

Abstract
Variation in PPP3CC, the gene that encodes the γ isoform of the calcineurin catalytic subunit, has been reported to be associated with schizophrenia. Because of its low expression level in most tissues, there has been little research devoted to the specific function of the calcineurin Aγ (CNAγ) versus the calcineurin Aα (CNAα) and calcineurin Aβ (CNAβ) catalytic isoforms. Consequently, we have a limited understanding of the role of altered CNAγ function in psychiatric disease. In this study, we demonstrate that CNAγ is present in the rodent and human brain and dephosphorylates a presynaptic substrate of calcineurin. Through a combination of immunocytochemistry and immuno-EM, we further show that CNAγ is localized to presynaptic terminals in hippocampal neurons. Critically, we demonstrate that RNAi-mediated knockdown of CNAγ leads to a disruption of synaptic vesicle cycling in cultured rat hippocampal neurons. These data indicate that CNAγ regulates a critical aspect of synaptic vesicle cycling and suggest that variation in PPP3CC may contribute to psychiatric disease by altering presynaptic function.
Keywords: calcineurin, neuron, schizophrenia, synapse, vesicles
Introduction
Calcineurin is a Ca2+/calmodulin-activated phosphatase involved in a variety of critical neuronal processes (1). It functions as a heterodimer with a 61-kDa catalytic and 19-kDa regulatory subunit. There are three isoforms of the catalytic subunit, CNAα,3 β, and γ, encoded by PPP3CA, PPP3CB, and PPP3CC, respectively (2). CNAα and β are known to be abundant in the brain and to modulate neuronal processes (3–6). In contrast, CNAγ expression has been thought to be restricted to the testis (7). Recent evidence suggests that PPP3CC mRNA is indeed present in the human brain (8, 9) and that CNAγ forms a functional phosphatase (10). However, there is no reported evidence that the CNAγ protein is expressed in the brain, and specific functions of CNAγ have not been identified.
Several lines of evidence implicate calcineurin dysfunction in the etiology of schizophrenia. A mouse with forebrain-specific knockout of the calcineurin regulatory subunit, calcineurin B1 (CNB1), has a behavioral phenotype that recapitulates several of the endophenotypes characteristic of schizophrenia, including a selective disruption of working memory (11, 12). Moreover, genetic and potential epigenetic variation in the PPP3CC gene has been reported to be associated with schizophrenia or its cognitive impairments in specific population-based or genome-wide methylation studies, respectively (9, 13–18), although genetic association has not been reported from large-scale, controlled genome-wide association studies performed to date. Finally, a reduction in PPP3CC mRNA levels was observed in the hippocampus of schizophrenia patients (8). Because the function of CNAγ in the brain has not been examined, the potential role of the PPP3CC gene in schizophrenia has remained unclear.
In this study, we show that CNAγ is expressed in adult rodent and human brains, dephosphorylates known presynaptic calcineurin substrates, and is present in presynaptic terminals in the hippocampus. We further show that RNAi-mediated knockdown of CNAγ results in a slowing of synaptic vesicle endocytosis. These data suggest that PPP3CC risk alleles may contribute to schizophrenia pathogenesis by altering synaptic vesicle cycling, adding to evidence implicating presynaptic dysfunction in central nervous system disorders (19, 20).
Experimental Procedures
Western Blotting
Rabbit polyclonal antisera were generated (Open Biosystems) against peptides unique to the individual calcineurin isoforms (Fig. 1A): CNAα, RRDAMPSDANLNSINK; CNAβ, RKDAVQQDGFNSLNTAH; human CNAγ 1, RKLDRFTEPPAFGPVCDLL; human CNAγ 2, PRKDSIHAGGPMKSVTS; mouse CNAγ, CHPQASRRTDHGKKAL; and rat CNAγ, SYHHDAGKTHPHPSKRSDHGKKAL. Affinity purification of antisera was performed (Open Biosystems). As specified in the text, purified antibodies or the original antisera were used for different assays.
FIGURE 1.

CNAγ is expressed in the brain and dephosphorylates synaptic calcineurin substrates. A, human (Hm) and mouse (Ms) CNAα, CNAβ, and CNAγ and rat (Rt) CNAγ recombinant proteins were generated in 293T cells. Protein extracts were probed on a Western blot with anti-human CNAα, CNAβ, and CNAγ antisera; anti-mouse CNAα, CNAβ, and CNAγ antisera; and anti-rat CNAγ antiserum to demonstrate antibody specificity. For the rat CNAγ antiserum analysis, mouse CNAα and CNAβ recombinant proteins were used because of complete amino acid sequence conservation. The higher molecular weight of the mouse relative to the rat CNAγ protein is due to an N-terminal cycle 3 GFP tag present on the recombinant mouse form of the protein. B, hippocampus protein extract from 5-week-old mice was probed on a Western blot with anti-mouse CNAα, CNAβ, and CNAγ antisera. C, Western blotting was performed on mouse tissue protein extracts using anti-mouse CNAα, CNAβ, and CNAγ antisera and a GAPDH antibody. The lack of a Western blot signal in the pancreas tissue extracts is likely due to protein degradation. D, immunoprecipitation (IP) of CNAγ from a human neocortex sample. Purified antibodies against different regions of the protein were used to immunoprecipitate (γ-1) and probe for human CNAγ (γ-2). E, Western blot showing the expression of CNAα, CNAβ, and CNAγ (using custom antisera) and GAPDH in neocortex protein samples from mice at the indicated ages. E, embryonic day; P, postnatal day. F, 293T cells were co-transfected with GFP-tagged constitutively active versions of CNAα, β, or γ (GFP-CNACA) and a dynamin I expression construct. Western blotting was performed on protein extracts for Ser(P)-778 and total dynamin I and for GFP.
