

Abstract
Objective:
To study rituximab in pediatric neuromyelitis optica (NMO)/NMO spectrum disorders (NMOSD) and the relationship between rituximab, B cell repopulation, and relapses in order to improve rituximab monitoring and redosing.
Methods:
Multicenter retrospective study of 16 children with NMO/NMOSD receiving ≥2 rituximab courses. According to CD19 counts, events during rituximab were categorized as “repopulation,” “depletion,” or “depletion failure” relapses (repopulation threshold CD19 ≥10 × 106 cells/L).
Results:
The 16 patients (14 girls; mean age 9.6 years, range 1.8–15.3) had a mean of 6.1 events (range 1–11) during a mean follow-up of 6.1 years (range 1.6–13.6) and received a total of 76 rituximab courses (mean 4.7, range 2–9) in 42.6-year cohort treatment. Before rituximab, 62.5% had received azathioprine, mycophenolate mofetil, or cyclophosphamide. Mean time from rituximab to last documented B cell depletion and first repopulation was 4.5 and 6.8 months, respectively, with large interpatient variability. Earliest repopulations occurred with the lowest doses. Significant reduction between pre- and post-rituximab annualized relapse rate (ARR) was observed (p = 0.003). During rituximab, 6 patients were relapse-free, although 21 relapses occurred in 10 patients, including 13 “repopulation,” 3 “depletion,” and 4 “depletion failure” relapses. Of the 13 “repopulation” relapses, 4 had CD19 10–50 × 106 cells/L, 10 had inadequate monitoring (≤1 CD19 in the 4 months before relapses), and 5 had delayed redosing after repopulation detection.
Conclusion:
Rituximab is effective in relapse prevention, but B cell repopulation creates a risk of relapse. Redosing before B cell repopulation could reduce the relapse risk further.
Classification of evidence:
This study provides Class IV evidence that rituximab significantly reduces ARR in pediatric NMO/NMOSD. This study also demonstrates a relationship between B cell repopulation and relapses.
Neuromyelitis optica (NMO) is an autoimmune inflammatory demyelinating disease of the CNS.1 Although previously considered a multiple sclerosis (MS) variant, IgG autoantibody targeting aquaporin-4 (AQP4) channel (NMO-IgG) has clearly demonstrated that NMO is a separate entity.2 NMO lesions are characterized by humoral inflammatory response and astrocytic cell death with AQP4 loss, followed by inflammatory demyelination and axonal damage.3
The course of NMO is characterized by a high relapse rate with accumulation of neurologic disability, potentially causing permanent blindness and paralysis. Therefore, relapse prevention is crucial. Differentiation from MS is important because some MS therapies fail to control or may aggravate NMO.4–6 Even though the optimal therapeutic regimen has not been established, acute NMO attacks are mainly treated with corticosteroids, plasma exchange, and IV immunoglobulin (IVIg); azathioprine, methotrexate, mycophenolate mofetil, rituximab, mitoxantrone, cyclophosphamide, and tocilizumab have been used to prevent relapses.7
Rituximab is an anti-CD20 chimeric monoclonal antibody that depletes B cells that is used in severe autoimmune and inflammatory CNS disorders despite the risk of infections, as recently demonstrated in a large pediatric study.8 One prospective and 3 retrospective adult NMO studies demonstrated reduced annualized relapse rate (ARR) and significantly improved Expanded Disability Status Scale (EDSS) score with rituximab.9–12 Pediatric data are more limited and retrospective.13–16 No study specifically addresses optimal rituximab monitoring and redosing to prevent relapses and reduce disability. To clarify these aspects, we retrospectively studied 16 children with NMO who received ≥2 rituximab courses in order to establish rituximab efficacy, the time from rituximab to B cell repopulation, and the relationship between B cell repopulation and relapses.
METHODS
Patients.
We identified 16 patients with NMO who received ≥2 rituximab courses (<18 years at first dose) from 9 international pediatric neuroimmunology centers. NMO was defined according to the revised Wingerchuk criteria for NMO17 and NMO spectrum disorders (NMOSD).18 In 13 of 16 patients, diagnosis of definite NMO was met based on the presence of both optic neuritis (ON) and transverse myelitis (TM).19 The remaining 3 children had NMOSD (1 had a single attack of isolated TM, 1 had an attack of TM and brainstem manifestations, and 1 had recurrent ON), and these patients were all NMO-IgG positive. Regarding serologic status, 15 of 16 patients were positive for NMO-IgG or AQP4 antibodies: 12 were tested and positive for NMO-IgG using immunofluorescence, and 3 were tested and positive for both NMO-IgG (using immunofluorescence) and anti-AQP4 antibodies (using cell-based assay). One patient was negative for NMO-IgG but positive for anti–myelin oligodendrocyte glycoprotein (MOG) antibodies using cell-based assay (not tested for anti-AQP4 antibodies) (patient 7).
