

Abstract
A newly identified pathway for selective degradation of the common mutant of the cystic fibrosis transmembrane conductance regulator (CFTR), F508del, is initiated by binding of the small heat shock protein, Hsp27. Hsp27 collaborates with Ubc9, the E2 enzyme for protein SUMOylation, to selectively degrade F508del CFTR via the SUMO-targeted ubiquitin E3 ligase, RNF4 (RING finger protein 4) (1). Here, we ask what properties of CFTR are sensed by the Hsp27-Ubc9 pathway by examining the ability of NBD1 (locus of the F508del mutation) to mimic the disposal of full-length (FL) CFTR. Similar to FL CFTR, F508del NBD1 expression was reduced 50–60% by Hsp27; it interacted preferentially with the mutant and was modified primarily by SUMO-2. Mutation of the consensus SUMOylation site, Lys447, obviated Hsp27-mediated F508del NBD1 SUMOylation and degradation. As for FL CFTR and NBD1 in vivo, SUMO modification using purified components in vitro was greater for F508del NBD1 versus WT and for the SUMO-2 paralog. Several findings indicated that Hsp27-Ubc9 targets the SUMOylation of a transitional, non-native conformation of F508del NBD1: (a) its modification decreased as [ATP] increased, reflecting stabilization of the nucleotide-binding domain by ligand binding; (b) a temperature-induced increase in intrinsic fluorescence, which reflects formation of a transitional NBD1 conformation, was followed by its SUMO modification; and (c) introduction of solubilizing or revertant mutations to stabilize F508del NBD1 reduced its SUMO modification. These findings indicate that the Hsp27-Ubc9 pathway recognizes a non-native conformation of mutant NBD1, which leads to its SUMO-2 conjugation and degradation by the ubiquitin-proteasome system.
Keywords: ABC transporter, chloride channel, post-translational modification (PTM), protein conformation, protein degradation, cystic fibrosis, cystic fibrosis transmembrane conductance regulator (CFTR), SUMO, SUMOylation, Prot
Introduction
The basis of the cAMP/PKA-regulated anion conductance at the apical membranes of secretory epithelial cells is the cystic fibrosis transmembrane conductance regulator (CFTR)3 (2). Similar to other ABC transporters, CFTR is comprised of two membrane-spanning domains (MSD1 and MSD2), two cytoplasmic nucleotide-binding domains (NBD1 and NBD2), and a central regulatory (R) domain. The latter harbors sites for protein kinase-mediated phosphorylation that enable the ATP-dependent gating of this unique ion channel. The most common mutation causing cystic fibrosis disease, F508del, results from the omission of a phenylalanine residue at position 508 in the first nucleotide-binding domain, NBD1. The deficient folding process of this mutant leads to its nearly complete ER-associated degradation (ERAD), and because of its complex folding pathway, a significant fraction of WT CFTR is also subjected to ERAD by most cells (3).
More than 90% of cystic fibrosis patients carry the F508del mutation on at least one allele; thus, correction of the folding defect imposed by this mutation offers the greatest potential for improving the quality of life and life expectancy of cystic fibrosis patients. Thus far, small molecules that can deliver a fraction of F508del CFTR to the cell surface (4) provide correction efficacy that achieves insufficient clinical improvement (5). Therefore, efforts to uncover the checkpoints in CFTR biogenesis where most F508del CFTR is lost to ERAD are expected to identify targets whose manipulation could provide therapy for this common disease mutation.
Whether a protein escapes ERAD is generally determined by its interaction with molecular chaperones, which may facilitate folding or degradation, depending on the conformational state of the protein (6, 7). CFTR biogenesis is monitored by multiple chaperone systems, including Hsp70, Hsp90, the Hsp40 co-chaperones, and calnexin. Chaperone interactions have been shown to decrease NBD1 aggregation in vitro and to assist with productive CFTR folding (8, 9). However, unstable conformations of CFTR remain bound to chaperones, and a prolonged association with Hsp70/Hsp90, for example, allows recruitment of the ubiquitin ligase CHIP (C terminus of HSC70-interacting protein), resulting in CFTR ubiquitylation and its degradation by the 26S proteasome (10–14).
In contrast to the ATP-dependent core chaperones, small heat shock proteins (sHsps) are ATP-independent chaperones that can hold proteins in non-native, but not denatured, protein conformations during cell stress, and these conformations have the potential to refold once the stress abates (15–17). We found previously that sHsps can distinguish between WT and F508del CFTR, selectively targeting the mutant protein to degradation (1, 18). Given the propensity of sHsps to associate with non-native protein conformations (15–17), we entertained the hypothesis that Hsp27 may recognize intermediate conformation(s) of CFTR during biogenesis, leading these proteins to degradation pathways when folding to the native conformation cannot be achieved, as for the F508del mutant.
Among the sHsps, we chose to study Hsp27 based on its expression level in airway epithelial cells (1), its role in diseases of protein folding/aggregation (19, 20), and its identification as a member of the CFTR interactome (21). Our prior work (1) showed that Hsp27 selectively binds and targets F508del CFTR for degradation via conjugation with the small ubiquitin-like modifier (SUMO). This modification arises from the strong physical interaction of Hsp27 with Ubc9, the single known E2 enzyme in the SUMO conjugation cascade (22). Modulating the expression of SUMO pathway components produced similar outcomes for mutant CFTR biogenesis/degradation as altering Hsp27 expression, consistent with linkage of the sHsp to CFTR SUMOylation. Of the three SUMO paralogs, Hsp27 promoted the modification of F508del CFTR by SUMO-2/3, paralogs of nearly identical sequence, which are capable of forming SUMO poly-chains because they contain consensus SUMOylation sites. SUMO-2/3 modification of CFTR recruited the SUMO-targeted ubiquitin ligase, RNF4, which can select poly-SUMOylated substrates for proteasome-mediated degradation. Experiments employing Hsp27 knockdown or a dominant-interfering RNF4 implicated this ubiquitin E3 in Hsp27-mediated F508del degradation (1). This SUMO- and SUMO-targeted ubiquitin ligase-mediated degradation pathway had previously been implicated only in the proteolysis of nuclear proteins (23).
In the present study, we asked what properties of F508del CFTR are sensed by the sHsp/SUMO pathway in promoting mutant protein degradation. Because CFTR is a large, polytopic protein, we simplified the system by focusing experimentally on NBD1, the domain bearing the F508del mutation that leads to Hsp27/Ubc9 incursion. The in vivo experiments were performed using a membrane-tethered NBD1 to evaluate its ability to mimic interactions of the SUMO pathway with full-length (FL) CFTR. The in vitro assays were performed using purified proteins; they allowed manipulation of the biophysical properties and stability of NBD1 to judge the structural basis underlying its SUMO modification.
Experimental Procedures
Antibodies and Reagents
Monoclonal antibodies targeting CFTR NBD1 (660 and 3G11) were obtained via Cystic Fibrosis Foundation Therapeutics (Bethesda, MD). Polyclonal antibodies against SUMO-1 or SUMO-2/3 were from Enzo Life Sciences (Ann Arbor, MI), anti-Hsp27 was from StressGen (Vancouver, Canada), and anti-β-actin was from Sigma-Aldrich. Horseradish peroxidase-conjugated secondary antibodies, anti-mouse and anti-rabbit, were obtained from Amersham Biosciences-Pharmacia Biotech (GE Healthcare Bio-Sciences), and anti-rat was from Jackson ImmunoResearch Laboratories. Complete protease inhibitor tablets were purchased from Roche. N-Ethylmaleimide was purchased from Thermo Scientific (Rockford, IL), and other chemicals were obtained from Sigma-Aldrich at the highest grade available.
Cell Culture and Transfection
HEK293 cells were cultured in DMEM (Invitrogen) with 10% fetal bovine serum (Hyclone, Thermo Scientific, Waltham, MA). The cells were maintained in a humidified incubator at 37 °C with 5% CO2. For protein overexpression, HEK293 cells grown in 60-mm dishes were transiently transfected with the indicated expression plasmids using Lipofectamine 2000 (Invitrogen). After 48 h, the cells were rinsed with phosphate-buffered saline and extracted in lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, and 20 mm N-ethylmaleimide plus protease inhibitors). Samples were incubated for 30 min in lysis buffer on ice and centrifuged at 16,000 × g for 10 min at 4 °C. Cell lysates were used for immunoblot analyses.
