

Abstract
Experimental evidence supports the role of mitochondrial ceramide accumulation as a cause of mitochondrial dysfunction and brain injury after stroke. Herein, we report that SIRT3 regulates mitochondrial ceramide biosynthesis via deacetylation of ceramide synthase (CerS) 1, 2, and 6. Reciprocal immunoprecipitation experiments revealed that CerS1, CerS2, and CerS6, but not CerS4, are associated with SIRT3 in cerebral mitochondria. Furthermore, CerS1, -2, and -6 are hyperacetylated in the mitochondria of SIRT3-null mice, and SIRT3 directly deacetylates the ceramide synthases in a NAD+-dependent manner that increases enzyme activity. Investigation of the SIRT3 role in mitochondrial response to brain ischemia/reperfusion (IR) showed that SIRT3-mediated deacetylation of ceramide synthases increased enzyme activity and ceramide accumulation after IR. Functional studies demonstrated that absence of SIRT3 rescued the IR-induced blockade of the electron transport chain at the level of complex III, attenuated mitochondrial outer membrane permeabilization, and decreased reactive oxygen species generation and protein carbonyls in mitochondria. Importantly, Sirt3 gene ablation reduced the brain injury after IR. These data support the hypothesis that IR triggers SIRT3-dependent deacetylation of ceramide synthases and the elevation of ceramide, which could inhibit complex III, leading to increased reactive oxygen species generation and brain injury. The results of these studies highlight a novel mechanism of SIRT3 involvement in modulating mitochondrial ceramide biosynthesis and suggest an important role of SIRT3 in mitochondrial dysfunction and brain injury after experimental stroke.
Keywords: brain, ceramide synthase, mitochondria, sirtuin, sphingolipid
Introduction
Insufficient blood flow to the brain, i.e. cerebral ischemia, initiates a complex cascade of molecular events, including excitotoxicity, dissipation of ion gradients, inflammation, cytosolic Ca2+ overload, and mitochondrial dysfunction leading to eventual cell death within the infarct core (1, 2). Peri-infarct penumbral tissue, a target for treatment strategies, is potentially salvageable because this region still exhibits cellular function and metabolic activity to various degrees (3). Mitochondrial dysfunction appears to be one essential step in penumbral tissue damage after ischemia/reperfusion (IR)2 brain injury (4).
Although IR-induced mitochondrial injury has been extensively studied (5–8), the molecular mechanisms of mitochondrial damage remain obscure. Several studies showed sphingolipid ceramide increases in the brain after experimental stroke, and reduction of ceramide generation was neuroprotective leading to smaller infarct sizes (9–11). Emerging evidence implicates sphingolipid ceramide, which is elevated in mitochondria after IR, as a causal factor of mitochondrial defects (12, 13). The restriction on mitochondrial respiratory chain function at the level of complex III has been shown in various rodent models of stroke (14). We and others have reported that ceramide could directly suppress respiratory chain complex III activity (15, 16).
Excessive formation of reactive oxygen species (ROS) occurs during cerebral IR (17–19). Baseline mitochondria generate very low amounts of ROS, but ROS generation is profoundly increased in the presence of complex I or III inhibitors (20). Complex III impairment observed in cerebral IR (14) could account for an increased ROS generation. Of note, exogenous ceramide has been shown to increase the ROS production in isolated mitochondria (21).
Vastly hydrophobic ceramides are generated inside the membranes, near their targets; therefore, sphingolipid metabolism is restricted to the membranes (22). The plasma membrane (23), lysosomes (24), and endoplasmic reticulum (25) are well characterized compartments of sphingolipid metabolism. Mitochondria, a less defined compartment of ceramide metabolism, contain a specific array of sphingolipid-metabolizing enzymes, including ceramide synthases (CerS) 1, 2, 4, and 6, whose expression and function we described in brain mitochondria (12). Each CerS displays a distinct substrate specificity profile for fatty acid acyl-CoA, CerS1 generates C18:0- and C18:1-ceramide; CerS2 and CerS4 produce C20:0-, C24:0-, and C24:1-ceramide; and CerS5 and CerS6 generate C14:0-, C16:0-, C18:0-, and C18:1-ceramide (26, 27). Mitochondrial ceramide engagement in apoptosis was unequivocally demonstrated using loss-of-function ceramide synthase mutants in the germ line of Caenorhabditis elegans (28).
Sirtuins (SIRT1–7), a family of NAD+-dependent protein deacetylases, are important regulators of metabolism and longevity (29). SIRT3 (30, 31), a primary mitochondrial deacetylase, coordinates the adaptive responses of several metabolic pathways in mitochondria (32–34). The role of SIRT3 in cell responses seems to be stimulus- and cell type-specific (35, 36). Thus, SIRT3 has been shown to operate in both stress resistance (anti-apoptosis) (37, 38) and tumor suppression (pro-apoptosis) (39, 40). In neuronal cells, SIRT3 overexpression protected cultured cortical neurons against excitotoxicity (41) and differentiated PC12 cells from oxidative stress-induced cell death (42).
In this study, we provide evidence for a novel role of SIRT3 in the regulation of mitochondrial ceramide synthase response to cerebral IR. Importantly, we show that Sirt3 gene ablation decreases ceramide, preserves mitochondrial respiratory chain function, reduces ROS generation and oxidative protein damage, and attenuates brain tissue injury in the experimental stroke mouse model. These studies emphasize a crucial role of SIRT3, a novel regulator of mitochondrial ceramide biosynthesis, in brain response to IR.
Experimental Procedures
Animals and Reagents
Male C57BL6 (8 weeks old) mice (Jackson Laboratory, Bar Harbor, ME) were acclimated for 1 week prior to experimentation. Sirt3 knock-out (KO) mice (C57BL6 genetic background) were originally developed by Dr. David B. Lombard (University of Michigan, Ann Arbor, MI) (30) and transferred to the animal facility at the Veterans Affairs Medical Center (Charleston, SC). Complete protease inhibitor mixture and PhosSTOP phosphatase inhibitor mixture were from Roche Applied Science. Fatty acyl-CoA and 17C-dihydrosphingosine were from Avanti Polar Lipids (Alabaster, AL). All other chemicals were purchased from Sigma.
Middle Cerebral Artery Occlusion (MCAO) Surgery and Induction of Ischemia
Experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Ralph H. Johnson Veterans Affairs Medical Center, Charleston SC, and followed the National Institutes of Health guidelines for experimental animal use. Under temporary anesthesia, mice were subjected to MCAO as described previously (43). Briefly, the left common carotid artery was exposed through a midline incision in the neck. Sham control animals were treated identically, but without MCAO. A microsurgical clip was placed around the origin of the internal carotid artery (ICA). The distal end of the ICA was ligated with 6-0 silk and transected. A 6-0 silk was tied loosely around the ICA stump. The clip was removed, and the fire-polished tip of a 5-0 nylon suture (poly-l-lysine-coated) was gently inserted into the ICA stump. The loop of the 6-0 silk was tightened around the stump and the nylon suture was advanced ∼11 mm (adjusted for body weight) into and through the ICA after removal of the aneurysm clip, until it rested in the anterior cerebral artery, thereby occluding the anterior communicating and middle cerebral arteries. After 1 h of MCAO, the suture was removed; blood flow was restored to normal; and the incision was closed. The wound was sutured followed by application of analgesic. Mice were placed on the heating pad until ambulatory and then returned to the cage.
Infarct Size Determination
Infarct size was measured by histological examination using 2,3,5-triphenyltetrazolium chloride (TTC) staining (12). Brains were dissected out and cut into 2-mm-thick coronal sections, which were stained with 2% TTC for 90 min at 37 °C. The TTC-stained sections were fixed with 10% neutral buffered formalin and kept in darkness at 4 °C for at least 24 h. The infarct area, identified by lack of TTC staining, was determined with a computer-assisted image analysis system, consisting of a computer equipped with a Quick Capture frame grabber card and a Hitachi CCD camera mounted on an Olympus microscope. ImageJ software (National Institutes of Health) was used for data analysis. A single operator blinded to treatment status performed all measurements. The areas of non-infarcted tissue ipsilateral and contralateral to the occluded side were measured. Infarct area was calculated using an indirect method, in which the non-infarcted region in the ischemic hemisphere was subtracted from the total surface area of the contralateral hemisphere to correct for edema (44). Infarct volume was calculated by summing up the infarct areas over all sections and multiplying by the distance between sections (2 mm). The infarct volume is presented in cubic millimeters (mm3).
Isolation of Mouse Brain Mitochondria
All procedures were performed at 4 °C as described (12, 45, 46). Briefly, tissue was placed immediately in ice-cold isolation medium containing 230 mm mannitol, 70 mm sucrose, 10 mm HEPES, and 1 mm EDTA, pH 7.4. Brain tissue (∼1 g) was homogenized in 10 ml of isolation medium using a Teflon-glass homogenizer. The homogenate was centrifuged at 900 × g for 10 min. The supernatant was then centrifuged at 12,000 × g for 10 min. The pellet was resuspended in 2 ml of 15% Percoll-Plus (GE Healthcare) and placed atop a discontinuous Percoll gradient consisting of a bottom layer of 4 ml of 40% Percoll and a top layer of 4 ml of 23% Percoll. The gradient was spun at 31,000 × g for 20 min in an SW41 rotor in a Beckman ultracentrifuge. The fraction at the 23–40% interface that contained mitochondria was washed three times with isolation medium by centrifugation at 12,000 × g for 10 min. Protein concentration was measured with a bicinchoninic acid assay (Sigma) using bovine serum albumin (BSA) as a standard. Typically, the contamination of mitochondria with ER was <1% by activity measurements of the ER-specific marker enzyme, NADPH-cytochrome c reductase (12, 45). Western blot analysis showed no contamination of mitochondria with Na +/K+-ATPase (plasma membrane marker), LAMP-2 (lysosomal marker), calnexin (ER marker), and myelin basic protein (myelin marker) (45, 46).
Mitochondrial Respiratory Chain Activity
Mitochondrial respiration was measured by recording oxygen consumption at 25 °C in a chamber equipped with a Clark-type oxygen electrode (Instech Laboratories, Plymouth Meeting, PA) as described previously (12, 45, 46). Briefly, mitochondria were incubated in the medium containing 125 mm KCl, 10 mm HEPES, 2 mm KH2PO4, 5 mm MgCl2, and 0.5 mg/ml mitochondrial protein supplemented with either complex I substrate (mixture of 5 mm glutamate and 5 mm malate) or complex II substrate (10 mm succinate) in the presence of 1 μm rotenone or complex IV substrate (1 mm ascorbate in the presence of 250 μm N,N,N′,N′-tetramethyl-p-phenylenediamine and 1 μm antimycin). Uncoupler-stimulated (state 3u) respiration was measured in the presence of 50 μm 2,4-dinitrophenol.