Data collection.
Data were retrospectively collected by the main investigator (R.C.D.) through telephone interviews to the physicians using a structured questionnaire created for this study. Information recorded included demographics, clinical characteristics of disease, immune therapies received besides rituximab, rituximab regimen, CD19 count measurements, and outcome. Data collection focused on the relationship between rituximab administration (timing, dose, number of courses, adverse reactions), CD19 counts, and relapses. First-line immune therapy was defined as corticosteroids, IVIg, and plasma exchange, whereas second-line immune therapy included rituximab, cyclophosphamide, azathioprine, and mycophenolate mofetil. Disease duration pre-rituximab was defined as the time between onset (first event) and initiation of rituximab treatment. Rituximab treatment duration was defined as the time between rituximab initiation and last follow-up (for patients with ongoing rituximab) or the date of final CD19 repopulation (for patients who stopped rituximab).
CD19 values and relationship to relapses.
The threshold for B cell repopulation was defined as CD19 count ≥10 × 106 cells/L, as previously proposed.16,20 In order to study the B cell status during relapses, we used the CD19 count closest to the clinical event (mean 4.6 days before or after the event, median 1, range 0–22). We categorized a relapse as a “repopulation” relapse when it was associated with B cell repopulation ≥10 × 106 cells/L, as a “depletion” relapse when it occurred despite B cell depletion <10 × 106 cells/L, or as a “depletion failure” relapse when it occurred following a rituximab course failing to deplete B cells despite conventional rituximab doses and adequate CD19 monitoring. In order to examine the timing of CD19 repopulation, we used data only from rituximab courses with evidence of both B cell depletion and subsequent repopulation (31 courses from 13 patients).
Therapeutic efficacy.
We used ARR as a clinical indicator of therapeutic efficacy by comparing the ARR pre-rituximab and during rituximab. ARR was calculated only when a time span of ≥6 months was available.12 One relapse (patient 13) occurred 14 days after the first rituximab course and was considered to occur before treatment effect because B cell depletion may take up to 1 month after rituximab administration.21 Pre- and post-rituximab ARR were compared using the Wilcoxon 2-sample test (only patients with both pre- and post-rituximab ARR were included). EDSS score was calculated retrospectively to assess the neurologic outcome at the last follow-up. We used Spearman correlation coefficient (nonparametric) for correlating relapse number with EDSS score at last follow-up.
Research questions and classification of evidence.
Our primary research objectives were to determine the efficacy of rituximab using ARR and to determine the relationship of relapses to B cell repopulation. Given the retrospective nature of our study and lack of a control group, our study represents Class IV evidence.
Standard protocol approvals, registrations, and patient consents.
Patient data were acquired after local ethical approval or using preexisting approved studies to collect deidentified clinical data.
RESULTS
Demographics.
Sixteen children (14 girls) with NMO or NMOSD treated with ≥2 rituximab courses were included in our study (mean age 9.6 years, median 10.9, range 1.8–15.3). The patient race was white (n = 8), black or African American (n = 5), Native Pacific Islander (n = 1), mixed white and Native Pacific Islander (n = 1), and mixed African, Asian, and white (n = 1).
Clinical presentation and disease course.
Disease onset was between 2000 and 2012. Ten patients had ON and/or TM at onset (ON: n = 4; TM: n = 4; both ON and TM: n = 2). The other presentations were brainstem disease only (n = 3), TM and brainstem disease (n = 2), and ON and brainstem disease (n = 1). The mean total duration of disease (time from onset to last follow-up) was 6.1 years (median 5.1, range 1.6–13.6). In the 16 children, 98 total events occurred (mean 6.1, median 5, range 1–11), most of which (71 of 98) were monosymptomatic attacks (isolated ON: n = 29; isolated TM: n = 38; isolated brainstem disease: n = 4). The remaining attacks were concurrent ON and TM (n = 13); TM and brainstem disease (n = 9); ON, TM, and brainstem disease (n = 2); ON and brainstem disease (n = 1); or other (n = 2). Figure 1 illustrates the clinical course of the 16 patients (clinical events, second-line immune therapies, and rituximab courses).
Figure 1. Clinical course of the 16 patients: Clinical events, second-line immune treatments, and rituximab courses.

AZA = azathioprine; CYC = cyclophosphamide; MMF = mycophenolate mofetil; RTX = rituximab.