For antiserum specificity analysis, human and mouse CNAα, CNAβ, and CNAγ cDNAs were subcloned into the pcDNA-DEST53 mammalian expression vector (Invitrogen), which includes an N-terminal cycle 3 GFP tag. The rat CNAγ construct was acquired in the pExpress-1 mammalian expression vector (Open Biosystems) without an epitope tag. Recombinant CNA proteins were generated by transfecting 293T cells in 6-well plates with the expression constructs using Lipofectamine 2000 (Invitrogen). Cells were lysed in radioimmune precipitation assay buffer (Sigma) and centrifuged at 15,000 × g for 15 min. Supernatants were harvested, and protein concentrations were determined with a bicinchoninic acid kit (Pierce). Protein (30 μg) was solubilized in NuPAGE running buffer and loaded onto 4–12% NuPAGE gels (Invitrogen). Proteins were transferred to nitrocellulose membranes, blocked in 10% milk, and incubated in custom rabbit anti-mouse CNAα, CNAβ, and CNAγ antisera (1:5000) overnight and in secondary antibodies for 1 h in 5% milk/Tris-buffered saline plus 0.1% Triton X-100. The secondary antibody was HRP-conjugated anti-rabbit IgG (1:10,000, Santa Cruz Biotechnology). Blots were developed with chemiluminescence (PerkinElmer Life Sciences) and imaged on a Biochemi (UVP).
For tissue Western blot analyses, tissue protein samples from adult male BALB/c mice were obtained from a commercial supplier (Biochain) and solubilized in NuPAGE running buffer. Western blot analyses were performed using custom rabbit anti-mouse CNAα, CNAβ, and CNAγ antisera (1:5000) as described above. For brain tissue Western blot analysis of 2-week-old or younger animals, brain tissue from three Swiss-Webster mice of either gender was harvested. For samples from mice older than 2 weeks, brain tissue from three male Swiss-Webster mice was harvested. Western blotting was performed as described above.
Immunoprecipitation
Human cerebral cortex samples (Asterand) were homogenized in radioimmune precipitation assay buffer plus protease inhibitors (Roche) and centrifuged for 15 min at 15,000 × g. Supernatants were incubated with affinity-purified antibody (5 μg) overnight at 4 °C with rocking. Protein A/G-agarose beads (20 μl, Thermo Scientific) were added, and samples were rocked for 1 h. Beads were pelleted and washed in radioimmune precipitation assay buffer, and protein was solubilized with 20 μl of NuPAGE sample buffer.
Co-transfection Assays
C terminus GFP-tagged constitutively active forms of human CNAα, β, and γ were produced using PCR to generate sequences encoding the catalytic and CNB1 binding domains in pcDNA-DEST53 (Invitrogen). 293T cells in 6-well plates were co-transfected with individual constitutively active calcineurin constructs and a dynamin I cDNA using Lipofectamine 2000 (Invitrogen). After overnight incubation, protein was extracted from transfected cells using radioimmune precipitation assay buffer, and Western blotting was performed. Additional primary antibodies were anti-GAPDH (1:200, Santa Cruz Biotechnology), anti-GFP (1:1000, Abcam), anti-Ser(P)-778 dynamin I (1:1000, Abcam), and anti-dynamin I (1:200, Santa Cruz Biotechnology). Additional secondary antibodies were HRP-conjugated anti-mouse IgG (1:10,000, Santa Cruz Biotechnology) and anti-sheep IgG (1:5000, Abcam).
Immunocytochemistry
Postnatal hippocampal cultures were generated as described previously (21). Neurons were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100, blocked with 5% BSA, and subsequently incubated with primary antibodies, purified rabbit anti-rat CNAγ (1 μg/ml) and mouse anti-vesicular glutamate transporter 1 (vGlut1, 1:1000, Synaptic Systems), overnight at 4 °C. After washing to remove primary antibodies, Alexa Fluor 488 anti-mouse- or Alexa Fluor 546 anti-rabbit-conjugated secondary antibodies were applied (1:1000). Images were acquired using a ×63 (numerical aperture 1.34) objective lens on a Leica epifluorescence microscope with a CoolSNAP HQ charge-coupled device camera driven by Metamorph. To quantify presynaptic CNAγ distribution, vGlut1-positive puncta were selected as a presynaptic marker, and the intensities of both vGlut1 and CNAγ were quantified using ImageJ.
Electron Microscopy
Vibratome sections of 4% paraformaldehyde and 0.1% glutaraldehyde perfusion-fixed mouse hippocampi were immunostained using rabbit anti-mouse CNAγ antiserum overnight at room temperature. Sections were incubated with biotinylated horse anti-rabbit secondary antibodies (1:250, Vector) for 2 h and in Avidin biotin peroxidase (1:200, Vector) for 2 h at room temperature. Tissue-bound peroxidase was visualized with a diaminobenzidine (5 mg/ml, H2O2 0.03%) reaction. Slices were osmicated, dehydrated, and embedded in durcupan (FLUKA, ACM). Ultrathin sections (60 nm thick) were prepared on an Ultracut UCT ultramicrotome (Leica) and analyzed in a Tecnai 10 electron microscope.
For post-embedding immuno-EM, sections were washed in phosphate buffer overnight and placed into sucrose solutions with increasing concentrations (0.5, 1, and 2 m sucrose for 0.5, 1, and 2 h, respectively). Sections were slammed onto copper blocks, cooled in liquid nitrogen, and, following low-temperature dehydration and freeze substitution, embedded in Lowicryl HM 20 resin (Chemische Werke Lowi). Post-embedding immunocytochemistry was performed on 60-nm serial sections on pioloform-coated nickel grids. Sections were incubated on drops of blocking solution for 1 h (20% BSA), followed by incubation on drops of anti-CNAγ antisera overnight at room temperature. The primary and secondary antibodies (15 nm gold-conjugated anti-rabbit IgG, 1:20, Amersham Biosciences) were diluted in TBS plus 0.03% Triton X-100 and 2% human serum albumin (Sigma). Following washes in TBS, the sections were washed in water and contrasted with saturated aqueous uranyl acetate and lead citrate.
