

Abstract
PaaI thioesterases are members of the TE13 thioesterase family that catalyze the hydrolysis of thioester bonds between coenzyme A and phenylacetyl-CoA. In this study we characterize the PaaI thioesterase from Streptococcus pneumoniae (SpPaaI), including structural analysis based on crystal diffraction data to 1.8-Å resolution, to reveal two double hotdog domains arranged in a back to back configuration. Consistent with the crystallography data, both size exclusion chromatography and small angle x-ray scattering data support a tetrameric arrangement of thioesterase domains in solution. Assessment of SpPaaI activity against a range of acyl-CoA substrates showed activity for both phenylacetyl-CoA and medium-chain fatty-acyl CoA substrates. Mutagenesis of putative active site residues reveals Asn37, Asp52, and Thr68 are important for catalysis, and size exclusion chromatography analysis and x-ray crystallography confirm that these mutants retain the same tertiary and quaternary structures, establishing that the reduced activity is not a result of structural perturbations. Interestingly, the structure of SpPaaI in the presence of CoA provides a structural basis for the observed substrate specificity, accommodating a 10-carbon fatty acid chain, and a large conformational change of up to 38 Å in the N terminus, and a loop region involving Tyr38-Tyr39. This is the first time PaaI thioesterases have displayed a dual specificity for medium-chain acyl-CoAs substrates and phenylacetyl-CoA substrates, and we provide a structural basis for this specificity, highlighting a novel induced fit mechanism that is likely to be conserved within members of this enzyme family.
Keywords: crystal structure, protein crystallization, protein purification, small-angle x-ray scattering (SAXS), x-ray crystallography, phenyl acetic acid, Streptococcus pneumoniae, thioesterase, fatty acyl-CoA
Introduction
Thioesterases (TEs)2 are a superfamily of enzymes responsible for the catalysis of thioester bonds between carbonyl and thiol groups. Due to their wide range of cellular functions, diverse substrate specificity, and structural differences, they have been classified into 23 families (1). TE families 1–13 hydrolyze thioester bonds between acyl moieties and CoA, TEs 14–19 exhibit preference to acyl groups linked to ACP, TEs 20–21 cleave thioester bonds between acyl groups and non-ACP proteins, and TEs 22–23 hydrolyze the glutathione and its derivatives from acyl groups. There are 5-folds within the TE superfamily, NagB, α/β-hydrolase, flavodoxin-like, hotdog, and lactamase, with the hotdog and α/β-hydrolase folds most prevalent (1).
The thioesterase characterized in this study is a member of the TE13 family. To date, members of this family include PaaI and PaaD, and have been structurally resolved from Thermus thermophilus (Protein Data Bank (PDB) 1J1Y) (2) and Escherichia coli (PDB 2FS2) (3). Biochemical and structural characterization of PaaI from these organisms has established that the enzymes form a tetrameric hotdog fold, with catalytic specificity toward the hydrolysis of phenylacetyl-CoA (3). The arrangement of the hotdog folds in these enzymes has been described as a back to back configuration, such that the double hotdog domain are arranged with the β-sheets “back to back” or facing inwards, and the central α-helices face the exterior. The active sites of double hotdog domains are commonly located at the cleft between two subunits, and there are generally two symmetry related active sites related within each double hotdog domain.
Regulation within this enzyme family has been characterized, including a structural basis involving a ligand-induced fit mechanism and intersubunit cooperativity. Interestingly, a half-of-sites cooperativity was described for the PaaI, and this appears to be somewhat conserved in other TE thioesterase families, including in eukaryotes, where hotdog domains that have fused through gene duplication, utilize only half of the potential active sites through mutation (4–9).
Because most of our current understanding within this class of enzyme is based on structural elucidations from two organisms, we have determined the structure and functional aspects of the PaaI thioesterase from Streptococcus pneumoniae, which possess 35 and 27% sequence identity to the reported E. coli and T. thermophilus enzymes, respectively. The tertiary structure is highly comparable with other PaaI thioesterase, and consistent with this, activity is exhibited for phenylacetyl-CoA. We also report the first small angle x-ray scattering assessment of the quaternary structure, providing evidence that the quaternary structure observed in solution is consistent with that in the crystal structure. Interestingly, higher activity was observed for medium-chain fatty acyl-CoA substrates, and this is the first report of a PaaI having specificity for these substrates. We provide a structural basis for this specificity, and moreover, the determination of the structures in the presence of CoA reveal a novel induced mechanism involving a large conformation switch in the N-terminal region to provide accessibility to the substrate binding site.
Experimental Procedures
Cloning and Mutagenesis
The SpPaaI encoding gene SP_1851 was amplified from genomic DNA of S. pneumoniae by PCR using specific primers; forward primer, TACTTCCAATCCAATGCCATGAAAGACTTTCATTTTGACGCTATATCTG and reverse primer TTATCCACTTCCAATGTTATTATATCCTTACCCGTCTTTCCTCAG, where the bold nucleotides are required for the ligation independent cloning procedure (10), and the underlined nucleotides are complementary to the gene sequence. The purified PCR product, cloned into the pMCSG-21expression vector encodes a His6 tag fusion at the N terminus of the protein and tobacco etch virus cleavage site for removal of the affinity tag. Three mutants of active site residues SpPaaI-N37A, SpPaaI-D52A, and SpPaaI-T68A were generated using specific primers and the QuikChange® II Site-directed mutagenesis kit (Stratagene).
Protein Expression and Purification
The fidelity of all recombinant clones was confirmed by PCR and DNA sequencing. Plasmids were transformed into the E. coli BL21(DE3) pLysS cell strains, and a single colony was used to inoculate 5 ml of Luria-Bertani broth containing 100 μg/ml of spectinomycin antibiotic. Cultures were grown overnight at 37 °C, 220 rpm, and used to inoculate 2 × 500-ml baffled flasks containing autoinduction media and spectinomycin to a final concentration of 100 μg/ml (11). Cultures were grown at 25 °C, 90 rpm, for 24 h, then harvested by centrifugation at 6,000 rpm for 30 min. Cell pellets were resuspended in His A buffer (100 mm sodium phosphate, pH 8.0, 300 mm sodium chloride, 20 mm imidazole) and stored at −80 °C for up to 1 month. To lyse the cells, three freezing-thawing cycles were performed in the presence of 1 ml of lysozyme (20 mg/ml). The cell debris was cleared by centrifugation at 12,000 rpm and 18 °C for 30 min and the supernatant was filtered through a 0.45-μm syringe filter (Millipore, USA) before applying to a 5-ml HisTrap affinity column (GE Healthcare, USA). Following loading, the column was washed with 10 column volumes of His A buffer to remove nonspecific proteins, and the protein was eluted using a gradient over 5 column volumes to 100% His buffer B (100 mm sodium phosphate, pH 8, 300 mm sodium chloride, 500 mm imidazole). Protein fractions eluted from the affinity column were pooled and treated with 100 μl of tobacco etch virus polymerase (5 mg/ml) overnight and further purified using size exclusion chromatography equilibrated in 50 mm Tris, pH 8.0, 150 mm NaCl. A total of 10 mg of wild-type SpPaaI (SpPaaI-WT) protein, 9 mg of SpPaaI-N37A, 7 mg of SpPaaI-D52A, and 12 mg of SpPaaI-T68A was produced per liter of bacterial culture, and each protein concentrated to 1 ml and stored at −80 °C in 50-μl aliquots.
