

Abstract
We developed a compact plane illumination plugin (PIP) device which enabled plane illumination and light sheet fluorescence imaging on a conventional inverted microscope. The PIP device allowed the integration of microscope with tunable laser sheet profile, fast image acquisition, and 3-D scanning. The device is both compact, measuring approximately 15 by 5 by 5 cm, and cost-effective, since we employed consumer electronics and an inexpensive device molding method. We demonstrated that PIP provided significant contrast and resolution enhancement to conventional microscopy through imaging different multi-cellular fluorescent structures, including 3-D branched cells in vitro and live zebrafish embryos. Imaging with the integration of PIP greatly reduced out-of-focus contamination and generated sharper contrast in acquired 2-D plane images when compared with the stand-alone inverted microscope. As a result, the dynamic fluid domain of the beating zebrafish heart was clearly segmented and the functional monitoring of the heart was achieved. Furthermore, the enhanced axial resolution established by thin plane illumination of PIP enabled the 3-D reconstruction of the branched cellular structures, which leads to the improvement on the functionality of the wide field microscopy.
OCIS codes: (170.3880) Medical and biological imaging, (170.2520) Fluorescence microscopy, (110.3010) Image reconstruction techniques, (110.2945) Illumination design, (170.6900) Three-dimensional microscopy
1. Introduction
Light Sheet Fluorescence Microscopy (LSFM) is a prominent imaging technique that uniquely enables multidimensional imaging over a wide range of scale from several microns to several millimeters [1–3]. Compared with the conventional wide field microscopy and confocal laser scanning microscopy, LSFM can significantly reduce photo-damage, enhance image contrast, and improve axial resolution when imaging multi-cellular organisms by optically sectioning the sample using a thin plane illuminating light sheet [1]. LSFM is currently on the verge of becoming a predominant visualization technique for life science research. In the past decade, several structure variants of LSFM, including Selective Plane Illumination Microscopy (SPIM), Multidirectional SPIM (mSPIM), Digitally Scanned Light sheet Microscopy (DSLM), Objective-Coupled Planar Microscopy (OCPI), Oblique Plane Microscope (OPM), Bessel-Beam-based Light sheet Microscopy, have been developed and widely used for various research topics, such as embryogenesis, tissue culture and brain mapping [4–17]. LSFM technology is characterized by its orthogonal illumination and detection setting which are comprised of four major components in the system: light sheet illumination, sample mounting, fluorescence detection, and data acquisition. The majority of current LSFM modalities, such as SPIM [1] and DSLM [2, 3], are based on completely new designs of all the four units and are therefore fully independent from conventional wide field microscopy or confocal scanning microscopy. A few other LSFM modalities, such as OCPI [12] and iSPIM [18], have been created on existing inverted microscopes. However, their LSFM functions remain independent from the microscope base. The inverted microscope in this case, complements the LSFM system by merely providing additional bright field observation or wide field fluorescence collection.
Despite a unique orthogonal illumination path, LSFM shares similar configurations of fluorescence detection and data acquisition parts with the widely used wide-field fluorescence microscopy. On one hand, constructing an entirely new LSFM system may not always be feasible due to the high requirements of resources as well as skills; on the other hand, the addition of an extra light-sheet illumination in the horizontal direction and utilization of delicate sample manipulation strategy can be accomplished on a common conventional microscope by combining the light-sheet illumination with existing optical path of the conventional microscope. However, several challenges in the implementation of this concept remain: (1) conventional wide-field microscope requires samples positioned on a glass slide, but the rough side-wall surface of the glass slide makes horizontal plane illumination nearly impossible; (2) the short working distance of the objective results in a limited space for integration with regular optics or mechanics; and (3) a majority of conventional microscopes has no z-translation apparatus on the sample stage; thus, rendering them incapable of performing light sheet scanning.
In this context, we addressed the aforementioned issues by using a simple, compact and cost-effective Plane illumination plugin (PIP) add-on device. The PIP was developed to be mounted onto the inverted microscope to enable the light sheet fluorescent imaging of a specimen. The PIP device has a compact structure that utilizes small and inexpensive electronics and optics elements. The lightweight bracing structure of the device was designed through Solidworks, produced by rapid 3-D printing, and easily mounted onto the stage of the inverted microscope. Three key parts were employed to generate the light sheet illumination: a laser diode module as the light source, an adjustable aperture for tuning the beam profile, and a cylindrical lens for line-focusing the beam. All of the components can be easily mounted onto a sliding rail with their center automatically aligned. As a result, this illumination arm of the PIP was able to generate a tunable Gaussian beam light sheet on the horizontal plane with a short optical path measuring only about 5 cm. The tunability of the cylindrical lens along the light propagation direction, allowed the waist of light sheet to be located properly on the sample. We simply mounted the sample tube onto the L shape adapting rod. The tube was then manually rotated on the rod until the sample’s least scattering side faces the light sheet and thereafter the sample can be vertically scanned (Z-scanning) through the light sheet with a steerable step size. Unlike other LSFM modalities, while the plane illumination on the sample takes place inside the PIP, the inverted microscope system maintains its functions for fluorescence detection and image acquisition. With the integration of PIP, superior in-focus excitation helps the conventional microscope to acquire sharp, haze-free sectional images. For example, the clear beating motion of a zebrafish embryo heart, which was otherwise difficult to visualize using either conventional microscope or confocal microscope, could be successfully captured using our PIP-added system. In addition, the axial confinement from PIP’s plane illumination significantly improved the axial resolution of the system, which was previously unsatisfactory due to the wide-field excitation. Thus, Z-scanning a multi-cellular branched structure was further performed to achieve the three-dimensional reconstruction of the entire cellular architecture and to resolve the complete intracellular structure. Unlike the common LSFM modalities that rely on expensive components and experienced operators, the PIP device makes full use of the existing microscopic platforms. Therefore, aside from being an inexpensive light sheet imaging solution and easy-to-use modality, PIP also avoids complicated optics alignment and imaging operation, for adaptation in resource-limited environments.