Presynaptic Functional Assays
Oligonucleotides of the rat Ppp3cc cDNA target sequence (CGGCTAACTTTGAAGGAAGTA, Invitrogen) were annealed and ligated into pSUPER. Constructs were transfected for 8 days, and experiments were performed 14–21 days after plating. To quantify the efficiency of the knockdown, immunocytochemistry was performed on neurons co-transfected with the Ppp3cc shRNA vector and vGlut1-pHluorin (22) using GFP (Invitrogen) and CNAγ antibodies and Alexa Fluor 488- or Alexa Fluor 546-conjugated secondary antibodies. Cells were imaged using an epifluorescence microscope with an electron multiplying charge-coupled device camera. Fluorescence levels of CNAγ were measured and corrected for background in surrounding regions at cell bodies in transfected knockdown neurons and in non-transfected control neurons, as described previously for related proteins (23). Transfected neurons were identified by vGlut1-pHluorin expression, which marks cell bodies when cells are fixed and permeabilized. For the shRNA-resistant construct, the rat Ppp3cc cDNA in the pExpress-1 expression vector was acquired (Open Biosystems). The shRNA target sequence was mutated using QuikChange site-directed mutagenesis (Stratagene). The sequence for the shRNA target sequence was changed to CGGCTTACGTTAAAAGAAGTA, leaving the amino acid sequence unchanged. For the inactive form of CNAγ, histidine 147 in the catalytic region of rat CNAγ was mutated to alanine as described previously (24) using QuikChange site-directed mutagenesis (Stratagene). Rat CNAα and CNAβ in the pcDNA3 vector were utilized for rescue experiments in CNAγ shRNA-transfected neurons.
Coverslips were mounted in a perfusion and stimulation chamber on a custom-built epifluorescence microscope. Cells were perfused at 75–250 μl/min with a solution containing 119 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 25 mm HEPES (pH 7.4), 30 mm glucose plus 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (Sigma) and 50 μm d,l-2-amino-5-phosphonovaleric acid (Sigma). Cells were imaged at 30 °C with a flexible resistive objective heater (Omega) utilizing an on/off controller (Minco). Cells were illuminated utilizing a 488-nm diode-pumped solid-state laser (Coherent), shuttered using an acousto-optic modulation. Fluorescence excitation and collection was through a ×40/1.3 numerical aperture Fluar Zeiss objective using 515- to 560-nm emission and 510-nm dichroic filters (Chroma) and a ×1.6 Optivar tube lens. Laser power at the back aperture was ∼1 milliwatt, imaging onto an iXon+ (Andor, DU-897E-BV) back-illuminated, electron-multiplying, charge-coupled device camera. Action potentials were evoked by passing 1-ms current pulses via platinum-iridium electrodes from an isolated current stimulator (World Precision Instruments). Fitting of endocytosis time constants with single exponential decays was done using OriginPro (OriginLab) with the Levenberg-Marquardt algorithm with a temporal offset for reacidification of ∼5 s (22).
The kinetics of synaptic vesicle reacidification were measured, with minor modifications, as described previously (25). Neurons transfected with vGlut1-pHluorin with or without CNAγ shRNA were stimulated at 50 Hz for 4 s. Surface vGlut1-pHluorin was acutely quenched by puffing MES-based Tyrode's buffer (pH 5.5), which permits the isolation of fluorescence from recently endocytosed synaptic vesicles. The decay of fluorescence was measured during this period, marking the reacidification of recently internalized vesicles.
Statistical Analyses
Data presented are mean ± S.E. Mann-Whitney U test or Student's t test was used for comparisons performed in GraphPad Prism 4.03, with p < 0.05 considered statistically significant.
Results
CNAγ Protein Is Expressed in the Brain and Forms a Functional Phosphatase
To examine the expression and localization of CNAγ relative to α and β, it was critical to develop antibodies specific to the individual isoforms. The C-terminal region of the proteins was chosen as the target for antibody generation because of its high level of amino acid sequence divergence (see “Experimental Procedures”). Custom rabbit polyclonal antibodies were developed against each isoform using peptides within this region as immunogens. The minimal sequence overlap between the corresponding peptides in the three calcineurin isoforms resulted in the absence of cross-reactivity of the antibodies (Fig. 1A).
Western blot analysis of mouse hippocampal protein extracts using these custom antisera demonstrated that CNAγ, as well as CNAα and CNAβ, are expressed in the brain (Fig. 1B). Tissue expression analysis demonstrated that, consistent with previous reports (3), CNAα and β proteins are enriched in the mouse brain. In addition, we found that, although CNAγ is expressed at the highest level in the testis, it is enriched in the brain relative to all other examined tissues (Fig. 1C). We confirmed the expression of CNAγ in the human neocortex by performing immunoprecipitation followed by Western blot analysis using purified custom anti-CNAγ antibodies (Fig. 1D).
A previous report on the basis of in situ hybridization analysis indicated that expression of Ppp3cc mRNA is regulated developmentally in specific rodent brain regions, with the highest expression during embryonic development, whereas expression of Ppp3ca and Ppp3cb peaks postnatally (26). To determine whether a similar regulation is observed at the protein level, we examined the developmental expression of the CNAγ protein in the mouse neocortex. We found that the protein levels for all three CNA isoforms increased postnatally, reaching a peak around 2 weeks of age and remaining higher than embryonic levels well into adulthood (Fig. 1D).