Plasmid Construction and Site-directed Mutagenesis
The Hsp27 plasmid was purchased from Origene (Rockville, MD). CD4T-NBD1 plasmids, WT and F508del, were as described (24). Briefly, the NBD1 domains, WT and F508del, were fused in frame to the C terminus of the transmembrane segment, CD4T; the domain boundaries are provided in supplemental Table S1 of Ref. 24. CD4T-WT-NBD1-K447R and CD4T-F508del-NBD1-K447R mutants were generated using the following primers: 5′-CCTGTCCTGAAAGATATTAATTTCAGGATAGAAAGAGGACAGTTG-3′ (forward) and 5′-CAACTGTCCTCTTTCTATCCTGAAATTAATATCTTTCAGGACAGG-3′ (reverse). Mutants were obtained using the QuikChange Lighting site-directed mutagensis kit (Agilent Technologies, Santa Clara, CA). All plasmid constructs were sequence-verified.
Immunoblot (IB) and Immunoprecipitation (IP) Analyses
For IBs, equal amounts of protein from HEK293 cell lysates were resolved by SDS-PAGE and transferred to PVDF membranes (PerkinElmer Life Sciences). Unbound sites were blocked for 1 h at room temperature with 5% nonfat milk powder in TBST (20 mm Tris, pH 8.0, 150 mm NaCl, 0.01% Tween 20). The blots were incubated with primary antibodies (above) at room temperature for 1 h; then washed three times, 10 min each, with TBST; and incubated for 1 h with horseradish peroxidase-conjugated secondary antibodies in TBST containing 5% nonfat milk, followed by three TBST washes. The reactive bands were visualized with enhanced chemiluminescence (PerkinElmer Life Sciences) and exposed to x-ray film.
For IPs, precleared HEK293 cell lysates (500 μg of protein) were mixed with the appropriate primary antibodies for 2 h at 4 °C in lysis buffer. Fifty microliters of washed protein G-Sepharose beads (Invitrogen) were added to each sample and incubated 4 h or overnight at 4 °C with gentle rotation. Immunocomplexes were washed three times with lysis buffer and precipitated by centrifugation at 12,000 × g for 10 s. The immune complexes were then suspended in SDS sample buffer and subjected to IB.
In Vitro SUMOylation Assays
The in vitro assay was routinely performed using reagents purchased from Enzo Life Sciences (Ann Arbor, MI), together with WT or F508del hNBD1, with quality control of the bacterially expressed proteins, as described (25). Variants that contain a single solubilizing mutation, F494N, were used in most experiments; this mutation enhances the yield of both NBDs from bacterial expression. Variants containing additional solubilizing and/or stabilizing mutations (25) were used to evaluate the relation of these NBD1 properties to their SUMO modification. For the standard assay, 15 ng of purified NBD1 was incubated in SUMOylation buffer (Enzo Life Sciences, Ann Arbor, MI) with a reaction mixture containing recombinant E1 (SAE1/SAE2, 0.4 μm), E2 (Ubc9, 4 μm), a SUMO paralog (3 μm), and Mg-ATP (2 mm), with or without purified recombinant human Hsp27 protein (15 ng), for 1 h at 27 °C. After the reaction was terminated with SDS sample buffer containing 2-mercaptoethanol, reaction products were resolved on 12% SDS-PAGE, and the gel shift resulting from SUMO modification was detected by immunoblot using anti-NBD1 (antibody 660), as indicated.
In Vivo SUMOylation Assays
For in vivo SUMOylation, a total of 1 mg of HEK293 cell lysate, prepared using lysis buffer (50 mm HEPES, 150 mm NaCl, 10% glycerol, 1 mm EDTA, 1% Nonidet P-40, pH 7.5, and 20 mm N-ethylmaleimide), was incubated with 1 μg of NBD1 antibody (660 or 3G11; Cystic Fibrosis Foundation Therapeutics) or CFTR R domain antibodies (217 plus 596; Cystic Fibrosis Foundation Therapeutics) overnight. Immunocomplexes were isolated by incubation with protein A-agarose beads (Invitrogen) for 4 h. Precipitates were isolated and washed three times with lysis buffer, and the proteins were resolved on 6% SDS-PAGE and subjected to IB analysis for SUMO modification.
NBD1 Stability and Structural Measurements
Thermal stability of NBD proteins was assessed as previously reported using SYPRO orange as a fluorescence marker of denaturation (25). Protein was diluted into buffers containing either 50 μm or 2 mm ATP, and the reaction was monitored on an ABI 7900 HT qPCR machine. A single hold at 25 °C was performed for 2 min, and the samples were ramped at 2% to 95 °C and held for 2 min.
NBD structural transitions were monitored by intrinsic tryptophan fluorescence using a BioTek Synergy 4 plate reader in kinetic mode. Protein was diluted into SUMO reaction buffer and excited at 290 nm. Excitation intensity was continuously monitored at 325 nm with the reaction temperature held at 27 °C.
Results
Membrane-tethered NBD1 Recapitulates the Impact of Hsp27 on Full-length CFTR
The F508del mutation destabilizes NBD1, makes it more susceptible to proteolysis, decreases its melting temperature and its thermal unfolding transition temperature, and accelerates its conversion to a less ordered, non-native conformation (26–28), thus reducing its folding yield (29, 30). Because our prior work indicated that the functional and physical interactions between Hsp27 and FL CFTR were directed at facilitating degradation of the F508del mutant (1, 18), we asked whether this selectivity of Hsp27 extended to WT versus F508del NBD1.
First, we expressed WT and mutant NBD1s tethered to a truncated CD4 membrane spanning segment, as used previously in studies of CFTR domain interactions (24). This approach produces membrane associated WT or F508del NBD1 that is largely retained in the ER, traffics poorly to the cell surface, and allows focus on ER-associated events. The potential for Hsp27 to identify NBD1 for degradation was evaluated in HEK293 cells by assessing the impact of its overexpression on the steady-state levels of CD4T-NBD1, WT or F508del. HEK293 cells were co-transfected with plasmids containing these fusion constructs, together with Hsp27 or empty vector as control. As shown in Fig. 1A, Hsp27 overexpression reduced the steady-state expression level of CD4T-F508del-NBD1 ∼50%, without significantly altering the expression of CD4T-WT-NBD1; this mutant selectivity was found previously for FL CFTR (1). The reduction in mutant NBD1 expression could be abrogated by preincubation of the cells with proteasome inhibitor (MG132) for 4–5 h prior to cell lysis (data not shown), indicating that Hsp27-induced CD4T-F508del-NBD1 degradation was mediated by the proteasome, also previously demonstrated for FL F508del CFTR.
FIGURE 1.

Selective Hsp27 impact on F508del NBD1 expression, binding, and SUMOylation in vivo. A, overexpression of Hsp27 promotes the degradation of CD4T-F508del-NBD1. HEK293 cells were transiently transfected with CD4T-WT-NBD1 or CD4T-F508del-NBD1 and with vector control or Hsp27 as described under “Experimental Procedures.” Whole cell lysates were extracted 48 h after transfection, and protein expression was detected by IB using the indicated antibodies. NBD1 protein levels were quantified and normalized to control values from four independent experiments. ***, p = 0.0003. B, Hsp27 preferentially interacts with CD4T-F508del-NBD1. CD4T-WT-NBD1 or CD4T-F508del-NBD1 was expressed in HEK293 cells as in A. The interaction of the NBDs with Hsp27 was evaluated by IP using anti-NBD1 followed by resolution of the co-IPed proteins by SDS-PAGE and IB with antibodies against Hsp27 or CD4. The densities of the Hsp27 blots were normalized to the amount of NBD1 in the IP. The quantitative data normalized to WT binding level are the averages of three independent experiments. *, p = 0.032. C, Hsp27 promotes NBD1 SUMOylation in vivo. CD4T-WT-NBD1 or CD4T-F508del-NBD1 and vector control or Hsp27 were expressed as above. After 48 h, the cells were lysed, and co-IP was performed using anti-NBD1 and resolved on SDS-PAGE and IB with antibodies against SUMO paralogs. The endogenous SUMO signals were normalized to the amount of NBD1 pulled down (CD4 IB) and then compared with their respective control values in three independent experiments. *, p = 0.023.