SAXS Data Collection
Small angle x-ray scattering (SAXS) data were collected at the Australian Synchrotron on the SAXS/WAXS beamline using the Pilatus 1M detector. Data were collected over concentration ranges of 0.3–5 mg/ml. For each sample, 50 μl of sample was drawn through a 1.5-mm quartz capillary and exposed to the x-ray beam. The scattering data were collected from q = 0.009 to 0.541 Å and reduced to remove background buffer and capillary noise/scattering. Detector images for each concentration were averaged using Scatterbrain to generate a number of SAXS data sets for subsequent analysis using ATSAS (version 2.4.3) software (12). PRIMUS (13) was used to subtract background scattering from data files and Guinier fits and P(r) distribution plots were generated using GNOM (14). CRYSOL (12) was used to generate theoretical curves and compare scattering data with crystal structure data (15).
Crystallization and X-ray Diffraction
SpPaaI-WT enzyme and mutants were screened using the hanging drop vapor diffusion method using the commercially available Hampton Research Screens, Crystal Screen, and PEG/Ion. From these screens, 6 conditions produced crystals (Crystal Screen Conditions 6, 9, and PEG/Ion 13, 20, 25, and 38), all of which contain a common precipitant of either PEG 3350 or PEG 4000. Crystals were optimized by varying pH, precipitant concentration, and protein concentration, to yield single diffraction quality crystals. The single diffraction quality crystal of SpPaaI-WT, SpPaaI-CoA, mutants SpPaaI-N37A, SpPaaI-D52A, and SpPaaI-T68A were obtained in conditions composed of 200 mm ammonium tartrate dibasic, 20% PEG 3350, 100 mm Tris, pH 7.5; 200 mm lithium sulfate, 100 mm Tris-HCl, 30% PEG 4000; 200 mm magnesium chloride, PEG 400, 100 mm HEPES, pH 7.5; 200 mm magnesium chloride, 14% PEG 3350, 100 mm Tris, pH 6.0, respectively. Diffraction data were collected at the Australian Synchrotron MX1 and MX2 beamlines, indexed and integrated using iMosflm (16), and scaled in Aimless (17–19). Molecular replacement was performed using Phaser (20) and PDB code 3LLB as a search model (data resulted in one top solution with an initial R-factor, and correlation coefficient of 0.32 and 0.78 for SpPaaI-WT, 0.37 and 0.69 for SpPaaI-CoA, 0.25 and 0.81 for SpPaaI-D52A, and 0.27 and 0.82 for SpPaaI-T68A). The structural refinement and model building was performed using Refmac version 5.7.0029 (21, 22), Phenix (23, 24), and Coot (25).
Activity Assays
Enzyme activity of SpPaaI-WT and mutants were measured spectrophotometrically using a coupled color change reaction with 5,5-dithiobis-(2-nitrobenzoate) as described previously (26–29). Each 100-μl assay volume contained 10 mm 5,5-dithiobis-(2-nitrobenzoate) in 100 mm sodium phosphate buffer, pH 7.5, 1.8 ng of protein, and decanoyl-CoA at various concentrations as specified. Release of CoA at 412 nm was measured over 20 min at 21 °C in a 96-well plate and activity was calculated using the extinction coefficient Σ412 = 13,600 m−1 cm−1 (29). GraphPad Prism (version 6) was used to plot the data and calculate enzyme kinetics.
Results and Discussion
Expression, Purification, and Structure Determination
The recombinant over-expression of SpPaaI-WT in E. coli BL21(DE3) pLysS using the autoinduction procedure (11) produced >10 mg of soluble protein per liter of culture medium, and purification by affinity and size exclusion chromatography resulted in >95% purity of SpPaaI-WT. The enzyme could be concentrated to >25 mg/ml (Fig. 1 and data not shown), and large protein crystals with dimensions of ∼0.3 × 0.3 × 0.05-mm were produced in 200 mm ammonium tartrate dibasic, 20% (w/v) PEG 3350, and 100 mm Tris, pH 7.5 (data not shown). Crystals were cryo-protected in a solution comprised of reservoir solution containing 25% glycerol and flash-cooled prior to data collection to minimize radiation damage during data collection. Crystals diffracted to 1.8 Å (data not shown) (Table 1), and reflections were integrated and scaled in C2221 with cell lengths of a = 46.18 Å, b = 118.44 Å, c = 86.64 Å using iMosflm (16) and Aimless (17–19) (Table 1, SpPaaI-WT; completeness >99%, Rpim 4.6%). The Matthews coefficient suggested that 2 protein monomers were present in the asymmetric unit (corresponding to a solvent content of 39%), and the structure was solved by molecular replacement (using PDB code 3LLB as search model) in Phaser (20). Model rebuilding and refinement in REFMAC (21, 22) produced a final model with Rcryst and Rfree of 21.6 and 25.4%, respectively, and coordinates have been deposited and validated and issued PDB code 4ZRF. Contiguous electron density enabled all residues of the enzyme to be modeled with the exception of the first 4 N-terminal residues, and 9 C-terminal residues. The final structure was comprised of 2 chains of 121 residues (5–125) in each chain, no Ramachandran outliers (30), and good stereochemistry (see Table 1 for the structure determination and refinement parameters of SpPaaI-WT). The r.m.s. deviation between the two chains was 0.3 Å, and due to the high structural similarity, all discussion relating to the monomer will be with reference to chain A.
FIGURE 1.