2. Method
2.1 Design of horizontal plane illumination
Our PIP design was initiated on the basis of two concepts. First, in light sheet microscopy, the comparatively low requirements on excitation source and shaping of hyperbola Gaussian light sheet enabled the miniature of the plane illumination path using a compact laser diode source and small optics. Second, state-of-art 3D printing techniques allowed for the fast and inexpensive prototyping of sophisticated structures, which were necessary for accommodating multiple components of a functional system. Based on these two concepts, we chose a compact laser diode as the excitation source, a tunable diaphragm or slit for beam control, and a cylindrical lens as the line-focusing generator to create an illuminating light sheet in the PIP. A sliding rail with several built-in racks was designed as the illumination part of PIP to house these optical elements. The laser holder fitted various types of laser diodes with a maximum diameter of 17.5mm by using appropriate laser adapters. A 450 nm wavelength blue laser diode module with 80 mW power was used in our experiment for the excitation of GFP samples. A tunable diaphragm with a 0.7 mm to 2 mm diameter clean aperture was used to control the vertical extent of the incident laser beam to further determine the axial confinement of the laser sheet as well as its confocal region, wherein the Gaussian laser sheet could be treated as uniform [13, 19, 20]. A 10mm x 20mm cylindrical lens with 30 mm focal-length lens was mounted at the end of the illumination path to generate a line-focused laser sheet on the horizontal plane. All the aforementioned elements were assembled onto the racks in the PIP. The optics housed racks allowed smooth sliding on the rail along the laser propagating direction (x-axis) for coarse tuning. The schematic of a working PIP device was shown in [Fig. 1(a)].
Fig. 1.

Schematic of the Plane Illumination Plugin (PIP) and its working principle. (a) The configuration of the PIP, which is based on fast 3D printing and several small optics. The key components include a laser diode module as light source, a cylindrical lens for light sheet generation, a micrometer drive for optical control, and a disposable cuvette as the sample holder. (b) Sample manipulation setup in PIP. A FEP tubing with sample mounted inside is connected to a micrometer via an L shape adapter. By tuning the micrometer, the sample together with the tubing can be vertically scanned through the light sheet, inside a water environment. (c) The working status of the PIP. The laser sheet propagates horizontally through the sample, generating a plane illumination on the fluorophores. The existing fluorescence detection system inside the microscope collects the sample signal and images it onto the camera. (d) The working principle of PIP with a Nikon Eclipse TE-2000 inverted microscope. A lens adapter is added on the objective lens to increase the working distance. Through mounting the PIP on the stage and inserting the sample, LSFM is enabled on a conventional wide-field microscope. (e), (f) show the photographs of the real PIP device with (e) the scene of PIP device working with a Nikon TE-2000 inverted microscope. (f) The image of PIP with ruler showing its dimensions.
2.2 Design of sample mounting and control
The PIP was designed to house a small chamber (12.5mm by 12.5mm by 37.5 mm) at the center of the device. A micro-actuator driven L shaped aluminum rod was inserted into this water chamber for adapting a piece of Fluorinated Ethylene Propylene (FEP) tube in which the samples were packaged inside using hydrogel [Fig. 1(b)]. We chose FEP tube because it has a refractive index of ~1.34, which is similar with the refractive index of both biological samples and the DI water (~1.33). This property minimized the light refraction on the curved water-FEP interface and significantly reduced the side effect of aberration while the excitation light and emission signal pass through this interface. The micro-actuator with 1 μm travel resolution was vertically fastened on top of the PIP to finely tune the z-position of the sample during 3D scanning. The sample could be positioned concurrently in the x-y plane into the center field of view by manipulating the microscope stage. For use, the PIP device could be easily mounted on the stage using the mounting holes. We transported samples into the FEP tube first, and then we attached the FEP tube to the aluminum rod, which was already inside the cuvette. We rotated the tube to adjust sample to the desired orientation. We prepared the cuvette by drilling two holes on one side of the wall, one for the L shape rod and another for filling water. At last, we filled the cuvette with DI water [Fig. 1(b)]. The aperture size of iris diaphragm, axial extent and confocal range of laser sheet sectioning were adjusted based on the sample dimensions. To optimize the plane illumination on the sample, we slid the cylindrical lens to roughly focus the light sheet into the sample region, approximately fitting the sample inside the confocal range of the Gaussian beam [21, 22]. However, since the confocal range could be ~300 μm in its shortest case, the precise tuning of the lens holder using bare hands became unpractical. Thus, we introduced a small rotation-to-translation mechanics to finely tune the cylindrical lens by rotating the screw-nut sets. The screws had one end attached into the threaded holes in the base track, and another end fixed with the lens holder. It enabled the fine movement of cylindrical lens along the laser propagation direction and the overlapping of light sheet’s confocal region with the sample’s region-of-interest [Fig. 1(c)]. In [Fig. 1(e)] and [Fig. 1(f)], we included the photographs of the real device in stand-alone mode and working status. More specific details of the PIP device are presented in the supplementary materials, including the CAD drawings with design specification and dimensions of all the customized parts, following by a step-by-step assembling protocal, which allows researchers to reproduce our device in a rapid and affordable way (see Visualization 1 (301.4KB, PDF) ), the exploded assembling animation of the whole device illustrating the assembling of PIP from independent parts (see Visualization 2 (1.1MB, AVI) ), and a part list of the device with vendors and prices (see Data File 1 (866B, CSV) ).
2.3 Image acquisition
The PIP generated a thin laser sheet propagating in the horizontal plane, which was orthogonal to the detection path of the inverted microscope. The laser sheet replaced the standard wide-field excitation of the inverted microscope and imposed a sharp plane illumination on the sample. The fluorescent signals emitted from the illuminated planes of the sample were collected by the inverted microscope and projected onto the camera via the microscope’s internal optical path. The confocal parameter of the laser sheet was widely tuned from hundreds of microns to several millimeters for imaging various multi-cellular organisms to match the dimension of the samples. The axial extent of the light sheet correspondingly varied from 5 to 10 microns, which represented the improved axial resolution compared to wide-field epi-fluorescence. When implementing 3D imaging, we used the micro-actuator to scan the sample through the steady light sheet along the z-direction with a step size at least half of the axial extent of the light sheet [23]. Normally, a low power objective such as 4X/0.13 and 10X/0.3, was used to cover the field-of-view (FOV) of the sample and a digital camera was mounted at the image plane to continuously record the sequence of the sectional images with high speed. The schematic diagram of entire PIP-aided light sheet microscopy is shown in [Fig. 1(d)].