Complex III Enzyme Activity
Complex III (ubiquinol:ferricytochrome c oxidoreductase; EC 1.10.2.2) activity was measured using decylubiquinol as a substrate (15, 47). Decylubiquinol was prepared by reducing decylubiquinone with borohydrate followed by extraction with organic solvent (15). The assay buffer consisted of 50 mm potassium phosphate, pH 7.4, 5 mm MgCl2, 2.5 mg/ml BSA, 60 μm cytochrome c, 3 mm sodium azide, 1 μg/ml rotenone, 0.6 mm dodecyl β-maltoside, 50 μg/ml mitochondrial protein, and 100 μm decylubiquinol. Activity was determined by measuring the increase in the absorbance of reduced cytochrome c at 550 nm with/without 2 μm antimycin A, a complex III activity inhibitor. Specific activity was calculated using an extinction coefficient of 19 mm−1 cm−1 for reduced cytochrome c and expressed as antimycin A-sensitive rate.
Mitochondrial Permeability Transition Pore (MPTP) Activity
MPTP opening was assayed by measurements of mitochondrial swelling using probe colorimeter (Brinkmann Instruments), as described previously (48).
Measurement of Mitochondrial Ca2+ Retention Capacity
The Ca2+-retention capacity of mitochondria was monitored using a Ca2+-selective electrode (Thermo Scientific/Orion, Rockford, IL) in a medium containing 250 mm sucrose, 10 mm HEPES, and 2 mm H3PO4, pH 7.4 (adjusted with Tris base). Mitochondria (0.5 mg/ml) were energized by 10 mm succinate with 1 μm rotenone and pulsed with 50 μm Ca2+ every 1.5 min. The increasing Ca2+ load caused a decline in Ca2+ uptake rates. Maximal Ca2+-retention capacity was defined as an amount of Ca2+ (per mg of protein) required to decrease the Ca2+ uptake rate by >90%. Simultaneously with Ca2+-retention capacity measurements, mitochondrial swelling was monitored using a probe colorimeter (Brinkmann Instruments) (45).
Antibodies
Rabbit monoclonal anti-voltage-dependent anion channel, rabbit monoclonal anti-acetylated lysine, rabbit monoclonal anti-SIRT3, rabbit monoclonal anti-SIRT5, rabbit monoclonal anti-IDH2, rabbit monoclonal anti-SOD2, rabbit monoclonal anti-cytochrome c, mouse monoclonal anti-acetylated lysine, and rabbit polyclonal anti-Bak antibodies were supplied by Cell Signaling Technology (Danvers, MA). Rabbit polyclonal anti-CerS6 and anti-NDUFA9 antibodies were purchased from Abcam (Cambridge, MA). Mouse monoclonal anti-SIRT5, rabbit polyclonal anti-CerS4, rabbit polyclonal anti-CerS2, rabbit polyclonal anti-CerS1, and mouse monoclonal anti-active Bax (6A7) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal anti-Bcl-xL antibody was from Sigma. Rabbit polyclonal anti-CerS2, rabbit polyclonal anti-CerS6, mouse polyclonal anti-CerS6, and mouse monoclonal anti-CerS6 antibodies were purchased from Novus (Littleton, CO). Secondary horseradish peroxidase-conjugated antibodies were supplied by Jackson ImmunoResearch (West Grove, PA).
Immunoprecipitation
Immunoprecipitations were performed as we described previously (45, 46, 49, 50). For immunoprecipitation, mitochondrial lysates (500 μg) were pre-cleared in buffer A (0.15 m NaCl, 0.5 mm EDTA, 0.5% Igepal CA-630, protease and phosphatase inhibitor mixture, 0.05 m Tris, pH 7.5, 0.2% BSA) by incubation with appropriate species-specific IgG-conjugated magnetic beads (Dynabeads, Invitrogen/Dynal) for 1 h. Antibodies then were added. After incubation at 4 °C overnight with gentle mixing, antibody-antigen complexes were captured with Dynabeads and washed two times with buffer A (without BSA), and then washed twice with Tris-buffered saline, pH 7.5. The immunoprecipitates were eluted by boiling in SDS sample buffer. As a control, the same immunoprecipitation procedure was performed except for the primary antibody application.
Western Blot
Proteins were analyzed by Western blot as described previously (45, 46, 49, 50). Proteins were separated by 4–15% SDS-PAGE, blotted to PVDF membrane, blocked with 5% non-fat dry milk (Bio-Rad) or 5% BSA in TBS-T buffer (10 mm Tris, 150 mm NaCl, and 0.2% Tween 20, pH 8.0), and subsequently probed with the appropriate primary antibody. Immunoreactive bands were visualized using a SuperSignal West Dura substrate (Thermo Scientific, Rockford, IL).
Analysis of Sphingolipids by Tandem Mass Spectrometry (MS)
Mitochondria and tissue samples were analyzed by reverse-phase high pressure liquid chromatography coupled to electrospray ionization followed by separation by MS as described previously (12, 49, 50). Sphingolipid analysis was performed in the Lipidomics Analytical Core Facility at Medical University of South Carolina using a Thermo Finnigan TSQ 7000 triple quadrupole mass spectrometer, operating in a multiple reaction monitoring positive-ionization mode. The peaks for the target analytes and internal standards were collected and processed with the Xcalibur software system. The target analyte peak area ratios from the samples were normalized to their respective internal standard and compared with the calibration curves using a linear regression model. Each sample was normalized to its respective total protein levels.
Ceramide Synthase Activity
The activity of ceramide synthase was determined as described previously (12). Briefly, mitochondria (500 μg) were incubated in 1 ml of medium containing 50 mm HEPES, pH 7.4, 2 mm KH2PO4, 0.5 mm DTT, 1 mm MgCl2, 5 mm n-octylglucoside, 10 μm 17C-dihydrosphingosine, and 10 μm C18:0-acyl-CoA (for CerS1) or 10 μm C16:0-acyl-CoA (for CerS6) or 10 μm C24:0-acyl-CoA (for CerS2) at 37 °C for 20 min with gentle stirring. The reaction was terminated; sphingolipids were extracted by addition of 1 ml of ethyl acetate/isopropyl alcohol/water (60:30:10% v/v/v) solvent system and subjected to analysis by tandem MS. Ceramide synthase-specific activity was expressed as picomoles of 17C-dihydroceramide/min per mg of protein.
Carbonyl Content Measurement
Protein carbonyls were quantified in mitochondria with the protein carbonyl content assay kit (Sigma) according to the manufacturer's instructions.
ROS Generation Measurement
ROS formation was assessed by oxidation of 2′,7′-dichlorodihydrofluorescein (H2-DCF). H2-DCF was obtained from H2-DCF diacetate (Molecular Probes) by chemical hydrolysis of the diacetate groups in 0.2 m KOH for 1 h before use (51). Mitochondria (0.25 mg/ml) were incubated at 25 °C in a basal incubation medium containing 250 mm sucrose, 10 mm HEPES, 2 mm phosphate, 10 μm H2-DCF, and a mixture of 5 mm glutamate/malate as a respiratory substrates. Fluorescence of oxidized dichlorofluorescein (filter wavelengths of 490 nm excitation and 520 nm emission) was measured using a Fluostar Optima plate reader (BMG Labtechnologies, Inc., Durham, NC) and expressed as arbitrary units/mg of protein/min.
Deacetylation Assay
Recombinant SIRT3 was purchased from Enzo Life Sciences (Farmingdale, NY). SIRT3 activity was validated using a SIRT3 Fluorimetric Drug Discovery kit (Enzo Life Sciences). CerS1, CerS2, and CerS6 were immunoprecipitated from Sirt3 KO brain mitochondria using specific antibodies. As a control, the same immunoprecipitation procedure was performed except for the primary antibody application (IgG). The immunoprecipitates were incubated for 2 h using a Thermomixer at 37 °C in a buffer (50 mm Tris-HCl, pH 8.0, 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2) in the presence or absence of recombinant SIRT3 enzyme, 1 mm NAD+, or 10 mm nicotinamide.
Statistical Analysis
All experiments and assays were performed three or more times. Data were collected, and the mean value of the treatment groups and the standard error were calculated. Data were analyzed for statistically significant differences between groups by one-way analysis of variance or Student's t test where appropriate (SigmaPlot software version 12.0). Statistical significance was ascribed to the data when p < 0.05.
Results
Ceramide Is Selectively Down-regulated in Cerebral Mitochondria of Sirt3 KO Mice
Brain tissue homogenate and purified non-synaptosomal mitochondria were prepared from wild type (WT) and Sirt3 KO mice. A sphingolipid profile of tissue and mitochondria was analyzed by tandem MS. There were no significant changes in ceramide, sphingosine, and sphingosine 1-phosphate (S1P) in tissue homogenate from SIRT3-deficient mouse brain compared with WT (Fig. 1A). The data are presented as a percentage change of WT mouse brain sphingolipid content shown in Table 1. Ceramide was significantly reduced in mitochondria of Sirt3 KO mice, whereas sphingosine and S1P did not change. The content of ceramide species characterized by long-chain fatty acid, including C16:0- and C18:0-ceramide, was decreased, whereas C18:1-ceramide was not changed (Fig. 1B). The data are presented as a percentage change of WT mouse brain mitochondria content shown in Table 2. Ceramide species containing very long-chain fatty acid such as C24:0- and C24:1-ceramide were attenuated. In contrast, C20:0- and C22:0-ceramide did not change. The data suggest that SIRT3 could control ceramide metabolism in mitochondria.
FIGURE 1.

Enhanced acetylation of ceramide synthases and reduced ceramide in cerebral mitochondria of Sirt3 KO mice. Sphingolipids were analyzed in brain tissue homogenate and purified mitochondria from WT and SIRT3-deficient mice as described under “Experimental Procedures.” Total ceramide (A) and ceramide species (B) were measured in cerebral mitochondria of Sirt3 KO mice. Data are means ± S.E., *, p < 0.05, n = 16. Each sample was normalized to its respective total protein levels. C, increased acetylation of ceramide synthases in cerebral mitochondria of Sirt3 KO mice. Mitochondria were immunoprecipitated (IP) with anti-acetyl-lysine antibody (Cell Signaling) and probed using anti-CerS1 (Santa Cruz Biotechnology), anti-CerS2 (Novus), anti-CerS4 (Santa Cruz Biotechnology), or anti-CerS6 (Novus) antibodies as described under “Experimental Procedures.” Input load, 20 μg/lane.
TABLE 1.
Sphingolipid content of a mouse brain tissue homogenate and mitochondria
Sphingolipid content (pmol/mg of protein) was determined in the WT mouse brain. Values are means ± S.E., n = 16.
| Homogenate | Mitochondria | |
|---|---|---|
| Ceramide | 1482.9 ± 98.4 | 814.4 ± 69.9 |
| Sphingosine | 39.6 ± 6.2 | 23.1 ± 6.4 |
| S1P | 24.3 ± 4.3 | 7.6 ± 0.9 |
TABLE 2.