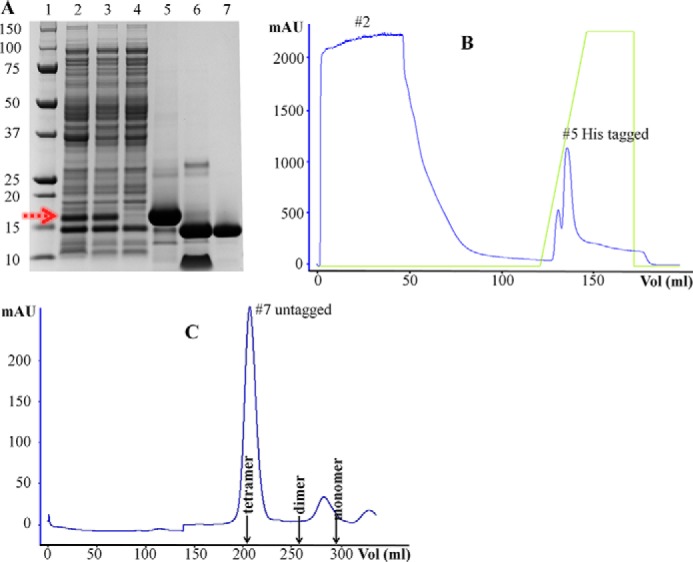
Purification of the SpPaaI by affinity and size exclusion chromatography yielding >95% pure protein. A, SDS-PAGE analysis of samples through the purification process: 1, size marker; 2, whole cell bacterial lysate showing over-expressed SpPaaI (red arrow); 3, soluble supernatant following centrifugation; 4, the flow-through of unbound proteins following loading of the supernatant onto the affinity column; 5, elution of His-tagged SpPaaI protein; 6, His tag removal from SpPaaI by tobacco etch virus protease; 7, SpPaaI following size exclusion chromatography. B, His tag affinity and C, size exclusion chromatography profiles with corresponding lane markers 2, 5, and 7 from A, showing a single, homogenous peak eluting from the size exclusion column at approximately molecular 60 kDa.
TABLE 1.
Data collection and refinement statistics of SpPaaI-WT, SpPaaI-CoA, SpPaaI-D52A, and SpPaaI-T68A
Statistics for the highest-resolution shell are shown in parentheses.
| SpPaaI-WT | SpPaaI-CoA | SpPaaI-D52A | SpPaaI-T68A | |
|---|---|---|---|---|
| Wavelength (Å) | 0.9537 | 0.9537 | 0.9196 | 0.9537 |
| Resolution range (Å) | 24.6–1.8 (1.90–1.80) | 44.5–2.2 (2.26–2.20) | 28.9–1.8 (1.90–1.80) | 25.6–2.0 (2.07–2.00) |
| Space group | C 2 2 21 | P1 | I 1 2 1 | C 2 2 2 |
| Unit cell | 46.3 118.5 86.6 | 50.1 71.8 82.4 82.6 81.4 79.6 | 45.0 45.6 112.4 90 94.7 90 | 46.4 119.2 48.2 |
| Unique reflections | 20,208 (1281) | 54,113 (4480) | 20,838 (2044) | 9,368 (913) |
| Multiplicity | 4.7 | 3.2 | 4.5 | 2.0 |
| Completeness (%) | 99.3 (99.5) | 95.7 (96.3) | 98.1 (98.1) | 99.7 (99.9) |
| Mean I/σ (I) | 7.7 (2.9) | 6.5 (2.3) | 21.6 (12.1) | 13.7 (8.7) |
| Wilson B-factor | 30.4 | 24.25 | 16.8 | 13.9 |
| Rpim | 0.046 (0.146) | 0.087 (0.459) | 0.025 (0.069) | 0.042 (0.071) |
| Model refinement | ||||
| Rwork | 0.218 (0.266) | 0.205 (0.237) | 0.185 (0.193) | 0.182 (0.197) |
| Rfree | 0.254 (0.315) | 0.236 (0.285) | 0.225 (0.281) | 0.220 (0.229) |
| Refined model | ||||
| Atoms | 1852 | 7703 | 1846 | 938 |
| Water molecules | 193 | 429 | 183 | 135 |
| No. of molecules in a.u. | 2 | 8 | 2 | 1 |
| Protein residues in absorbance units | 242 | 1001 | 242 | 122 |
| R.m.s. deviation | ||||
| R.m.s. (bonds) | 0.008 | 0.006 | 0.007 | 0.008 |
| R.m.s. (angles) | 1.05 | 1.00 | 1.02 | 0.97 |
| Ramachandran plot | ||||
| Ramachandran favored (%) | 98 | 98 | 97 | 98 |
| Ramachandran allowed (%) | 2 | 2 | 3 | 2 |
| Ramachandran outliers (%) | 0 | 0 | 0 | 0 |
| Average B factor (Å2) | 22.1 | 32.0 | 21.7 | 17.3 |
| Macromolecules | 21.4 | 31.6 | 20.8 | 16.2 |
| Solvent | 28.4 | 36.4 | 30.7 | 24.9 |
| PDB code | 4ZRF | 4ZRB | 4XY5 | 4XY6 |
The SpPaaI monomer contained a six-stranded anti-parallel β-sheet that half encloses a central α-helix (Fig. 2A). The overall topology of the secondary structure elements, β-β-α-β-β-β-β fits the classic “hotdog” fold, with the α-helix representing the “sausage” and the anti-parallel β-sheet representing the “bun.” The length of the β-strands vary between 6 to 13 residues and are comprised of Glu16-Arg21, His24-Lys30, Asp65-Tyr75, Val84-Gln94, Thr98-Thr106, and Asn112-Gly124.
FIGURE 2.

Structure of SpPaaI. A, monomer of SpPaaI comprised a 6-stranded anti-parallel β sheet that cradles a central α-helix; B, two SpPaaI monomers in the asymmetric unit of the crystal; and C, quaternary structure and biological unit comprising a dimer of double hotdog domains orientated back to back with respect to the central α-helices. The salt bridge interactions within the interfaces are shown for D, interface I involving interactions between His43 and Asp52 within the double hotdog dimer (chain A:B and C:D); and E, interface II salt bridge interactions between Asp102 and Lys115 within the biological tetramer (chains A:D and chains B:C).