2.4 Sample preparation
2.4.1 3D culture of human aortic endothelial cells
Human aortic endothelial cells (HAEC) (Cell Applications) were cultured in endothelial growth medium (Cell Applications) supplemented with 5% Fetal Bovine Serum (FBS) (Gibco). HAEC were treated with recombinant GFP-LC3 adenovirus, kindly provided by Dr. Junichi Sadoshima from Rutgers New Jersey Medical School, at a multiplicity of infection (MOI) 1:100 to induce GFP expression and allow for fluorescent visualization of the cells. The transfected HAEC cells were grown in a 96-well plate at 20,000 cells/well on a 100 μL Matrigel Growth-factor-reduced matrix (BD Biosciences) that was allowed to gel before seeding cells in DMEM + 5% FBS + 25 ng/mL VEGF. Tube formation was visualized following 6 hours incubation at 37°C.
2.4.2 Zebrafish embryo culture
Adult Tg (fli1a: EGFP) and Tg (cmlc2: GFP) transgenic zebrafish were raised in the UCLA zebrafish core facility. All the experiments were performed in compliance with the approval of GLA Institutional Animal Care and Use Committee (IACUC) protocol (Zebrafish IACUC Protocol number: 101004-14). The fish were maintained with filtered fresh water under 14 hours of incandescent light and 10 hours of dark conditions. In Tg(fli1a:EGFP) fish, the fli1a promoter-driven enhanced green fluorescent protein (EGFP) was expressed predominantly in vascular endothelial and endocardial cells. In Tg(cmlc2:GFP) fish, the cmlc promoter-driven green fluorescent protein (GFP) was expressed exclusively in myocardia cells. The embryos were transferred to a petri dish and incubated in 28.5°C. To maintain transparency of zebrafish embryos, embryo medium was supplemented with 0.003% phenylthiourea (PTU) to suppress pigmentation at 10 hpf.
2.4.3 Sample preparation for PIP
Samples, such as trans-genetic zebrafish embryo or GFP-labeled cell macro-structures, were mounted using low melt point agarose at 0.5% to 1% concentration in 2 mm diameter FEP tubing, which has a refraction index (1.34) very similar to water (1.33) and minimized the index mismatch with surrounding phosphate buffered solution in PIP’s sample cuvette during imaging [1].
3. Result
3.1 Light sheet calibration
The tunable light sheet profiles generated by PIP were characterized to verify the feasibility of the device. A Sony monochrome CCD profiler with 2.2 μm pixel size was mounted on a motorized translation stage to sequentially acquire the projections of the light sheet along its propagation direction (Figure not shown). Two representative light sheets generated at the iris diaphragm size of both 1.8 mm and 1 mm were profiled for imaging the embryonic zebrafish hearts and HAEC tube formations. The smallest Full-Width-at-Half-Maximum (FWHM) of the light sheet at each diaphragm setting could be directly obtained from the projection image recorded at the focus [Fig. 2(a)] and the range showing FWHM variation could be further reconstructed and measured through stacking the projections [Fig. 2(b)]. In Fig. 2(b), the confocal parameter, within which the light sheet thickness could be considered as constant, were labeled with double-headed arrows to show the optimal region for sectioning. The theoretical calculation of the confocal parameter and Rayleigh range, which determined the region of an ideal Gaussian beam [21, 22, 24], were detailed and compared with the experimental data (see Data File 2 (278B, CSV) ). The 1.8 mm diaphragm size generated a sharper light sheet with thickness of ~5.5 μm and a shorter confocal parameter of ~300 μm, suitable for small samples, such as embryonic zebrafish heart or C. elegans. Meanwhile, the 1 mm diaphragm offered a thicker light sheet of ~10μm and longer confocal parameter of ~1mm, to enable the sectioning on macroscopic objects, such as early-stage zebrafish embryos and 3D-cultured cellular branches, etc. The intensity distribution of the light sheets was further mapped in 3-D space [25] [data not shown].
Fig. 2.

Characterization of the tunable laser sheet of PIP. (a) Tuning the laser sheet thickness simply by adjusting the diaphragm. As the beam width decreases through reducing the aperture size of diaphragm from 1.8 mm (top) to 1 mm (bottom), the laser sheet becomes less focused and linearly thicker at the waist. The scale bars are 200 μm in i to ii, and 10 μm in inserts, respectively. (b) The corresponding change of the confocal region, which represents the available area for light sectioning. The double-headed arrow line shows the length of the confocal range (x direction), within which the light sheet can be considered uniform. The length of the confocal range is inversely proportional to the square of the light sheet thickness. The scare bars are 100 μm.
3.2 System resolution measurement
A micro-sphere (~400 nm in diameter) was used as the point source to compare the resolving power among 3 different imaging configurations, including the inverted microscope, PIP-mounted inverted microscope with 1 mm aperture and PIP-mounted inverted microscope with 1.8 mm aperture, shown in Fig. 3(a)-3(c), respectively. The normalized line intensity profiles of the resolved bead images along the lateral (x) and axial (z) directions were further plotted in Fig. 3(d) and 3(e). The 50% peak intensity levels were labeled correspondingly in cyan contour lines to display the FWHM intensities that indicated the lateral and axial extents of the point source under 3 settings [5, 8, 14, 17]. For these three cases, FWHMs at lateral direction were measured all around 1.7 μm. The lateral resolutions were similar due to the use of identical detection objectives. However, the axial FWHMs of the resolved point source were significantly improved from ~23 μm to ~6 μm by replacing the original wide-field excitation with plane illumination of the plugin device.