Ceramide species content of WT mouse brain mitochondria
Ceramide species (pmol/mg of protein) were determined in WT mouse brain mitochondria. Values are means ± S.E., n = 16.
| Ceramide | Mitochondria |
|---|---|
| C16:0 | 12.8 ± 2.9 |
| C18:0 | 226.1 ± 33.4 |
| C18:1 | 117.6 ± 18.8 |
| C20:0 | 91.5 ± 16.1 |
| C22:0 | 13.8 ± 3.3 |
| C24:0 | 118.7 ± 15.5 |
| C24:1 | 236.9 ± 31.1 |
SIRT3 Regulates Mitochondrial CerS1, -2, and -6 Acetylation
Although the majority of ceramides are synthesized de novo at the cytosolic side of the endoplasmic reticulum (52, 53), some ceramides could be also produced in cerebral mitochondria by CerS1, -2, -4, and -6 (45). Selective attenuation of ceramide in mitochondria of SIRT3-deficient mice suggests that SIRT3 could regulate the activity of mitochondrial CerS by deacetylation. The acetylation state of mitochondrial CerS was elucidated in immunoprecipitation experiments using anti-acetyl-lysine antibodies followed by Western blotting with specific anti-CerS antibodies (Fig. 1C). In WT mitochondria, CerS1, -2, and -6 were acetylated but not CerS4. Furthermore, CerS1, -2, and -6 were hyperacetylated in mitochondria from SIRT3-deficient mice. SIRT3 knockdown did not change the expression of CerS1, -2, -4, and -6 in mitochondria (Input). The data suggest that mitochondrial CerS1, -2, and -6 are substrates of SIRT3-mediated deacetylation.
SIRT3 Is a Part of Multiprotein Complex Containing Mitochondrial CerS1, -2, and -6 but Not CerS4
SIRT3 has been demonstrated to deacetylate some of its substrates such as SOD2 (54) and complex I subunit NDUFA9 (33) via direct interaction, whereas association with the others, cyclophilin D for example (55), has not been reported. Given that SIRT3 appears to regulate mitochondrial CerS acetylation, we examined a possible physical association between the deacetylase and ceramide synthases. Co-immunoprecipitation studies were performed using anti-CerS6 antibodies. Fig. 2A shows that CerS6 is associated with SIRT3 in baseline cerebral mitochondria. Further analysis of the immunoprecipitates revealed that CerS1 and CerS2 are also complexed with CerS6 and SIRT3. However, CerS4 was not detected in the immunoprecipitates. The association of SIRT3 with ceramide synthases was confirmed in reciprocal immunoprecipitation experiments using anti-SIRT3 antibodies (Fig. 2B). CerS1, CerS2, and CerS6 were found in a complex with SIRT3, but CerS4 was not present. Of note, CerS1, -2, and -6 selectively complexed with SIRT3 because there was no association of ceramide synthases with another mitochondrial sirtuin SIRT5 (Fig. 2C). The association of SIRT3 with mitochondrial CerS1, -2, and -6, but not with CerS4, was corroborated in co-immunoprecipitation experiments using anti-CerS2 antibodies (Fig. 2D). The results of the immunoprecipitation studies indicate that CerS1, -2, and -6 are complexed with SIRT3, which could regulate the ceramide synthase deacetylation in cerebral mitochondria. Moreover, the data suggest that mitochondrial CerS1, -2, and -6 could form a complex to coordinate the synthesis of various ceramide species in mitochondria. This is in line with previously reported regulation of CerS activity by heterodimer formation. Thus, CerS2 activity was enhanced by co-expression with a catalytically active form of CerS5 or CerS6 (56).
FIGURE 2.
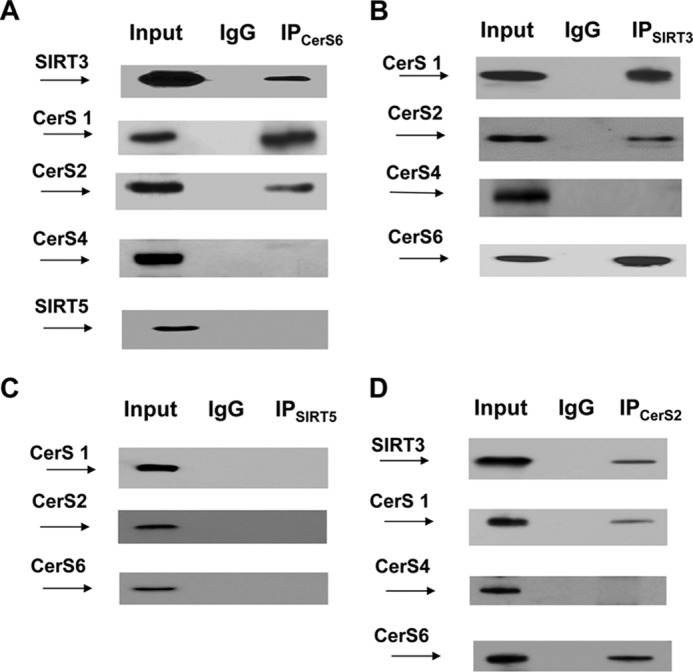
SIRT3 is associated with mitochondrial CerS1, -2, and -6. SIRT3 association with CerS1, CerS2, and CerS6 was detected in reciprocal immunoprecipitation (IP) experiments. Mitochondria were immunoprecipitated with anti-CerS6 antibody (Novus) and were probed using anti-SIRT3 (Cell Signaling), anti-CerS1 (Santa Cruz Biotechnology), anti-CerS2 (Santa Cruz Biotechnology), anti-CerS4 (Santa Cruz Biotechnology), or anti-SIRT5 (Cell Signaling) antibodies (A); with anti-SIRT3 antibody (Cell Signaling) and probed using anti-CerS1 (Santa Cruz Biotechnology), anti-CerS2 (Novus), anti-CerS4 (Santa Cruz Biotechnology), or anti-CerS6 (Novus) antibodies (B); with anti-SIRT5 antibody (Santa Cruz Biotechnology) and probed using anti-CerS1 (Santa Cruz Biotechnology), anti-CerS2 (Santa Cruz Biotechnology), or anti-CerS6 (Abcam) antibodies (C); and with anti-CerS2 antibody (Novus) and probed using anti-SIRT3 (Santa Cruz Biotechnology), anti-CerS1 (Santa Cruz Biotechnology), anti-CerS4 (Santa Cruz Biotechnology), or anti-CerS6 (Abcam) antibodies (D). Input load: 20 μg/lane. As a control, the same immunoprecipitation procedure was performed except for primary antibody application (IgG).
SIRT3 Directly Deacetylates Mitochondrial CerS1, -2, and -6
Having demonstrated that mitochondrial CerS1, -2, and -6 are acetylated SIRT3-associated proteins, we sought to determine whether SIRT3 could regulate the level of ceramide synthase acetylation in vitro. CerS1, -2, and -6 were immunoprecipitated from the cerebral mitochondria of Sirt3 KO mice using specific antibodies. The immunocomplexes were incubated in the presence or absence of active recombinant SIRT3, NAD+, or nicotinamide (NAM), a sirtuin inhibitor. Fig. 3 shows that that SIRT3 deacetylated CerS1, -2, and -6 in a NAD+-dependent manner, although the deacetylation was blocked by NAM, an inhibitor of SIRT3 activity (Fig. 3, A–C). The corresponding ceramide synthase activity measurements indicated that SIRT3-mediated deacetylation stimulated CerS1, -2, and -6 activity, although blocking the SIRT3-mediated deacetylation of ceramide synthases with NAM attenuated the increase in ceramide synthase enzyme activity (Fig. 3D).
FIGURE 3.

SIRT3 deacetylates and activates CerS1, -2, and -6. CerS1, -2, and -6 were immunoprecipitated from baseline cerebral mitochondria of Sirt3 KO mice. CerS1 (A), CerS2 (B), and CerS6 (C) containing immunocomplexes (IC) were incubated with active recombinant SIRT3 in the presence or absence of NAD+ and NAM, a sirtuin inhibitor. In all panels, the same immunoprecipitation procedure was performed except for the primary antibody application as a control (IgG). After deacetylation, ICs were analyzed by Western blot using anti-CerS1 (Santa Cruz Biotechnology), anti-CerS2 (Santa Cruz Biotechnology), anti-CerS6 (Novus), anti-acetyl-lysine (Cell Signaling), and anti-SIRT3 (Cell Signaling) antibodies or by tandem MS (D) to determine the corresponding ceramide synthase activity as described under “Experimental Procedures.” Data are means ± S.E., *, p < 0.05, n = 4.
To rule out a potential inhibitory effect of NAM on CerS activity, CerS1, -2, and -6 activity was measured in WT cerebral mitochondria with/without 10 mm NAM. There was no change in specific activity in the presence of NAM for CerS1 (16.9 ± 1.9 versus 17.3 ± 1.8 pmol of 17C-dihydro-C18:0-ceramide/min/mg of protein, n = 4), CerS2 (8.7 ± 0.9 versus 8.9 ± 1.1 pmol of 17C-dihydro-C24:0-ceramide/min/mg of protein, n = 4), and CerS6 (6.9 ± 0.7 versus 6.7 ± 0.8 pmol of 17C-dihydro-C16:0-ceramide/min/mg of protein, n = 4). The results provide biochemical evidence that SIRT3 deacetylates and activates CerS1 and -2 in mitochondria.
IR Decreases Ceramide Synthase Acetylation Resulting in Augmented Enzyme Activity
To elucidate the impact of SIRT3-mediated deacetylation on mitochondrial ceramide synthase response to cerebral IR, the acetylation status of ceramide synthases was examined in WT cerebral mitochondria after experimental stroke. Mitochondria were purified from contralateral and ipsilateral (IR) brain hemisphere of a mouse exposed to 1 h of MCAO followed by 24 h of reperfusion. Immunoprecipitation studies were conducted using anti-acetyl-lysine antibodies. Fig. 4A shows that IR attenuated the acetylation of CerS1, -2, and -6 suggesting increased SIRT3 activity. There was no change in CerS4 acetylation after IR. The lack of change in SIRT3 protein expression levels after IR suggests a post-translational mechanism of its activation. To determine whether IR-induced deacetylation is specific to ceramide synthases, the acetylation state of known targets of SIRT3, such as NDUFA9 (33), SOD2 (54), and IDH2 (57), was determined in brain mitochondria after IR (Fig. 4A). In IR-damaged mitochondria, there were less AcNDUFA9, AcSOD2, and AcIDH2 compared with control. The data indicate that IR triggered deacetylation of multiple SIRT3 target proteins, including CerS1, -2, and -6.
FIGURE 4.