Despite being divergent from CNAα and β (2), CNAγ forms a functional phosphatase (10). However, it is not known whether it dephosphorylates calcineurin substrates involved in neuronal function. To address this question, we examined whether CNAγ can dephosphorylate the calcineurin substrate dynamin I, a protein critical for presynaptic function (27). We first generated expression constructs for constitutively active forms of the human CNA isoforms lacking the autoinhibitory domain and for dynamin I. We used these constructs to co-express each isoform with dynamin I in 293T cells and analyzed the level of phosphorylation of the dynamin I residue Ser-778, a known calcineurin substrate (28). Expression of all three isoforms of CNA, including CNAγ, resulted in a decreased level of phosphorylation of dynamin I Ser-778 (Fig. 1E), indicating that CNAγ forms a functional phosphatase capable of modifying known synaptic calcineurin substrates.
CNAγ Is Present in Presynaptic Terminals
Calcineurin is known to modulate neuronal processes occurring in multiple subcellular compartments (1). To investigate the neuronal functional role of CNAγ, we first analyzed whether it is localized to a specific neuronal subcellular compartment. Fluorescence immunocytochemistry analysis of CNAγ performed on cultured rat hippocampal neurons revealed a punctate staining pattern colocalized with vGlut1, a marker of excitatory presynaptic terminals (Fig. 2A). Densitometry measurements performed for CNAγ and vGlut1 on 504 vGlut1 puncta showed that most excitatory synapses within hippocampal cultures contain measurable levels of CNAγ and that the level of CNAγ signal correlates with vGlut1 levels (Fig. 2B, R2 = 0.38, p < 0.0001). These data indicate that CNAγ is localized to synaptic contacts.
FIGURE 2.
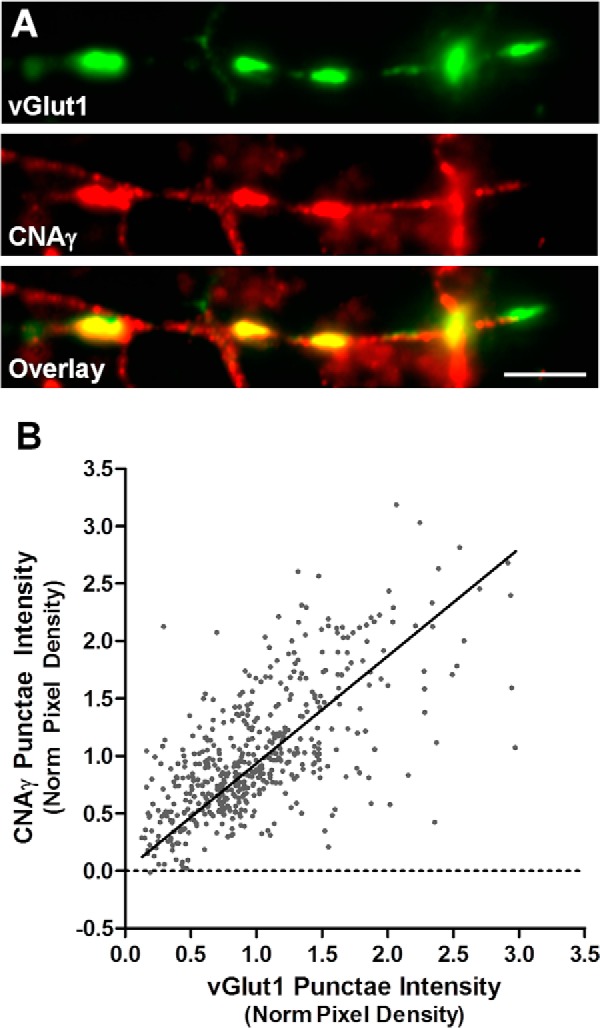
CNAγ is localized to synapses in hippocampal cultures. A, immunocytochemistry was performed on cultured rat hippocampal neurons with antibodies against rat CNAγ (affinity purified antisera) and vGlut1. Scale bar = 10 μm. B, mean pixel intensities of vGlut1 and CNAγ labeling were measured from 504 vGlut1-positive puncta from five neurons. Data were fit with linear regression (R2 = 0.39, p < 0.0001). Norm, normalized.
We next performed immuno-EM analysis to further resolve the subcellular localization of CNAγ. Pre-embedding immuno-EM labeling for CNAα and CNAβ in the mouse hippocampus showed a postsynaptic enrichment (Fig. 3, A and B), consistent with previous reports on the localization of calcineurin (29, 30). In contrast, CNAγ labeling was observed most prominently in the vicinity of synaptic vesicle clusters (Fig. 3, C–E). This subcellular localization of CNAγ was supported using post-embedding immunogold labeling in mouse hippocampal sections (Fig. 3, F–H). These results indicate that CNAγ is present in presynaptic terminals.
FIGURE 3.

CNAγ is present in presynaptic terminals in the hippocampus. A—E, pre-embedding immuno-EM micrographs were performed on mouse hippocampus sections using anti-mouse CNAα (A), CNAβ (B), or CNAγ (C–E) antisera. F—H, post-embedding immunogold micrographs on mouse hippocampus sections using anti-mouse CNAγ antisera. Arrows indicate CNA labeling. Scale bars = 500 nm. S, spine; T, presynaptic terminal; P, postsynaptic density; M, mitochondria.
CNAγ Modulates Synaptic Vesicle Cycling
The localization of CNAγ to presynaptic terminals suggests that it may have a role in modulating presynaptic function. Therefore, we examined the effects of shRNA-mediated CNAγ knockdown on synaptic vesicle cycling in primary neurons using the vGlut1-pHluorin system (22). Rat hippocampal neurons were co-transfected with a Ppp3cc-specific shRNA and the vGlut1-pHluorin construct, and immunocytochemistry analysis for CNAγ was performed to confirm knockdown of the CNAγ protein (Fig. 4, A and B; Mann-Whitney U test, U = 2289, p < 0.0001).