All possible quaternary structures were assessed by characterizing the interfaces both within the asymmetric unit, and possible oligomers formed by symmetry operations. The most probable biological quaternary structure was a tetramer of SpPaaI monomers, with D2 symmetry, formed by a dimer of the dimers present in the asymmetric unit (see below for confirmation of the quaternary structure) (Fig. 2C). Two types of intermolecular interfaces dominate within the tetramer, interface I comprising contacts that mediate interactions between the double hotdog domain (see Fig. 2C, chains A:B and C:D), and interface II, the binding region between two double hotdog domains associating in a back to back conformation (Fig. 2C, chains A:D and B:C). The contacts that form the double hotdog domain at interface I include an extensive array of hydrogen bonds and two salt bridges as summarized in Table 2, and comprise a buried interface area of 1,329 Å2 (or ∼20% total surface area). The two salt bridge interactions are formed between residues His43 and Asp52 and the corresponding symmetry related interaction (Fig. 2D). Interactions at interface II are much weaker, with a total interface area of only 430 Å2 (6% total surface area), mediated through six hydrogen bonds (Table 2) and four salt-bridge interactions, the latter interactions involving Lys115 and Asp102 (Fig. 2E). A full description of the interactions is summarized in Table 2.
TABLE 2.
SpPaaI interactions present within interface I and interface II
| H-bonds | Chain | Distance | Chain |
|---|---|---|---|
| Å | |||
| Interface I | |||
| 1 | A: His5 (ND1) | 3.17 | B: Tyr15 (OH) |
| 2 | A: Phe6 (N) | 3.21 | B: Tyr15 (OH) |
| 3 | A: Ile9 (N) | 2.99 | B: Ala11 (O) |
| 4 | A: Ser10 (OG) | 3.48 | B: Ile9 (O) |
| 5 | A: Ser10 (OG) | 3.67 | B: Ser 10 (OG) |
| 6 | A: Ala11 (N) | 2.73 | B: Ile9 (O) |
| 7 | A: Tyr15 (OH) | 2.88 | B: Phe6 (O) |
| 8 | A: Tyr39 (OH) | 3.49 | B: Asp65 (OD1) |
| 9 | A: Gly45 (N) | 3.63 | B: Asp52 (OD2) |
| 10 | A: Gln53 (NE2) | 2.75 | B: Tyr38 (OH) |
| 11 | A: Gln70 (NE2) | 3.37 | B: Ser72 (OG) |
| 12 | A: Ser71 (N) | 2.93 | B: Ile73 (O) |
| 13 | A: Ser72 (OG) | 2.95 | B: Ser72 (OG) |
| 14 | A: Ile73 (N) | 2.88 | B: Ser71 (O) |
| 15 | A: Tyr75 (N) | 3.1 | B: Leu69 (O) |
| 16 | A: Tyr15 (OH) | 3.17 | B: His5 (ND1) |
| 17 | A: Tyr15 (OH) | 3.21 | B: Phe6 (N) |
| 18 | A: Ala11 (O) | 2.99 | B: Ile9 (N) |
| 19 | A: Ile9 (O) | 3.48 | B: Ser10 (OG) |
| 20 | A: Ile9 (O) | 2.73 | B: Ala11 (N) |
| 21 | A: Phe6 (O) | 2.88 | B: Tyr15 (OH) |
| 22 | A: Asp65 (OD1) | 3.49 | B: Tyr39 (OH) |
| 23 | A: Asp52 (OD2) | 3.63 | B: Gly45 (N) |
| 24 | A: Tyr38 (OH) | 2.75 | B: GLN53 (NE2) |
| 25 | A: Ser72 (OG) | 3.37 | B: GLN70 (NE2) |
| 26 | A: Ile73 (O) | 2.93 | B: Ser71 (N) |
| 27 | A: Ser71 (O) | 2.88 | B: Ile73 (N) |
| 28 | A: Leu69 (O) | 3.1 | B: Tyr75 (N) |
| Salt bridges | |||
| 1 | A: His43 (NE2) | 3.53 | B: Asp52 (OD2) |
| 2 | A: Asp52 (OD2) | 3.53 | B: His43 (NE2) |
| Interface II | |||
| 1 | A: Ser72 (OG) | 2.5 | D: Ser72 (OG) |
| 2 | A: Thr119 (OG1) | 3.07 | D: Asn74 (OD1) |
| 3 | A: Lys115 (NZ) | 2.66 | D: Asp102 (OD1) |
| 4 | B: Lys115 (NZ) | 3.77 | C: Thr117 (OG1) |
| 5 | B: Asn74 (OD1) | 2.8 | C: Thr119 (OG1) |
| 6 | B: Asp102 (OD2) | 3.18 | C: Lys115 (NZ |
| Salt bridges | |||
| 1 | A: Lys115 (NZ) | 2.66 | D: Asp102 (OD1) |
| 2 | A: Lys115 (NZ) | 2.97 | D: Asp102 (OD2) |
| 3 | B: Asp102 (OD1) | 3.84 | C: Lys115 (NZ) |
| 4 | B: Asp102 (OD2) | 3.18 | C: Lys115 (NZ) |
Confirmation of Quaternary Structure
Although the interactions of interface II appear rather weak, the proposed tetrameric arrangement of hotdog domains observed in the crystal is supported by both experimental data obtained in solution, including small angle x-ray scattering and analytical size exclusion chromatography, as well as other crystal forms obtained for the mutant SpPaaI (see below). Confirming that the overall quaternary structure is not concentration dependent, the SAXS data showed similar scattering profiles over a range of concentrations (0.3–5 mg/ml) (Fig. 3) (see also Table 3 for overview of SAXS data). Comparison of the theoretical scattering profiles of different possible oligomeric states with the experimental SAXS scattering profile clearly establish that the best fit was achieved with a back to back tetramer arrangement. This experimentally determined tetrameric quaternary structure of SpPaaI is likely be conserved in other PaaI structures, with two deposited structures (PDB codes 1J1Y and 2FS2) showing similar quaternary structures (see Fig. 4A; quaternary structures of SpPaaI, E. coli-PaaI (PDB 2FS2), T. thermophilus-PaaI (PDB code 1J1Y)).
FIGURE 3.

SAXS data supporting the quaternary arrangement of hotdog domains observed from the crystallographic data. A, scattering profiles of SpPaaI at concentrations from 0.3 to 5 mg/ml from PRIMUS. B, SpPaaI scattering data (red dots) superimposed on the theoretical scattering curves (blue line) for the SpPaaI monomer, dimer, arrangement in the asymmetric unit, and the proposed tetramer using CRYSOL.
TABLE 3.