IR triggers SIRT3-mediated deacetylation of ceramide synthase and increased enzyme activity. Mitochondria were prepared from the WT and Sirt3 KO mouse brain at baseline (BL) or WT and Sirt3 KO mouse brain hemisphere ipsilateral (IR) and contralateral (CON) to the damage following 1 h of MCAO/24 h of reperfusion. A, WT control (CON) and ischemic (IR) brain mitochondria were immunoprecipitated (IP) with anti-acetyl-lysine antibody (Cell Signaling) and probed using anti-CerS1 (Santa Cruz Biotechnology), anti-CerS2 (Novus), anti-CerS4 (Santa Cruz Biotechnology), anti-CerS6 (Novus), anti-NDUFA9 (Abcam), anti-SOD2 (Cell Signaling), or anti-IDH2 (Cell Signaling) antibodies. Input load: 20 μg/lane. B, specific CerS1, CerS2, and CerS6 activity was determined using a 17C-dihydro-sphingosine as a substrate. The enzyme activity is expressed as picomoles of 17C-dihydro-C18:0-ceramide/min per mg of protein (CerS1), picomoles of 17C-dihydro-C24:0-ceramide/min/mg of protein (CerS2), and picomoles of 17C-dihydro-C16:0-ceramide/min/mg of protein (CerS6). Data are means ± S.E., *, p < 0.05, n = 8.
To elucidate how the decreased acetylation of ceramide synthase affected the enzyme activity, CerS1, -2, and -6 specific activities were measured in WT baseline cerebral mitochondria and mitochondria from WT control and ischemic brain. The catalytic activities of ceramide synthases were significantly augmented in WT ischemic mitochondria compared with WT control (Fig. 4B). The data indicate that a decreased acetylation of ceramide synthase enhanced the enzyme activity. To confirm that SIRT3 is required for the increased activity of ceramide synthases, Sirt3 KO mice were employed. Consistent with the decreased ceramide levels in baseline mitochondria of Sirt3 KO mice (Fig. 1A), the activities of CerS1, -2, and -6 were lower in baseline and control mitochondria of Sirt3 KO mice compared with WT. Moreover, SIRT3 knockdown prevented the activation of CerS1, -2, and -6 in cerebral mitochondria after IR (Fig. 4B). The data suggest that IR-induced activation of SIRT3 is responsible for the reduced acetylation of ceramide synthases and increased enzyme activities.
Mitochondrial Ceramide Accumulation Is Reduced in SIRT3-deficient Mice after IR
Emerging evidence indicates cerebral IR-induced ceramide accumulation in brain tissue (9, 11) and specifically in cerebral mitochondria (12). To ascertain the role of SIRT3 in mitochondrial ceramide accumulation after cerebral IR, the ceramide species profile was determined by tandem MS in WT and Sirt3 KO mice. Consistent with the increased activities of mitochondrial CerS1, -2, and -6, the content of the most of ceramide species and total ceramide was greater in WT mitochondria after cerebral IR (Fig. 5, A and B). Sirt3 gene ablation significantly attenuated the IR-induced mitochondrial ceramide species, including C16:0-, C18:0-, C22:0-, C24:0-, and C24:1 ceramide, and total ceramide accumulation (Fig. 5, A and B). However, SIRT3 deficiency did not affect the accumulation of C18:1- and C20:0-ceramide in cerebral mitochondria after IR. The data suggest that SIRT3-mediated deacetylation of ceramide synthases results in increased ceramide accumulation within cerebral mitochondria after experimental stroke.
FIGURE 5.

IR-induced mitochondrial ceramide accumulation is attenuated in SIRT3-deficient mice. Mitochondria were purified from the WT and Sirt3 KO mouse brain hemisphere ipsilateral (IR) and contralateral (Con) to the damage following 1 h of MCAO/24 h of reperfusion. Ceramide species (A) and total ceramide (B) were measured as described under “Experimental Procedures.” Data are means ± S.E., *, p < 0.05, n = 16. Each sample was normalized to its respective total protein levels.
Respiratory Chain Complex I Activity Is Reduced in Baseline SIRT3-deficient Cerebral Mitochondria
Of the three mitochondrial sirtuins (SIRT3–5), only Sirt3 KO mice display robust hyperacetylation of mitochondrial proteins, indicating that SIRT3 is a major mitochondrial deacetylase (30). SIRT3 has been demonstrated to directly deacetylate and activate numerous mitochondrial proteins involved in energy production, including subunits of mitochondrial respiratory chain complex I (33) (complex II and F-ATP synthase (58). To elucidate how the increased acetylation of the respiratory chain subunits modulate the functional activity of the respiratory chain, the oxidative phosphorylation was assessed in baseline cerebral mitochondria of WT and SIRT3 null mice. The respiratory chain enzymes transport electrons from electron donors at complex I or complex II to complex III and then to complex IV where the electrons reach their acceptor oxygen. The oxygen consumption rates supported by substrates of complex I (glutamate), complex II (succinate), and complex IV (ascorbate) were measured in state 2 (ADP-limited) and in the presence of the uncoupler of oxidative phosphorylation, 2,4-dinitrophenol (state 3u). The respiration rate in the presence of uncoupler (state 3u) represents a maximal electron transport chain activity. Similar state 2 respiration rates supported by the substrate of either complex I, complex II, or complex IV were observed in WT and Sirt3 KO mitochondria (Fig. 6A) indicating that lack of SIRT3 did not cause an additional proton leak across the inner mitochondrial membrane. With complex I substrates glutamate plus malate, the maximal respiration rates determined in the presence of uncoupler (state 3u) were about 35% lower in cerebral mitochondria purified from the Sirt3 KO brain compared with WT (Fig. 6A). The maximal respiration rates (state 3u) supported by either substrate of complex II or complex IV were not different between WT and SIRT3-deficient mitochondria indicating that absence of SIRT3 resulted in a decreased activity of complex I. The data suggest that, despite the numerous acetylation sites on mitochondrial respiratory chain enzymes affected by Sirt3 knockdown, only complex I activity was compromised in baseline cerebral mitochondria of Sirt3 KO mice.
FIGURE 6.

Respiratory chain activity in baseline and ischemic cerebral mitochondria from WT and Sirt3 KO mice. Mitochondria were purified from WT and Sirt3 KO mouse brain at baseline (A) and from the WT and Sirt3 KO mouse brain hemispheres ipsilateral (IR) and contralateral (con) to damage following 1 h of MCAO/24 h of reperfusion (B). Mitochondrial respiration was measured by recording oxygen consumption with a Clark-type oxygen electrode in the presence of complex I substrate 5 mm glutamate plus 5 mm malate (glutamate), complex II substrate 10 mm succinate (succinate), and complex IV substrate 1 mm ascorbate plus 250 μm N,N,N′,N′-tetramethyl-p-phenylenediamine (ascorbate) without (state 2) or with 50 μm 2,4-dinitrophenol (state 3u). Data are means ± S.E., *, p < 0.05, n = 16.
Sirt3 Gene Ablation Rescues the IR-induced Defect in Respiratory Complex III Activity
To investigate the role of SIRT3 in mitochondrial respiratory chain response to cerebral IR, the respiratory chain activity (state 3u) was determined in WT and SIRT3-deficient cerebral mitochondria after experimental stroke (Fig. 6B). The respiration rates supported by complex IV substrate (ascorbate) were not affected by IR in either WT or Sirt3 KO mouse mitochondria. In line with previous reports (8, 12), IR reduced the oxygen consumption rate supported by complex I substrate (glutamate) by 33% in WT mitochondria, which could be attributed to the inhibition of complex I or complex III. However, SIRT3 knockdown did not have any effect on glutamate-supported respiration rate after IR, which is consistent with the reduced complex I activity in baseline SIRT3-deficient mitochondria (Fig. 6A, state 3u). In contrast, IR reduced the respiration supported by succinate by 37% in WT mitochondria, which was rescued in SIRT3-deficient mitochondria (Fig. 6B). This indicates that SIRT3 mediates the IR-induced inhibition of complex III activity.
To investigate further the impact of Sirt3 gene ablation on the respiratory chain complex III, the enzymatic activity of complex III was determined in WT and SIRT3-deficient mitochondria after IR. Fig. 7A shows that IR decreased complex III activity by 41% in WT mitochondria, which was rescued in SIRT3-null mitochondria. The results of these studies suggest that IR triggered SIRT3-mediated deacetylation of ceramide synthases that increased their activity and caused ceramide accumulation in mitochondria. Consistent with a previous report (12), accumulated ceramide inhibited respiratory chain activity at the level of complex III in ischemic mitochondria.
FIGURE 7.

Sirt3 gene ablation rescued IR-induced complex III activity defect and diminished ROS formation and protein carbonyls in cerebral mitochondria. Mitochondria were purified from the WT and Sirt3 KO mouse brain hemispheres ipsilateral (IR) and contralateral (Con) to damage following 1 h of MCAO/24 h of reperfusion. A, respiratory chain complex III enzyme activity was determined by measuring the reduction of cytochrome c (cyt c) as described under “Experimental Procedures.” Data are means ± S.E., *, p < 0.05, n = 6. B, ROS generation was measured in the presence of respiratory chain complex I substrate 5 mm glutamate plus 5 mm malate (glutamate + malate) and complex II substrate 10 mm succinate (succinate). Data are means ± S.E., *, p < 0.05, n = 8. Bottom panel, protein carbonyls were assessed as described under “Experimental Procedures.” Data are means ± S.E., *, p < 0.05, n = 8.
IR-induced ROS and Protein Carbonyls Were Attenuated in SIRT3-deficient Cerebral Mitochondria
SIRT3 has been implicated in strengthening the cellular antioxidant defense by activating mitochondrial SOD2 to scavenge ROS. Thus, in response to caloric restriction, SIRT3 reduced cellular ROS levels by deacetylating mitochondrial SOD2, which increased its antioxidative activity in hepatocytes (59). However, in cerebral IR, SIRT3-mediated ceramide accumulation and inhibition of complex III activity could result in an increased ROS generation. It has been reported that exogenous ceramide enhanced ROS generation in isolated mitochondria by interrupting the electron flow at the level of complex III (15, 21).
To determine how SIRT3 impacts the mitochondrial ROS generation in response to IR, the ROS production was measured in cerebral mitochondria purified from WT and Sirt3 KO mouse brain. Mitochondrial ROS were quantified using H2-DCF, which rapidly reacts with ROS to form the highly fluorescent compound dichlorofluorescein. Fig. 7B shows increased ROS generation in ischemic WT mitochondria respiring either on glutamate or succinate, which is consistent with IR-induced blockade of complex III. Importantly, SIRT3 knockdown prevented the IR-induced ROS generation. The data suggest that SIRT3-mediated activation of ceramide synthases leading to accumulation of ceramide is the primary cause of increased ROS production in mitochondria after cerebral IR. To elucidate whether SIRT3-induced ROS generation leads to augmented protein oxidative modification, protein carbonyl levels were determined in WT and Sirt3 KO mitochondria after cerebral IR. The protein carbonyls were higher in WT mitochondria after IR (Fig. 7, bottom panel). SIRT3 knockdown precluded the IR-triggered increase in protein carbonyl levels. The data indicate that SIRT3 is required for IR-induced generation of ROS leading to enhanced oxidative protein modification.