FIGURE 4.

CNAγ knockdown alters synaptic vesicle cycling. A, immunocytochemistry for GFP (left panel) and CNAγ (right panel) performed on primary hippocampal neurons co-transfected with vGlut1-pHluorin and a shRNA targeting CNAγ. White arrows, transfected neurons; yellow arrows, untransfected control neurons. Scale bar = 20 μm. B, normalized histograms of the CNAγ fluorescence (Fluo) intensities from cultured rat hippocampal neurons co-transfected with vGlut1-pHluorin and CNAγ shRNA (gamma-KD, n = 20) or untransfected neurons from the same fields of view (n = 970). C, representative waveform showing the recovery phase of vGlut1-pHluorin fluorescence responses from a control, a CNAγ shRNA-transfected neuron ((gamma-KD), and a CNAγ shRNA- and shRNA-resistant CNAγ co-transfected neuron (gamma-Rescue) in response to a 10-Hz, 10-s stimulation normalized to waveform peaks. Inset, representative non-normalized raw waveforms from neurons as described above. Red line, stimulus period. Scale bars = 0.025 ΔF/F (vertical) and 10 s (horizontal). D, mean ± S.E. peak vGlut1-pHluorin fluorescence intensities in response to a 10-Hz, 10-s stimulation normalized to the peak NH4Cl fluorescence levels from control (n = 8) neurons, CNAγ shRNA-transfected (gamma-KD, n = 6) neurons, and neurons co-transfected with CNAγ shRNA and the shRNA-resistant CNAγ expression vector (gamma-Res, n = 5). E, vGlut1-pHluorin fluorescence recovery time constants (τ) were calculated as described under “Experimental Procedures.” Shown are the mean ± S.E. recovery τ values from waveforms as shown in C from control neurons (n = 11), CNAγ shRNA-transfected neurons (n = 17), and neurons co-transfected with CNAγ shRNA and the shRNA-resistant CNAγ expression vector (gamma-Res, n = 9). F, mean ± S.E. synaptic vesicle reacidification kinetics measured in neurons transfected with either vGlut1-pHluorin alone (control, n = 7) or vGlut1-pHluorin and CNAγ shRNA (gamma-KD, n = 6). G, mean ± S.E. recovery τ values from control neurons (n = 10), CNAγ shRNA-transfected neurons (gamma-KD, n = 9), and neurons co-transfected with CNAγ shRNA and the shRNA-resistant CNAγ expression vector (gamma-Res, n = 10), a CNAα expression vector (alpha, n = 8), a CNAβ expression vector (beta, n = 13), or an inactivated CNAγ expression vector (gammaH147A, n = 6). **, p < 0.01; ***, p < 0.001. Norm., normalized; A.U., arbitrary units.
We next analyzed the effects of CNAγ knockdown on synaptic vesicle release and recycling. First, we measured vGlut1-pHluorin responses to a 10-Hz, 100-action potential stimulation from individual neurons transfected with Ppp3cc shRNA plus vGlut1-pHluorin or vGlut1-pHluorin alone. The fluorescence signals induced by the stimulation at each bouton were normalized to those obtained by rapid alkalization of the entire labeled vesicle pool using NH4Cl (23). These experiments showed that reduction of CNAγ expression led to a modest reduction in vesicle exocytosis, but this difference did not achieve statistical significance (Fig. 4, C and D; Student's t test, t(25) = 1.25, p = 0.24). In contrast, when we examined the kinetics of vesicle endocytosis by fitting the fluorescence recovery with a single exponential curve (Fig. 4, C and E), we found that there was a significant slowing of the time constant of fluorescence recovery (Fig. 3E; Student's t test, t(26) = 3.91, p < 0.001). Critically, restoration of the expression of CNAγ by co-transfection with a shRNA-resistant expression vector resulted in a restoration of synaptic vesicle endocytosis (Fig. 4E; Student's t test, t(18) = 0.43, p = 0.7 relative to the control). These data indicate that CNAγ modulates synaptic vesicle recovery. The recovery kinetics result from a combination of two processes: vesicle endocytosis and vesicle reacidification. Analysis of synaptic vesicle reacidification showed no effect of CNAγ knockdown on this process (Fig. 4F; Student's t test, t(9) = 0.13, p = 0.90), demonstrating that the loss of CNAγ specifically affects synaptic vesicle endocytosis.
Because there are three isoforms of CNA, it is possible that this alteration in synaptic vesicle cycling following CNAγ knockdown results from a reduction in total CNA levels and not from disruption of a specific role of CNAγ in this process. Moreover, the observed alteration in synaptic vesicle endocytosis does not establish an enzymatic role for CNAγ in this process because it is possible that a structural role could underlie its involvement in presynaptic function. To address these questions, we performed further synaptic functional analyses. As shown in Fig. 4G, we replicated the original observation that CNAγ knockdown slows synaptic vesicle recovery (Fig. 4G; Student's t test, t(17) = 4.8, p < 0.001 relative to the control) and that this effect could be rescued by expression of an shRNA-resistant CNAγ construct (Fig. 4G; Student's t test, t(18) = 0.95, p = 0.35 relative to the control). In parallel, we coexpressed CNAα or CNAβ with the CNAγ shRNA, and, in both cases, synaptic vesicle recovery remained significantly slowed relative to control neurons (Fig. 4G; Student's t test CNAα: t(16) = 3.9, p = 0.001; Student's t test CNAβ: t(21) = 4.4, p < 0.001 relative to the control), indicating that the effect of CNAγ shRNA is specific for CNAγ. Lastly, we co-expressed an inactive version of CNAγ that has a single amino acid point mutation in the catalytic domain shown previously to inactivate calcineurin (24). Co-expression of this construct with CNAγ shRNA also failed to rescue the impairment in synaptic vesicle cycling (Fig. 4G; Student's t test, t(14) = 4.1, p = 0.001 relative to the control). In sum, these data indicate that CNAγ has a unique enzymatic role relative to the other CNA isoforms in synaptic vesicle cycling.