Fig. 3.

The comparison of resolving power measured before and after the PIP was mounted. (a) Visualization of the 400 nm point source by regular inverted microscope using a 10X/0.3 air objective. The subsets from the left to the right correspondingly show the x-y, x-z, y-z planes and the volumetric rendering. The axial extent is obviously larger than the lateral extents, due to the relatively low numerical aperture. (b) - (c), Visualization of the 400 nm point source by the same optical setting of the inverted microscope, but with PIP mounted. In (b), the aperture of the PIP are set to be 1 mm while in (c), this value is 1.8 mm. With PIP mounted, the axial extents become significantly smaller due to the sharp plane illumination. Scale bars in (a) to (c) are 5 microns. (d) - (e), normalized line intensity plots of the resolved point source images, along lateral and axial directions, respectively. 1/2 intensity levels are drawn in the cyan lines, to show the FWHMs that reflect the resolutions of the systems. 1/e2 intensity level is drawn in green line to indicate the actually resolved diameters of the nano-particle.
3.3 Haze-free motion capture of beating embryonic zebrafish heart in vivo
Imaging dynamic structures with fast cardiac contraction presents technical challenges to the traditional light microscopy. Conventional microscopy is limited by out-of-focus excitation, which prevents the system from yielding sharp sectional images. In addition, confocal microscopy has a limited scanning speed and thus lacks the capability of freezing the fast motion. Light-sheet microscopy could outperform both conventional microscopy and confocal microscopy to image highly dynamic structures, such as a beating embryonic zebrafish heart, by including both sharp, in-focus sectioning and rapid plane acquisition [23, 26–30]. The advantages of the PIP were demonstrated by imaging the zebrafish embryonic heart [Fig. 4(a)] in a multi-step approach. First, regular epi-fluorescence images of the embryonic heart were captured by an inverted microscope (Nikon Eclipse TE-2000) using 10X/0.3 objective and sCMOS camera (Hamamatsu ORCA Flash 4.0). The serial time frames were shown in Fig. 4(b) at 100 ms intervals. Due to the limited axial resolution [measured ~28 μm in Fig. 3(a)] and extensive out-of-focus excitation [Fig. 4(b)] from the wide-field fluorescence illumination, the images were poor in quality to demarcate cardiac structure from out-of-focus blurs. Since PIP device did not block either the wide-field illumination or epi-fluorescence illumination, the experiment was done with sample mounting on PIP, while the external PIP laser was turned off.
Fig. 4.

Contrast-enhanced light sheet imaging of beating embryonic zebrafish heart by PIP mounted microscope. (a) Light sheet sectioning inside a live embryonic zebrafish heart (4 d.p.f., Tg: (cmlc: gfp)). PIP enables the selective plane illumination on specific region of interest of the beating heart. (b) and (c) show the image comparison between regular inverted microscope and PIP mounted inverted microscope. The dynamic inner and outer boundaries of the beating heart can be clearly resolved when PIP is added. (d) shows the control heart images (same stage embryo) from a home-built, standard SPIM system with identical illumination and detection settings of PIP imaging. Scale bars in all images are 50 μm.
After turning on the laser diode and activating the plane illumination, we re-imaged the beating heart using the additional light-sheet mode with ~5.5 micron axial sectioning. The resulting optical power density at the focus was around 7 μJ / μm2. The PIP significantly improved the image contrast by greatly confining the axial excitation and reducing the image contamination by the out-of-focus fluorescing [Fig. 4(c)]. As a result, the selective plane of the cardiac motion could be digitally visualized with both high temporal and spatial resolution, revealing the dynamic change in the endo- and epicardial boundaries. We further compared the performance of PIP with a standard Selective Plane Illumination Microscopy (SPIM) system that we assembled on an optical bench. Our SPIM system was a home-built SPIM system consisted of several costly components and at the same time required considerable space and highly skillful people to align the optics (data not shown). Figure 4(d) showed the control heart images (same stage embryo) from the SPIM system containing identical illumination (a laser sheet of ~6 μm FWHM) and detection (10X/0.3 detection objective plus Hamamatsu Flash 4.0 camera) configurations of PIP imaging. With an integrated PIP system, a conventional wide-field microscope could perform optical sectioning with sharp contrast comparable to that of a professional light-sheet microscopy. Though in term of light sheet imaging, the standard SPIM was more powerful than PIP-integrated inverted microscope with higher tunability, because of some advanced functions, such as multi-view fusion, we did prove that the PIP is eligible in assisting the inverted microscope with superior light sheet imaging. In addition, with the improved axial resolution, reconstruction of the complete cardiac cycle from the 3-D beating heart could be further accomplished using either a high speed camera or retrospective cardiac synchronization algorithm [31].
3.4 Image-based quantification of beating embryonic zebrafish heart
The cardiac mechanics of a beating embryonic zebrafish heart was further characterized by quantifying changes in volume and strain rate during a cardiac cycle based on the improved image results. For the computation of the heart dynamics, a clear segmentation of the myocardium boundary was substantially required as input for the analysis. Due to the low image contrast caused by excessive out-of-focus fluorescence contamination, only the outer boundary of the ventricle could be distinguished and segmented in conventional microscope result [Fig. 5(a), 1st row]. The inner boundary of the ventricle remained vague. However, light sheet illumination with either PIP (2nd row) or SPIM (3rd row) enabled a significant contrast enhancement in resolving the endocardial boundary. As a result, the clear segmentation of the fluid domain was established to perform Computational Fluid Dynamics (CFD) analysis over a time scale [Fig. 5(a), 2nd row and 3rd row].
Fig. 5.