Lack of SIRT3 Did Not Affect MPTP Activity and Mitochondrial CRC
It is widely accepted that MPTP, a high conductance channel, plays a major role in determining the extent of the IR-induced brain injury, which is reflected in the size of the necrotic area or infarct core. In vitro, MPTP opening due to Ca2+ overload and increased ROS causes dissipation of transmembrane potential, ATP hydrolysis, Ca2+ release, pyridine nucleotide depletion, and matrix swelling that results in the outer membrane rupture and the release of pro-apoptotic proteins such as cytochrome c, apoptosis-inducing factor, and endonuclease G (60). CypD is an important modulator that sensitizes MPTP to Ca2+ and confers sensitivity to cyclosporin A, but it is not an essential pore component (61, 62). Recent studies indicate that MPTP forms from the F-ATP synthase, and CypD binding to the oligomycin sensitivity-conferring protein subunit of F-ATP synthase regulates the enzyme's catalytic activity (63). SIRT3 has been shown to deacetylate CypD at lysine 145 diminishing its peptidyl-prolyl cis-trans isomerase activity. Ethanol seems to sensitize the MPTP to opening, in part by inhibiting the SIRT3 activity leading to increased acetylation of CypD in tumor cells (64). In aging, SIRT3-mediated deacetylation of CypD at lysine 166 suppresses the MPTP opening in cardiac mitochondria from 13 months but not from 3- to 6-month-old mice (65).
To investigate the potential role of SIRT3 in regulating MPTP activity, Ca2+-induced MPTP opening was measured in WT and SIRT3-deficient cerebral mitochondria after IR (Fig. 8, A and B). In WT control mitochondria energized by the complex II substrate succinate, while complex I was inhibited by rotenone, an addition of 300 μm Ca2+ resulted in increased mitochondrial swelling indicative of MPTP opening. The addition of pore-forming peptide alamethicin permitted quantification of the maximal mitochondrial swelling response. Fig. 8A shows that IR did not affect the MPTP activity in WT mitochondria. SIRT3 knockdown did not alter the MPTP activity either in control or ischemic mitochondria (Fig. 8B).
FIGURE 8.

Lack of change in MPTP activity and mitochondrial CRC after cerebral IR. Mitochondria were purified from the ipsilateral (IR) and contralateral (CON) hemispheres of WT and Sirt3 KO mouse brain following 1 h of MCAO/24 of h reperfusion. A and B, arrows indicate the addition of mitochondria (MITO). MPTP opening was induced by addition of 300 μm Ca2+ followed by addition of pore-forming peptide alamethicin (ALA) to determine the maximal response. Data are representative of three independent experiments. C, sequential Ca2+ (50 μm each) caused gradual decreases in Ca2+ uptake rates. The arrows indicate the addition of mitochondria (MITO) and Ca2+. Data are representative of four independent experiments. D, quantitative assessment of CRC (nmol of Ca2+/mg of protein). Data are means ± S.E. n = 4.
To confirm the findings, CRC was measured in WT and SIRT3-null cerebral mitochondria (Fig. 8, C and D). Mitochondria energized by succinate in the presence of rotenone were subjected to sequential pulses of 50 μm Ca2+ that resulted in a gradual decrease in Ca2+ uptake rates until the Ca2+ uptake was almost completely inhibited (Fig. 8C). Consistent with lack of cerebral IR effect on MPTP activity, quantification of CRC revealed no changes in the ability of WT cerebral mitochondria to retain Ca2+ due to IR (Fig. 8D). The CRC of SIRT3-deficient mitochondria was similar to the WT mitochondria. The data suggest that cerebral IR-induced SIRT3-mediated elevation of ceramide is not involved in modulation of Ca2+-dependent MPTP activity and mitochondrial Ca2+ homeostasis.
The MPTP appears to serve as a switch between apoptosis and necrosis. Cell death proceeds through necrosis when the MPTP opens causing dissipation of the trans-membrane potential and an inhibition of ATP production. If the MPTP does not open, cellular ATP can be maintained to support the energy demand of apoptosis (61). Of note, the mitochondria used for the experiments were purified from brain peri-infarct tissue where the IR-damaged cells are undergoing apoptotic cell death (66). The results of these studies support the notion that cerebral IR-triggered mechanisms of mitochondrial injury in the peri-infarct tissue do not involve MPTP opening and disturbance of mitochondrial Ca2+ homeostasis.
SIRT3 Enhanced Active Bax Association with Mitochondria after IR
Although MPTP opening plays an important role in necrotic cell death, the mitochondrial outer membrane (MOM) permeabilization has been implicated as a key mechanism of apoptotic cell death. In response to apoptotic stimuli, two members of the Bcl-2 family, proapoptotic proteins Bak and Bax, could form large MOM pores permitting translocation of proapoptotic factors such as cytochrome c from inter-mitochondrial membrane space to the cytoplasm. In non-apoptotic cells, Bax continually binds to mitochondria and retrotranslocates back to the cytoplasm through interaction with Bcl-xL (67). An inhibition of retro-translocation to the cytosol following apoptotic insults causes Bax to accumulate in MOM. Both identified (Bid, Bim, and Puma) and unidentified MOM resident proteins have been proposed to activate Bax and Bak facilitating MOM permeabilization (68). In contrast to Bax, Bak is constitutively inserted in the MOM.
To elucidate a possible role of SIRT3 in regulating the MOM permeabilization, the levels of proapoptotic (Bax and Bak) and anti-apoptotic Bcl-xL were analyzed in cerebral mitochondria of WT and SIRT3-deficient mice after IR. The expression levels of active Bax, Bak and Bcl-xL in baseline cerebral mitochondria were similar in WT and SIRT3-deficient mice (Fig. 9A). Western blot analysis revealed IR-induced accumulation of active Bax in WT mitochondria, which was rescued by SIRT3 knockdown (Fig. 9B). There were no significant changes in Bak after IR. Although Bcl-xL was slightly attenuated after IR, the lack of SIRT3 did not have any effect. In line with the increased MOM permeabilization, cytochrome c level was reduced in WT mitochondria after IR, although there was no change in cytochrome c expression in SIRT3-deficient mitochondria (Fig. 9B). The data suggest that SIRT3-dependent ceramide accumulation contributes to Bax-mediated MOM permeabilization and pro-apoptotic protein release from cerebral mitochondria after IR.
FIGURE 9.
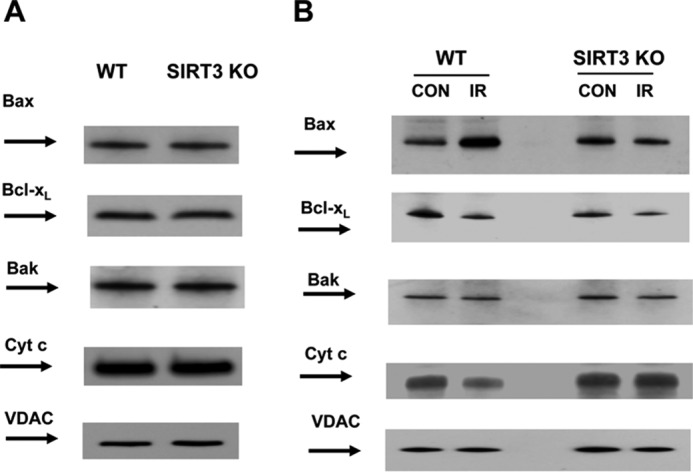
IR-enhanced mitochondrial active Bax is attenuated in SIRT3-deficient mice. Mitochondria were purified from the brain of WT and Sirt3 KO mice at the baseline (A) or from the ipsilateral (IR) and contralateral (CON) hemispheres of WT and Sirt3 KO mouse brain following 1 h of MCAO/24 h of reperfusion (B). Equal amount of mitochondrial sample (20 μg) was loaded into the lane. The expression of Bcl-2 family proteins and cytochrome c (Cyt c) was characterized by Western blotting using anti-active Bax (6A7), anti-Bak, anti-Bcl-xL and anti-cytochrome c-specific antibodies. To confirm equal loading of samples, the membranes were stripped and probed with anti-voltage-dependent anion channel (VDAC) antibody. Data are representative of three independent experiments.
SIRT3 Deficiency Attenuated Brain Damage after IR
To assess the role of SIRT3 in promoting brain injury after stroke, the infarct volume was measured in WT and Sirt3 KO mice after cerebral IR. Brain injury after IR was significantly attenuated in SIRT3-deficient mice, indicating the important role of the mitochondrial SIRT3 in promoting brain injury (Fig. 10, A and B). The data suggest a critical role for SIRT3-mediated ceramide accumulation in promoting mitochondrial dysfunction and brain injury after stroke.
FIGURE 10.

Sirt3 gene ablation or MR treatment reduced brain damage after cerebral IR. A, representative images of WT and SIRT3KO mice brain damage after 1 h of MCAO/24 h of reperfusion. B, infarct volume was determined using TTC staining of brain slices and image analysis as described under “Experimental Procedures.” Data are means ± S.E., *, p < 0.05, n = 8. C and D, WT mice were given myriocin via gavage (2 mg/kg) following 1 h of MCAO/3 h of reperfusion. WT mice control group received a vehicle control. Representative images of brain damage in vehicle control (WT IR) and MR treatment (WT IR + MR) group at 24 h of reperfusion (C). Infarct volume was determined after 1 h of MCAO/24 h of reperfusion by TTC staining of brain slices and image analysis (D). Data are means ± S.E., *, p < 0.05, n = 8.