Discussion
PPP3CC encodes CNAγ, one of three isoforms of the catalytic subunit of calcineurin (2). Specific haplotypes of this gene have been associated with schizophrenia (9, 13–17), and the expression of its mRNA has been found to be reduced in the hippocampus of human schizophrenia patients (8). Because CNAγ has long been thought to be a testis-specific protein (7), there has been no reported information on functions of CNAγ relevant to its role in schizophrenia. In this study, we show that CNAγ is present in presynaptic terminals, dephosphorylates known presynaptic calcineurin substrates, and modulates synaptic vesicle endocytosis. These findings raise the possibility that variations in PPP3CC contribute to psychiatric disease through altering presynaptic function.
Calcineurin and Presynaptic Function
Calcineurin is involved in multiple neuronal processes, including postsynaptic receptor trafficking, intracellular signaling, cytoskeletal regulation, synaptic vesicle release and recovery, and activity-dependent gene transcription (1). Presynaptically, calcineurin regulates multiple aspects of the synaptic vesicle cycle. Knockdown of the essential CNB1 regulatory subunit results in changes in recycling vesicle pool size and release kinetics (23). The change in release kinetics could be attributed to a modulation of calcium influx via N-type calcium channels by opposing activities of cyclin-dependent kinase 5 and CNAα (5). Furthermore, a set of phosphoproteins involved in vesicle endocytosis are substrates of calcineurin, and blockade of their dephosphorylation results in disruption of this process (6, 28, 31). Although calcineurin-mediated dephosphorylation of these proteins has been reported to play a role in bulk endocytosis (32), recently, dephosphorylation of dynamin has been pinpointed in controlling the acceleration of endocytosis during brief action potential trains (33).
Despite the importance of calcineurin in presynaptic function, relatively little is known about the specific roles of the calcineurin catalytic subunit isoforms in this process. CNAα has been implicated in controlling release probability via modulation of calcium influx (5), and a recent study has reported that knockout of the CNAβ protein impairs synaptic vesicle endocytosis (6). The data presented in this report suggest that CNAγ plays a key enzymatic role in the regulation by calcineurin of synaptic vesicle endocytosis. Because expression of either CNAα or CNAβ did not overcome the effects of CNAγ deficiency on synaptic vesicle endocytosis, it appears that the role of CNAγ in this process can be separated from those of the other CNA isoforms. Precise definition of the specific roles of each isoform in presynaptic function will require further study. In addition, a modest decrease in the rate of exocytosis, which did not achieve significance, was observed following CNAγ knockdown. It is possible that more pronounced effects of CNAγ knockdown on exocytosis would occur in response to higher-frequency stimulation, as was observed for CNB1 knockdown (19). In this regard, further experiments to examine the effects of CNAγ knockdown across a range of stimuli will be informative. Moreover, it will be of great interest, in future studies, to define the precise functional roles of the different calcineurin isoforms in synaptic vesicle cycling and to determine whether there is a distinction between structural and catalytic functions for the calcineurin subunits in this process. Last, our data do not preclude the localization of CNAγ to additional subcellular compartments in which it may play additional roles in neuronal function.
A study of mRNA expression in the developing rat brain suggests a prenatal role of PPP3CC in schizophrenia pathogenesis (26). Here we show that CNAγ protein levels in the cerebral cortex increase during postnatal development and remain relatively high into adulthood. This discrepancy may be due to species differences or developmental regulation of CNAγ protein levels. Regardless, our data suggest that alterations in CNAγ function could play a role in schizophrenia by altering cortical function during postnatal development or adulthood. In addition, given the high level of CNAγ expression in adult testis, it is possible that it may affect schizophrenia pathogenesis through effects in the paternal germline (34).
Presynaptic Dysfunction and Psychiatric Disease
There is an expanding body of research linking alterations of presynaptic function with schizophrenia. A number of genes involved in this process have been reported to be associated with schizophrenia (35–37). The expression of the mRNA or protein encoded by presynaptic genes is altered in human schizophrenia brain samples (38–40), and a number of known schizophrenia risk genes modulate presynaptic function (41, 42). Moreover, a recent study of mice with forebrain-specific deletion of calcineurin that have behavioral abnormalities relevant to schizophrenia provided evidence for a presynaptic mechanism for working memory impairments (19). Given these findings, it will be of considerable interest to study the behavioral function of mice following the specific deletion of CNAγ. In summary, the findings presented here suggest that alterations in PPP3CC may contribute to schizophrenia pathogenesis by modulating synaptic vesicle cycling and further implicate presynaptic dysfunction in psychiatric disease (19, 20).
Author Contributions
J. R. C., D. J. G., S. H. K., and T. A. R. conceived the study design. S. H. K. performed the immunocytochemistry experiments. S. H. K. and J. W. K. performed the synaptic vesicle cycling assays. B. L., C. J. A., and J. J. M. performed the biochemistry experiments. T. L. H. performed the electron microscopy experiments. J. R. C. and D. J. G. wrote the manuscript. All authors edited and approved the manuscript.