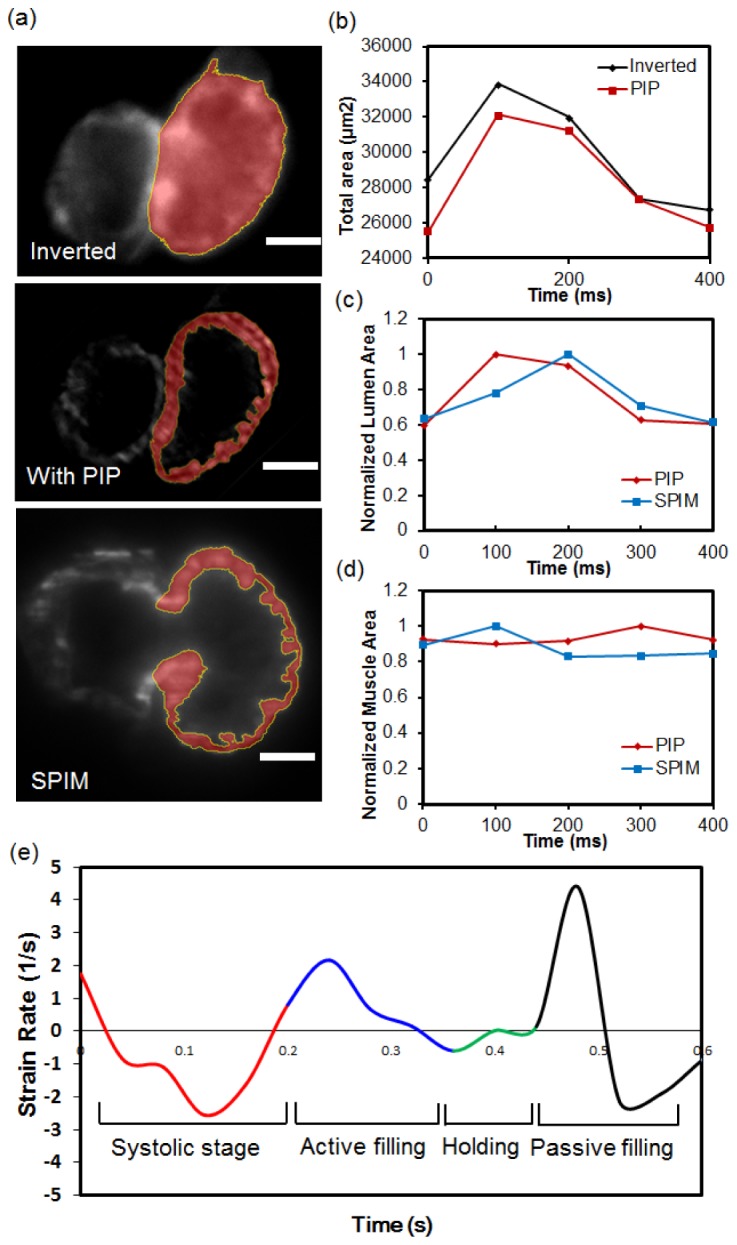
Quantifying the area dynamics of beating embryonic zebrafish heart. (a) Segmentation of the ventricle’s inner and outer boundary. In the wide-field fluorescent image, due to the out-of-focus blur, only the outer boundary of the ventricle can be identified (yellow line). In PIP and SPIM images, by significantly enhancing image contrast, the inner boundary as well as ventricle’s myocardium structure (red area) can be accurately segmented. Scale bars in all sub-graphs are 50 μm. (b) The calculated area variation of the complete ventricle area in 400 millisecond time. (c) The normalized area change of the inside fluid domain, which is only enabled in PIP and SPIM image results. (d) The corresponding area change of the segmented ventricle muscle during heart beating. (e) The strain rate of the beating embryonic zebrafish heart during a cardiac cycle. The strain under each time point was calculated based on the girth variation of the ventricle inner boundary.
With segmenting the outer (epicardial) boundary, the changes in volume of the entire heart were calculated and further plotted with respect to the 400-millisecond time span [Fig. 5(b)]. More importantly, a resolvable inner boundary acquired from the PIP image further enabled the segmentation of the fluid domain and complete myocardium architecture for future investigation of cardiac mechanics. The changes in volume in the aforementioned 400 ms time were shown in Fig. 5(c) and Fig. 5(d), respectively. The strain rate of the beating heart could also be computed based on the girth variation of the outer segmented boundary within a cardiac cycle [32] [Fig. 5(e)]. As a result, the successive occurrence of cardiac contraction (systole), active and passive filling during cardiac relaxation (diastole), was identified as red, blue and green lines showed.
3.5 3D visualization of branched cells column using PIP
We set the PIP aperture size to 1 mm, formulating a 10 μm-thick light sheet (Fig. 2) to optically section an entire branched HAEC cells column with hundreds of micron length in each dimension. The aforementioned 10X/0.3 objective and Hamamatsu ORCA Flash 4.0 sCMOS camera were used to acquire the images. The cellular branching structure was further scanned through the light sheet with a step size of 3 μm, to obtain a Z-stack for reconstructing the complete 3-D architecture. A stack of wide-field fluorescent images were obtained with the same detection objective and Z-scanning parameters. Selected 2-D plane images from regular wide-field fluorescence mode and light-sheet mode were compared in Figs. 6(a), 6(b), in which adjacent frames had a 40 μm interval of step size. In the wide-field group [Fig. 6(a)], due to the blunt sectioning and severe out-of-focus excitation, several cells at different depth were illuminated together with excessive out-of-focus blurs overlapping, resulting that the adjacent frames showed inconspicuous difference. After PIP laser was switched on, through the sharp axial sectioning, the cells with Z depth diverging greater than 10 μm could be easily sensed, so that fewer cells displayed on each Z frame, making significant difference among the frames. At the same time, the side illumination of the PIP provided superior confinement of the excitation along z direction, and substantially reduced the out-of-focus blurs. Therefore, in Fig. 6(b), clean and haze-free cells could be visualized in each frame of the stack. Figure 6(c) and 6(d) further compared the projections of the x-y, x-z and y-z planes, in wide-field mode and light sheet mode respectively [14, 18, 33]. With remarkably enhanced axial resolution and contrast, a clear volumetric reconstruction of complete branched structure was achieved exclusively under light sheet mode [Fig. 6(d), rightmost subgraph]. It could potentially benefit a variety of image-based analysis, such as precisely locating the cells and quantifying the formed patterns, which are both valuable for understanding the mechanisms underlying branch morphogenesis. Therefore, in addition to handling a small region-of-interest within live animals, PIP also demonstrated its superior performance on imaging larger organisms. We further validated this feature by three dimensionally visualizing the vessel system of zebrafish tail labelled with GFP [28, 34, 35], and significant quality enhancement were also obtained (data not shown).