Second, we evaluated the possibility that Hsp27 selects F508del NBD1 for degradation via a preferential interaction with the mutant. For these experiments, the CD4T-NBD1s were subjected to IP, followed by Hsp27 IB. When corrected for the Hsp27-induced reduction in mutant protein expression, the co-IP data (Fig. 1B) show that approximately twice as much Hsp27 interacted with F508del as with WT NBD1. The bar graph plots data in which the amount of Hsp27 in the IP was normalized to the amount of NBD1 pulled down. The data of Fig. 1 (A and B) validate the use of membrane-tethered NBD1s to mimic the impact of Hsp27 on FL CFTR degradation (1). Quantitatively, the effects of Hsp27 here were not as great as those observed for the full-length protein: mutant NBD1 expression was reduced 50% versus 70% for FL F508del CFTR, and its interaction was ∼2-fold greater with F508del NBD1 versus WT, whereas 6-fold more Hsp27 co-IPed with FL F508del versus the WT protein. However, the selectivity of Hsp27 for mutant protein degradation and binding was preserved qualitatively. In addition, the retention of the mutant and WT CD4T-NBDs in the ER indicates that the selective degradation of F508del NBD1 was not due to the escape of WT NBD1 to the plasma membrane but is due to the ability of Hsp27 to distinguish the mutant and WT NBD1 conformations.
Third, we evaluated the modification of CD4T-WT-NBD1 and CD4T-F508del-NBD1 by SUMO paralogs and the possibility that Hsp27 facilitated this process. NBD1 was IPed from HEK293 cells expressing the CD4T-NBD1 fusions and co-expressing Hsp27 or empty vector. The NBD1 precipitates were blotted for SUMO-1 or SUMO-2/3 to detect modification by endogenous SUMO paralogs. As shown in Fig. 1C, single SUMO-1 bands were observed in both the WT and F508del precipitates at a position above the molecular masses of the CD4T-NBD fusions (which are ∼75 kDa); these bands would be indicative of conjugation with a single SUMO-1. In contrast, the SUMO-2/3 blot shows evidence of higher molecular mass conjugates that likely reflect the addition of SUMO poly-chains (31). The laddering pattern observed here is consistent with Hsp27-induced modification of FL F508del CFTR by SUMO-2/3; the latter led to our identification of RNF4 as a poly-SUMO-targeted ubiquitin ligase that directs FL mutant CFTR to proteasomal degradation (1). For quantitation of the aggregate data (bar graph), the Hsp27-induced SUMO-1 or SUMO-2/3 intensities for F508del-NBD1 were normalized to their respective control signals. The modification of CD4T-F508del-NBD1 in the IP by SUMO-2/3 was increased 3-fold by overexpressed Hsp27, similar to the results obtained for FL F508del CFTR. In contrast, Hsp27 did not significantly alter SUMO modification WT NBD1. Thus, the CD4T-NBD1 fusions mirror the action of Hsp27 on expression level, Hsp27 binding, and SUMOylation of WT and mutant FL CFTRs, impacting primarily the mutant NBD1.
In Vitro SUMOylation of NBD1
WT or F508del NBD1 SUMOylation was performed in vitro using purified human 1S-NBD1, a variant containing a single solubilizing mutation (F494N) that was introduced into the NBD during crystallization trials (32). This modification provides sufficient solubility to bacterially expressed F508del NBD1 to enhance the yield of folded protein to levels required for the biophysical measurements and SUMO modification studies performed here. The 1S mutation raises the melting temperature of NBD1 and its thermally induced unfolding by only 1–2 °C, while preserving the difference between WT and F508del NBD1 thermal destabilization (∼9 °C) (25). For the SUMO conjugation assay, purified WT and F508del 1S-NBDs were incubated with purified SUMOylation components: the SUMO E1 heterodimer (SAE1/SAE2), the SUMO E2 (Ubc9), plus Mg-ATP. As modifier, different experiments employed the paralogs SUMO-1, -2, or -3. The conjugation reaction was allowed to proceed at 27 °C, usually for 1 h, before being terminated by addition of sample buffer (see “Experimental Procedures” for details). After SDS-PAGE and transfer, NBD1 and SUMO-modified NBD1 were detected by NBD1 or SUMO immunoblot.
The time course of SUMOylation and its dependence on NBD1 protein concentration were monotonic relations, leading to standard assay parameters that produced readily detected signals: 15 ng of NBD1 protein/assay in a reaction volume of 6 μl, with 1 h of incubation at 27 °C. Any deviations from these conditions are noted in the text and figure legends. The data used to establish the standard assay conditions are provided in Figs. 5 and 6, because these findings apply in later studies of the relation of NBD1 SUMOylation to its conformation. Identification of the SUMOylated NBD1 was verified using secondary antibodies labeled with different infrared fluorophores for simultaneous detection of NBD1 and SUMO (Fig. 2A). From the orange band observed at ∼47 kDa, it is evident that a single SUMO conjugation site is identified most prominently, although additional, weaker signals are visible when the level of modification is high (see below).
FIGURE 5.
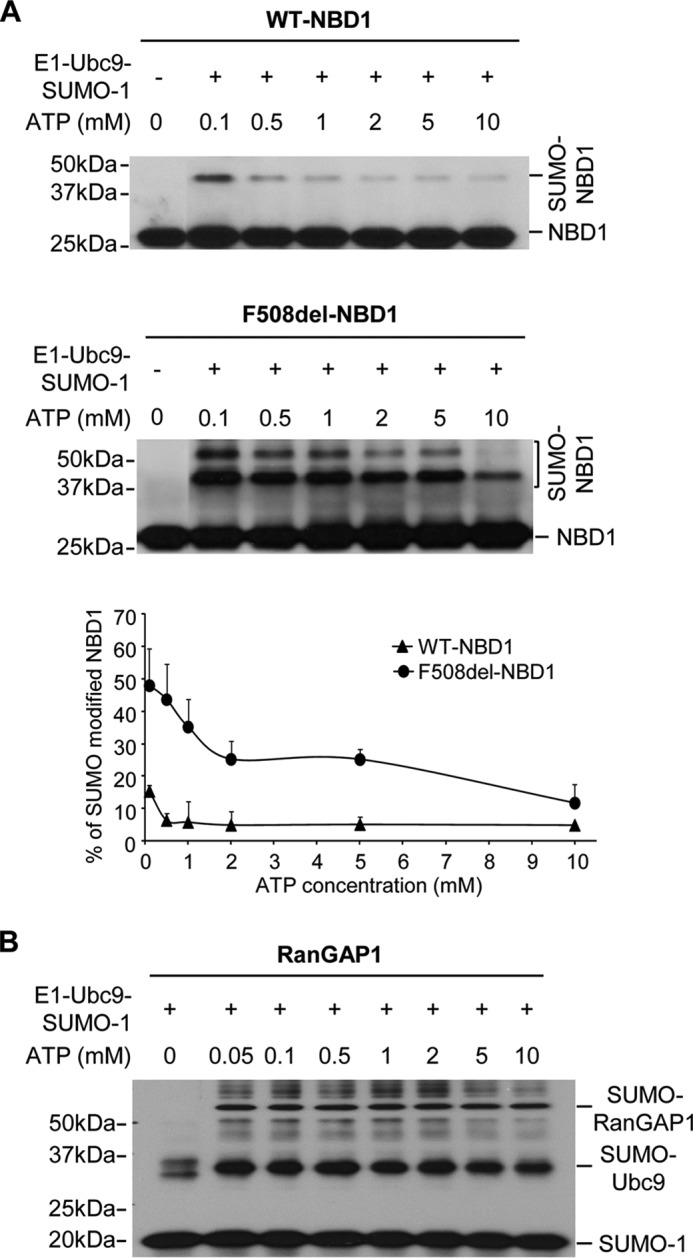
Dependence of WT- and F508del-NBD1 SUMOylation on ATP concentration in vitro. A, ATP concentration was varied as indicated under otherwise standard in vitro assay conditions. The graph plots the percentages of total NBD1 signal detected at higher molecular mass, as indicated; the data are from three independent experiments. B, ATP concentration dependence of in vitro SUMOylation of the model substrate, RanGAP1, run under the standard assay conditions and detected by SUMO-1 IB.
FIGURE 6.

SUMOylation kinetics and [ATP] dependence. These data were used to establish the standard assay conditions and for subsequent experiments relating to NBD1 conformation. A, time courses of WT and F508del NBD1 modification by SUMO-1 at 0.1 or 2 mm ATP at 27 °C. Immunoblots were performed using NBD1 (antibody 660) antibody. Quantitation of the data is provided to the right, where density values were normalized to values at 5 min; the mean data are derived from three independent experiments. B, in vitro SUMOylation of WT or F508del NBD1 at three levels of purified protein, run for 1 h at 27 °C. The indicated reactions included preheating to 30 °C for 30 min prior to adding the SUMOylation reagents for the standard assay. Controls were performed without added E1, E2, or ATP (left and right lanes).
FIGURE 2.

