

Abstract
TRAP1 (tumor necrosis factor receptor-associated protein 1), a mitochondrial Hsp90 family chaperone, has been identified as a critical regulator of cell survival and bioenergetics in tumor cells. To discover novel signaling networks regulated by TRAP1, we generated Drosophila TRAP1 mutants. The mutants successfully developed into adults and produced fertile progeny, showing that TRAP1 is dispensable in development and reproduction. Surprisingly, mutation or knockdown of TRAP1 markedly enhanced Drosophila survival under oxidative stress. Moreover, TRAP1 mutation ameliorated mitochondrial dysfunction and dopaminergic (DA) neuron loss induced by deletion of a familial Parkinson disease gene PINK1 (Pten-induced kinase 1) in Drosophila. Gamitrinib-triphenylphosphonium, a mitochondria-targeted Hsp90 inhibitor that increases cell death in HeLa and MCF7 cells, consistently inhibited cell death induced by oxidative stress and mitochondrial dysfunction induced by PINK1 mutation in mouse embryonic fibroblast cells and DA cell models such as SH-SY5Y and SN4741 cells. Additionally, gamitrinib-triphenylphosphonium also suppressed the defective locomotive activity and DA neuron loss in Drosophila PINK1 null mutants. In further genetic analyses, we showed enhanced expression of Thor, a downstream target gene of transcription factor FOXO, in TRAP1 mutants. Furthermore, deletion of FOXO almost nullified the protective roles of TRAP1 mutation against oxidative stress and PINK1 mutation. These results strongly suggest that inhibition of the mitochondrial chaperone TRAP1 generates a retrograde cell protective signal from mitochondria to the nucleus in a FOXO-dependent manner.
Keywords: Drosophila, genetics, mitochondria, neurobiology, signal transduction, mitochondria
Introduction
Mitochondria, the cellular power plants that provide ATP through oxidative phosphorylation, have a critical role in cell survival and death. Diverse stress and death signals converge on these organelles and release mitochondrial death proteins to activate cell death pathways in the cytosol (1). Consistent with this, dysfunctional mitochondria have been heavily implicated in various human diseases, including Parkinson disease (PD)4 (2). Langston et al. (3) discovered that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a specific inhibitor of mitochondrial complex I, causes chronic parkinsonism in primates. Two other mitochondrial toxins, rotenone and paraquat, also induce parkinsonism in various model animals (4).
Recently, Drosophila genetic analyses successfully elucidated a familial PD gene PINK1 (Pten-induced kinase 1) that encodes a mitochondrial kinase PINK1 as the molecular link between mitochondrial quality control and parkinsonism (5–7). Further genetic and cell biological studies revealed that PINK1 translocates Parkin, an E3 ubiquitin ligase encoded by another familial PD gene parkin, to mitochondria and regulates mitochondrial remodeling processes such as mitochondrial fusion/fission and mitophagy. Moreover, PINK1 also regulates mitochondrial trafficking, mitochondrial protective gene expression, and complex I activity through various partners, suggesting PINK1 as a molecular checkpoint in the maintenance of mitochondrial function and integrity (8).
Mitochondrial dysfunction in tumors was first discovered by Otto Warbug. He found that cancerous cells generate ATP mainly through glycolysis (9). His finding, called the “Warbug Effect,” and the fundamental role of mitochondria in cell survival and death suggested that mitochondrion is an important target in developing specific anti-cancer agents (10). In investigating a mitochondrial protein network specific to tumor cells, Kang et al. (11) found that disabling mitochondrial heat shock protein 90 (Hsp90) family proteins, including TRAP1, causes cell death specifically in tumor cells. TRAP1 was initially identified as a novel protein binding to the intracellular domain of tumor necrosis factor receptor 1 and thus named “TRAP1” (12). This initial finding suggested its localization in the cytoplasm, but the following analyses demonstrated that TRAP1 mostly localizes in mitochondria via its N-terminal mitochondria targeting sequence (13, 14). It shares 34% sequence identity and 60% overall homology with other Hsp90 family members, and Hsp90 inhibitors like geldanamycin and radicol also inhibit TRAP1 activity in vitro (13). Interestingly, TRAP1 is highly expressed in mitochondria of various tumor cells and human tumor specimens, but it is expressed at low levels in mitochondria of corresponding normal tissues (11). When cells were treated with mitochondria-targeted Hsp90 inhibitors or when TRAP1 was down-regulated by RNAi, extensive cell death was observed only in tumor cells, and the sensitivity to anti-cancer agents was substantially increased (11). Further biochemical analyses revealed that TRAP1 can directly interact with cyclophilin D and inhibits its activity for opening mitochondrial permeability transition pore to induce cell death (11). Additionally, Pridgeon et al. (15) reported that phosphorylation of TRAP1 by PINK1 is responsible for protecting neuroendocrine tumor-derived PC-12 cells from reactive oxygen species (ROS). These data suggest that TRAP1 is an important cell-protective protein in mitochondria, especially in tumor cells. However, in recent cell metabolic studies, TRAP1 directly binds to and inhibits the complex II of the mitochondrial respiratory chain (16), and TRAP1 deficiency promotes mitochondria respiration (17, 18), suggesting additional roles of TRAP1 in the cell.
In this study, we found that loss of TRAP1 function in Drosophila markedly enhances survival rate under oxidative stress and rescues mitochondrial dysfunction and dopaminergic (DA) neuronal loss induced by PINK1 mutation. Consistent with these genetic data, the mitochondrial Hsp90 inhibitor gamitrinib also protected various mammalian cell models from oxidative stress and ameliorated PINK1 null mutation-induced defects in both Drosophila and mammalian systems. Further genetic analyses demonstrated that the cell protective effect induced by TRAP1 down-regulation is mediated by FOXO (Forkhead box O) transcription factors.
Experimental Procedures
Drosophila Strains
da-GAL4, hs-GAL4, and TRAP1P (TRAP1EY21851) strains were obtained from the Bloomington Stock Center. TRAP1P mutants were backcrossed for six generations into w1118 controls to remove genetic background effects. The insertion sites of P-element in TRAP1P are located at +1,955 of TRAP1 ORF. A revertant (TRAP1RV) and a deletion mutant (TRAP1D6) were generated from P-element excision of TRAP1P. In DNA sequencing analyses, TRAP1RV showed a precise excision of the P-element with no insertion or deletion of nucleotides. By contrast, 2.9 kb (base pairs 6,632,775–6,635,658, according to the Drosophila melanogaster chromosome sequence release 6), including most of TRAP1 ORF (amino acids 86–691), was deleted in TRAP1D6. TRAP1 cDNA was subcloned into the pUAST vector and microinjected into w1118 embryos. PINK1B9 was generated as previously described (6). The FOXO21 and FOXO25 lines were from E. Hafen. The TRAP1KK102212 RNAi line was purchased from the Vienna Drosophila RNAi Center.
Climbing Assays
Groups of fifteen 3-day-old males were transferred into climbing ability test vials and incubated for 1 h at room temperature for environmental acclimatization. After tapping the flies down to the bottom, the number of climbing flies in 10 s were counted. For each group, ten trials were performed, and the climbing score (percentage ratio of the number of climbed flies against the total number) was obtained. The average climbing score with standard deviation was calculated for four independent tests.
Oxidative Stress Assays
30 male flies (3-day-old) were starved for 6 h and transferred to a vial containing a gel of PBS, 5% sucrose, and an oxidative stress agent (20 mm paraquat or 5 mm rotenone) as indicated in figure legends. Dead flies were counted at the indicated time points. We repeated at least four times with 30 flies per genotype (n ≥ 120) to obtain the average survival rate with standard deviation.