This work was supported by a research and development collaboration with Otsuka Pharmaceutical Co. Ltd. J. R. C., J. J. M., and D. J. G. are employees and stockholders of Galenea Corp. B. L. and C. J. A. are stockholders of Galenea Corp. T. A. R. is a consultant and stockholder of Galenea Corp.
- CNA
- calcineurin catalytic subunit.
References
- 1. Groth R. D., Dunbar R. L., and Mermelstein P. G. (2003) Calcineurin regulation of neuronal plasticity. Biochem. Biophys. Res. Commun. 311, 1159–1171 [DOI] [PubMed] [Google Scholar]
- 2. Rusnak F., and Mertz P. (2000) Calcineurin: form and function. Physiol. Rev. 80, 1483–1521 [DOI] [PubMed] [Google Scholar]
- 3. Jiang H., Xiong F., Kong S., Ogawa T., Kobayashi M., and Liu J. O. (1997) Distinct tissue and cellular distribution of two major isoforms of calcineurin. Mol. Immunol. 34, 663–669 [DOI] [PubMed] [Google Scholar]
- 4. Kayyali U. S., Zhang W., Yee A. G., Seidman J. G., and Potter H. (1997) Cytoskeletal changes in the brains of mice lacking calcineurin A α. J. Neurochem. 68, 1668–1678 [DOI] [PubMed] [Google Scholar]
- 5. Kim S. H., and Ryan T. A. (2013) Balance of calcineurin Aα and CDK5 activities sets release probability at nerve terminals. J. Neurosci. 33, 8937–8950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun T., Wu X. S., Xu J., McNeil B. D., Pang Z. P., Yang W., Bai L., Qadri S., Molkentin J. D., Yue D. T., and Wu L. G. (2010) The role of calcium/calmodulin-activated calcineurin in rapid and slow endocytosis at central synapses. J. Neurosci. 30, 11838–11847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muramatsu T., and Kincaid R. L. (1992) Molecular cloning and chromosomal mapping of the human gene for the testis-specific catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin A). Biochem. Biophys. Res. Commun. 188, 265–271 [DOI] [PubMed] [Google Scholar]
- 8. Eastwood S. L., Burnet P. W., and Harrison P. J. (2005) Decreased hippocampal expression of the susceptibility gene PPP3CC and other calcineurin subunits in schizophrenia. Biol. Psychiatry 57, 702–710 [DOI] [PubMed] [Google Scholar]
- 9. Gerber D. J., Hall D., Miyakawa T., Demars S., Gogos J. A., Karayiorgou M., and Tonegawa S. (2003) Evidence for association of schizophrenia with genetic variation in the 8p21.3 gene, PPP3CC, encoding the calcineurin γ subunit. Proc. Natl. Acad. Sci. U.S.A. 100, 8993–8998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kilka S., Erdmann F., Migdoll A., Fischer G., and Weiwad M. (2009) The proline-rich N-terminal sequence of calcineurin Aβ determines substrate binding. Biochemistry 48, 1900–1910 [DOI] [PubMed] [Google Scholar]
- 11. Miyakawa T., Leiter L. M., Gerber D. J., Gainetdinov R. R., Sotnikova T. D., Zeng H., Caron M. G., and Tonegawa S. (2003) Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 100, 8987–8992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zeng H., Chattarji S., Barbarosie M., Rondi-Reig L., Philpot B. D., Miyakawa T., Bear M. F., and Tonegawa S. (2001) Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell 107, 617–629 [DOI] [PubMed] [Google Scholar]
- 13. Horiuchi Y., Ishiguro H., Koga M., Inada T., Iwata N., Ozaki N., Ujike H., Muratake T., Someya T., and Arinami T. (2007) Support for association of the PPP3CC gene with schizophrenia. Mol. Psychiatry 12, 891–893 [DOI] [PubMed] [Google Scholar]
- 14. Kyogoku C., Yanagi M., Nishimura K., Sugiyama D., Morinobu A., Fukutake M., Maeda K., Shirakawa O., Kuno T., and Kumagai S. (2011) Association of calcineurin A γ subunit (PPP3CC) and early growth response 3 (EGR3) gene polymorphisms with susceptibility to schizophrenia in a Japanese population. Psychiatry Res. 185, 16–19 [DOI] [PubMed] [Google Scholar]
- 15. Liu Y. L., Fann C. S., Liu C. M., Chang C. C., Yang W. C., Hung S. I., Yu S. L., Hwang T. J., Hsieh M. H., Liu C. C., Tsuang M. M., Wu J. Y., Jou Y. S., Faraone S. V., Tsuang M. T., Chen W. J., and Hwu H. G. (2007) More evidence supports the association of PPP3CC with schizophrenia. Mol. Psychiatry 12, 966–974 [DOI] [PubMed] [Google Scholar]
- 16. Sacchetti E., Scassellati C., Minelli A., Valsecchi P., Bonvicini C., Pasqualetti P., Galluzzo A., Pioli R., and Gennarelli M. (2013) Schizophrenia susceptibility and NMDA-receptor mediated signalling: an association study involving 32 tagSNPs of DAO, DAOA, PPP3CC, and DTNBP1 genes. BMC Med. Genet. 14, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi J., Gershon E. S., and Liu C. (2008) Genetic associations with schizophrenia: meta-analyses of 12 candidate genes. Schizophr. Res. 104, 96–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wockner L. F., Noble E. P., Lawford B. R., Young R. M., Morris C. P., Whitehall V. L., and Voisey J. (2014) Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 4, e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cottrell J. R., Levenson J. M., Kim S. H., Gibson H. E., Richardson K. A., Sivula M., Li B., Ashford C. J., Heindl K. A., Babcock R. J., Rose D. M., Hempel C. M., Wiig K. A., Laeng P., Levin M. E., Ryan T. A., and Gerber D. J. (2013) Working memory impairment in calcineurin knock-out mice is associated with alterations in synaptic vesicle cycling and disruption of high-frequency synaptic and network activity in prefrontal cortex. J. Neurosci. 33, 10938–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waites C. L., and Garner C. C. (2011) Presynaptic function in health and disease. Trends Neurosci. 34, 326–337 [DOI] [PubMed] [Google Scholar]