Myriocin Treatment Decreased Mitochondrial Ceramide and Brain Injury after IR
In response to cerebral IR, ceramide increase could be due to activation of SM hydrolysis by acid sphingomyelinase and/or stimulation of de novo ceramide biosynthesis. Thus, there was less injury in the brain of acid sphingomyelinase-deficient mice, which was accompanied by decreased accumulation of ceramide in brain tissue; however, mitochondrial ceramide levels were not investigated (9). Of note, IR-induced activation of de novo ceramide biosynthesis in the brain tissue that was associated with stimulation of ceramide synthase activity, accumulation of ceramide, and respiratory chain damage in cerebral mitochondria was decreased in the JNK3-deficient mice. The infarct size was significantly lower in the JNK3-deficient mice suggesting that JNK3 is an upstream regulator of the mitochondrial ceramide biosynthesis, which could play an important role in IR brain response (12). An inhibitor of serine palmitoyltransferase, a key enzyme of de novo ceramide biosynthesis, myriocin (MR) has been shown to reduce ceramide tissue levels in various disease models (69–71). To shed more light on mitochondrial ceramide involvement in promoting brain injury after IR, WT mice were treated with MR dissolved in DMSO and diluted with 50% Tween 20 in PBS. Mice received 0.2 ml of MR (2 mg/kg) through a gavage after 1 h of MCAO followed 3 h of reperfusion. Control group received the vehicle control. Importantly, MR did not affect physiological parameters in mice, including a mean arterial blood pressure, heart rate, arterial pH levels, PaCO2 and PaO2. Brains were subjected to analysis at 24 h post-MCAO. Importantly, there was reduced brain injury in mice treated with MR (Fig. 10, C and D). Ceramide species were significantly attenuated in mitochondria from MR-treated mice after IR (Fig. 11). The data provide further support for the essential role of mitochondrial ceramide in brain injury after IR.
FIGURE 11.

Myriocin treatment reduced mitochondrial ceramide after IR. WT mice were given MR via gavage (2 mg/kg) following 1 h of MCAO/3 h of reperfusion. WT mice control group received a vehicle control. Mitochondria were isolated from the ipsilateral (IR) and contralateral (Con) hemispheres after 1 h of MCAO/24 h of reperfusion, and ceramide species were determined by tandem MS. Data are means ± S.E., *, p < 0.05, n = 12.
Discussion
The present studies provide evidence for a novel SIRT3-dependent mechanism furthering mitochondrial dysfunction and brain injury after experimental stroke. Our data suggest that cerebral IR triggers SIRT3-mediated deacetylation of CerS1, -2, and -6, which results in the accumulation of ceramide, mitochondrial dysfunction, and generation of ROS promoting brain injury (Fig. 12). This is the first demonstration of the essential role of SIRT3 in cerebral mitochondrial dysfunction after stroke. Given the significance of mitochondria in regulation of neural cell function and survival, the characterization of SIRT3-mediated mitochondrial impairment triggered by cerebral IR is a valuable contribution to our understanding of the pathophysiological mechanisms of brain damage after stroke.
FIGURE 12.
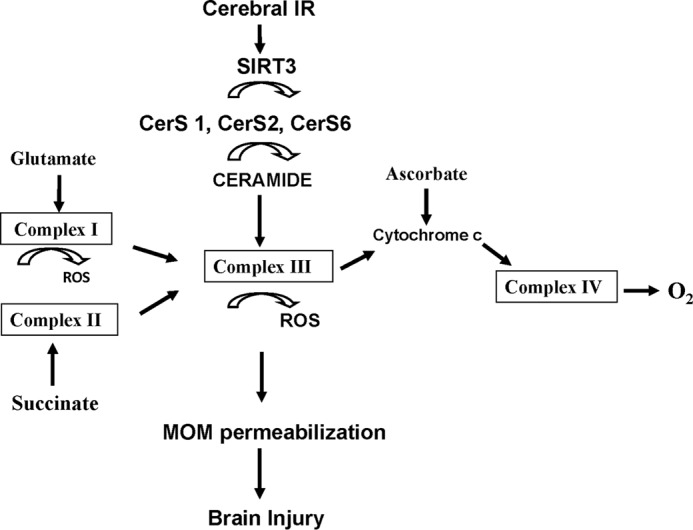
Proposed model of SIRT3 involvement in promoting mitochondrial dysfunction and brain injury after experimental stroke. Cerebral IR leads by an as yet unidentified mechanism to SIRT3 activation resulting in enhanced deacetylation of mitochondrial ceramide synthases 1, 2, and 6 that increases their activity leading to accumulation of ceramide and mitochondrial dysfunction. Accumulated ceramide inhibits respiratory chain activity at the level of complex III that is accompanied by increased ROS and augmented expression of activated Bax that contributes to MOM permeabilization and brain injury.
Our studies identify a novel mechanistic link between SIRT3 and mitochondrial sphingolipid metabolism that is important for mitochondrial response to cerebral IR. Here, we show that ceramide is selectively down-regulated, whereas sphingosine and S1P are not changed in cerebral mitochondria of SIRT3-deficient mice (Fig. 1, A and B). Furthermore, our studies indicate that mitochondrial ceramide synthases CerS1, CerS2, and CerS6, but not CerS4, are the substrates and binding partners of SIRT3 (Figs. 1C and 2). Importantly, SIRT3-mediated deacetylation increased the catalytic activity of the enzymes (Fig. 4), which resulted in accumulation of ceramide in mitochondrial after cerebral IR (Fig. 5). Importantly, SIRT3 deacetylated and activated mitochondrial CerS1, -2, and -6 in vitro (Fig. 3). Based on rapid changes of CerS activity to various stimuli, post-translational mechanisms of CerS regulation have been postulated, but the experimental evidence is scarce (56, 72). Thus, CerS1, CerS2, and CerS5 were predicted to be phosphorylated by a large scale phosphorylation analysis (73). Our studies are the first demonstration of post-translational regulation of CerS activity by acetylation/deacetylation.
These studies provide further insight into spatial organization of ceramide synthesis showing that three mitochondrial ceramide synthases CerS1, -2, and -6 are forming a tripartite complex associated with SIRT3. It has been previously reported that CerS2, -5, and -6 might exist as heterodimers in HeLa cells (74). Furthermore, the activity of CerS seems to be modulated by the formation of both homo- and heterodimers in the overexpression system (56). The results of these studies draw attention to the mitochondrial multi-CerS complex, in which activity is modulated by the primary mitochondrial deacetylase, SIRT3. Given a dual intracellular localization of CerS (12) in the ER and in mitochondria, SIRT3-mediated deacetylation of mitochondrial ceramide synthases suggests a possible involvement of another SIRT family member in the regulation of CerS acetylation in the ER. Thus, it has been shown that SIRT1 deacetylates AceCS1 and HMGCS1 in the cytoplasm, whereas SIRT3 deacetylates the homologous AceCS2 and HMGCS2 in the mitochondria (75).
Our studies point to a critical role of SIRT3 as a facilitator of ceramide generation in mitochondria leading to reduced respiratory complex III activity after cerebral IR. One major function of SIRT3 is regulation of mitochondrial electron transport chain activity to maintain energy homeostasis. SIRT3 interacts with complex I component NDUFA9 and both subunits of complex II that in the context of SIRT3 knockdown correlates with the decreased respiratory chain complex activity (33, 58). In contrast to previous reports showing the reduced complex II activity in liver and brown adipose tissue mitochondria from Sirt3 KO mice (35, 58), there was no reduction of complex II activity in cerebral mitochondria of Sirt3 KO mice (Fig. 6A). However, consistent with previous findings in liver mitochondria (33), complex I activity was significantly reduced in cerebral mitochondria purified from Sirt3 KO mice (Fig. 6A). The results of our studies further support the concept that SIRT3-mediated deacetylation regulates its target activity in a tissue- and stimulus-specific manner (58). Overall, these studies reveal SIRT3 as a novel determinant of mitochondrial dysfunction in cerebral IR.
In response to cerebral IR, Sirt3 gene ablation rescued the IR-induced defect at the level of complex III (Figs. 6B and 7A). The data suggest that SIRT3-mediated activation of CerS and accumulation of ceramide are responsible for cerebral IR-induced impairment of complex III. This is in line with our previous studies showing that IR triggered JNK3-dependent accumulation of mitochondrial ceramide that selectively inhibited respiration at the level of complex III (12). Furthermore, it suggests that JNK3 could be a potential upstream regulator of SIRT3 function in response to cerebral IR.
Our studies extend the experimental evidence of an important role of ROS in mitochondrial damage after experimental stroke. Here, we show that IR stimulated ROS generation during the oxidation of the substrates by respiratory chain in cerebral mitochondria (Fig. 7B). Direct interaction of proteins with ROS can lead to formation of protein derivatives or peptide fragments possessing carbonyl groups; therefore, the level of carbonyl groups in proteins has been widely used as a marker of oxidative protein damage. As expected, cerebral IR augmented the protein carbonyl levels in mitochondria (Fig. 7, bottom panel). Remarkably, SIRT3 knockdown reduced the ROS generation and protein carbonyls in mitochondria after stroke. This is consistent with the hypothesis that cerebral IR triggers SIRT3-dependent ceramide-mediated inhibition of complex III leading to increased generation of ROS and protein carbonylation.
The results of our studies demonstrating a critical role of SIRT3 in mitochondrial ROS generation after cerebral IR do not contradict the evidence of SIRT3 involvement in cellular antioxidant defense system (76). SIRT3 interacts and directly deacetylates hepatic SOD2, a primary mitochondrial antioxidant enzyme, enhancing its ability to scavenge ROS in response to caloric restriction (59). In addition, SIRT3 deacetylates an isocitrate dehydrogenase 2 (IDH2), a tricarboxylic acid cycle enzyme producing NADPH, due to caloric restriction. Enhanced activity of IDH2 by SIRT3-mediated deacetylation produced increased levels of NADPH that resulted in increased activity of glutathione reductase and augmented generation of glutathione, a major cellular antioxidant (57).
The results of our studies suggest that IR-induced SIRT3-mediated deacetylation activates both the antioxidant defense pathways (SOD2 and IDH2) and the CerS-dependent ROS-generating pathway. The data are consistent with the notion that IR-induced ROS production exceeds the availability of endogenous antioxidants and provides a mechanistic insight into the pivotal role of ROS in cerebral IR. This is in line with results of studies showing robust neuroprotection in animals modified to bolster endogenous mitochondrial antioxidants (77, 78). Altogether, these studies provide further support for the concept that the SIRT3-mediated outcome is produced by a dynamic interplay of multiple physiological cascades, emphasizing the importance of defining the stimulus- and tissue-specific context.
Our studies showed that SIRT3-dependent excessive accumulation of mitochondrial ceramide did not have any impact on MPTP activity and mitochondrial Ca2+ handling after cerebral IR. The results of these studies are consistent with the previously demonstrated role of MPTP opening as a cause of the neural cell demise by necrosis in the infarct core after stroke, although it remains closed during apoptotic cell death in the penumbra (61). The potential role of Ca2+-dependent MPTP opening in IR-induced mitochondrial injury has been recently challenged. In mice lacking mitochondrial calcium uniporter, where Ca2+ overload does not occur during reperfusion, the extent of necrosis was the same as that observed in the hearts from WT littermates (79). These findings raise many questions such as what the mechanism of IR-induced necrotic cell death is and what the mechanism of MPTP opening in the hearts of mitochondrial calcium uniporter-null mice is. Certainly, investigation into the mechanisms of MPTP opening after cerebral IR in mitochondrial calcium uniporter-deficient mice is warranted.