Muscle Section and TUNEL Assay
The thoraces from 3-day-old flies were embedded in Spurr's resin and sectioned as previously described (6). The serial sections were then stained with toluidine blue dye and observed with BX-50 microscope (Olympus). For the TUNEL assay, apoptosis in the thoraces of 3-day-old flies was detected using the in situ cell death detection kit (Roche). DAPI (Sigma) was used to visualize the nucleus of muscle cells. Fluorescence images were obtained with BX-50 microscope (Olympus).
mtDNA PCR and ATP Assay
For mtDNA PCR, total DNA from five thoraces of 3- or 30-day-old flies was extracted. Then quantitative real time PCR was performed as previously described (6). Genomic DNA levels of rp49 were measured for internal controls. The results were expressed as fold changes relative to the control. For ATP assay, five thoraces from 3-day-old flies were dissected, and ATP concentration was measured as previously described (6). The relative ATP level was calculated by dividing the measured ATP concentration by the total protein concentration. Protein concentration was determined by a bicinchoninic acid assay (Sigma). In the mtDNA PCR and ATP assays, the average values with standard deviation were obtained from three independent experiments.
Immunostaining
Adult brain was fixed with 4% paraformaldehyde and stained with anti-tyrosine hydroxylase (TH) rabbit antibody (1:50, Pel-Freez) as previously described (6). Brains were observed and imaged by LSM 700 confocal microscope (Zeiss) and BX-50 microscope (Olympus).
ROS Measurements in Adult Flies
To measure whole body ROS levels, ten 3-day-old male flies were dissected in 250 μl of distilled water containing aminotriazol (2 mg/ml). Samples were centrifuged to obtain supernatants, and ROS concentration was measured as previously described (19). The relative ROS level was calculated by dividing the measured ROS concentration by the total protein concentration. The average ROS level with standard deviation was obtained from three independent experiments. For imaging ROS production in fly tissues, the indirect flight muscles were dissected in Schneider's medium (Sigma) and incubated for 5 min in Schneider's medium containing 30 μm dihydroethidium (Invitrogen). Muscles were observed and imaged by BX-50 microscope (Olympus).
Mammalian Cell Culture and Transfection
NIH 3T3, MEF, COS-1, HeLa, MCF-7, 293E, and SH-SY5Y cells were grown in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) at 37 °C in a humidified atmosphere with 5% CO2. SN4741 cells were grown in RF medium-containing DMEM supplemented with 10% fetal bovine serum, 1% glucose, and 2 mm l-glutamine at 33 °C in a humidified atmosphere with 5% CO2. Wild type and PINK1−/− MEFs were provided by Drs. Un Jung Kang and Xiaoxi Zhuang. siRNAs for control (Bioneer; catalog no. SN-1003), mouse FOXO1 (Bioneer; catalog no. 1359213), or mouse FOXO3 (Bioneer; catalog no. 1359223) were transfected to MEF cells using the RNAiMAX reagent (Invitrogen) according to the manufacturer's protocol. shRNAs for control (Sigma; catalog no. SHC016) or TRAP1 (Sigma; catalog no. TRCN0000112172) were co-transfected with lentivirus packaging plasmids into 293T cells according to the manufacturer's protocol. The resulting lentiviral particles were used to infect MEF cells. After 24 h of infection, cells were incubated in puromycin (2.5 mg/ml)-containing media for 72 h to select infected cells.
MTT Assay
Cells were seeded in 12-well plates at a density of 1.5 × 105 cells/well. After pretreatment of G-TPP (LegoChem Biosciences) or 17-allylamino-demethoxygeldanamycin (17-AAG) at the indicated concentrations for 4 h, the cells were treated with 1 mm paraquat. (For lentivirus-infected MEF cells, 0.5 mm paraquat was treated.) After 20 h of incubation, the culture medium was removed and replaced with a medium containing 0.5 mg/ml of MTT dissolved in PBS (pH 7.2). After 4 h, the formed formazan crystals were dissolved in 400 μl of DMSO, and the absorbance intensity was measured at a wavelength of 595 nm using EL-312e microplate reader (BIOTEK). The relative cell viability was expressed as a percentage relative to the untreated control cells. The average viability with standard deviation was obtained from three independent experiments.
Annexin V Staining
MEF cells were seeded in 60-mm plates with cell density of 1 × 106 cells/plate. Treatment of G-TPP (5 μm), 17-AAG (5 μm), and paraquat (1 mm) were performed as described above. The cells were stained using the annexin V-FITC apoptosis detection kit (BD Biosciences) according to the manufacturer's protocol. Stained cells were analyzed by flow cytometry using EPICS XL cytometer (Beckman Coulter Inc.). A total of 10,000 events were analyzed for each sample, and the necrotic cell death rates obtained from three independent experiments were presented as the mean values with standard deviations.
Measurement of Intracellular ROS Levels
MEF, SH-SY5Y, and SN4741 cells were pretreated with G-TPP (5 μm) or 17-AAG (5 μm) for 4 h. Following 7 h of treatment of 1 mm paraquat (6 h of treatment of 0.5 mm paraquat to lentivirus-infected MEF cells), the cells were incubated with 5 μm 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA; Invitrogen) for 30 min at 37 °C. The cells were trypsinized, washed with PBS, suspended in PBS, and analyzed with EPICS XL cytometer (Beckman Coulter). A total of 10,000 events were analyzed for each sample, and the results obtained from three independent experiments were presented as the mean values with standard deviations.
Measurement of Mitochondrial Membrane Potential
MEF, SH-SY5Y, and SN4741 cells were pretreated with G-TPP (5 μm) or 17-AAG (5 μm) for 4 h. Following 6 h of treatment of 1 mm paraquat (4 h treatment of 0.5 mm paraquat to lentivirus-infected MEF cells), cells were incubated with 1 μg/ml of JC-1 dye (Invitrogen) for 20 min and analyzed by microscopy. Fluorescence images were captured by LSM-700 confocal microscope (Carl Zeiss). To quantify mitochondrial membrane potential, cells were trypsinized and stained with 1 μg/ml of JC-1 dye for 20 min. The red and green fluorescence intensities of JC-1 dye were measured by flow cytometry using EPICS XL cytometer (Beckman Coulter). A total of 10,000 events were analyzed for each sample, and the results obtained from three independent experiments were presented as the mean values with standard deviations.
Treatment of Galactose Media to MEF Cells
Wild type or PINK1−/− MEFs were seeded in 6-well plates at a density of 4 × 105 cells/well. After 24 h of incubation, the cells were changed into glucose or galactose culture medium with indicated reagents as described previously (20). Twenty hours later, cells were trypsinized, and their mitochondrial membrane potential was checked using flow cytometry as described above.
Immunoblot
His-tagged full-length Drosophila TRAP1 protein was purified by nickel affinity column and injected into rabbits to generate anti-dTRAP1 antibody. 3-day-old flies were homogenized with lysis buffer as described previously (21). For detection of mammalian TRAP1, FOXO1 or FOXO3 protein, NIH 3T3, MEF, COS-1, HeLa, MCF7, and 293E cells were lysed with lysis buffer. The lysates were purified by centrifugation and boiled in SDS sample buffer. The samples were subjected to SDS-PAGE, and proteins were transferred to nitrocellulose membranes. Membranes were incubated for 30 min in blocking solution and further incubated with anti-dTRAP1 antibody, anti-mouse TRAP1 antibody (BD Biosciences), anti-FOXO1 antibody (Cell Signaling Technology), anti-FOXO3 antibody (Cell Signaling Technology), anti-actin antibody (Santa Cruz), or anti-tubulin antibody (Developmental Studies Hybridoma Bank) as described previously (21). Membrane-bound antibodies were detected with ImageQuant LAS 4000 system (GE Healthcare Life Sciences).
S2 Cell Culture, Transfection, and Immunocytochemistry
The plasmid constructs for S2 cell transfection were generated by subcloning cDNA for full-length (WT) or N-terminal deleted (ΔN) TRAP1 with C-terminal HA tag into pUAST vector. The following primers were used to amplify WT TRAP1 cDNA (5′-GCG GAA TTC GCC ACC ATG TCT GTA CGA GCG ATG GG-3′, 5′-GCG CTC GAG GTA TTT CTC CAG GGC CCG CGA TAG-3′) and ΔN TRAP1 cDNA (5′-CGC GAA TTC GCC ACC ATG ACG GAG ACC AAG CAG GCA TC-3′, 5′-GCG CTC GAG GTA TTT CTC CAG GGC CCG CGA TAG-3′). S2 cells were cultured and transiently transfected as described previously (21). To induce TRAP1 expression on pUAST vector, we co-transfected pMTGAL4 plasmids that contain GAL4 gene with metallothionein promoter. Twenty-four hours before cell staining, CuSO4 was treated to induce expression of GAL4 and TRAP1. The cells were preincubated with 5 μg/ml MitoTracker Red CMXRos (Molecular Probes) for 1 h at 25 °C and then subjected to the standard immunocytochemistry using anti-HA antibody (Invitrogen).