Fig. 6.

Visualization of 3D-cultured cell branches using PIP-integrated conventional microscope. (a) Sequential image slices acquired by a conventional inverted microscope using 10X/0.3 air objective. Out-of-focus excitation caused drastic image deterioration. (b) Sequential image slices acquired at the same z depths using identical wide-field detection plus 10 μm optical sectioning of PIP. (c) - (d) The maximum intensity projections (MIPs) and the volume renderings obtained without and with PIP, respectively. y-z, x-z projections of the 3D reconstructed images were also shown. The incorporation of PIP achieved significant axial resolution enhancement and background noise reduction. The cells geometry and intracellular morphology were also exclusively revealed by clear volume rendering in PIP group (rightmost). Scale bars in all images are 100 μm.
4. Discussion
While conventional epi-fluorescence microscopy is currently still the backbone of most life science research and medical diagnosis despite its imperfect axial performance and inefficiencies of 3D imaging, light sheet microscopy provides significantly improved axial performance and greatly reduced photo-toxicity. The similarities between conventional epi-fluorescence microscopes and LSFM provided motivation for the development of the plane illumination plugin (PIP) to enhance the performance of conventional epi-fluorescence microscopes in a simple and cost-effective way.
We first simplified the generation of plane illumination, using a compact laser diode and a cylindrical lens to form the light sheet. A tunable aperture was designed to tune the light sheet properties, such as thickness and usable region. To create plane illumination horizontally proximal to the microscope’s objective, we then designed a bracing structure for PIP, with a flat rail structure holding the optical elements and aligning the illuminating light path. Alongside the rail and illumination path, we designed an L shape hollow rod, dipping it into a customized cuvette filled with water, to hold a FEP tube with sample mounted inside and deliver the sample precisely at the intersection of sheet illumination and fluorescence detection [36–38]. To enable the Z-scanning of sample which is necessary for 3D imaging, the rod was designed to be controlled by a vertical micrometer fixed on the top of PIP. By tuning the micrometer, the sample could be scanned vertically through the light sheet in the water chamber, together with the tube. In current proof-of-principle study, we used manual micrometer for scanning, which could be inconvenient when the experiment setup requires hundreds of steps to image larger specimens. In future designs, the micrometer could be further replaced by a miniature motorized actuator, to carry out automated 3D scanning with higher precision. In our work, we used Nikon CFI Plan Fluor 10X objective as the detection lens most. It has a convenient working distance of 16 mm and comparatively small NA of 0.3, with which we can easily make PIP work well with the microscope and at the same time, the spherical aberration could be kept minor during our 3D scanning (Fig. 6(b)). At the same time, we realized that we cannot always rely on the low-to-middle magnification objective if we want to scale up this technique to help epi-fluorescence microscope with even higher resolution (subcellular resolution) 3D imaging, in which the spherical aberration will become much more significant and water-dipping lens is always required. This problem can be smoothly solve by customizing the cuvette with thinner walls and designing a larger and flatter bottom space that allows the objective to move really close to the cuvette with aqueous medium sandwiched in between.
Some of the previous research, such as OpenSPIM [39, 40], had already developed step-by-step guides on how to build in-house LSFM systems. These user-friendly instructions were aimed for the spread of advanced light sheet imaging in a way of standardized design, moderate cost, and easy operation. Likewise, PIP shares the similar spirit of these methods, while it goes further on simplifying the design and reducing a significant amount of cost via fully utilizing the traditional inverted microscopy, which many of research labs have already owned. The total expense of the prototype of PIP we demonstrated in this work was merely ~400 USD. It is noteworthy that the capability of PIP can be strengthened and upgraded for more advanced light sheet imaging, such as dual side illumination after including precise vertical motion control, multi-color imaging after the integration of fiber guided light source, and fast automatic scanning after the integration of motorized actuator. It could be roughly estimated that even with the upgrades, the cost of PIP can still be well below 5K.
Although current version of PIP remains insufficient to achieve the same powerful performance of those high-cost light sheet modalities, such as Zeiss Light Sheet Z.1, it has the advantages of compact add-on mode, simple design, easy fabrication and significantly low cost, which offer a promising solution for conducting advanced imaging in a resource-limited environment.
5. Conclusion
In summary, as an alternative to stand-alone systems, which are currently the dominant way to access cutting-edge light sheet microscope, we have developed a simple and cost-effective device, called plane illumination plugin (PIP), to turn a conventional inverted microscope into a light sheet microscope. PIP has integrated several compact optical and mechanical elements into a small form factor which is rapidly prototyped by 3D printing technology. The elements that form the illumination path were self-aligned in PIP, and generated a tunable light sheet to illuminate the sample horizontally from the side. The sample-mounting aspect of PIP allows the precise manipulation of the sample, locating it at the intersection of plane illumination path and fluorescence detection path, with a water environment surrounded and 3D Z-scan enabled. PIP was designed to be highly compatible with the conventional inverted microscope, which has been ubiquitously used for a broad range of applications. We have made full use of the inverted microscope’s fluorescent detection system. By combining PIP with inverted microscope, we have shown the conventional inverted microscope is capable of harvesting superior images on various multi-cellular organisms, with greatly suppressed out-of-focus blurs in 2D plane images and significantly enhanced axial resolution in stacked 3D image. The palm-size PIP device could be an excellent enhancement to the conventional microscopy and provide an affordable solution to conduct advanced light sheet imaging under various resource-limited environments, such as ordinary laboratory, and for various research fields such as developmental biology, regenerative medicine and tissue engineering.