Data collection and scattering-derived parameters for SpPaaI SAXS data
| SpPaaI | |
|---|---|
| Data collection parameters | |
| Instrument | Pilatus 1 m |
| Beam geometry | 250 × 150 μm |
| Wavelength (Å) | 1.54 |
| q range (Å−1) | 0.01–0.5 |
| Exposure time (s) | 21.0 |
| Concentration range (mg ml−1) | 0.3 - 5 |
| Temperature (C) | 26 |
| Structural parameters | |
| I (0) (cm−1) (from P (r)) | 0.03 |
| Rg (Å) (from P (r)) | 24.83 |
| I (0) (cm−1) (from Guinier) | 0.03 |
| Rg (Å) (from Guinier) | 24.93 |
| Dmax (Å) | 67 |
| Porod volume estimate (Å3) | 89, 294 |
| Dry volume calculated from sequence (Å3) | 18, 413 |
| Molecular mass determination | |
| Partial specific volume (cm3 g−1) | 0.75 |
| Molecular mass (Da) (from I (0)) | 63,000 |
| Calculated monomeric molecular mass from sequence (Da) | 60,000 |
FIGURE 4.
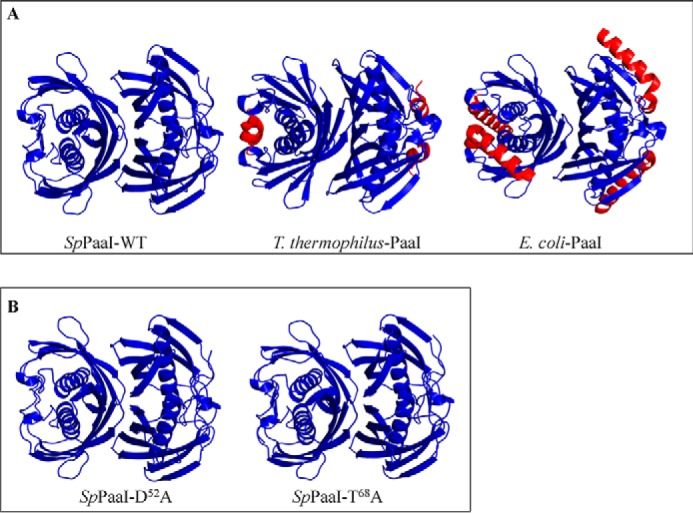
Structures of: A, SpPaaI-WT, T. thermophilus-PaaI, and E. coli-PaaI showing similar tertiary and quaternary arrangements (blue color) and minor secondary structural differences (red color); B, SpPaaI-D52A and SpPaaI-T68A mutant enzymes solved in different space groups demonstrating high structural conservation in both tertiary and quaternary structures.
Substrate Specificity, Specific Activity, and Active Site Determination
Because the substrate specificity of SpPaaI has yet to be determined, we assessed the activity for a range of thioester-CoA substrates (4, 29). We found that whereas SpPaaI-WT displayed a greater activity for phenylacetyl (PA)-CoA than very short and long chain saturated fatty acyl-CoA substrates, the greatest activity was observed for medium-chain fatty acyl-CoA substrates (Fig. 5A). This was confirmed through enzyme kinetic comparisons of SpPaaI against decanoyl (C10)-CoA and PA-CoA, revealing a kcat of 6.5 s−1 and specificity constant of 7.2 × 104 (m−1 s−1) for PA-CoA, and respective values of 32.8 s−1 and 1.8 × 105 (m−1 s−1) for C10-CoA (Fig. 5, B and C, Table 4). Interestingly, the enzyme kinetic data presented evidence of a multistate model rather than a simple Michaelis-Menten kinetic model. Accordingly, the data best fitted a nonlinear allosteric sigmoidal curve, exhibiting a Hill coefficient (H) of two, for both C10-CoA and PA-CoA substrates (Fig. 5, B and C, Table 4). This is consistent with the structural data (described below) revealing that half the active sites of SpPaaI, when crystallized in the presence of PA-CoA, were occupied by the reaction product, CoA. Despite a relatively high degree of structural homology within hotdog containing thioesterases, a diverse range of active site configurations has been reported (see Table 5). To better understand both the active site configuration within the SpPaaI thioesterase and establish a more general understanding of active sites within thioesterases, we set out to characterize the active site through mutagenesis of putative active site residues. Examination of the active site region determined from the location of the terminal S atom within CoA, cocrystallized with a number of structures (described below), revealed three putative active site residues. These were Asn37 (positioned in the middle of the loop between β2 and the central α helix), Asp52 (located on the middle of the central α helix), and Thr68 (positioned within β3, Fig. 6). To investigate the role of these residues in catalysis, we generated N37A, D52A, and T68A mutants and tested for activity against C10-CoA and PA-CoA. The activity of both N37A and D52A mutants was completely abolished in both substrates, whereas the activity of T68A showed a marked reduction (Fig. 5, B and C). This active site is most similar to that described for human thioesterase superfamily member 2 (hTHEM2: PDB code 3F5O, see Table 5), suggesting that this is a highly conserved active site from prokaryotes to humans (5, 32, 33).
FIGURE 5.

Activity of SpPaaI and SpPaaI mutants against acyl-CoA substrates. A, screening of substrates (330 μm of each substrate per reaction, acetyl-CoA (C2-CoA), malonyl-CoA (C3-CoA), butyryl-CoA (C4-CoA), hexanoyl-CoA (C6-CoA), octanoyl-CoA (C8-CoA), decanoyl-CoA (C10-CoA), lauroyl-CoA (C12-CoA), myristoyl CoA (C14-CoA), palmitoyl-CoA (C16-CoA), stearoyl-CoA (C18-CoA), and arachidonoyl-CoA (C20-CoA)) against SpPaaI-WT. B, specific activity of SpPaaI-WT and mutants for decanoyl-CoA. C, specific activity comparison of SpPaaI-WT and SpPaaI mutants for phenylacetyl-CoA (PA-CoA).
TABLE 4.
Kinetic parameters of SpPaaI-WT and mutants
| Enzyme | Km | Kcat | Kcat/Km | Hill coefficient |
|---|---|---|---|---|
| μm | s−1 | m−1 s−1 | ||
| Decanoyl-CoA | ||||
| SpPaaI-WT | 183 ± 7 | 32.8 ± 1.1 | 1.8 ± 105 | 2.2 |
| SpPaaI-N37A | NDa | ND | ND | ND |
| SpPaaI-D52A | ND | ND | ND | ND |
| SpPaaI-T68A | ND | ND | ND | ND |
| Phenylacetyl-CoA | ||||
| SpPaaI-WT | 90 ± 5.2 | 6.5 ± 0.3 | 7.2 × 104 | 2.2 |
| SpPaaI-N37A | ND | ND | ND | ND |
| SpPaaI-D52A | ND | ND | ND | ND |
| SpPaaI-T68A | ND | ND | ND | ND |
a ND, not determined due to low activity.