Synthesis of Double-stranded RNA
For synthesis of TRAP1 double-stranded RNA (dsRNA), we used oligonucleotides containing a T7 polymerase binding site (5′-TAA TAC GAC TCA CTA TAG GG-3′) at the 5′ of the following primers, 5′-CCG ACT TGG AGG ATT CAA AAC-3′ and 5′-ACC GGT GTG GCT CTT TAC AC-3′. The primers were designed to produce dsRNA to mimic TRAP1KK102212 RNAi fly. Control dsRNA was synthesized as previously described (21). The purified PCR products were subjected to an in vitro T7 transcription reaction using the MEGAscriptTM kit (Ambion).
Luciferase Assay
To measure transactivation activity of FOXO, S2 cells were transfected with pUAST FOXO, p8XFK1tkLuc FOXO reporter (22), pRL-TK Renilla reporter, and pMTGAL4 plasmids. Control or TRAP1 dsRNA was also co-transfected with DNA plasmids. Two days later, FOXO expression was induced by CuSO4 treatment. After 24 h, luciferase assays were performed using Dual-LuciferaseTM reporter assay kit (Promega) according to the manufacturer's instructions. The average luciferase activity with standard deviation was obtained from three independent experiments.
Quantitative RT-PCR
Total RNA from five thoraces of 3-day-old flies was extracted and reversely transcribed as previously described (23). Then quantitative real time PCR was performed using SYBR Premix Ex Taq (Takara) on Prism 7000 real time PCR System (ABI). rp49 levels were measured for internal control. The results were expressed as fold changes relative to the control. The average mRNA level with standard deviation was obtained from three independent experiments. For primer pairs, we used rp49-F (GCT TCA AGA TGA CCA TCC GCC C) and rp49-R (GGT GCG CTT GTT CGA TCC GTA AC), TRAP1-F (GCA GCG TTC AAT ATC ACC ATT G) and TRAP1-R (GAC CTC GTG GTC GGA GTC TAA GG), CG3161-F (GTC TTC TGA AGT GAG CAG CGA C) and CG3161-R (CAT CAC TGA CAT GGC AGC AAT ACC), and Thor-F (GAA GGT TGT CAT CTC GGA TCC) and Thor-R (CAT GAA AGC CCG CTC GTA G).
Genotypes
The genotypes used were TRAP1RV (TRAP1RV/TRAP1RV); TRAP1P (TRAP1P/TRAP1P); TRAP1D6 (TRAP1D6/TRAP1D6); hs>TRAP1 (hs-GAL4/UAS-TRAP1); da/+ (da-GAL4/+); da>TRAP1i (TRAP1KK102212/+; da-GAL4/+); WT (+/Y); B9 (PINK1B9/Y); B9, TRAP1P (PINK1B9/Y; TRAP1P/(26)TRAP1P); B9, TRAP1D6 (PINK1B9/Y; TRAP1D6/TRAP1D6); TRAP1P, FOXO− (TRAP1P/TRAP1P; FOXO21/FOXO25); FOXO− (FOXO21/FOXO25); B9, TRAP1P, FOXO−/+ (PINK1B9/Y; TRAP1P/TRAP1P; FOXO21/+); and B9, FOXO− (PINK1B9/Y; FOXO21/FOXO25).
Quantification and Statistical Analyses
For quantification of wing and thorax phenotypes, the percentage of defective thorax and wing phenotypes of 3-day-old males was measured (n > 200). For quantification of DA neurons, four major DA neuron clusters from 15 brains of each genotype were observed in a blind fashion to eliminate bias (n = 30). To compare three or more groups, we used a one-way ANOVA with Sidak correction. For two-group comparison, we used Student's two-tailed t test. The Kaplan-Meier estimator and the log rank test were conducted on the pooled cumulative survival data to determine whether each treatment had any effect on the longevity of individuals using Online Application Survival Analysis Lifespan Assays.
Results
Generation and Characterization of Drosophila TRAP1 Mutants
The Drosophila TRAP1 gene encodes a polypeptide of 691 amino acids with a molecular mass of ∼70 kDa (Fig. 1, A and D). In structural analysis, Drosophila TRAP1 shows an overall similarity of 66% with its human homolog and contains all of the canonical motifs of human TRAP1, such as ATP-binding domain and mitochondrial targeting motif (13) (Fig. 1A). Although a mutant TRAP1 protein without its N-terminal mitochondrial targeting motif was dispersed throughout the cytosol of Drosophila S2 cells, WT TRAP1 protein was specifically localized to mitochondria (Fig. 1B), confirming that Drosophila TRAP1 is a mitochondrial protein.
FIGURE 1.

Characterization of TRAP1 mutants. A, amino acid sequence similarities (%) of Drosophila TRAP1 with its human homolog. Black, mitochondrial targeting sequence; white, ATP-binding domain; gray, middle domain; dotted, dimerization domain. B, mitochondrial localization of TRAP1. Subcellular localization of wild type (TRAP1WT) and N-terminal deleted (TRAP1ΔN) C-terminally HA-tagged TRAP1 in S2 cells was determined by co-staining with anti-HA antibody (green) and MitoTracker (red). C, schematic genomic organization of the TRAP1 locus. Black rectangles, coding sequences; gray rectangles, untranslated regions. Genomic structures of TRAP1P and TRAP1D6 are described under “Experimental Procedures.” D, immunoblot analyses of revertant (RV), TRAP1 mutants (TRAP1P and TRAP1D6), and TRAP1-overexpressing flies (hs [heat shock] > TRAP1). Actin (Act) was used as a loading control. E and F, comparison of TRAP1 (E) and CG3161 (F) mRNA levels in the whole body of flies (n = 3). G, RV and TRAP1P flies showed normal development to adult stages. H, toluidine blue-stained longitudinal sections of the indirect flight muscle from 30-day-old male flies. I, comparison of the ATP contents in fly thoraces from 3- or 30-day-old males (n = 3). J, quantification of the mtDNA in fly thoraces (n = 3). aa, amino acids; Cox I, cytochrome c oxidase subunit I; Cox III, cytochrome c oxidase subunit III; Cyt B, cytochrome b. Significance was determined by one-way ANOVA with Sidak correction. ***, p < 0.001; NS, not significant. Error bars indicate S.D. Scale bars, yellow, 5 μm. Details of all the indicated genotypes in this and other figures are described under “Experimental Procedures.”
From the Bloomington Drosophila Stock Center, we obtained a TRAP1 mutant (TRAP1P) with a P-element insertion in the coding sequence of the gene (Fig. 1C). By expression of delta 2–3 transposase, TRAP1D6 allele was generated through imprecise excision of the P-element, and the revertant TRAP1RV was obtained through precise excision to be used as a control (Fig. 1C). In immunoblot analysis, P-element insertion in the TRAP1 gene (TRAP1P) almost completely blocked TRAP1 protein expression, and deletion of the TRAP1 gene (TRAP1D6) resulted in the complete loss of TRAP1 protein expression (Fig. 1D). Moreover, quantitative RT-PCR analysis demonstrated severe and complete reduction of TRAP1 transcript in TRAP1P and TRAP1D6 mutants, respectively (Fig. 1E). By contrast, these two mutants demonstrated no meaningful changes in the transcript level of CG3161, the adjacent gene to the P-element insertion site (Fig. 1F), confirming a specific inhibition of TRAP1 gene expression in the mutants.
