics (Gross M. L,. ed), American Society For Mass Spectrometry, Baltimore, MD [Google Scholar]
- 31. Coales S. J., E S. Y., Lee J. E., Ma A., Morrow J. A., and Hamuro Y. (2010) Expansion of time window for mass spectrometric measurement of amide hydrogen/deuterium exchange reactions. Rapid Commun. Mass. Spectrom. 24, 3585–3592 [DOI] [PubMed] [Google Scholar]
- 32. Bai Y., Milne J. S., Mayne L., and Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hvidt A. (1964) A discussion of the pH dependence of the hydrogen-deuterium exchange of proteins. C. R. Trav. Lab. Carlsberg 34, 299–317 [PubMed] [Google Scholar]
- 34. Skinner J. J., Lim W. K., Bédard S., Black B. E., and Englander S. W. (2012) Protein hydrogen exchange: testing current models. Protein Science 21, 987–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Skinner J. J., Lim W. K., Bédard S., Black B. E., and Englander S. W. (2012) Protein dynamics viewed by hydrogen exchange. Protein Sci. 21, 996–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krishna M. M., Hoang L., Lin Y., and Englander S. W. (2004) Hydrogen exchange methods to study protein folding. Methods 34, 51–64 [DOI] [PubMed] [Google Scholar]
- 37. Bai Y., Englander J. J., Mayne L., Milne J. S., and Englander S. W. (1995) Thermodynamic parameters from hydrogen exchange measurements. Methods Enzymol. 259, 344–356 [DOI] [PubMed] [Google Scholar]
- 38. Englander S. W., and Kallenbach N. R. (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16, 521–655 [DOI] [PubMed] [Google Scholar]
- 39. Schoch A., Kettenberger H., Mundigl O., Winter G., Engert J., Heinrich J., and Emrich T. (2015) Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc. Natl. Acad. Sci. U.S.A. 112, 5997–6002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.