TABLE 5.
Variation among active site residues of closely related thioesterases to SpPaaI
| R.m.s. deviation with SpPaaI | PDB | TE subfamily | Active site residues |
|---|---|---|---|
| 0a | 4I82-A | 13 | Asn37, Asp52, Thr68 |
| 1 | 3F5O-A | 8 | Asn50, Asp65, Ser83, Gly57 |
| 1.2 | 2DSL-A | 13 | Gly40, Asp48 |
| 1.6 | 3BJK-A | 6 | Asp44 |
| 1.6 | 3D6L-A | 6 | Asp33 |
| 1.7 | 2PZH-A | 9 | Tyr7, Asp11, His18 |
| 1.9 | 3B6E-A | 11 | Gly55, Glu63 |
a This study.
FIGURE 6.

Structural alignment of SpPaaI. A, primary and secondary structure alignment of SpPaaI using the ENDscript/ESPript web server: helices are represented by α, strands by β, and turns with T. The predicated active site amino acid residues are indicated by green triangles and blue boxes. Two highly conserved Gly residues that precede the central helix are highlighted in red. B, active site residues of SpPaaI are contributed from residues on both chains of the dimer: Asn37 of chain A and Asp52 and Thr68 of chain B in dimer AB are shown.
The Structures of SpPaaI-D52A and SpPaaI-T68A
To confirm that the loss of activity observed in the mutants were due to loss of active site residues rather than loss of structure, as well as to confirm the tertiary and quaternary structures in different space groups, we solved the structures of two of the mutant proteins in alternate crystal forms, and confirmed that all three enzymes form the same tetrameric association by size exclusion chromatography (data not shown). SpPaaI-D52A and SpPaaI-T68A mutants were crystallized in I121 and C222 space groups, and contained 2 and 1 molecules in the asymmetric unit, respectively. The refined structure of the SpPaaI-WT was used for molecular replacement. Refinement to an R/Rfree (%) of 18.52/22.54 and 18.21/22.07, respectively, revealed clear density, including those residues containing mutations. The structures were highly similar to the wild-type enzyme, and comparison of the crystallographic tetramers in the three crystal structures revealed r.m.s. deviations values of 0.38 Å between SpPaaI-WT and SpPaaI-D52A and 0.48 Å between SpPaaI-WT and SpPaaI-T68A (see Table 1 for the structure determination and refinement parameters of SpPaaI-WT and mutants, confirming similar tertiary quaternary structures (Fig. 4)). Similarly, the positioning of the active site residues within the mutant enzymes were structurally conserved and highly similar to that observed within SpPaaI-WT (data not shown).
SpPaaI:CoA-bound Structure Reveals a Novel Induced Fit Conformational Change
To assess whether the structural elucidation of SpPaaI could provide insights into the substrate specificity determined in the activity assays, modeling of CoA-bound ligands from superimposed structures (residues 5–123 of PDB code 4ZRF and residues 2–139 of PDB code 3F5O) was performed, but revealed clashes in the adenosine moiety of CoA, and a large occlusion in the substrate binding pocket. We therefore set out to determine the structure of SpPaaI in the presence of both PA-CoA and C10-CoA. Crystals were obtained in the presence of both substrates, however, only conditions containing PA-CoA led to a complete data set. The asymmetric unit of the crystal structure contained 8 double hotdog domains, arranged as two tetrameric quaternary configurations as determined in the apo and mutant forms (r.m.s. deviations 0.38 and 0.48 Å). Interestingly, CoA, the end product of the reaction, was bound at half the possible binding sites (Fig. 7). A number of other structures have also exhibited a half-of-sites reactivity, whereby only one of two domains/protomers are participating in the given reaction (34–36). There are four proposed mechanisms that result in a half-of-the-sites reactivity. In the first mechanism, the domains/protomers are not identical, for example, isozymes having a single amino acid change in the binding site that alters ligand binding capacity. A second mechanism includes dimerization of identical polypeptide chains in an asymmetric fashion, introducing non-identical conformations of two active sites (37). A third mechanism occurs when the active sites are sufficiently close together, and the binding of one molecule sterically blocks or electrostatically repels the second molecule. Finally, the fourth mechanism includes a ligand-induced conformational change, upon ligand binding, in one of the two identical sites, an induced a conformational change in the other active site (2, 36). In our structure, we observed a large 34-Å conformational change within the first 11 N-terminal residues, in half of the thioesterase domains (Fig. 8). Thus, in each of the two tetramers within the asymmetric unit, half the hotdog domains contained the N-terminal residues positioned in a β-strand conformation, traversing the other hotdog domain in the protomer (similar to all the apo- and mutant forms crystallized across three space groups), whereas within the other half the hotdog domains contained double hotdog protomers where the N-terminal residues are kinked back at a pitch of ∼135° to form an α-helix, which starts at Ala11. Significantly, these two different conformations moderate the spatial position of the loop connecting β-strand 2 with the central α-helix. This displaces the two tyrosine residues, Tyr38-Tyr39, by up to 12 Å from the ligand binding site (Fig. 8 and data not shown) (see Table 6 for a list of interacting residues at these interfaces). Because the crystal structures did not reveal electron density for the fatty acyl molecules, we modeled a complex of SpPaaI bound with decanoyl-CoA (obtained by superimposing PDB 3F05) and phenylacetyl-CoA (obtained by superimposing PDB code 4QD8). This revealed well accommodated binding sites for both a 10-carbon fatty acid chain as well as phenylacetate (data not shown). These binding sites are formed largely from hydrophobic residues contributed from both protomers within the double hotdog fold, including Met1, Phe4, Ile60, Gly66, Val67, Thr68, Phe121, and Thr123 from one protomer, and Asn37, Tyr39, Ala42, Tyr75, Leu76, and Ala78 from protomer 2 binding to decanoyl-CoA; and Asp52, Val67, Thr68, Leu69, and Phe121 from one protomer, and Gly44, Tyr75, Leu76, and Lys77 from protomer 2 binding phenylacetyl-CoA.
FIGURE 7.

Quaternary structure of SpPaaI in the presence of CoA. The CoA ligand was bound within chains A and B, as supported by an Fo − Fc annealed omit map contoured at 3σ (green mesh, left panels), but not in chains C and D (green mesh, right panels).
FIGURE 8.