TRAP1 mutants successfully developed into adults and showed no apparent defects in reproduction (Fig. 1G and data not shown), indicating dispensable roles of TRAP1 in Drosophila development and reproduction. Based on its specific localization to mitochondria, we analyzed mitochondrial function and morphology in the indirect flight muscle from TRAP1 mutants. Through muscle section staining with toluidine blue, dark blue mitochondria became visible between light blue muscle fibers as described previously (6). Cross-thoracic sections of TRAP1 mutants showed well organized and intact mitochondrial structures of the indirect flight muscle compared with the revertant (RV) control (Fig. 1H). Moreover, biochemical studies revealed that the ATP level of the indirect flight muscle, an important index of mitochondrial function, was not decreased in both 3- and 30-day-old TRAP1 mutants (Fig. 1I). mtDNA content, indicating mitochondria abundance in tissues, was also not changed in 3- or 30-day-old TRAP1 mutants (Fig. 1J), suggesting that general mitochondrial functions and abundance are properly maintained in TRAP1 mutants.
Mutation or Knockdown of TRAP1 Induces Oxidative Stress Resistance
To further investigate the phenotypes of TRAP1 mutants, we analyzed the survival of TRAP1 mutants after exposure to oxidative stress induced by mitochondrial toxins. On the standard media, TRAP1 mutants showed no significant defect in life span (Fig. 2A) with weak mortality (∼10%) in early time points. In biochemical analyses and dihydroethidium staining, they showed significantly increased in vivo ROS level (Fig. 2, B and C). Surprisingly, when placed on a medium containing paraquat, a free radical inducer using electrons from the mitochondrial respiratory chain, TRAP1 mutants showed significantly increased survival compared with RV and WT controls (Fig. 2D). Moreover, TRAP1 mutants were also resistant to rotenone, a specific inhibitor of mitochondrial respiratory chain complex I (Fig. 2E). Down-regulation of TRAP1 using RNAi expression (Fig. 2F) also strongly increased the resistance to rotenone (Fig. 2G), further confirming that inhibition of TRAP1 expression induces resistance to mitochondrial toxins and oxidative stress.
FIGURE 2.

Suppression of TRAP1 enhances oxidative stress resistance. A, lifespan of revertant (RV) and TRAP1 mutants (TRAP1P). The number of surviving males was counted at the indicated days, and the survival ratios were presented as percentile values (log rank test: p = 0.33, groups with the same letter do not differ significantly). B, relative levels of whole body ROS (n = 3) between RV, TRAP1P, and TRAP1D6. C, dihydroethidium (DHE) staining of the indirect flight muscle from fly thoraces. D, survival curves of revertant (RV), wild type (WT), and TRAP1 mutant (TRAP1P and TRAP1D6) male flies on paraquat-containing food (log rank test: p < 0.001, groups with the same letter do not differ significantly). E, survival curves of male flies on rotenone-containing food (log rank test: p < 0.001, groups with the same letter do not differ significantly). F, comparison of TRAP1 mRNA levels in control (da/+) and TRAP1 RNAi expressing (da>TRAP1i) male flies. G, survival curves of male (♂) and female (♀) flies on paraquat-containing food (log rank test: p < 0.05, groups with the same letter do not differ significantly). Significance was determined by one-way ANOVA with Sidak correction. ***, p < 0.001. Error bars indicate S.D. Scale bar, white, 10 μm.
TRAP1 Mutations Ameliorate PINK1 Mutant Phenotypes
Resistance to oxidative stress is often closely related to mitochondrial function and integrity (4), and ROS sensitivity is dramatically increased in flies lacking a familial PD gene PINK1 (5, 8). Under rotenone treatment, PINK1 null mutants (B9) showed decreased survival rates compared with WT controls as previously reported (Fig. 3A). When we mutated TRAP1 in PINK1 null mutants, the decreased survival rates of PINK1 null mutants were dramatically rescued (Fig. 3A), indicating that TRAP1 mutations can restore mitochondrial dysfunction induced by PINK1 deletion. Indeed, the crushed thoraces and downturned wings of PINK1 null mutants were markedly rescued by TRAP1 mutations (Fig. 3, B and C). Muscle sections demonstrated that TRAP1 mutations inhibit mitochondria disruption and apoptotic cell death induced by loss of PINK1 (Fig. 3D). In subsequent biochemical analyses, PINK1 and TRAP1 double mutants showed significant recovery of mtDNA content and ATP level in the indirect flight muscle (Fig. 3, E and F, respectively). Moreover, TRAP1 mutations also successfully ameliorated the decreased locomotor activities of PINK1 mutants (Fig. 3G).
FIGURE 3.

TRAP1 mutation rescues PINK1 null mutant phenotypes. A, survival curves of wild type controls (WT), PINK1 null mutants (B9), and PINK1 and TRAP1 double mutants (B9, TRAP1P and B9, TRAP1D6) on rotenone-containing food (log rank test: p < 0.05, groups with the same letter do not differ significantly). B, percentages of the flies with defective thorax or wing phenotypes. C, light stereomicrographs of the fly thoraces. White arrow, collapsed-thorax phenotype. D, toluidine blue-stained longitudinal sections of the indirect flight muscle (top panels) and merged images of TUNEL (red) and DAPI (blue) staining of the indirect flight muscle in fly thoraces (bottom panels). E, quantification of the mtDNA of thoraces (n = 3). F, comparison of the ATP content of thoraces (n = 3). G, comparison of the climbing ability (n = 4). Significance was determined by one-way ANOVA with Sidak correction. **, p < 0.01; ***, p < 0.001. Error bars indicate S.D. Scale bars, yellow, 10 μm; white, 20 μm.
To further study TRAP1 mutation-induced changes in PINK1 mutant phenotypes, we stained adult Drosophila brains with TH antibody to check DA neurons. In Drosophila brain, most DA neurons are located in four major DA neuron clusters: dorsolateral clusters 1 (DL1), dorsomedial clusters (DM), posteriomedial clusters, and dorsolateral clusters 2 (DL2) (6). 30-day-old PINK1 mutants showed a significant decrease in the number of DA neurons specifically in DL1 and DM clusters (Fig. 4, A and B), as previously reported (6). TRAP1 mutations successfully inhibited the DA neuron degeneration in both clusters of PINK1 mutants (Fig. 4, A and B). These results confirmed that TRAP1 mutations can rescue mitochondrial dysfunction and all the PD-related phenotypes induced by PINK1 null mutation.
FIGURE 4.
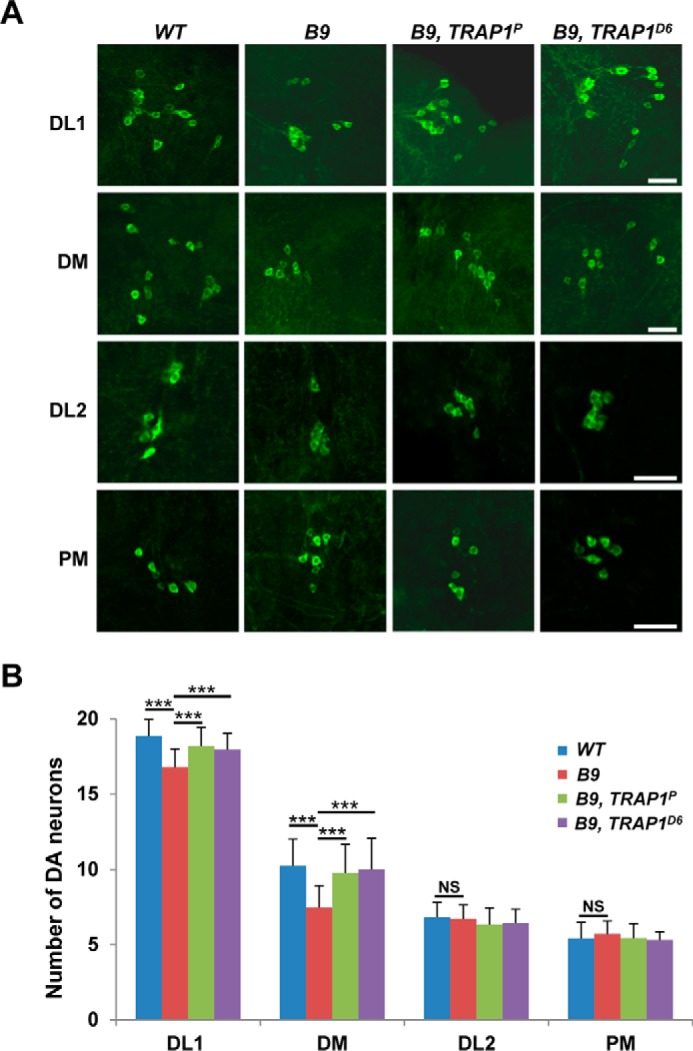
TRAP1 mutation ameliorates the DA neuronal degeneration in PINK1 null mutants. A, images of the DA neurons within DL1, DM, DL2, and posteriomedial clusters (PM) of the adult brains from wild type controls (WT), PINK1 null mutants (B9), and PINK1 and TRAP1 double mutants (B9, TRAP1P and B9, TRAP1D6). DA neurons were stained with anti-TH antibody (green). B, graph showing the average number of DA neurons in each cluster (n = 30). Significance was determined by one-way ANOVA with Sidak correction. ***, p < 0.001; NS, not significant. Error bars indicate S.D. Scale bars, white, 20 μm.