In vitro SUMOylation and dependence on Hsp27. A, identification of SUMO-1 modified WT and F508del NBD1. Immunoblots were performed using monoclonal anti-NBD1 (antibody 660) and polyclonal anti-SUMO-1 antibodies, detected with IRDye 680RD donkey anti-mouse or IRDye 800CW donkey anti-rabbit secondary antibodies using a Li-Cor Odyssey. B, selective SUMO-1 modification of F508del-NBD1 requires the SUMO E1 and E2-conjugating enzymes and ATP and is reversed by the SUMO protease, SENP-1. Purified 1S-NBD1, WT or F508del, was incubated with SUMOylation components for 60 min at 27 °C. The reaction mixture was resolved by SDS-PAGE, and NBD1 was detected by antibody (antibody 660). C, preferential modification of F508del-NBD1 by SUMO-2 in vitro is augmented by Hsp27. SUMOylation reactions were run with purified paralogs, and NBD1 was detected as in B. SUMOylated F508del-NBD1 densities were quantified and normalized to their respective control values from three independent experiments. *, p = 0.023; **, p = 0.008.
The requirements for in vitro SUMOylation of WT and F508del NBD1 are illustrated in Fig. 2B, where SUMO conjugation was detected from the gel shift in the signal detected by an NBD1 antibody. Omission of the enzymatic components (SUMO E1 and E2) SUMO-1 and ATP did not lead to generation of a modified NBD1 signal, a necessary negative control. Other experiments (not shown) indicated that each of these components is required to generate SUMO-conjugated NBD1, and this is illustrated in the second and sixth lanes, where SUMO was omitted. As noted previously for FL CFTR in vivo (1), in vitro SUMO modification of F508del NBD1, was greater than that of the WT NBD (also detected in Fig. 2A), which likely reflects a change in conformational state that is more readily modified. Earlier studies (25) indicate that the conformational defect in F508del NBD1, reflected in its thermodynamic instability, augments its in vitro ubiquitylation, and the SUMOylationof NBD1 has a similar basis, as shown below. Finally, including the SUMO protease, SENP-1, in the reaction mix eliminated the SUMO-NBD1 signal for the WT and mutant proteins, as expected for cleavage of the isopeptide bond linking SUMO to substrate.
Next, we asked whether the SUMO paralog dependence of Hsp27-induced NBD1 modification was preserved in the in vitro assay. These data are illustrated in Fig. 2C. As controls, omission of the components for conjugation or the inclusion of SENP-1 in the reaction mix abrogated NBD1 modification. F508del NBD1 was more highly modified by all paralogs than WT NBD, as observed above in vivo and previously (1). Again, the SUMO signals were normalized to their respective control intensities (bar graph). In the presence of Hsp27, the modification of F508del NBD1 by SUMO-2 was greater than that by the other paralogs. In addition, the sHsp had no significant effect on modification of the WT NBD1. Its impact was largely confined to F508del NBD1 conjugation by SUMO-2. Thus, even in this simplified system, SUMO paralog selectivity mimics that observed for Hsp27-induced SUMOylation in vivo.
Together, these observations recapitulate the in vivo SUMOylation assays, in which FL WT and F508del CFTRs were expressed with or without Hsp27, showing preferential SUMO-2 modification and degradation of F508del CFTR by Hsp27, without significantly facilitating the SUMOylation or degradation of WT CFTR (1). The findings are consistent with the concept that conformational differences between WT and mutant CFTR lead to a selective interaction of F508del with Hsp27/Ubc9 and its modification by SUMO-2 and that these processes are preserved in the in vitro assays.
Mutation at Lys447 Abrogates Hsp27-mediated NBD1 Degradation
Several prediction algorithms identify the lysine at amino acid position 447 of NBD1 as a strong consensus SUMOylation site (ψKXD/E), where ψ is a hydrophobic residue, X is any amino acid, and the conjugated lysine is followed by an acidic residue. Given the Lys447 site prediction, we determined the ability of Hsp27 to promote the degradation of F508del NBD1 in HEK293 cells by expressing CD4T-F508del-NBD1 containing the conservative mutation, K447R. The results are shown in Fig. 3A. Similar to the data presented in Fig. 1A, Hsp27 co-expression reduced the steady-state level of F508del NBD1; however, the impact of Hsp27 was lost in the K447R mutant of CD4T-F508del-NBD1. In addition, mutation of the Lys447 site led to a consistent ∼30% increase in the expression level of F508del NBD1, reflecting basal activity of this endogenous degradation pathway.
FIGURE 3.

Hsp27-mediated degradation and SUMOylation of F508del-NBD1 in vivo requires Lys447. A, the SUMO consensus site, Lys447, was mutated to generate CD4T-F508del-NBD1-K447R; this mutant and its control were expressed in HEK293 cells with Hsp27 or vector control, as above. After 48 h, F508del-NBD1 expression was assessed by IB using the indicated antibodies. Protein levels were quantified and normalized to the vector control from three independent experiments. **, p = 0.001; *, p < 0.05. B, HEK293 cells were transfected with the plasmids used in A. NBD1 SUMOylation was detected by NBD1 IP followed by IB with antibodies against NBD1 or endogenous SUMO-2/3. HC, antibody heavy chain. The SUMO signal was normalized to the amount of NBD1 pulled down and then normalized to control; data from three independent experiments. *, p = 0.007. C, K447R does not abrogate the impact of Hsp27 on FL F508del degradation. F508del CFTR or F508del CFTR-K447R and Hsp27 or control plasmids were transfected into HEK293 cells as above. Protein expression was detected by IB using the indicated antibodies. F508del CFTR levels in Hsp27 co-expressing cells were normalized to their respective controls from four independent experiments. **, p = 0.008.
Next, we determined the ability of Hsp27 to induce SUMOylation of the membrane-tethered F508del NBD1 bearing the K447R mutation in HEK293 cells expressing CD4T-F508del-NBD1 with and without the K447R mutation. Fig. 3B illustrates the results obtained from IP/IB experiments in which we pulled down the CD4T fusion proteins, followed by IB for SUMO-2/3. The degree of SUMO-2/3 modification of F508del-NBD1 was increased with co-expression of Hsp27, but the sHsp did not further modify the K447R variant. Thus, the data of Fig. 3 (A and B) indicate that Lys447 is the predominant SUMOylation site that mediates Hsp27-induced degradation of F508del NBD1 via its conjugation with SUMO-2/3. Next, we asked whether the introduction of the K447R mutation into the full-length protein would reduce the impact of Hsp27 on the expression of F508del CFTR. As shown in Fig. 3C, this variant remained susceptible to Hsp27-induced degradation, which most likely indicates that additional SUMOylation sites are present that can mediate this action of Hsp27. Alternatively, other sites may be recruited when Lys447 is lost, as observed for other target proteins (33).
Finally, we evaluated the accessibility of position Lys447 for modification by Hsp27-Ubc9 based on modeling of CFTR (34) and the crystal structure of NBD1 (35). In the NBD1 depiction shown in Fig. 4A, the Lys447 residue is shown as a red sphere, and it is exposed at the surface of the solved NBD1 structure. This is true also in the full-length homology model of CFTR (34) (image not shown), suggesting that Lys447 is accessible for modification. SUMOylation sites are often followed by a larger acidic region, and although the consensus sequence in NBD1, FKIE, is not followed by a larger downstream region of electronegativity in the linear sequence, there is an adjacent acidic area in the crystal structure of the NBD (Fig. 4B) that may serve this purpose. We have not yet determined whether this region is important for NBD1 modification.
FIGURE 4.