Conformational changes within the N-terminal and loop regions harboring Tyr38-Tyr39 between the two double hotdog protomers observed in the quaternary structure of SpPaaI bound to CoA. In the left panel, the N-terminal residues form an α-helix (magenta, schematic), and are kinked at 135° compared with the conformation of these residues in the other double hotdog protomer, where these residues bind through a β-strand that traverse across hotdog domains within the protomer (right panels). In the left panel, these conformational changes result in the loop containing Tyr38-Tyr39 to be positioned in a manner that results in an opened substrate binding site, whereas in the right panel, this loop and positioning of the these residues occlude the substrate site.
TABLE 6.
Interactions of N-terminal residues 1–11 present in the CoA (top, chains A: B) and unliganded (bottom, chains C:D) chains of SpPaaI structure
| H-bonds | Chain | Distance | Chain |
|---|---|---|---|
| Å | |||
| CoA bound | |||
| 1 | A: Phe4 (O) | 3.61 | B: Tyr15 (OH) |
| 2 | A: Phe4 (O) | 3.41 | B: Ile17 (CD1) |
| 3 | A: Phe4 (CE2) | 3.41 | B: Leu57 (CD2) |
| 4 | A: His5 (CA) | 3.54 | B: Tyr15 (OH) |
| 5 | A: His5 (ND1) | 3.73 | B: Tyr15 (OH) |
| 6 | A: Phe6 (CE2) | 3.57 | B: Tyr15 (CE1) |
| 7 | A: Phe6 (CZ) | 3.65 | B: Phe12 (CD1) |
| 8 | A: Ala8 (CB) | 3.52 | B: Ala11 (O) |
| 9 | A: Ala8 (CB) | 3.59 | B: Phe12 (O) |
| 10 | A: Ile9 (CD1) | 3.69 | A: His43 (NE2) |
| 11 | A: Ser10 (CB) | 3.79 | A: Ser34 (OG) |
| 12 | A: Ala11 (CB) | 3.63 | A: Thr49 (CG2) |
| Unbound enzyme | |||
| 1 | C: Asp3 (OD1) | 3.11 | D: Tyr39 (OH) |
| 2 | C: Asp3 (CB) | 3.40 | D: Tyr38 (CD2) |
| 3 | C: Phe4 (CE1) | 3.76 | D: Gln53 (NE2) |
| 4 | C: Phe4 (CZ) | 3.65 | D: Leu57 (CD1) |
| 5 | C: Phe6 (CE2) | 3.77 | D: Asp7 (OD1) |
| 6 | C: Asp7 (OD2) | 3.58 | D: Tyr38 (N) |
| 7 | C: Asp7 (OD2) | 3.40 | D: Asn37 (CB) |
| 8 | C: Ala11 (CB) | 3.51 | D: Thr49 (CG2) |
Comparison of SpPaaI with Structurally Related Enzymes
A comparison with other related thioesterases was performed using DALI to assess active site configurations and substrate binding sites between the most structurally related proteins (38). Overall, SpPaaI displayed quite low sequence homology to other thioesterases (6–27% identity) with the exception of a protein from Streptococcus mutans UA159 (PDB code 3LBB; sequence identity 67%), solved by the structural genomics consortium, but remains uncharacterized and unpublished. This enzyme was solved in apo (PDB code 3LBB) and CoA (PDB code 3LBE) bound forms, and revealed similar conformational changes noted in our structures, but with some major exceptions. In the apo structure, all N-terminal residues were positioned in the β-strand configuration, similar to those observed in our apo- and mutant enzymes, however, in the PDB 3LBE deposition, the CoA-bound structure contained CoA bound at all the sites, and all hotdog domains were induced into an active α-helix conformation.
It is therefore likely that these enzymes may exist in different CoA-bound states, which correlate directly with the number of domains that have induced N-terminal conformational changes and exposed substrate binding sites. In this study, we have characterized structural conformations where none of the thioesterase domains are bound by CoA, and correspondingly, these thioesterase domains display N-terminal conformations that solely exist in a β-strand configuration (PDB codes 4ZRF, 4XY5, and 4XY6). We have also determined a structure where half the thioesterase domains are bound by CoA, and therefore only half the thioesterase domains are induced in the β-strand configuration, whereas the other half are in the α-helix conformation (PDB code 4ZRB). We have also identified through homologous structural alignment, a closely related structure where the thioesterase domains are all bound to CoA, and concomitantly, all domains exist in the α-helix configuration (PDB code 3LBE). Interestingly, other thioesterases have been shown to display induced conformational changes, but through different mechanisms. For example, the conformational changes observed in the PaaI structures of T. thermophilus shows CoA exhibits a half-of-sites reactivity through small, rigid body changes in the rearrangement of hotdog subunits. Moreover, it was shown in this system that CoA was bound at two of the four hotdog domains within the biological unit of the crystal structure. Similarly, half-of-sites reactivity has also been noted in other families of thioesterases including those exhibiting a hexameric quaternary structure, but also through different mechanisms (4, 9). Thus, it appears that half-of-sites is conserved in this family of thioesterases, and it is possible that the structure determined in our study represents a transition state along the pathway in becoming fully loaded with CoA. The precise biological role for the half-of-sites reactivity in thioesterase domains remains to be elucidated, however, that it appears to be present in different thioesterase families including prokaryotes and eukaryotes, and that different mechanisms have evolved to maintain these features within thioesterases, is indicative they are important for function. That the active site of nearly all thioesterase members is achieved through a double hotdog association, and yet most thioesterases are reported to exhibit tetrameric and hexameric quaternary structures, may in part be related to this half-of-sites reactivity and a potential mechanism of regulation, although this needs to be confirmed experimentally.
Conclusion
SpPaaI is a tetrameric hot-dog thioesterase, with domains arranged in a back to back configuration. This tetrameric configuration has been confirmed in solution for the first time using SAXS, and activity has been determined for both phenylacetyl-CoA and medium-chain acyl-CoA substrates. Residues Asn37, Asp52, and Thr68 were shown to be crucial for activity, and the structural integrity of these mutants were confirmed by crystallography. CoA occupies only half of the potentially active binding sites, and is associated with a large conformational change in the N terminus of the protein of up to 38 Å. This is associated with conformational changes in a loop region containing Tyr38-Tyr39 that modulates the accessibility of the substrate binding site. The functional role of half-of-sites reactivity remains to be elucidated, however, is conserved among members of the thioesterase family.