A TRAP1 Inhibitor G-TPP Specifically Protects Mammalian Cells under Paraquat-induced Stress
Recent correlations between TRAP1 and multidrug-resistant cancers initiated development of TRAP1 inhibitors (24). Kang et al. (25) developed gamitrinibs, the small molecules that contain a mitochondrial targeting module and a prototype structure of Hsp90 inhibitor geldanamycin. As expected, gamitrinibs successfully inhibited TRAP1 in mitochondria with no effect on cytosolic Hsp90 (25). To investigate the effect of TRAP1 inhibition on oxidative stress in mammalian cells, we tested paraquat-induced cytotoxicity in several TRAP1-expressing mammalian cell lines pretreated with gamitrinib-triphenylphosphonium (G-TPP), the most recently developed gamitrinib (24, 25). G-TPP effectively protected NIH 3T3, MEF, and COS-1 cells from paraquat-induced cell death in a dose-dependent manner (Fig. 5A). However, in HeLa and MCF7 cells that are highly sensitive to TRAP1 inhibitors (11, 25), G-TPP treatment dose-dependently increased paraquat-induced cell death (Fig. 5B). G-TPP also augmented paraquat-induced cell death in 293E cells (Fig. 5B). Interestingly, these two completely different effects of G-TPP in inducing cell death on different cell lines were accompanied by different expression levels of TRAP1 protein (Fig. 5C); those cells sensitive to G-TPP expressed higher levels of TRAP1, but those resistant to G-TPP expressed lower levels. Although we could not understand the underlying molecular mechanisms for this interesting result, we speculated that TRAP1 concentration might be critically related to determining cell fate in ROS-induced cell death.
FIGURE 5.

G-TPP specifically inhibits paraquat-induced cytotoxicity in mammalian cells. A and B, cell viability of paraquat-treated cells with increasing concentrations of G-TPP. 17-AAG was used as a negative control. Cell viability was measured by MTT assay as described under “Experimental Procedures” (n = 3). C, immunoblot analyses of TRAP1 in mammalian cells. Actin (Act) was used as loading control. D, cell viability of paraquat-treated MEF cells infected with control (Con) or TRAP1-specific shRNA (TRAP1i) lentivirus. Cell viability was measured by MTT assay (n = 3). The inset shows immunoblots for TRAP1 protein (TRAP1) in MEF cells. Tubulin (Tub) was used as a loading control. E, propidium iodide (PI) and annexin V FITC (FITC) staining of control (Con), G-TPP-treated (G-TPP), paraquat-treated (PQ), G-TPP and paraquat-treated (PQ G-TPP), and 17-AAG- and paraquat-treated (PQ 17-AAG) MEF cells. i, ii, iii, and iv denote viable, early apoptotic, late apoptotic, and necrotic regions, respectively. F, necrotic cell death rates (n = 3). G, confocal images of JC-1-stained MEF cells. JC-1 exhibits a fluorescence emission shift from green to red, induced by its membrane potential-dependent accumulation in mitochondria (26). H, quantification of relative mitochondrial membrane potentials. The red and green fluorescence ratios of JC-1-stained cells were analyzed using flow cytometry (n = 3). I, measurement of mitochondrial membrane potentials in control (Con) or TRAP1 shRNA expressing (TRAP1i) MEF cells. The red and green fluorescence ratios of JC-1-stained cells were analyzed using flow cytometry (n = 3). J, flow cytometric analysis of CM-H2DCFDA-stained MEF cells. Black line, control cells; red line, CM-H2DCFDA treated cells. K, quantification of relative fluorescence intensities in CM-H2DCFDA flow cytometric analyses (n = 3). L, measurement of relative ROS levels in control (Con) or TRAP1 shRNA expressing (TRAP1i) MEF cells. The fluorescence intensities of CM-H2DCFDA-stained cells were analyzed using flow cytometry (n = 3). Significance was determined by one-way ANOVA with Sidak correction. *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant. Error bars indicate S.D. Scale bar, white, 20 μm.
To understand how TRAP1 functions in cell protection against ROS, we stained MEF cells with propidium iodide and annexin V. The MEF cells treated with paraquat induced propidium iodide uptake only, and G-TPP pretreatment almost completely inhibited it (Fig. 5, E and F), demonstrating that G-TPP prevents paraquat-induced necrotic cell death. We also monitored mitochondrial membrane potential, a reliable parameter for mitochondrial function, using JC-1 fluorescent dye. JC-1 exhibits a fluorescence emission shift from green to red, induced by its membrane potential-dependent accumulation in mitochondria (26). Consequently, a decrease in red/green fluorescence ratio indicates mitochondrial membrane depolarization (26). Control and only G-TPP-treated cells showed strong red florescence (Fig. 5G), which indicates a normal mitochondrial membrane potential (Fig. 5H), whereas paraquat-treated cells developed intense green florescence with weak red signal (Fig. 5G), which indicates a collapse of mitochondrial membrane potential (Fig. 5H). Notably, the paraquat-induced decrease in red/green florescence ratio was strongly suppressed by pretreatment with G-TPP, indicating that G-TPP blocks mitochondrial membrane potential decrease induced by paraquat treatment (Fig. 5, G and H). Moreover, the ROS indicator CM-H2DCFDA clearly showed that the paraquat-induced increase of intracellular ROS level was successfully suppressed by G-TPP (Fig. 5, J and K). In contrast to G-TPP, 17-AAG, a geldanamycin derivative without a mitochondrial targeting module, failed to inhibit the paraquat-induced cell death (Fig. 5, A, E, and F), a decrease in mitochondrial potential (Fig. 5, G and H), and an increase in ROS level (Fig. 5, J and K). In addition, suppression of TRAP1 expression using TRAP1-specific shRNA inhibited the cell death (Fig. 5D), mitochondrial membrane depolarization (Fig. 5I), and ROS generation (Fig. 5L) induced by paraquat treatment at the very similar level as G-TPP did. Furthermore, G-TPP treatment could not significantly enhance this TRAP1 shRNA-induced cell protection (Fig. 5, D, I, and L). These data further confirmed that the G-TPP-induced cell protection resulted from a specific inhibition of TRAP1.
G-TPP Inhibits Paraquat-induced Cytotoxity in DA Neuron Cell Models
We next examined SH-SY5Y cell, a popular DA neuron cell model derived from human neuroblastoma (27) to further test the cell protective role of G-TPP. G-TPP pretreatment successfully protected SH-SY5Y cells from paraquat treatment (Fig. 6A). Further analyses showed that G-TPP inhibited a collapse of mitochondrial membrane potential (Fig. 6B) and intracellular ROS increase (Fig. 6C) induced by paraquat in SH-SY5Y cells. Moreover, G-TPP also suppressed loss of mitochondrial membrane potential (Fig. 6, D and E) and ROS generation (Fig. 6F) in paraquat-treated SN4741 cells, a mouse DA neuronal cell line expressing TH (28). Overall, these data demonstrated that G-TPP can protect various mammalian cells including DA neuron models from the paraquat-induced cytotoxicity. These mammalian results are highly consistent with the protective phenotypes against oxidative stress shown in Drosophila TRAP1 mutants (Fig. 2).