Having demonstrated the lack of MPTP response to cerebral IR, we investigated a potential SIRT3 role in modulating the permeability of the MOM. Most apoptotic signaling pathways converge on mitochondria to initiate MOM permeabilization that results in the release of pro-apoptotic proteins such as cytochrome c from the inter-membrane space. Bcl-2 family proteins, Bax, Bak, and Bcl-xL, function through a complex set of heterodimeric interactions regulating MOM permeabilization and apoptotic cell death (68). Bax/Bak oligomerization within the MOM is thought to be a major mechanism of formation of the pores permitting the release of pro-apoptotic proteins. Recently, the ability of monomeric Bax to initiate the formation of large pores in the MOM was demonstrated (80). Our studies point to an important role of SIRT3 in modulation of Bax-mediated permeabilization of MOM. Cerebral IR resulted in elevated active monomeric Bax association with mitochondria, which was attenuated in SIRT3-deficient mitochondria (Fig. 9B). The results from this study implicate SIRT3 as an upstream regulator of Bax-dependent MOM permeabilization in response to cerebral IR.
Our studies also provide experimental evidence for critical involvement of sphingolipids in the regulation of MOM permeabilization and apoptosis (81). Recently, sphingolipid S1P and its breakdown product, hexadecenal, have been identified as cofactors that can promote Bak and Bax-mediated MOM permeabilization, respectively (82). The results of our studies support the notion that another sphingolipid ceramide is an essential determinant of Bax-dependent MOM permeabilization (83) after cerebral IR. These studies suggest that SIRT3-dependent accumulation of mitochondrial ceramide could facilitate Bax-dependent pore formation. This is concordant with a previous report showing that inhibiting CerS activity with fumonisin B1 prevented radiation-induced elevation of ceramide, accumulation of Bax, and MOM permeabilization in HeLa cells (84).
Our studies are the first demonstration of the pathophysiological role of SIRT3 in promoting mitochondrial dysfunction (Fig. 6B) and brain damage after experimental stroke (Fig. 10, A and B). In light of the oxidative stress-ameliorative effects assigned to SIRT3 function in caloric restriction studies and the protective effect of overexpressed SIRT3 in neural cells (41, 42), stimulation of SIRT3 activity has been proposed to reduce mitochondrial deficits associated with cerebral oxidative stress and neurodegeneration (76). However, our studies showing that Sirt3 gene ablation protected mitochondria against cerebral IR-induced damage suggest that small molecule inhibitors of SIRT3 activity could be beneficial in the stroke treatment. Overall, our studies support the notion that SIRT3-dependent deacetylation of mitochondrial proteins functions in a target substrate- and tissue-specific manner to fine-tune multiple metabolic programs.
In summary, this study provides experimental evidence that cerebral IR triggers SIRT3-mediated deacetylation and activation of ceramide synthases 1, 2, and 6 leading to accumulation of mitochondrial ceramide. The accumulated ceramide blocks respiratory complex III function, which results in the elevation of ROS and protein carbonyls promoting MOM permeabilization and brain damage. The data shed more light on the role and regulation of the mitochondrial ceramide synthases and accentuate the function of sphingolipid-metabolizing enzymes in the pathobiology of stroke.
Author Contributions
S. A. N. and T. I. G. conceived and coordinated the study and wrote the paper. D. B. L. and W. B. M. contributed to the design of the study and interpretation of the results. S. A. N. and T. I. G. performed and analyzed the experiments in Figs. 5, 6, 8, and 11. C. L. R. and J. A. K. performed and analyzed the experiments in Figs. 1–4, 7, and 9. M. S. K. and J. Y. designed, performed, and analyzed the experiments in Fig. 10. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Alexander S. Novgorodov for help with preparation of the manuscript.
This work was supported in part by National Institutes of Health Grants R01NS083544 (to T. I. G.), R01GM101171 (to D. B. L.), and P30 GM103339 (to Lipidomics Core Facility. Medical University of South Carolina) and Veterans Affairs Merit Award I01BX001104 (to T. I. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IR
- ischemia/reperfusion
- CRC
- Ca2+-retention capacity
- CerS
- ceramide synthase
- CypD
- cyclophilin D
- H2-DCF
- 2′,7′-dichlorodihydrofluorescein
- ER
- endoplasmic reticulum
- MCAO
- middle cerebral artery occlusion
- MPTP
- mitochondrial permeability transition pore
- MOM
- mitochondrial outer membrane
- MR
- myriocin
- NAM
- nicotinamide
- ROS
- reactive oxygen species
- SIRT
- sirtuin
- S1P
- sphingosine 1-phosphate
- TTC
- 2,3,5-triphenyltetrazolium chloride
- ICA
- internal carotid artery
- TTC
- 2,3,5-triphenyltetrazolium chloride.
References
- 1. Hertz L. (2008) Bioenergetics of cerebral ischemia: a cellular perspective. Neuropharmacology 55, 289–309 [DOI] [PubMed] [Google Scholar]
- 2. Vandenabeele P., Galluzzi L., Vanden Berghe T., and Kroemer G. (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 [DOI] [PubMed] [Google Scholar]
- 3. Watts L. T., Lloyd R., Garling R. J., and Duong T. (2013) Stroke neuroprotection: targeting mitochondria. Brain Sci. 3, 540–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanderson T. H., Reynolds C. A., Kumar R., Przyklenk K., and Hüttemann M. (2013) Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 47, 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fiskum G. (2000) Mitochondrial participation in ischemic and traumatic neural cell death. J. Neurotrauma 17, 843–855 [DOI] [PubMed] [Google Scholar]
- 6. Polster B. M., and Fiskum G. (2004) Mitochondrial mechanisms of neural cell apoptosis. J. Neurochem. 90, 1281–1289 [DOI] [PubMed] [Google Scholar]
- 7. Chan P. H. (2004) Future targets and cascades for neuroprotective strategies. Stroke 35, 2748–2750 [DOI] [PubMed] [Google Scholar]
- 8. Sims N. R., and Anderson M. F. (2002) Mitochondrial contributions to tissue damage in stroke. Neurochem. Int. 40, 511–526 [DOI] [PubMed] [Google Scholar]
- 9. Yu Z. F., Nikolova-Karakashian M., Zhou D., Cheng G., Schuchman E. H., and Mattson M. P. (2000) Pivotal role for acidic sphingomyelinase in cerebral ischemia-induced ceramide and cytokine production, and neuronal apoptosis. J. Mol. Neurosci. 15, 85–97 [DOI] [PubMed] [Google Scholar]
- 10. Herr I., Martin-Villalba A., Kurz E., Roncaioli P., Schenkel J., Cifone M. G., and Debatin K. M. (1999) FK506 prevents stroke-induced generation of ceramide and apoptosis signaling. Brain Res. 826, 210–219 [DOI] [PubMed] [Google Scholar]
- 11. Ohtani R., Tomimoto H., Kondo T., Wakita H., Akiguchi I., Shibasaki H., and Okazaki T. (2004) Upregulation of ceramide and its regulating mechanism in a rat model of chronic cerebral ischemia. Brain Res. 1023, 31–40 [DOI] [PubMed] [Google Scholar]
- 12. Yu J., Novgorodov S. A., Chudakova D., Zhu H., Bielawska A., Bielawski J., Obeid L. M., Kindy M. S., and Gudz T. I. (2007) JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J. Biol. Chem. 282, 25940–25949 [DOI] [PubMed] [Google Scholar]
- 13. Novgorodov S. A., and Gudz T. I. (2011) Ceramide and mitochondria in ischemic brain injury. Int. J. Biochem. Mol. Biol. 2, 347–361 [PMC free article] [PubMed] [Google Scholar]
- 14. Anderson M. F., and Sims N. R. (1999) Mitochondrial respiratory function and cell death in focal cerebral ischemia. J. Neurochem. 73, 1189–1199 [DOI] [PubMed] [Google Scholar]
- 15. Gudz T. I., Tserng K. Y., and Hoppel C. L. (1997) Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J. Biol. Chem. 272, 24154–24158 [DOI] [PubMed] [Google Scholar]
- 16. Di Paola M., Cocco T., and Lorusso M. (2000) Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 39, 6660–6668 [DOI] [PubMed] [Google Scholar]
- 17. Ste-Marie L., Vachon P., Vachon L., Bémeur C., Guertin M. C., and Montgomery J. (2000) Hydroxyl radical production in the cortex and striatum in a rat model of focal cerebral ischemia. Can. J. Neurol. Sci. 27, 152–159 [PubMed] [Google Scholar]
- 18. Mori T., Asano T., Matsui T., Muramatsu H., Ueda M., Kamiya T., Katayama Y., and Abe T. (1999) Intraluminal increase of superoxide anion following transient focal cerebral ischemia in rats. Brain Res. 816, 350–357 [DOI] [PubMed] [Google Scholar]
- 19. Dirnagl U., Lindauer U., Them A., Schreiber S., Pfister H. W., Koedel U., Reszka R., Freyer D., and Villringer A. (1995) Global cerebral ischemia in the rat: online monitoring of oxygen free radical production using chemiluminescence in vivo. J. Cereb. Blood Flow Metab. 15, 929–940 [DOI] [PubMed] [Google Scholar]
- 20. Kushnareva Y., Murphy A. N., and Andreyev A. (2002) Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 368, 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. García-Ruiz C., Colell A., Marí M., Morales A., and Fernández-Checa J. C. (1997) Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 272, 11369–11377 [DOI] [PubMed] [Google Scholar]
- 22. Hannun Y. A., and Obeid L. M. (2011) Many ceramides. J. Biol. Chem. 286, 27855–27862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stancevic B., and Kolesnick R. (2010) Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 584, 1728–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kitatani K., Idkowiak-Baldys J., and Hannun Y. A. (2008) The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 20, 1010–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tidhar R., and Futerman A. H. (2013) The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2511–2518 [DOI] [PubMed] [Google Scholar]
- 26. Spassieva S., Seo J. G., Jiang J. C., Bielawski J., Alvarez-Vasquez F., Jazwinski S. M., Hannun Y. A., and Obeid L. M. (2006) Necessary role for the Lag1p motif in (dihydro)ceramide synthase activity. J. Biol. Chem. 281, 33931–33938 [DOI] [PubMed] [Google Scholar]
- 27. Mizutani Y., Kihara A., and Igarashi Y. (2005) Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem. J. 390, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deng X., Yin X., Allan R., Lu D. D., Maurer C. W., Haimovitz-Friedman A., Fuks Z., Shaham S., and Kolesnick R. (2008) Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science 322, 110–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Imai S., and Guarente L. (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lombard D. B., Alt F. W., Cheng H. L., Bunkenborg J., Streeper R. S., Mostoslavsky R., Kim J., Yancopoulos G., Valenzuela D., Murphy A., Yang Y., Chen Y., Hirschey M. D., Bronson R. T., Haigis M., et al. (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang J. Y., Hirschey M. D., Shimazu T., Ho L., and Verdin E. (2010) Mitochondrial sirtuins. Biochim. Biophys. Acta 1804, 1645–1651 [DOI] [PubMed] [Google Scholar]