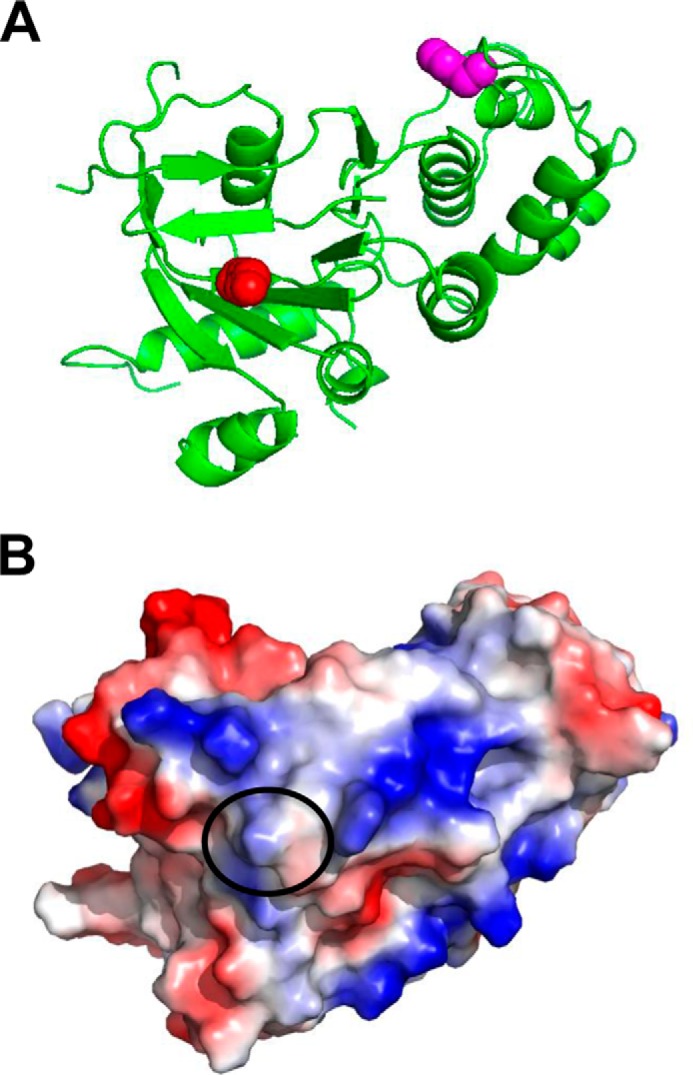
Structural analysis of the location of Lys447 using experimental x-ray structures of NBD1. A and B, the experimentally determined structure of NBD1 is shown as a ribbon (A) or the calculated electrostatic surface (B). The Lys447 residue is shown as a red sphere in the ribbon, and its contribution to the surface on NBD1 is circled in the surface model. The location of the Phe508 residue is shown as magenta spheres for reference in A. The images were rendered using Protein Data Bank code 2BBO for NBD1 (35). As noted in the text, surface exposure of Lys447 is also predicted in the full-length homology model of Serohijos et al. (34).
ATP Dependence of NBD1 SUMOylation
Next, we returned to in vitro SUMOylation assays to ask what physical properties of F508del NBD1 promote its SUMO modification. The interaction of ATP with NBD1 is known to stabilize its folded conformation, and this plays a role in the development of FL CFTR structure (26, 27, 36, 37). This behavior is similar to other proteins in which ligand binding induces stability and escape from ERAD (38, 39). We examined the ATP dependence of NBD1 SUMOylation in vitro over the range 0.1–10 mm ATP; these results are illustrated in Fig. 5. NBD1 conformation has been shown to be sensitive to ATP binding, and it is stabilized by saturating [ATP]; thus, ATP can be used to regulate structural transitions between native and non-native conformers (26) and assess which are most amenable to SUMO conjugation.
Our standard in vitro assay conditions include 2 mm ATP, a nucleotide concentration where SUMO modification of F508del NBD1 is favored over WT NBD1 (Figs. 1 and 2 and Ref. 1). As shown in Fig. 5A, this difference in modification persists over the entire range of [ATP] examined: SUMO conjugation of F508del NBD1 increased progressively as ATP was reduced, whereas modification of WT NBD1 was minimal and did not increase until ATP was below 0.5 mm.
Because charging of the SUMO E1 enzyme is an ATP-dependent process, we asked whether the low ATP concentrations employed may influence the SUMO conjugation reactions themselves. To evaluate this possibility, we determined SUMOylation of the model substrate, RanGAP1 (Ran GTPase-activating protein 1), the first known SUMOylation target protein (40). SUMO modification of RanGAP1 was not affected over this range of [ATP], as illustrated in Fig. 5B. In addition to the RanGAP1 data, the rise in SUMO modification of NBD1 at reduced ATP was opposite to that expected if the energy requiring step in SUMO transfer were compromised. Rather, these findings indicate that NBD1 modification increases as ligand binding is reduced, consistent with the idea that NBD1 destabilization at low [ATP] promotes its SUMO conjugation. These findings suggested that a non-native conformation of NBD1 may be the target of SUMO modification, given the stability difference between WT and F508del NBD1 and the ATP dependence of their SUMOylation.
Next, SUMOylation of the NBD1 proteins was assessed kinetically under conditions mirroring those that will evaluate NBD1 conformation. In the presence of 0.1 or 2 mm ATP, both the WT and F508del protein demonstrated time-dependent modification by SUMO in which the F508del protein again showed increased SUMOylation as compared with WT NBD1 (Fig. 6A). Changes in the apparent sensitivity of NBD1 SUMOylation to ATP, especially of the F508del variant, suggested that stabilization of the native state might protect the NBD from modification. This conclusion is supported also by the influence of an elevated preincubation temperature on the SUMOylation of NBD1, as shown in Fig. 6B. Here, the WT and F508del NBDs were subjected to preheating at 30 °C for 30 min prior to running the standard SUMOylation assay. At all NBD1 concentrations, SUMO modification of the WT and mutant NBDs was increased by thermal destabilization of the native state, particularly for F508del NBD1.
SUMOylation Targets a Non-native Conformation of NBD1
Intrinsic fluorescence and differential calorimetry, as well as other biophysical methods, have been used to monitor the unfolding of NBD1 as it transitions from the native conformation to the denatured state (26, 27). As the WT or F508del NBDs are exposed to increasingly denaturing conditions, they undergo an increase in intrinsic fluorescence characteristic of a partial unfolding step, producing an intermediate conformation, i.e. a transitional phase, in which NBD1 loses its native secondary structure characteristics. This state is followed by formation of the fully denatured random coil at high denaturant (26, 27). The significance of the initial transition in NBD1 unfolding is reflected by the influence of the F508del mutation, which transits to the non-native state at lower denaturant, as well as the influence of ATP and revertant mutations, which stabilize the NBD and suppress the initial conformational transition (41, 42).
To further assess the thermodynamic stability of purified WT and F508del NBD1 proteins, thermal melts were performed (Fig. 7A). SYPRO orange binding and fluorescence was followed as a function of increasing temperature and protein unfolding using a qPCR machine as previously described (43). As the protein unfolds, the SYPRO orange dye binds exposed hydrophobic regions of the protein, producing an increase in fluorescence. The change in fluorescence as a function of temperature then provides an indirect measure of the conformational properties of the protein and its stability.
FIGURE 7.

SUMOylation is inversely proportional to NBD1 thermal stability. A, temperature-dependent unfolding of NBD1. Purified WT and F508del 1S-NBDs were diluted into buffers, and the fluorescence of SYPRO orange, which interacts with exposed hydrophobic regions, was monitored. The apparent T½ values for the unfolding transitions of WT NBD1 at 2 mm ATP (closed circles) and 0.1 mm ATP (open circles) and for F508del NBD1 at 2 mm ATP (triangles) are provided in the text and in C and are in agreement with previously determined values (25). B, solubilizing and revertant mutations (25) reduce the extent of F508del NBD1 SUMOylation in vitro. C, relation between relative in vitro SUMOylation (Fig. 7B) and thermal stability data for 1S, 3S, and 7S F508del NBD1s (25) (***, p = 0.0003, 3S; p = 0.0002, 7S). D, SUMOylation follows the kinetics of NBD1 transition to a non-native conformation. WT and F508del NBD1 SUMO modification (upper panels) were determined at 0.1 mm ATP and 27 °C at the times indicated (data from Fig. 6A). Immunoblots were performed using NBD1 (660) antibody. Intrinsic fluorescence values at 325 nm were normalized to the values at 5 min for WT and F508del 1S-NBD1.
The WT and F508del 1S-NBD proteins were purified and diluted into buffers containing either 2 or 0.1 mm ATP. Under both low and high ATP conditions, the WT protein showed a single fluorescence transition, consistent with its unfolding. In the presence of 0.1 mm ATP, the WT protein showed an apparent T½ of 35 °C. This value increased to 39 °C in the presence of 2 mm ATP. The F508del NBD protein showed a similar single unfolding transition in 2 mm ATP, with an apparent T½ of 28.5 °C. In 0.1 mm ATP, the F508del protein showed elevated baseline fluorescence and no observable unfolding transition, suggesting that the protein had begun to denature prior to the fluorescence measurement at low ATP. These data are consistent with previous reports of reduced F508del NBD1 thermal stability (27). Further, the stabilization of both WT and F508del proteins in the presence of saturating ATP is consistent with ligand-induced conformational stabilization of the native state of this domain, as suggested earlier.