FIGURE 6.

G-TPP protects DA neuron model cells under paraquat-induced stress. Control (Con), G-TPP-treated (G-TPP), paraquat-treated (PQ), G-TPP- and paraquat-treated (PQ G-TPP), and 17-AAG- and paraquat-treated (PQ 17-AAG) DA neuron model cells were analyzed to measure following features. A, cell viability of SH-SY5Y cells. Cell viability was measured by MTT assay (n = 3). The inset shows immunoblots for TRAP1 protein (TRAP1) in SH-SY5Y, SN4741, and MEF cells. Tubulin (Tub) was used as a loading control. B, quantification of relative mitochondrial membrane potentials in SH-SY5Y cells. The red and green fluorescence ratios of JC-1-stained cells were analyzed using flow cytometry (n = 3). C, quantification of relative fluorescence intensities in CM-H2DCFDA-stained SH-SY5Y cells using flow cytometric analyses (n = 3). D, confocal images of JC-1-stained SN4741 cells. E, quantification of relative mitochondrial membrane potentials in SN4741 cells. The red and green fluorescence ratios of JC-1-stained cells were analyzed using flow cytometry (n = 3). F, quantification of relative fluorescence intensities in CM-H2DCFDA-stained SN4741 cells using flow cytometric analyses (n = 3). Significance was determined by one-way ANOVA with Sidak correction. *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant. Error bars indicate S.D. Scale bar, white, 20 μm.
G-TPP Rescues PINK1 Mutant Phenotypes in Drosophila and Mammalian Systems
Based on the recovery of PINK1 null phenotypes by TRAP1 mutations (Figs. 3 and 4), we tested whether G-TPP administration also rescues loss of function defects in PINK1. We raised 1-day-old flies on either normal fly food supplemented with G-TPP (1 mm or 5 mm) or vehicle alone for 3 days and checked their locomotor activity using climbing assay. Wild type controls showed no difference in locomotor activity under G-TPP treatment compared with vehicle alone controls (Fig. 7A), indicating no significant side effect of G-TPP on fly models. In PINK1 null mutants, G-TPP markedly restored locomotor activity in a dose-dependent manner (Fig. 7A). After 30 days of G-TPP administration, we counted the number of DA neurons in DL1 (Fig. 7, B and C) and DM (Fig. 7, D and E) clusters. Consistent with the motor activity results (Fig. 7A), loss of DA neurons in PINK1 null mutants was successfully rescued by G-TPP (Fig. 7, B–E).
FIGURE 7.

G-TPP treatment rescues PINK1 null mutant phenotypes. A, climbing assay of G-TPP-treated wild type (WT) and PINK1 null mutant (B9) flies (n = 4). B, images of the DA neurons within DL1 cluster of the adult brain from 30-day-old flies incubated in food-containing G-TPP at indicated concentrations. DA neurons were stained with anti-TH antibody (green). C, average numbers of the DA neurons in DL1 clusters (n = 30). D, images of the DA neurons within DM cluster of the adult brain from 30-day-old flies incubated in food containing G-TPP at indicated concentrations. DA neurons were stained with anti-TH antibody (green). E, average numbers of the DA neurons in DM clusters (n = 30). F, measurement of mitochondrial membrane potentials in wild type (PINK1+/+) or PINK1 knock-out (PINK1−/−) MEF cells incubated in glucose or galactose medium. G-TPP or 17-AAG was treated at indicated concentrations (n = 3). Significance was determined by one-way ANOVA with Sidak correction. *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant. Error bars indicate S.D. Scale bar, white, 20 μm.
We next treated PINK1 null MEFs incubated in galactose medium with G-TPP. Because cells grown in galactose medium mainly rely on oxidative phosphorylation to generate ATP, PINK1-deficient MEF cells showed a substantial decrease in mitochondrial membrane potential in galactose media, whereas they maintained normal membrane potential in glucose media (Fig. 7F). Strikingly, G-TPP restored the decreased mitochondrial membrane potential in galactose media in a dose-dependent manner, although 17-AAG had no effect in the concentration that G-TPP can induce almost complete recovery (Fig. 7F). These pharmacological data confirmed that TRAP1 inhibition can rescue PINK1 null phenotypes in both fruit fly and MEF, suggesting that G-TPP has the potential to suppress PINK1-linked pathogenesis including PD.
FOXO Mediates TRAP1 Mutation-induced Cell Protective Signals
We previously discovered that FOXO transcription factor complements PINK1 null mutant phenotypes (23). Additionally, Jünger et al. (29) reported that FOXO null mutants are sensitive to oxidative stress. Based on these roles of FOXO in oxidative stress and mitochondria, we investigated the function of FOXO in TRAP1 mutants to uncover the molecular mechanism underlying the cell protection induced by TRAP1 inhibition. Surprisingly, deletion of FOXO gene nullified the increased survival of TRAP1 mutants grown on paraquat- (Fig. 8A) and rotenone-containing media (Fig. 8B). Furthermore, a heterozygous FOXO mutation aggravated the climbing ability rescued by TRAP1 mutation in PINK1 mutants (Fig. 8C). The FOXO heterozygous mutation also inhibited TRAP1 mutation to rescue the decreased ATP level of PINK1 mutants (Fig. 8D). Moreover, FOXO deletion almost completely blocked G-TPP to rescue the locomotor defect in PINK1 null mutants (Fig. 8E). Based on these fly data, we checked the roles of FOXO transcription factors in G-TPP-treated mammalian cells. Consistent with the fly data, G-TPP strongly enhanced the cell viability of paraquat-treated MEF cells (Fig. 8F). However, when we suppressed FOXO1 or FOXO3 expression using siRNA technology, the increased viability of G-TPP-treated MEF cells was significantly down-regulated (Fig. 8F), suggesting that FOXO transcription factors mediate a conserved role in cell protection induced by TRAP1 suppression in both mammalian cells and Drosophila. All of these results consistently support that TRAP1 inhibition suppresses oxidative stress and rescues PINK1 mutant phenotypes in a FOXO-dependent manner.
FIGURE 8.

FOXO mediates cell-protective signals induced by TRAP1 mutation. A, survival curves of wild type control (WT), TRAP1 mutant (TRAP1P), TRAP1 and FOXO double mutant (TRAP1P, FOXO−), and FOXO mutant (FOXO−) male flies on paraquat-containing food (log rank test: p < 0.05, groups with the same letter do not differ significantly). B, survival curves of flies on rotenone-containing food (log rank test: p < 0.001, groups with the same letter do not differ significantly). C, climbing ability test of PINK1 mutants (B9), PINK1 and TRAP1 double mutants (B9, TRAP1P), and PINK1 and TRAP1 double mutants with a heterozygous FOXO mutation (B9, TRAP1P FOXO−/+) (n = 4). D, comparison of the ATP content of thoraces (n = 3). E, comparison of climbing ability between G-TPP-treated PINK1 mutants (B9) and PINK1 and FOXO double mutants (B9, FOXO−) (n = 4). F, FOXO transcription factors mediate the increased cell viability of the paraquat-treated MEF cells by G-TPP treatment. To down-regulate FOXO gene expression, control (Con), FOXO1-specific (FOXO1i), or FOXO3-specific (FOXO3i) siRNA was transfected as described under “Experimental Procedures.” Cell viability was measured by MTT assay (n = 3). The inset shows immunoblots for FOXO1 and FOXO3 protein in MEF cells. Tubulin (Tub) was used as a loading control. G, comparison of Thor mRNA level in wild type (WT), TRAP1 mutants (TRAP1P), and TRAP1 and FOXO double mutants (TRAP1P, FOXO−) (n = 3). H, transactivation activity of FOXO in control (Con) or TRAP1 dsRNA transfected (TRAP1i) S2 cells (n = 3). 8×FK1tkLuc was used as a reporter to quantitatively measure FOXO transcriptional activity (22). I, survival curves of wild types (WT) and TRAP1 mutants (TRAP1P) on paraquat-containing food. Before paraquat treatment, flies were incubated in normal or NAC (5 mg/ml)-containing standard fly food for 3 days (log rank test: p < 0.05, groups with the same letter do not differ significantly). J, comparison of Thor mRNA levels. Flies were incubated in normal or NAC (5 mg/ml)-containing standard fly food for 3 days. Significance was determined by one-way ANOVA with Sidak correction. *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant. Error bars indicate S.D.