- 21. Ryan T. A. (1999) Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J. Neurosci. 19, 1317–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balaji J., and Ryan T. A. (2007) Single-vesicle imaging reveals that synaptic vesicle exocytosis and endocytosis are coupled by a single stochastic mode. Proc. Natl. Acad. Sci. U.S.A. 104, 20576–20581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim S. H., and Ryan T. A. (2010) CDK5 serves as a major control point in neurotransmitter release. Neuron 67, 797–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mondragon A., Griffith E. C., Sun L., Xiong F., Armstrong C., and Liu J. O. (1997) Overexpression and purification of human calcineurin α from Escherichia coli and assessment of catalytic functions of residues surrounding the binuclear metal center. Biochemistry 36, 4934–4942 [DOI] [PubMed] [Google Scholar]
- 25. Atluri P. P., and Ryan T. A. (2006) The kinetics of synaptic vesicle reacidification at hippocampal nerve terminals. J. Neurosci. 26, 2313–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eastwood S. L., Salih T., and Harrison P. J. (2005) Differential expression of calcineurin A subunit mRNA isoforms during rat hippocampal and cerebellar development. Eur. J. Neurosci. 22, 3017–3024 [DOI] [PubMed] [Google Scholar]
- 27. Ferguson S. M., and De Camilli P. (2012) Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anggono V., Smillie K. J., Graham M. E., Valova V. A., Cousin M. A., and Robinson P. J. (2006) Syndapin I is the phosphorylation-regulated dynamin I partner in synaptic vesicle endocytosis. Nat. Neurosci. 9, 752–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Halpain S., Hipolito A., and Saffer L. (1998) Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J. Neurosci. 18, 9835–9844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sík A., Hájos N., Gulácsi A., Mody I., and Freund T. F. (1998) The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 95, 3245–3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cousin M. A., and Robinson P. J. (2001) The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci. 24, 659–665 [DOI] [PubMed] [Google Scholar]
- 32. Clayton E. L., and Cousin M. A. (2009) The molecular physiology of activity-dependent bulk endocytosis of synaptic vesicles. J. Neurochem. 111, 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Armbruster M., Messa M., Ferguson S. M., De Camilli P., and Ryan T. A. (2013) Dynamin phosphorylation controls optimization of endocytosis for brief action potential bursts. eLife 2, e00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Milekic M. H., Xin Y., O'Donnell A., Kumar K. K., Bradley-Moore M., Malaspina D., Moore H., Brunner D., Ge Y., Edwards J., Paul S., Haghighi F. G., and Gingrich J. A. (2015) Age-related sperm DNA methylation changes are transmitted to offspring and associated with abnormal behavior and dysregulated gene expression. Mol. Psychiatry 20, 995–1001 [DOI] [PubMed] [Google Scholar]
- 35. Wong A. H., Trakalo J., Likhodi O., Yusuf M., Macedo A., Azevedo M. H., Klempan T., Pato M. T., Honer W. G., Pato C. N., Van Tol H. H., and Kennedy J. L. (2004) Association between schizophrenia and the syntaxin 1A gene. Biol. Psychiatry 56, 24–29 [DOI] [PubMed] [Google Scholar]
- 36. Chen Q., He G., Wang X. Y., Chen Q. Y., Liu X. M., Gu Z. Z., Liu J., Li K. Q., Wang S. J., Zhu S. M., Feng G. Y., and He L. (2004) Positive association between synapsin II and schizophrenia. Biol. Psychiatry 56, 177–181 [DOI] [PubMed] [Google Scholar]
- 37. Lee H. J., Song J. Y., Kim J. W., Jin S. Y., Hong M. S., Park J. K., Chung J. H., Shibata H., and Fukumaki Y. (2005) Association study of polymorphisms in synaptic vesicle-associated genes, SYN2 and CPLX2, with schizophrenia. Behav. Brain Funct. 1, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Behan A. T., Byrne C., Dunn M. J., Cagney G., and Cotter D. R. (2009) Proteomic analysis of membrane microdomain-associated proteins in the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder reveals alterations in LAMP, STXBP1 and BASP1 protein expression. Mol. Psychiatry 14, 601–613 [DOI] [PubMed] [Google Scholar]
- 39. Maycox P. R., Kelly F., Taylor A., Bates S., Reid J., Logendra R., Barnes M. R., Larminie C., Jones N., Lennon M., Davies C., Hagan J. J., Scorer C. A., Angelinetta C., Akbar M. T., Akbar T., Hirsch S., Mortimer A. M., Barnes T. R., and de Belleroche J. (2009) Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol. Psychiatry 14, 1083–1094 [DOI] [PubMed] [Google Scholar]
- 40. Faludi G., and Mirnics K. (2011) Synaptic changes in the brain of subjects with schizophrenia. Int. J. Dev. Neurosci. 29, 305–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kvajo M., McKellar H., Drew L. J., Lepagnol-Bestel A. M., Xiao L., Levy R. J., Blazeski R., Arguello P. A., Lacefield C. O., Mason C. A., Simonneau M., O'Donnell J. M., MacDermott A. B., Karayiorgou M., and Gogos J. A. (2011) Altered axonal targeting and short-term plasticity in the hippocampus of Disc1 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 108, E1349–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jentsch J. D., Trantham-Davidson H., Jairl C., Tinsley M., Cannon T. D., and Lavin A. (2009) Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology 34, 2601–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]