These relationships can be assessed also by examining the influence of solubilizing and revertant mutations that increase F508del NBD1 solubility and stability. Mutations that make NBD1 more soluble were introduced during crystallization trials to provide for its structural resolution (36). Revertant mutations enhance NBD1 stability and can partially correct processing of FL F508del CFTR (41, 42, 44). Their influence on the in vitro SUMOylation of F508del NBD1 is illustrated in Fig. 7B. The reference signal was provided by 1S-NBD1, used in other modification assays. As shown here, introduction of the 3S (F508del mutations F494N, Q637R, and F429S) and 7S (F508del mutations F494N, Q637R, F429S, G550E, R553Q, and R555K) stabilizing mutations leads to a marked reduction in NBD1 SUMOylation. Comparison of the data of Fig. 7B with previously determined thermal melt T½ values for these revertants (25) shows an inverse correlation between thermal stability and SUMO modification (Fig. 7C).
In a related approach, the structural transitions of the purified NBD1 proteins were followed kinetically by intrinsic fluorescence and then correlated with pseudo-kinetic studies of SUMOylation at specific reaction time points. SUMOylation of the WT and F508del NBD1 proteins was determined as a function of incubation time in the presence of 100 μm ATP (data from Fig. 6A).
For the fluorescence measurements, NBD proteins were diluted into the SUMO reaction buffer and incubated at 27 °C (Fig. 7D). The initial fluorescence of both the WT and F508del NBDs was similar under these conditions, suggesting that both proteins started in similar structural states. The fluorescence of the F508del NBD1 increased rapidly at 27 °C and low [ATP], consistent with a change in protein structure and loss of the native state. This rapid rise in fluorescence slowed after 10–15 min, perhaps influenced by NBD1 aggregation (26, 27). Under these conditions, the fluorescence of the WT protein remained relatively stable over this time course, suggesting that this domain showed minimal structural change. For F508del NBD1, the transition to a non-native conformation, reported by its intrinsic fluorescence, was followed by increases in NBD1 SUMOylation. These data provide further evidence that SUMO modification is associated with the formation of a non-native NBD1 protein conformation.
Discussion
NBD1 Mimics Full-length CFTR Interactions with the SUMO Pathway
For nearly two decades, it has been appreciated that the yield of folded NBD1 is impaired by the F508del mutation (45). In the full-length protein, this mutation produces unstable intermediates, conformations that can be stabilized by chemical chaperones (46) or by the introduction of revertant mutations (41, 47). The loop structure surrounding the F508 locus was perturbed in crystal structures of the mutant NBD1 (30, 32), but this structural change on its own was relatively minor. Later, the conformational instability of mutant NBD1 was recognized to propagate to other CFTR domains and domain interfaces, thus affecting the conformations of MSD1, MSD2, and NBD2 (24, 25, 28, 48–51).
These findings were consistent with the concept that the F508del mutation produces kinetically and energetically trapped intermediate conformation(s) during CFTR folding that cannot progress to the native state. Whereas CFTR domain folding takes place co-translationally (52), domain assembly is a protracted process that may take 30 min or more (51). Based on these results, our current concept of therapeutic F508del correction implies that small molecules should suppress both the energetic defects in NBD1 folding and the conformational mismatches at domain interfaces to provide sufficient stability and function for F508del CFTR to yield a mature protein that conducts anions across epithelial cell apical membranes (25, 50, 53, 54).
Another important consideration in therapeutic correction of the basic defect in F508del CFTR is whether perturbation of the extrinsic chaperone complexes that monitor and facilitate CFTR folding can be manipulated to augment the folding yield of the mutant. The selective physical interaction of the sHsp, Hsp27, with F508del CFTR and its collaboration with the SUMO E2, Ubc9, has highlighted a novel quality control pathway that distinguishes between the conformations of the WT and F508del proteins, processing the mutant for proteasome-mediated disposal via SUMO modification. The present studies show that this discrimination capability extends to the isolated NBD1 domain, in which the common cystic fibrosis mutation lies. Indeed, the selectivity of Hsp27-Ubc9 for F508del NBD1 SUMOylation that leads to its degradation is evident both in vivo and in vitro.
Cytosolic domains of CFTR tethered to membrane anchors ensure their surveillance by ER quality control (24). CD4T-tethered F508del NBD1 showed selective Hsp27 binding, SUMOylation, and degradation when compared with CD4T-WT-NBD1. In addition, Hsp27 preferentially augmented the modification of CD4T-F508del-NBD1 by SUMO-2/3, the SUMO paralogs that are capable of forming poly-SUMO chains. These properties mirror those of the full-length mutant protein (1), and they lead to the recognition of F508del by the Ring domain ubiquitin E3 ligase, RNF4 (55). Its ability to recognize the SUMO poly-chains formed by SUMO-2 or -3 is conferred by the presence of tandem SUMO interacting motifs at the RNF4 N terminus. In extending these studies, the use of CD4T-tethered domains and of membrane-spanning domains linked to the cytosolic domains of CFTR should yield a more complete assessment of the ability of the Hsp27-Ubc9 pathway and the SUMO-2/3 modification it evokes to sense the conformations of individual CFTR domains and domain combinations. This could include domain combinations containing WT or F508del NBD1 and those harboring less common disease mutations that impact CFTR folding, permitting the diversity of this quality control pathway to be assessed.
A predicted SUMOylation site at Lys447 was verified as a critical locus of SUMO modification in F508del NBD1 using the charge-preserving mutation, K447R. F508del NBD1 containing this mutation was expressed at a higher level, and it obviated the ability of Hsp27 to reduce the expression level of CD4T-F508del-NBD1, as well as reducing its modification by endogenous SUMO-2/3. Preliminary studies (Fig. 3C) showed that introduction of the K447R mutation into full-length F508del CFTR did not alter the ability of this sHsp to reduce its expression. This finding suggests that the full-length protein can be modified at other sites or that there may be a hierarchy of site modification. This appears to be true for F508del NBD1, which showed additional bands at high levels of modification (e.g. Figs. 2C, 5A, and 7B); however, mutation of the site at Lys447 was sufficient to abrogate the effect of Hsp27 on expression of the mutant NBD and its modification by SUMO-2/3 (Fig. 3, A and B). Future studies will be directed at the goal of identifying the full complement of SUMOylation sites in FL CFTR and evaluating their significance.
In Vitro SUMOylation: Requirements and Relation to NBD1 Stability
The results of experiments to modify WT or F508del NBD1 proteins with SUMO in vitro using purified components complemented the in vivo studies that utilized CD4T-NBD1 or full-length CFTR (1). In addition, this approach provided a means of ruling out off target effects that may influence CFTR because of the effect of SUMOylation on other proteins or pathways. The in vitro modification assay can be used also to evaluate the involvement of additional components of the modification pathway, such as SUMO proteases, Ubc9, or E3 enzymes and their post-translational modifications. Prior results from co-expression of SENP-1 suggested that an interplay between SUMO proteases and SUMO-conjugating enzymes could modulate the impact of Hsp27-mediated F508del degradation, similar to the interplay between ubitquitin E3s and deubiquitylating enzymes (56, 57).
Notably, the SUMO paralog dependence of NBD1 modification paralleled that of FL F508del CFTR, previously examined in vivo (1). SUMO-2 modification was augmented by the inclusion of Hsp27 protein in the reaction mixture, similar to the action of Hsp27 from in vivo overexpression studies. Quantitatively, Hsp27-induced modification of F508del NBD1 in vitro was not as great as that observed for F508del full-length or CD4T-NBD1, a finding that may reflect differences in accessibility of enzymes to the modification site or the need for other factors present within cells.
SUMOylation Targets a Non-native Conformation of NBD1
F508del NBD1 was more strongly SUMOylated than the WT protein under a variety of conditions. Increased SUMO modification, particularly of F508del NBD1, as [ATP] was reduced is consistent with destabilization of the native NBD1 conformation, because ATP serves as a ligand that shifts the equilibrium between native and non-native conformations by stabilizing the native state. This finding suggested that a transitional conformation of the destabilized NBD might serve as the substrate for SUMO conjugation, and several findings supported this interpretation for the differences in WT and F508del modification: (a) SUMOylation of F508del NBD1 was greater than that of WT NBD1, paralleling its stability defect; (b) SUMO conjugation of both the WT and mutant NBD1 was enhanced by preincubation at 30 °C, which initiates unfolding of the domain; (c) SUMO modification was augmented at low [ATP], removing a ligand that is known to stabilize the domain conformation (38, 39); (d) time-dependent increases in intrinsic fluorescence, which monitors formation of the non-native conformation (26, 27), were followed by F508del NBD1 SUMOylation in vitro; and (e) the extent of F508del NBD1 SUMO modification decreased with the introduction of solubilizing or revertant mutations. These variants enhance the stability of the mutant NBD, generating a conformation that has more native-like properties and is less subject to SUMO modification.