Acknowledgment
The authors would like to thank Tyler Beebe, Jonathan Law and Qiming Zhang for their insightful comments and discussion that help on improving manuscript and discussion. This study was supported by Ben Rich-Lockheed Martin Professor Endowment (C.M.H), The Ministry of Science and Technology of China (2015AA042602, P.F.), National Science Foundation (NSF) (1263236, 0968895, 1102301), National Institutes of Health (HL118650 (T.K.H.), HL083015 (T.K.H.), HD069305 (T.K.H.), HL111437 (T.K.H.)), and AHA Pre-Doctoral Fellowship 15PRE21400019 (J.L.).
References and links
- 1.Huisken J., Swoger J., Del Bene F., Wittbrodt J., Stelzer E. H., “Optical sectioning deep inside live embryos by selective plane illumination microscopy,” Science 305(5686), 1007–1009 (2004). 10.1126/science.1100035 [DOI] [PubMed] [Google Scholar]
- 2.Keller P. J., Schmidt A. D., Wittbrodt J., Stelzer E. H., “Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy,” Science 322(5904), 1065–1069 (2008). 10.1126/science.1162493 [DOI] [PubMed] [Google Scholar]
- 3.Keller P. J., Schmidt A. D., Santella A., Khairy K., Bao Z., Wittbrodt J., Stelzer E. H., “Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy,” Nat. Methods 7(8), 637–642 (2010). 10.1038/nmeth.1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahrens M. B., Orger M. B., Robson D. N., Li J. M., Keller P. J., “Whole-brain functional imaging at cellular resolution using light-sheet microscopy,” Nat. Methods 10(5), 413–420 (2013). 10.1038/nmeth.2434 [DOI] [PubMed] [Google Scholar]
- 5.Breuninger T., Greger K., Stelzer E. H., “Lateral modulation boosts image quality in single plane illumination fluorescence microscopy,” Opt. Lett. 32(13), 1938–1940 (2007). 10.1364/OL.32.001938 [DOI] [PubMed] [Google Scholar]
- 6.Chen B.-C., Legant W. R., Wang K., Shao L., Milkie D. E., Davidson M. W., Janetopoulos C., Wu X. S., Hammer J. A., 3rd, Liu Z., English B. P., Mimori-Kiyosue Y., Romero D. P., Ritter A. T., Lippincott-Schwartz J., Fritz-Laylin L., Mullins R. D., Mitchell D. M., Bembenek J. N., Reymann A. C., Böhme R., Grill S. W., Wang J. T., Seydoux G., Tulu U. S., Kiehart D. P., Betzig E., “Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution,” Science 346(6208), 1257998 (2014). 10.1126/science.1257998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelbrecht C. J., Greger K., Reynaud E. G., Kržic U., Colombelli J., Stelzer E. H., “Three-dimensional laser microsurgery in light-sheet based microscopy (SPIM),” Opt. Express 15(10), 6420–6430 (2007). 10.1364/OE.15.006420 [DOI] [PubMed] [Google Scholar]
- 8.Engelbrecht C. J., Stelzer E. H., “Resolution enhancement in a light-sheet-based microscope (SPIM),” Opt. Lett. 31(10), 1477–1479 (2006). 10.1364/OL.31.001477 [DOI] [PubMed] [Google Scholar]
- 9.Huisken J., Stainier D. Y., “Selective plane illumination microscopy techniques in developmental biology,” Development 136(12), 1963–1975 (2009). 10.1242/dev.022426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Planchon T. A., Gao L., Milkie D. E., Davidson M. W., Galbraith J. A., Galbraith C. G., Betzig E., “Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination,” Nat. Methods 8(5), 417–423 (2011). 10.1038/nmeth.1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmid B., Shah G., Scherf N., Weber M., Thierbach K., Campos C. P., Roeder I., Aanstad P., Huisken J., “High-speed panoramic light-sheet microscopy reveals global endodermal cell dynamics,” Nat. Commun. 4, 2207 (2013). 10.1038/ncomms3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holekamp T. F., Turaga D., Holy T. E., “Fast three-dimensional fluorescence imaging of activity in neural populations by objective-coupled planar illumination microscopy,” Neuron 57(5), 661–672 (2008). 10.1016/j.neuron.2008.01.011 [DOI] [PubMed] [Google Scholar]
- 13.Huisken J., Stainier D. Y., “Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM),” Opt. Lett. 32(17), 2608–2610 (2007). 10.1364/OL.32.002608 [DOI] [PubMed] [Google Scholar]
- 14.Ritter J. G., Veith R., Siebrasse J.-P., Kubitscheck U., “High-contrast single-particle tracking by selective focal plane illumination microscopy,” Opt. Express 16(10), 7142–7152 (2008). 10.1364/OE.16.007142 [DOI] [PubMed] [Google Scholar]
- 15.Swoger J., Verveer P., Greger K., Huisken J., Stelzer E. H., “Multi-view image fusion improves resolution in three-dimensional microscopy,” Opt. Express 15(13), 8029–8042 (2007). 10.1364/OE.15.008029 [DOI] [PubMed] [Google Scholar]
- 16.Turaga D., Holy T. E., “Miniaturization and defocus correction for objective-coupled planar illumination microscopy,” Opt. Lett. 33(20), 2302–2304 (2008). 10.1364/OL.33.002302 [DOI] [PubMed] [Google Scholar]
- 17.Verveer P. J., Swoger J., Pampaloni F., Greger K., Marcello M., Stelzer E. H., “High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy,” Nat. Methods 4(4), 311–313 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Wu Y., Ghitani A., Christensen R., Santella A., Du Z., Rondeau G., Bao Z., Colón-Ramos D., Shroff H., “Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans,” Proc. Natl. Acad. Sci. U.S.A. 108(43), 17708–17713 (2011). 10.1073/pnas.1108494108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y., Wawrzusin P., Senseney J., Fischer R. S., Christensen R., Santella A., York A. G., Winter P. W., Waterman C. M., Bao Z., Colón-Ramos D. A., McAuliffe M., Shroff H., “Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy,” Nat. Biotechnol. 31(11), 1032–1038 (2013). 10.1038/nbt.2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cella Zanacchi F., Lavagnino Z., Perrone Donnorso M., Del Bue A., Furia L., Faretta M., Diaspro A., “Live-cell 3D super-resolution imaging in thick biological samples,” Nat. Methods 8(12), 1047–1049 (2011). 10.1038/nmeth.1744 [DOI] [PubMed] [Google Scholar]