FOXO transcription factors induce expression of crucial target genes to regulate important cellular processes. When we checked the mRNA level of Thor, a representative FOXO target gene encoding Drosophila 4E-binding protein (29), in TRAP1 mutants, it was significantly increased (Fig. 8G). In previous studies, ectopic expression of Thor rescued oxidative stress-related phenotypes of FOXO null mutants (30), as well as mitochondrial dysfunction and DA neuron loss phenotypes of PINK1 mutants (23), indicating that FOXO induces Thor gene expression in our TRAP1 mutants. Further supporting this idea, deletion of FOXO in TRAP1 mutants suppressed Thor expression to control levels (Fig. 8G). Moreover, TRAP1 knockdown also increased the transcription activity of FOXO in Drosophila S2 cells (Fig. 8H), suggesting FOXO as a critical mediator of cell-protective signal induced by TRAP1 inhibition.
Owusu-Ansah et al. (31) found that FOXO relays signals from ROS generated by mitochondria to the nucleus, and we observed that mutation of TRAP1 increases the ROS level in Drosophila (Fig. 2, B and C). These data suggest that TRAP1 inhibition can induce FOXO-mediated stress response signaling through ROS. To test this possibility, we grew flies on standard media containing the ROS scavenger N-acetylcysteine (NAC) for 3 days and transferred them to paraquat-containing media for oxidative stress test. This NAC pretreatment had no effect on the survival of wild type flies (Fig. 8I). By contrast, NAC significantly suppressed the enhanced survival induced by TRAP1 mutation (Fig. 8I), demonstrating that oxidative stress resistance induced in TRAP1 mutants is dependent on ROS generated by TRAP1 mutation. The NAC treatment also inhibited the induction of Thor gene expression in TRAP1 mutants (Fig. 8J), suggesting that ROS mediates the FOXO-dependent cell protective signaling in TRAP1 mutants.
Discussion
In this study, we generated and characterized Drosophila TRAP1 mutants. Our TRAP1 mutants successfully developed into adults and showed no significant defects in their life span. Although TRAP1 has been regarded as a mitochondrial protective protein, we did not observe any meaningful defects in mitochondrial morphology, ATP level, and mtDNA content in 3- or 30-day-old TRAP1 mutants (Fig. 1). However, under treatment of free radical inducers, such as rotenone or paraquat, loss of TRAP1 significantly increased survival rate (Fig. 2). Moreover, TRAP1 mutation ameliorated oxidative stress sensitivity, mitochondrial dysfunction, and DA neuronal loss in Drosophila PINK1 null mutants (Figs. 3 and 4). Consistent with these fruit fly data, TRAP1 KO mice showed reduced age-associated tissue degeneration with activated oxidative chain complex activities (18).
These genetic data were fully supported by the following pharmacological analyses. G-TPP inhibited cell death and restored the decreased mitochondrial membrane potential in various paraquat-treated mammalian cells, such as MEFs (Fig. 5) and DA neuron model cell lines (Fig. 6). Moreover, G-TPP treatment ameliorated decreased motor activity and DA neuron degeneration in Drosophila PINK1 null mutants and rescued mitochondrial dysfunction in PINK1 null MEFs (Fig. 7). Overall, our genetic and pharmacological data clearly demonstrated that TRAP1 inhibition can induce resistance against oxidative stress and rescue PINK1 null defects in both Drosophila and mammalian systems.
These results raised important questions: How does TRAP1 suppression induce oxidative stress resistance although it increases ROS levels (Fig. 2)? What is the molecular mechanism underlying the cell protection induced by TRAP1 inhibition? ROS has been regarded as detrimental to many biological processes. However, recent reports showed that ROS can activate beneficial signals especially from mitochondria (32). When Schulz et al. (33) restricted glucose availability in Caenorhabditis elegans, they observed life span extension and oxidative stress resistance accompanied by increased ROS production. Pretreatment of anti-oxidants, such as NAC, inhibited elevated expression of cell protective enzymes in glucose-restricted worms and subsequently blocked the extension of life span and the resistance against oxidative stress. Consistently, in TRAP mutants, NAC treatment suppressed the enhancement in survival on paraquat-containing media (Fig. 8I), suggesting that ROS generated by TRAP1 mutation is not detrimental but beneficial as shown in previous studies (31, 32). Then what makes ROS beneficial? Yang and Hekimi (34) investigated types of ROS from mitochondria in long-lived C. elegans mutants. They observed that mitochondrial superoxide, which was also detected in dihydroethidium staining of TRAP1 mutants (Fig. 2C), was critical to the life span extension induced by several mitochondrial protein mutations. In other analyses, low doses of paraquat, which generate various types of ROS from mitochondria (34), successfully prolonged life span, whereas higher concentrations shortened it (35). These results suggest that certain types or amounts of ROS are critical to its beneficial roles and indicate that TRAP1 down-regulation potentially induces appropriate types or amounts of ROS for cell protection.
In genetic analyses to find a molecular link between TRAP1 inhibition and cell protection, FOXO loss of function nullified the oxidative stress resistance induced by TRAP1 mutation or TRAP1 inhibition (Fig. 8, A and B). Consistently, loss of function mutations of FOXO reaggravated the rescued phenotypes of PINK1 null mutants by TRAP1 mutation or G-TPP treatment and also suppressed TRAP1 mutation-induced gene expression of Thor, a FOXO target gene that has a critical role in mitochondrial protection and oxidative stress resistance (23, 30) (Fig. 8, C–E and G). We also observed that TRAP1 inhibition requires FOXO transcription factors to induce cell protection against oxidative stress in mammalian cells (Fig. 8F). These data consistently demonstrated that FOXO transcription factors mediate cell protection and survival signal induced by TRAP1 inhibition. Moreover, NAC suppressed the enhanced Thor expression and the resistance against oxidative stress in TRAP1 mutants, suggesting that ROS generated by TRAP1-inhibited mitochondria induces FOXO-mediated gene expression to protect cells and animals from oxidative stress and PINK1 mutation (Fig. 8, I and J). Owusu-Ansah et al. (31) also reported that ROS from mitochondria activates nuclear gene expression through FOXO.
We observed that G-TPP successfully protects various mammalian cells, such as NIH 3T3, MEF, SH-SY5Y, and SN4741, from oxidative stress. Contrarily, it also potentiated oxidative stress-induced cell toxicity in HeLa and MCF7 cells that are very sensitive to TRAP1 inhibitors (11, 25) (Fig. 5, A and B). In biochemical analyses, G-TPP caused toxicity in cells with elevated expression of TRAP1, whereas G-TPP protects cells expressing TRAP1 at relatively low levels (Fig. 5C). These results raise a possibility that TRAP1 expression level reflects different cellular contexts such as amounts of stress on mitochondria. In cells under heavy mitochondrial stress, TRAP1 is overexpressed to protect mitochondria. In this case, TRAP1 inhibition by G-TPP treatment abruptly stops mitochondrial protection mechanisms and subsequently induces cell death. However, it is possible that, in cells not mainly dependent on TRAP1-mediated protection, TRAP1 is weakly expressed, and its inhibition can generate weak and beneficial mitochondrial stress to induce cell protective signals. Testing this hypothesis and finding the molecular mechanism underlying the correlation between TRAP1 expression levels and the sensitivity to G-TPP will be our future topics.
In this report, we showed that genetic and pharmacological inhibition of TRAP1 protects cells from oxidative stress and mitochondrial dysfunction. Furthermore, they can generate a compensatory retrograde signal from mitochondria, also known as mitohormesis (32), to up-regulate cell protective gene expression. These unexpected results raise the possibility that TRAP1 inhibitors developed for anti-cancer therapy might be used to treat human pathology induced by mitochondrial disorders, including PD.