Efforts to identify folding intermediates of proteins have revealed transitional conformations that form as a variety of globular proteins denature, and these intermediates share some similarities (58). Non-native intermediates that form as proteins unfold were found to have native-like secondary structure, but they lacked the specific conformations characteristic of native, tertiary structures. However, their relation to the intermediates forming during the reverse process, that of protein folding, is not clear. Ohgushi and Wada (59) first characterized these intermediates as molten globules, and for α-lactalbumen, this molten globule state was found to be identical to the intermediate that forms transiently during folding of the protein (60).
Transitional conformations can be generated using mild denaturing conditions, such as raised temperature, moderate concentrations of denaturants, or for inherently unstable proteins, by lack of ligand binding, e.g. our ATP data. As implied above, the relation between the structure of NBD1 that is targeted by SUMOylation and that of an intermediate that forms along the folding pathway remains to be determined. Whether there are transitions during CFTR biogenesis that make the protein available for SUMO modification is not yet known. As noted above, CFTR domains fold co-translationally, but the interfaces between domains are resolved slowly (37, 51, 52). To the extent that these are reversible processes, our data suggest that the formation of a transitional NBD1 structure could lead to its SUMO modification as an early step in CFTR biogenesis, perhaps co-translationally, as has been observed for ubiquitylation (61). On the other hand, the protracted domain association process also suggests the possibility that transitional intermediates could be targeted for SUMO conjugation and degradation as domain assembly proceeds. Thus, interfering with this post-translational modification could make more immature CFTR available to improve the efficacy of existing or developing correctors of the folding process.
Author Contributions
R. A. F. and P. H. T. conceived and coordinated the study and wrote the paper. X. G. and A. A. designed, performed, and analyzed the experiments, and X. G. and P. H. T. prepared the figures. G. L. L. conceived and designed, and A. R. purified the NBD1 constructs used in this study.
This work was supported by National Institutes of Health Grants R01-DK068196 (to R. A. F.) and R01-DK083284 (to P. H. T.), by Cystic Fibrosis Foundation Therapeutics Grants FRIZZE05X0 (to R. A. F.) and THIBOD09XX0 (to P. H. T.), and by funds from the Cystic Fibrosis Canada and Canadian Institute of Health Research (to G. L. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CFTR
- cystic fibrosis transmembrane conductance regulator
- F508del
- delta-F508
- NBD
- nucleotide-binding domain
- SUMO
- small ubiquitin-like modifier
- Hsp
- Heat shock protein
- sHsp
- small heat shock protein
- Ubc9
- SUMO E2-conjugating enzyme
- FL CFTR
- full-length CFTR
- ER
- endoplasmic reticulum
- ERAD
- ER associated degradation
- MSD
- membrane-spanning domain
- E1
- SUMO-activating enzyme
- E2
- SUMO-conjugating enzyme
- STUbL
- SUMO-targeted ubiquitin ligase
- CD4T
- cluster of differentiation 4 truncated
- IP
- immunoprecipitation
- IB
- immunoblot
- SENP-1
- sentrin-specific protease 1.
References
- 1. Ahner A., Gong X., Schmidt B. Z., Peters K. W., Rabeh W. M., Thibodeau P. H., Lukacs G. L., and Frizzell R. A. (2013) Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell 24, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frizzell R. A., and Hanrahan J. W. (2012) Physiology of epithelial chloride and fluid secretion. Cold Spring Harb. Perspect. Med. 2, a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Farinha C. M., and Amaral M. D. (2005) Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell Biol. 25, 5242–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rowe S. M., and Verkman A. S. (2013) Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb. Perspect. Med. 3, a009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clancy J. P., Rowe S. M., Accurso F. J., Aitken M. L., Amin R. S., Ashlock M. A., Ballmann M., Boyle M. P., Bronsveld I., Campbell P. W., De Boeck K., Donaldson S. H., Dorkin H. L., Dunitz J. M., Durie P. R., Jain M., Leonard A., McCoy K. S., Moss R. B., Pilewski J. M., Rosenbluth D. B., Rubenstein R. C., Schechter M. S., Botfield M., Ordoñez C. L., Spencer-Green G. T., Vernillet L., Wisseh S., Yen K., and Konstan M. W. (2012) Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67, 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cyr D. M., Höhfeld J., and Patterson C. (2002) Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem. Sci. 27, 368–375 [DOI] [PubMed] [Google Scholar]
- 7. Amaral M. D. (2004) CFTR and chaperones: processing and degradation. J. Mol. Neurosci. 23, 41–48 [DOI] [PubMed] [Google Scholar]
- 8. Strickland E., Qu B. H., Millen L., and Thomas P. J. (1997) The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 272, 25421–25424 [DOI] [PubMed] [Google Scholar]
- 9. Meacham G. C., Lu Z., King S., Sorscher E., Tousson A., and Cyr D. M. (1999) The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 18, 1492–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., and Riordan J. R. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 11. Meacham G. C., Patterson C., Zhang W., Younger J. M., and Cyr D. M. (2001) The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105 [DOI] [PubMed] [Google Scholar]
- 12. Sun F., Zhang R., Gong X., Geng X., Drain P. F., and Frizzell R. A. (2006) Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J. Biol. Chem. 281, 36856–36863 [DOI] [PubMed] [Google Scholar]
- 13. Ward C. L., Omura S., and Kopito R. R. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 14. Younger J. M., Chen L., Ren H. Y., Rosser M. F., Turnbull E. L., Fan C. Y., Patterson C., and Cyr D. M. (2006) Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126, 571–582 [DOI] [PubMed] [Google Scholar]
- 15. Rajaraman K., Raman B., and Rao C. M. (1996) Molten-globule state of carbonic anhydrase binds to the chaperone-like α-crystallin. J. Biol. Chem. 271, 27595–27600 [DOI] [PubMed] [Google Scholar]
- 16. Treweek T. M., Lindner R. A., Mariani M., and Carver J. A. (2000) The small heat-shock chaperone protein, α-crystallin, does not recognize stable molten globule states of cytosolic proteins. Biochim. Biophys. Acta 1481, 175–188 [DOI] [PubMed] [Google Scholar]
- 17. Lindner R. A., Treweek T. M., and Carver J. A. (2001) The molecular chaperone α-crystallin is in kinetic competition with aggregation to stabilize a monomeric molten-globule form of α-lactalbumin. Biochem. J. 354, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahner A., Nakatsukasa K., Zhang H., Frizzell R. A., and Brodsky J. L. (2007) Small heat-shock proteins select DeltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol. Biol. Cell 18, 806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Evgrafov O. V., Mersiyanova I., Irobi J., Van Den Bosch L., Dierick I., Leung C. L., Schagina O., Verpoorten N., Van Impe K., Fedotov V., Dadali E., Auer-Grumbach M., Windpassinger C., Wagner K., Mitrovic Z., Hilton-Jones D., Talbot K., Martin J. J., Vasserman N., Tverskaya S., Polyakov A., Liem R. K., Gettemans J., Robberecht W., De Jonghe P., and Timmerman V. (2004) Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 36, 602–606 [DOI] [PubMed] [Google Scholar]
- 20. Dierick I., Irobi J., De Jonghe P., and Timmerman V. (2005) Small heat shock proteins in inherited peripheral neuropathies. Ann. Med. 37, 413–422 [DOI] [PubMed] [Google Scholar]
- 21. Wang X., Venable J., LaPointe P., Hutt D. M., Koulov A. V., Coppinger J., Gurkan C., Kellner W., Matteson J., Plutner H., Riordan J. R., Kelly J. W., Yates J. R. 3rd, and Balch W. E. (2006) Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815 [DOI] [PubMed] [Google Scholar]
- 22. Gareau J. R., and Lima C. D. (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sriramachandran A. M., and Dohmen R. J. (2014) SUMO-targeted ubiquitin ligases. Biochim. Biophys. Acta 1843, 75–85 [DOI] [PubMed] [Google Scholar]