- 21.Self S. A., “Focusing of spherical Gaussian beams,” Appl. Opt. 22(5), 658–661 (1983). 10.1364/AO.22.000658 [DOI] [PubMed] [Google Scholar]
- 22.Siegman A., Lasers (University Science, Mill Valley, Calif., (1986) Chap 13, pp. 663–680. [Google Scholar]
- 23.Lorbeer R.-A., Heidrich M., Lorbeer C., Ramírez Ojeda D. F., Bicker G., Meyer H., Heisterkamp A., “Highly efficient 3D fluorescence microscopy with a scanning laser optical tomograph,” Opt. Express 19(6), 5419–5430 (2011). 10.1364/OE.19.005419 [DOI] [PubMed] [Google Scholar]
- 24.Boyd G., Kleinman D., “Parametric interaction of focused Gaussian light beams,” J. Appl. Phys. 39(8), 3597–3639 (1968). 10.1063/1.1656831 [DOI] [Google Scholar]
- 25.Andersen D. R., Regan J. J., “Reflection and refraction of a three-dimensional Gaussian beam at a nonlinear interface,” J. Opt. Soc. Am. A 6(9), 1484–1492 (1989). 10.1364/JOSAA.6.001484 [DOI] [Google Scholar]
- 26.Arnaout R., Ferrer T., Huisken J., Spitzer K., Stainier D. Y., Tristani-Firouzi M., Chi N. C., “Zebrafish model for human long QT syndrome,” Proc. Natl. Acad. Sci. U.S.A. 104(27), 11316–11321 (2007). 10.1073/pnas.0702724104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi T. Y., Kim J. H., Ko D. H., Kim C. H., Hwang J. S., Ahn S., Kim S. Y., Kim C. D., Lee J. H., Yoon T. J., “Zebrafish as a new model for phenotype-based screening of melanogenic regulatory compounds,” Pigment Cell Res. 20(2), 120–127 (2007). 10.1111/j.1600-0749.2007.00365.x [DOI] [PubMed] [Google Scholar]
- 28.Kaufmann A., Mickoleit M., Weber M., Huisken J., “Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope,” Development 139(17), 3242–3247 (2012). 10.1242/dev.082586 [DOI] [PubMed] [Google Scholar]
- 29.Swoger J., Muzzopappa M., López-Schier H., Sharpe J., “4D retrospective lineage tracing using SPIM for zebrafish organogenesis studies,” J. Biophotonics 4(1-2), 122–134 (2011). 10.1002/jbio.201000087 [DOI] [PubMed] [Google Scholar]
- 30.Weber M., Huisken J., “Light sheet microscopy for real-time developmental biology,” Curr. Opin. Genet. Dev. 21(5), 566–572 (2011). 10.1016/j.gde.2011.09.009 [DOI] [PubMed] [Google Scholar]
- 31.Taylor J. M., Girkin J. M., Love G. D., “High-resolution 3D optical microscopy inside the beating zebrafish heart using prospective optical gating,” Biomed. Opt. Express 3(12), 3043–3053 (2012). 10.1364/BOE.3.003043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J., Moghadam M. E., Kung E., Cao H., Beebe T., Miller Y., Roman B. L., Lien C.-L., Chi N. C., Marsden A. L., Hsiai T. K., “Moving domain computational fluid dynamics to interface with an embryonic model of cardiac morphogenesis,” PLoS One 8(8), e72924 (2013). 10.1371/journal.pone.0072924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panier T., Romano S. A., Olive R., Pietri T., Sumbre G., Candelier R., Debrégeas G., “Fast functional imaging of multiple brain regions in intact zebrafish larvae using Selective Plane Illumination Microscopy,” Front. Neural Circuits 7, 65 (2013). 10.3389/fncir.2013.00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herbert S. P., Huisken J., Kim T. N., Feldman M. E., Houseman B. T., Wang R. A., Shokat K. M., Stainier D. Y., “Arterial-venous segregation by selective cell sprouting: an alternative mode of blood vessel formation,” Science 326(5950), 294–298 (2009). 10.1126/science.1178577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ntziachristos V., “Going deeper than microscopy: the optical imaging frontier in biology,” Nat. Methods 7(8), 603–614 (2010). 10.1038/nmeth.1483 [DOI] [PubMed] [Google Scholar]
- 36.Chang T.-Y., Pardo-Martin C., Allalou A., Wählby C., Yanik M. F., “Fully automated cellular-resolution vertebrate screening platform with parallel animal processing,” Lab Chip 12(4), 711–716 (2012). 10.1039/C1LC20849G [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koo B. K., Development and Optimization of High-Throughput Zebrafish Screening Platform (Massachusetts Institute of Technology, 2010). [Google Scholar]
- 38.Petzold A. M., Bedell V. M., Boczek N. J., Essner J. J., Balciunas D., Clark K. J., Ekker S. C., “SCORE imaging: specimen in a corrected optical rotational enclosure,” Zebrafish 7(2), 149–154 (2010). 10.1089/zeb.2010.0660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pitrone P. G., Schindelin J., Stuyvenberg L., Preibisch S., Weber M., Eliceiri K. W., Huisken J., Tomancak P., “OpenSPIM: an open-access light-sheet microscopy platform,” Nat. Methods 10(7), 598–599 (2013). 10.1038/nmeth.2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chardès C., Mélénec P., Bertrand V., Lenne P.-F., “Setting up a simple light sheet microscope for in toto imaging of C. elegans development,” J. Vis. Exp. 87, 51342 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]