Author Contributions
Y. K. participated in the research. All authors participated in writing and editing the final manuscript.
The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 4ZRF, 4ZRB, 4XY5, and 4XY6) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TE
- thioesterase
- PDB
- Protein Data Bank
- SAXS
- small-angle x-ray scattering
- r.m.s.
- root mean square
- PA
- phenylacetyl.
References
- 1. Cantu D. C., Chen Y., Lemons M. L., and Reilly P. J. (2011) ThYme: a database for thioester-active enzymes. Nucleic Acids Res. 39, D342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kunishima N., Asada Y., Sugahara M., Ishijima J., Nodake Y., Sugahara M., Miyano M., Kuramitsu S., Yokoyama S., and Sugahara M. (2005) A novel induced-fit reaction mechanism of asymmetric hotdog thioesterase PAAI. J. Mol. Biol. 352, 212–228 [DOI] [PubMed] [Google Scholar]
- 3. Song F., Zhuang Z., Finci L., Dunaway-Mariano D., Kniewel R., Buglino J. A., Solorzano V., Wu J., and Lima C. D. (2006) Structure, function, and mechanism of the phenylacetate pathway hotdog-fold thioesterase PaaI. J. Biol. Chem. 281, 11028–11038 [DOI] [PubMed] [Google Scholar]
- 4. Swarbrick C. M., Roman N., Cowieson N., Patterson E. I., Nanson J., Siponen M. I., Berglund H., Lehtiö L., and Forwood J. K. (2014) Structural basis for regulation of the human acetyl-CoA thioesterase 12 and interactions with the steroidogenic acute regulatory protein-related lipid transfer (START) domain. J. Biol. Chem. 289, 24263–24274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantu D. C., Ardèvol A., Rovira C., and Reilly P. J. (2014) Molecular mechanism of a hotdog-fold acyl-CoA thioesterase. Chemistry 20, 9045–9051 [DOI] [PubMed] [Google Scholar]
- 6. Rodríguez-Guilbe M., Oyola-Robles D., Schreiter E. R., and Baerga-Ortiz A. (2013) Structure, activity, and substrate selectivity of the Orf6 thioesterase from Photobacterium profundum. J. Biol. Chem. 288, 10841–10848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhuang Z., Latham J., Song F., Zhang W., Trujillo M., and Dunaway-Mariano D. (2012) Investigation of the catalytic mechanism of the hotdog-fold enzyme superfamily Pseudomonas sp. strain CBS3 4-hydroxybenzoyl-CoA thioesterase. Biochemistry 51, 786–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song F., Thoden J. B., Zhuang Z., Latham J., Trujillo M., Holden H. M., and Dunaway-Mariano D. (2012) The catalytic mechanism of the hotdog-fold enzyme superfamily 4-hydroxybenzoyl-CoA thioesterase from Arthrobacter sp. strain SU. Biochemistry 51, 7000–7016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Forwood J. K., Thakur A. S., Guncar G., Marfori M., Mouradov D., Meng W., Robinson J., Huber T., Kellie S., Martin J. L., Hume D. A., and Kobe B. (2007) Structural basis for recruitment of tandem hotdog domains in acyl-CoA thioesterase 7 and its role in inflammation. Proc. Natl. Acad. Sci. U.S.A. 104, 10382–10387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eschenfeldt W. H., Lucy S., Millard C. S., Joachimiak A., and Mark I. D. (2009) A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Studier F. W. (2005) Protein production by auto-induction in high-density shaking cultures. Protein Exp. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 12. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 14. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 15. Svergun D., Barberato C., and Koch M. H. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 16. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 20. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 23. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Wei J., Kang H. W., and Cohen D. E. (2009) Thioesterase superfamily member 2 (Them2)/acyl-CoA thioesterase 13 (Acot13): a homotetrameric hotdog fold thioesterase with selectivity for long-chain fatty acyl-CoAs. Biochem. J. 421, 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tilton G. B., Shockey J. M., and Browse J. (2004) Biochemical and molecular characterization of ACH2, an acyl-CoA thioesterase from Arabidopsis thaliana. J. Biol. Chem. 279, 7487–7494 [DOI] [PubMed] [Google Scholar]
- 28. Hunt M. C., Solaas K., Kase B. F., and Alexson S. E. (2002) Characterization of an acyl-CoA thioesterase that functions as a major regulator of peroxisomal lipid metabolism. J. Biol. Chem. 277, 1128–1138 [DOI] [PubMed] [Google Scholar]
- 29. Yamada J., Furihata T., Tamura H., Watanabe T., and Suga T. (1996) Long-chain acyl-CoA hydrolase from rat brain cytosol: purification, characterization, and immunohistochemical localization. Arch. Biochem. Biophys. 326, 106–114 [DOI] [PubMed] [Google Scholar]
- 30. Ramachandran G. N., Ramakrishnan C., and Sasisekharan V. (1963) Stereochemistry of polypeptides chain configurations. J. Mol. Biol. 7, 95–99 [DOI] [PubMed] [Google Scholar]
- 31. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapidab-initioshape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marfori M., Kobe B., and Forwood J. K. (2011) Ligand-induced conformational changes within a hexameric acyl-CoA thioesterase. J. Biol. Chem. 286, 35643–35649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Willis M. A., Zhuang Z., Song F., Howard A., Dunaway-Mariano D., and Herzberg O. (2008) Structure of YciA from Haemophilus influenzae (HI0827), a hexameric broad specificity acyl-coenzyme A thioesterase. Biochemistry 47, 2797–2805 [DOI] [PubMed] [Google Scholar]
- 34. Koshland D. E., Jr. (1996) The structural basis of negative cooperativity: receptors and enzymes. Curr. Opin. Struct. Biol. 6, 757–761 [DOI] [PubMed] [Google Scholar]
- 35. Levitzki A., Stallcup W. B., and Koshland D. E. (1971) Half-of-the-sites reactivity and conformational states of cytidine triphosphate synthetase. Biochemistry 10, 3371–3378 [DOI] [PubMed] [Google Scholar]
- 36. Stallcup W. B., and Koshland D. E. Jr. (1973) Half-of-the sites reactivity and negative co-operativity: the case of yeast glyceraldehyde 3-phosphate dehydrogenase. J. Mol. Biol. 80, 41–62 [DOI] [PubMed] [Google Scholar]
- 37. Adams M. J., Blundell T. L., Dodson E. J., Dodson G. G., Vijayan M., Baker E. N., Harding M. M., Hodgkin D. C., Rimmer B., and Sheat S. (1969) Structure of rhombohedral 2 zinc insulin crystals. Nature 224, 491–495 [Google Scholar]
- 38. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]