- 24. Du K., and Lukacs G. L. (2009) Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell 20, 1903–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rabeh W. M., Bossard F., Xu H., Okiyoneda T., Bagdany M., Mulvihill C. M., Du K., di Bernardo S., Liu Y., Konermann L., Roldan A., and Lukacs G. L. (2012) Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell 148, 150–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Protasevich I., Yang Z., Wang C., Atwell S., Zhao X., Emtage S., Wetmore D., Hunt J. F., and Brouillette C. G. (2010) Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 19, 1917–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang C., Protasevich I., Yang Z., Seehausen D., Skalak T., Zhao X., Atwell S., Spencer Emtage J., Wetmore D. R., Brouillette C. G., and Hunt J. F. (2010) Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci. 19, 1932–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thibodeau P. H., Richardson J. M. 3rd, Wang W., Millen L., Watson J., Mendoza J. L., Du K., Fischman S., Senderowitz H., Lukacs G. L., Kirk K., and Thomas P. J. (2010) The cystic fibrosis-causing mutation deltaF508 affects multiple steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 285, 35825–35835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qu B. H., and Thomas P. J. (1996) Alteration of the cystic fibrosis transmembrane conductance regulator folding pathway. J. Biol. Chem. 271, 7261–7264 [DOI] [PubMed] [Google Scholar]
- 30. Thibodeau P. H., Brautigam C. A., Machius M., and Thomas P. J. (2005) Side chain and backbone contributions of Phe508 to CFTR folding. Nat. Struct. Mol. Biol. 12, 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruderer R., Tatham M. H., Plechanovova A., Matic I., Garg A. K., and Hay R. T. (2011) Purification and identification of endogenous polySUMO conjugates. EMBO Rep. 12, 142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lewis H. A., Zhao X., Wang C., Sauder J. M., Rooney I., Noland B. W., Lorimer D., Kearins M. C., Conners K., Condon B., Maloney P. C., Guggino W. B., Hunt J. F., and Emtage S. (2005) Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J. Biol. Chem. 280, 1346–1353 [DOI] [PubMed] [Google Scholar]
- 33. Matic I., van Hagen M., Schimmel J., Macek B., Ogg S. C., Tatham M. H., Hay R. T., Lamond A. I., Mann M., and Vertegaal A. C. (2008) In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol. Cell Proteomics 7, 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Serohijos A. W., Hegedus T., Aleksandrov A. A., He L., Cui L., Dokholyan N. V., and Riordan J. R. (2008) Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U.S.A. 105, 3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lewis H. A., Wang C., Zhao X., Hamuro Y., Conners K., Kearins M. C., Lu F., Sauder J. M., Molnar K. S., Coales S. J., Maloney P. C., Guggino W. B., Wetmore D. R., Weber P. C., and Hunt J. F. (2010) Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J. Mol. Biol. 396, 406–430 [DOI] [PubMed] [Google Scholar]
- 36. Lewis H. A., Buchanan S. G., Burley S. K., Conners K., Dickey M., Dorwart M., Fowler R., Gao X., Guggino W. B., Hendrickson W. A., Hunt J. F., Kearins M. C., Lorimer D., Maloney P. C., Post K. W., Rajashankar K. R., Rutter M. E., Sauder J. M., Shriver S., Thibodeau P. H., Thomas P. J., Zhang M., Zhao X., and Emtage S. (2004) Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23, 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lukacs G. L., Mohamed A., Kartner N., Chang X. B., Riordan J. R., and Grinstein S. (1994) Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 13, 6076–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Devaraneni P. K., Martin G. M., Olson E. M., Zhou Q., and Shyng S. L. (2015) Structurally distinct ligands rescue biogenesis defects of the KATP channel complex via a converging mechanism. J. Biol. Chem. 290, 7980–7991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loo T. W., Bartlett M. C., and Clarke D. M. (2005) Rescue of folding defects in ABC transporters using pharmacological chaperones. J. Bioenerg. Biomembr. 37, 501–507 [DOI] [PubMed] [Google Scholar]
- 40. Matunis M. J., Coutavas E., and Blobel G. (1996) A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 135, 1457–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Teem J. L., Berger H. A., Ostedgaard L. S., Rich D. P., Tsui L. C., and Welsh M. J. (1993) Identification of revertants for the cystic fibrosis delta F508 mutation using STE6-CFTR chimeras in yeast. Cell 73, 335–346 [DOI] [PubMed] [Google Scholar]
- 42. DeCarvalho A. C., Gansheroff L. J., and Teem J. L. (2002) Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator delta f508. J. Biol. Chem. 277, 35896–35905 [DOI] [PubMed] [Google Scholar]
- 43. Layton C. J., and Hellinga H. W. (2010) Thermodynamic analysis of ligand-induced changes in protein thermal unfolding applied to high-throughput determination of ligand affinities with extrinsic fluorescent dyes. Biochemistry 49, 10831–10841 [DOI] [PubMed] [Google Scholar]
- 44. Pissarra L. S., Farinha C. M., Xu Z., Schmidt A., Thibodeau P. H., Cai Z., Thomas P. J., Sheppard D. N., and Amaral M. D. (2008) Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem. Biol. 15, 62–69 [DOI] [PubMed] [Google Scholar]
- 45. Qu B. H., Strickland E. H., and Thomas P. J. (1997) Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J. Biol. Chem. 272, 15739–15744 [DOI] [PubMed] [Google Scholar]
- 46. Brown C. R., Hong-Brown L. Q., Biwersi J., Verkman A. S., and Welch W. J. (1996) Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1, 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang X. M., Wang X. T., Yue H., Leung S. W., Thibodeau P. H., Thomas P. J., and Guggino S. E. (2003) Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 278, 51232–51242 [DOI] [PubMed] [Google Scholar]
- 48. Cui L., Aleksandrov L., Chang X. B., Hou Y. X., He L., Hegedus T., Gentzsch M., Aleksandrov A., Balch W. E., and Riordan J. R. (2007) Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 365, 981–994 [DOI] [PubMed] [Google Scholar]
- 49. Rosser M. F., Grove D. E., Chen L., and Cyr D. M. (2008) Assembly and misassembly of cystic fibrosis transmembrane conductance regulator: folding defects caused by deletion of F508 occur before and after the calnexin-dependent association of membrane spanning domain (MSD) 1 and MSD2. Mol. Biol. Cell 19, 4570–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mendoza J. L., Schmidt A., Li Q., Nuvaga E., Barrett T., Bridges R. J., Feranchak A. P., Brautigam C. A., and Thomas P. J. (2012) Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell 148, 164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Du K., Sharma M., and Lukacs G. L. (2005) The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 12, 17–25 [DOI] [PubMed] [Google Scholar]
- 52. Kleizen B., van Vlijmen T., de Jonge H. R., and Braakman I. (2005) Folding of CFTR is predominantly cotranslational. Mol. Cell 20, 277–287 [DOI] [PubMed] [Google Scholar]
- 53. Okiyoneda T., Veit G., Dekkers J. F., Bagdany M., Soya N., Xu H., Roldan A., Verkman A. S., Kurth M., Simon A., Hegedus T., Beekman J. M., and Lukacs G. L. (2013) Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 9, 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Patrick A. E., and Thomas P. J. (2012) Development of CFTR Structure. Front. Pharmacol. 3, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tatham M. H., Geoffroy M. C., Shen L., Plechanovova A., Hattersley N., Jaffray E. G., Palvimo J. J., and Hay R. T. (2008) RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 10, 538–546 [DOI] [PubMed] [Google Scholar]
- 56. Hassink G. C., Zhao B., Sompallae R., Altun M., Gastaldello S., Zinin N. V., Masucci M. G., and Lindsten K. (2009) The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 10, 755–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bomberger J. M., Barnaby R. L., and Stanton B. A. (2010) The deubiquitinating enzyme USP10 regulates the endocytic recycling of CFTR in airway epithelial cells. Channels 4, 150–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matsuo K., Sakurada Y., Yonehara R., Kataoka M., and Gekko K. (2007) Secondary-structure analysis of denatured proteins by vacuum-ultraviolet circular dichroism spectroscopy. Biophys. J. 92, 4088–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ohgushi M., and Wada A. (1983) “Molten-globule state”: a compact form of globular proteins with mobile side-chains. FEBS Lett. 164, 21–24 [DOI] [PubMed] [Google Scholar]
- 60. Dobson C. M. (1991) NMR spectroscopy and protein folding: studies of lysozyme and α-lactalbumin. Ciba Found. Symp. 161, 167–181 [DOI] [PubMed] [Google Scholar]
- 61. Sato S., Ward C. L., and Kopito R. R. (1998) Cotranslational ubiquitination of cystic fibrosis transmembrane conductance regulator in vitro. J. Biol. Chem. 273, 7189–7192 [DOI] [PubMed] [Google Scholar]