- 32. Hirschey M. D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D. B., Grueter C. A., Harris C., Biddinger S., Ilkayeva O. R., Stevens R. D., Li Y., Saha A. K., Ruderman N. B., Bain J. R., et al. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ahn B. H., Kim H. S., Song S., Lee I. H., Liu J., Vassilopoulos A., Deng C. X., and Finkel T. (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 14447–14452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schlicker C., Gertz M., Papatheodorou P., Kachholz B., Becker C. F., and Steegborn C. (2008) Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 382, 790–801 [DOI] [PubMed] [Google Scholar]
- 35. Verdin E., Hirschey M. D., Finley L. W., and Haigis M. C. (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem. Sci. 35, 669–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pillai V. B., Sundaresan N. R., Jeevanandam V., and Gupta M. P. (2010) Mitochondrial SIRT3 and heart disease. Cardiovasc. Res. 88, 250–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pillai V. B., Sundaresan N. R., Kim G., Gupta M., Rajamohan S. B., Pillai J. B., Samant S., Ravindra P. V., Isbatan A., and Gupta M. P. (2010) Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 285, 3133–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sundaresan N. R., Gupta M., Kim G., Rajamohan S. B., Isbatan A., and Gupta M. P. (2009) Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 119, 2758–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim H. S., Patel K., Muldoon-Jacobs K., Bisht K. S., Aykin-Burns N., Pennington J. D., van der Meer R., Nguyen P., Savage J., Owens K. M., Vassilopoulos A., Ozden O., Park S. H., Singh K. K., Abdulkadir S. A., et al. (2010) SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17, 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Allison S. J., and Milner J. (2007) SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle 6, 2669–2677 [DOI] [PubMed] [Google Scholar]
- 41. Kim S. H., Lu H. F., and Alano C. C. (2011) Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS ONE 6, e14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shulyakova N., Sidorova-Darmos E., Fong J., Zhang G., Mills L. R., and Eubanks J. H. (2014) Over-expression of the Sirt3 sirtuin protects neuronally differentiated PC12 cells from degeneration induced by oxidative stress and trophic withdrawal. Brain Res. 1587, 40–53 [DOI] [PubMed] [Google Scholar]
- 43. Gary D. S., Bruce-Keller A. J., Kindy M. S., and Mattson M. P. (1998) Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J. Cereb. Blood Flow Metab. 18, 1283–1287 [DOI] [PubMed] [Google Scholar]
- 44. Swanson R. A., Morton M. T., Tsao-Wu G., Savalos R. A., Davidson C., and Sharp F. R. (1990) A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab. 10, 290–293 [DOI] [PubMed] [Google Scholar]
- 45. Novgorodov S. A., Chudakova D. A., Wheeler B. W., Bielawski J., Kindy M. S., Obeid L. M., and Gudz T. I. (2011) Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis. J. Biol. Chem. 286, 4644–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Novgorodov S. A., Riley C. L., Yu J., Borg K. T., Hannun Y. A., Proia R. L., Kindy M. S., and Gudz T. I. (2014) Essential roles of neutral ceramidase and sphingosine in mitochondrial dysfunction due to traumatic brain injury. J. Biol. Chem. 289, 13142–13154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dutta R., McDonough J., Yin X., Peterson J., Chang A., Torres T., Gudz T., Macklin W. B., Lewis D. A., Fox R. J., Rudick R., Mirnics K., and Trapp B. D. (2006) Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 59, 478–489 [DOI] [PubMed] [Google Scholar]
- 48. Novgorodov S. A., Gudz T. I., and Obeid L. M. (2008) Long-chain ceramide is a potent inhibitor of the mitochondrial permeability transition pore. J. Biol. Chem. 283, 24707–24717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Novgorodov A. S., El-Alwani M., Bielawski J., Obeid L. M., and Gudz T. I. (2007) Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J. 21, 1503–1514 [DOI] [PubMed] [Google Scholar]
- 50. Chudakova D. A., Zeidan Y. H., Wheeler B. W., Yu J., Novgorodov S. A., Kindy M. S., Hannun Y. A., and Gudz T. I. (2008) Integrin-associated Lyn kinase promotes cell survival by suppressing acid sphingomyelinase activity. J. Biol. Chem. 283, 28806–28816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Malley Y., Fink B. D., Ross N. C., Prisinzano T. E., and Sivitz W. I. (2006) Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. J. Biol. Chem. 281, 39766–39775 [DOI] [PubMed] [Google Scholar]
- 52. Merrill A. H., Jr. (2002) De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J. Biol. Chem. 277, 25843–25846 [DOI] [PubMed] [Google Scholar]
- 53. Mandon E. C., Ehses I., Rother J., van Echten G., and Sandhoff K. (1992) Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 267, 11144–11148 [PubMed] [Google Scholar]
- 54. Chen Y., Zhang J., Lin Y., Lei Q., Guan K. L., Zhao S., and Xiong Y. (2011) Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 12, 534–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shulga N., Wilson-Smith R., and Pastorino J. G. (2010) Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J. Cell Sci. 123, 894–902 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56. Laviad E. L., Kelly S., Merrill A. H. Jr., and Futerman A. H. (2012) Modulation of ceramide synthase activity via dimerization. J. Biol. Chem. 287, 21025–21033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Someya S., Yu W., Hallows W. C., Xu J., Vann J. M., Leeuwenburgh C., Tanokura M., Denu J. M., and Prolla T. A. (2010) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Finley L. W., Haas W., Desquiret-Dumas V., Wallace D. C., Procaccio V., Gygi S. P., and Haigis M. C. (2011) Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 6, e23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Qiu X., Brown K., Hirschey M. D., Verdin E., and Chen D. (2010) Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667 [DOI] [PubMed] [Google Scholar]
- 60. Bernardi P., and Di Lisa F. (2015) The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 78, 100–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., and Tsujimoto Y. (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 [DOI] [PubMed] [Google Scholar]
- 62. Schinzel A. C., Takeuchi O., Huang Z., Fisher J. K., Zhou Z., Rubens J., Hetz C., Danial N. N., Moskowitz M. A., and Korsmeyer S. J. (2005) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 102, 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Giorgio V., Bisetto E., Soriano M. E., Dabbeni-Sala F., Basso E., Petronilli V., Forte M. A., Bernardi P., and Lippe G. (2009) Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 284, 33982–33988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shulga N., and Pastorino J. G. (2010) Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. J. Cell Sci. 123, 4117–4127 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65. Hafner A. V., Dai J., Gomes A. P., Xiao C. Y., Palmeira C. M., Rosenzweig A., and Sinclair D. A. (2010) Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li Y., Powers C., Jiang N., and Chopp M. (1998) Intact, injured, necrotic and apoptotic cells after focal cerebral ischemia in the rat. J. Neurol. Sci. 156, 119–132 [DOI] [PubMed] [Google Scholar]
- 67. Edlich F., Banerjee S., Suzuki M., Cleland M. M., Arnoult D., Wang C., Neutzner A., Tjandra N., and Youle R. J. (2011) Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Volkmann N., Marassi F. M., Newmeyer D. D., and Hanein D. (2014) The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ. 21, 206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Park T. S., Rosebury W., Kindt E. K., Kowala M. C., and Panek R. L. (2008) Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol. Res. 58, 45–51 [DOI] [PubMed] [Google Scholar]
- 70. Kurek K., Wiesiol̸ek-Kurek P., Piotrowska D. M., L̸ukaszuk B., Chabowski A., and Żendzianendzian-Piotrowska M. (2014) Inhibition of ceramide de novo synthesis with myriocin affects lipid metabolism in the liver of rats with streptozotocin-induced type 1 diabetes. Biomed. Res. Int. 2014, 980815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chun L., Junlin Z., Aimin W., Niansheng L., Benmei C., and Minxiang L. (2011) Inhibition of ceramide synthesis reverses endothelial dysfunction and atherosclerosis in streptozotocin-induced diabetic rats. Diabetes Res. Clin. Pract. 93, 77–85 [DOI] [PubMed] [Google Scholar]
- 72. Park J. W., Park W. J., and Futerman A. H. (2014) Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochim. Biophys. Acta 1841, 671–681 [DOI] [PubMed] [Google Scholar]
- 73. Villén J., Beausoleil S. A., Gerber S. A., and Gygi S. P. (2007) Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A. 104, 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mesicek J., Lee H., Feldman T., Jiang X., Skobeleva A., Berdyshev E. V., Haimovitz-Friedman A., Fuks Z., and Kolesnick R. (2010) Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell. Signal. 22, 1300–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hirschey M. D., Shimazu T., Capra J. A., Pollard K. S., and Verdin E. (2011) SIRT1 and SIRT3 deacetylate homologous substrates: AceCS1,2 and HMGCS1,2. Aging 3, 635–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kincaid B., and Bossy-Wetzel E. (2013) Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Niizuma K., Yoshioka H., Chen H., Kim G. S., Jung J. E., Katsu M., Okami N., and Chan P. H. (2010) Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta 1802, 92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Keller J. N., Kindy M. S., Holtsberg F. W., St Clair D. K., Yen H. C., Germeyer A., Steiner S. M., Bruce-Keller A. J., Hutchins J. B., and Mattson M. P. (1998) Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 18, 687–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pan X., Liu J., Nguyen T., Liu C., Sun J., Teng Y., Fergusson M. M., Rovira I. I., Allen M., Springer D. A., Aponte A. M., Gucek M., Balaban R. S., Murphy E., and Finkel T. (2013) The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gillies L. A., Du H., Peters B., Knudson C. M., Newmeyer D. D., and Kuwana T. (2015) Visual and functional demonstration of growing Bax-induced pores in mitochondrial outer membranes. Mol. Biol. Cell 26, 339–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Patwardhan G. A., Beverly L. J., and Siskind L. J. (2015) Sphingolipids and mitochondrial apoptosis. J. Bioenerg. Biomembr. 10.1007/s10863-015-9602-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chipuk J. E., McStay G. P., Bharti A., Kuwana T., Clarke C. J., Siskind L. J., Obeid L. M., and Green D. R. (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kashkar H., Wiegmann K., Yazdanpanah B., Haubert D., and Krönke M. (2005) Acid sphingomyelinase is indispensable for UV light-induced Bax conformational change at the mitochondrial membrane. J. Biol. Chem. 280, 20804–20813 [DOI] [PubMed] [Google Scholar]
- 84. Lee H., Rotolo J. A., Mesicek J., Penate-Medina T., Rimner A., Liao W. C., Yin X., Ragupathi G., Ehleiter D., Gulbins E., Zhai D., Reed J. C., Haimovitz-Friedman A., Fuks Z., and Kolesnick R. (2011) Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS ONE 6, e19783. [DOI] [PMC free article] [PubMed] [Google Scholar]