Author Contributions
H. K. designed, performed, and analyzed the fly experiments. J. Y. designed, performed, and analyzed the mammalian cell experiments. M. J. K. designed, performed, and analyzed the experiments shown in Fig. 4. S. C., J.-R. C., J.-M. K., and Y. H. Y. provided technical assistance or contributed to the preparation of the figures and text. All authors reviewed the results and approved the final version of the manuscript. H. K. and J. C. designed the study and wrote the paper.
Acknowledgments
We are grateful to Drs. E. Hafen, U. J. Kang, and X. Zhuang for flies and MEFs. We also thank to the members of the Chung laboratory and the Mitochondria Hub Regulation Center for discussions and encouragement.
This work was supported by National Research Foundation of Korea Grants NRF-2012-R1A1A1012482 and NRF-2009-0093188 funded by the Korean government (Ministry of Education, Science, and Technology) (to H. K.) and by the National Creative Research Initiatives grant through the National Research Foundation of Korea funded by Ministry of Science, Information/Communication Technology, and Future Planning (Ministry of Science, ICT, and Future Planning) Grant 2010-0018291 and BK21 Plus Program from Ministry of Education (to J. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- PD
- Parkinson disease
- G-TPP
- gamitrinib-triphenylphosphonium
- MEF
- mouse embryonic fibroblast
- Hsp90
- heat shock protein 90
- DA
- dopaminergic
- TH
- tyrosine hydroxylase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- CM-H2DCFDA
- 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- RV
- revertant
- 17-AAG
- 17-allylamino-demethoxygeldanamycin
- NAC
- N-acetylcysteine
- ROS
- reactive oxygen species
- mtDNA
- mitochondrial DNA
- dsRNA
- double-stranded RNA
- ANOVA
- analysis of variance
- DL1
- dorsolateral clusters 1
- DL2
- dorsolateral clusters 2
- DM
- dorsomedial clusters.
References
- 1. Kroemer G., Galluzzi L., and Brenner C. (2007) Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163 [DOI] [PubMed] [Google Scholar]
- 2. Schapira A. H., Cooper J. M., Dexter D., Clark J. B., Jenner P., and Marsden C. D. (1990) Mitochondrial complex I deficiency in Parkinson's disease. J. Neurochem. 54, 823–827 [DOI] [PubMed] [Google Scholar]
- 3. Langston J. W., Ballard P., Tetrud J. W., and Irwin I. (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 [DOI] [PubMed] [Google Scholar]
- 4. Henchcliffe C., and Beal M. F. (2008) Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 4, 600–609 [DOI] [PubMed] [Google Scholar]
- 5. Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., and Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 6. Park J., Lee S. B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J. M., and Chung J. (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161 [DOI] [PubMed] [Google Scholar]
- 7. Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J. W., Yang L., Beal M. F., Vogel H., and Lu B. (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U.S.A. 103, 10793–10798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koh H., and Chung J. (2012) PINK1 as a molecular checkpoint in the maintenance of mitochondrial function and integrity. Mol. Cells 34, 7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 10. Fulda S., Galluzzi L., and Kroemer G. (2010) Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, 447–464 [DOI] [PubMed] [Google Scholar]
- 11. Kang B. H., Plescia J., Dohi T., Rosa J., Doxsey S. J., and Altieri D. C. (2007) Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 131, 257–270 [DOI] [PubMed] [Google Scholar]
- 12. Song H. Y., Dunbar J. D., Zhang Y. X., Guo D., and Donner D. B. (1995) Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J. Biol. Chem. 270, 3574–3581 [PubMed] [Google Scholar]
- 13. Felts S. J., Owen B. A., Nguyen P., Trepel J., Donner D. B., and Toft D. O. (2000) The Hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 275, 3305–3312 [DOI] [PubMed] [Google Scholar]
- 14. Cechetto J. D., and Gupta R. S. (2000) Immunoelectron microscopy provides evidence that tumor necrosis factor receptor-associated protein 1 (TRAP-1) is a mitochondrial protein which also localizes at specific extramitochondrial sites. Exp. Cell Res. 260, 30–39 [DOI] [PubMed] [Google Scholar]
- 15. Pridgeon J. W., Olzmann J. A., Chin L. S., and Li L. (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 5, e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sciacovelli M., Guzzo G., Morello V., Frezza C., Zheng L., Nannini N., Calabrese F., Laudiero G., Esposito F., Landriscina M., Defilippi P., Bernardi P., and Rasola A. (2013) The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 17, 988–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoshida S., Tsutsumi S., Muhlebach G., Sourbier C., Lee M. J., Lee S., Vartholomaiou E., Tatokoro M., Beebe K., Miyajima N., Mohney R. P., Chen Y., Hasumi H., Xu W., Fukushima H., Nakamura K., Koga F., Kihara K., Trepel J., Picard D., and Neckers L. (2013) Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. U.S.A. 110, E1604–E1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lisanti S., Tavecchio M., Chae Y. C., Liu Q., Brice A. K., Thakur M. L., Languino L. R., and Altieri D. C. (2014) Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Reports 8, 671–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee Y. N., Shim Y. J., Kang B. H., Park J. J., and Min B. H. (2012) Over-expression of human clusterin increases stress resistance and extends lifespan in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 420, 851–856 [DOI] [PubMed] [Google Scholar]
- 20. Mortiboys H., Thomas K. J., Koopman W. J., Klaffke S., Abou-Sleiman P., Olpin S., Wood N. W., Willems P. H., Smeitink J. A., Cookson M. R., and Bandmann O. (2008) Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 64, 555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee J. H., Koh H., Kim M., Kim Y., Lee S. Y., Karess R. E., Lee S. H., Shong M., Kim J. M., Kim J., and Chung J. (2007) Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 447, 1017–1020 [DOI] [PubMed] [Google Scholar]
- 22. Biggs W. H. 3rd, Meisenhelder J., Hunter T., Cavenee W. K., and Arden K. C. (1999) Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. U.S.A. 96, 7421–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koh H., Kim H., Kim M. J., Park J., Lee H. J., and Chung J. (2012) Silent information regulator 2 (Sir2) and Forkhead box O (FOXO) complement mitochondrial dysfunction and dopaminergic neuron loss in Drosophila PTEN-induced kinase 1 (PINK1) null mutant. J. Biol. Chem. 287, 12750–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kang B. H. (2012) TRAP1 regulation of mitochondrial life or death decision in cancer cells and mitochondria-targeted TRAP1 inhibitors. BMB Reports 45, 1–6 [DOI] [PubMed] [Google Scholar]
- 25. Kang B. H., Plescia J., Song H. Y., Meli M., Colombo G., Beebe K., Scroggins B., Neckers L., and Altieri D. C. (2009) Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Invest. 119, 454–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reers M., Smith T. W., and Chen L. B. (1991) J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 30, 4480–4486 [DOI] [PubMed] [Google Scholar]
- 27. Biedler J. L., Roffler-Tarlov S., Schachner M., and Freedman L. S. (1978) Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 38, 3751–3757 [PubMed] [Google Scholar]
- 28. Son J. H., Chun H. S., Joh T. H., Cho S., Conti B., and Lee J. W. (1999) Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J. Neurosci. 19, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jünger M. A., Rintelen F., Stocker H., Wasserman J. D., Végh M., Radimerski T., Greenberg M. E., and Hafen E. (2003) The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J. Biol. 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tettweiler G., Miron M., Jenkins M., Sonenberg N., and Lasko P. F. (2005) Starvation and oxidative stress resistance in Drosophila are mediated through the eIF4E-binding protein, d4E-BP. Genes Dev. 19, 1840–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Owusu-Ansah E., Yavari A., Mandal S., and Banerjee U. (2008) Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 40, 356–361 [DOI] [PubMed] [Google Scholar]
- 32. Yun J., and Finkel T. (2014) Mitohormesis. Cell Metab. 19, 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schulz T. J., Zarse K., Voigt A., Urban N., Birringer M., and Ristow M. (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293 [DOI] [PubMed] [Google Scholar]
- 34. Yang W., and Hekimi S. (2010) A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 8, e1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee S. J., Hwang A. B., and Kenyon C. (2010) Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 20, 2131–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]