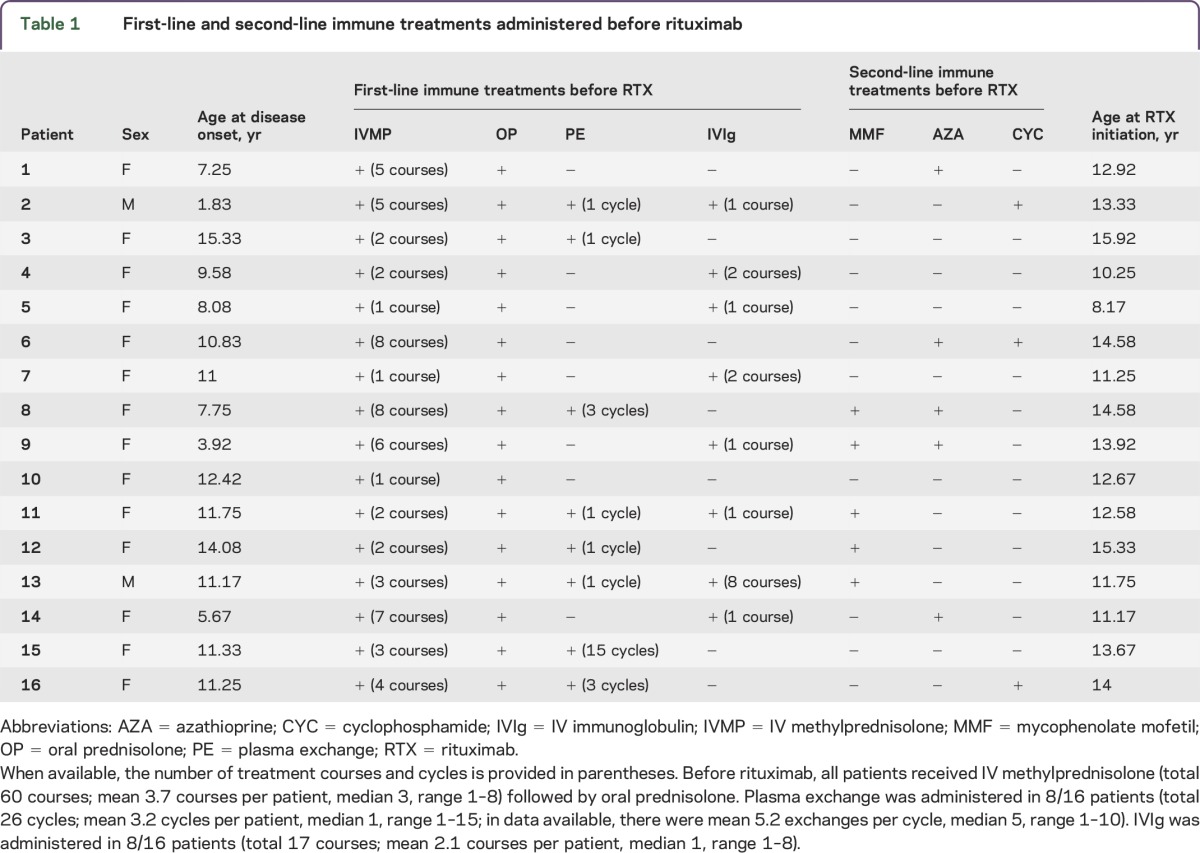
Immune therapies before rituximab.
Before rituximab, all patients received IV methylprednisolone followed by oral prednisolone tapers; 8 patients received plasma exchange and 8 received IVIg. Ten patients received other second-line immune treatments before rituximab (figure 1 and table 1): mycophenolate mofetil (n = 5; 2 of 5 also received azathioprine), azathioprine (n = 5; 2 of 5 also received mycophenolate mofetil and 1 of 5 also received cyclophosphamide), and cyclophosphamide (n = 3; 1 of 3 also received azathioprine).
Table 1.
First-line and second-line immune treatments administered before rituximab
Rituximab administration.
A total of 76 rituximab courses were administered in the 16 patients (mean 4.7, median 4.5, range 2–9) (figure 1). The mean time between the first infusion of the first and last rituximab courses was 29.8 months (median 23.5, range 5.7–93). The protocols for administration (in descending order of frequency) were as follows: 1,000 mg × 2 infusions 2–4 weeks apart (n = 31 courses), 375 mg/m2 × 4 weekly infusions (n = 19 courses), 375 mg/m2 × 1 infusion (n = 10 courses), 375 mg/m2 × 2 infusions 2 weeks apart (n = 9 courses), 750 mg/m2 × 2 infusions 2 weeks apart (n = 5 courses), and 500 mg/m2 × 2 infusions 2 weeks apart (n = 2 courses). Rituximab was redosed at a mean of 7.9 months (median 7.6, range 2.2–28.3). In some patients it was redosed after a relapse, in others after detection of B cell repopulation, and in a minority at regular intervals regardless of B cell status. At last available follow-up, rituximab was ongoing in 13 patients. Rituximab was discontinued in the 3 remaining patients because of difficulty coming to the hospital (patient 1), treatment failure (patient 2), and relapse freedom for ≥2 years (patient 10).
Infusion reactions and adverse events.
Data on infusion reactions to rituximab and adverse events were available in 14 of 16 patients. Infusion reactions occurred in 6 of 14 children (dyspnea: n = 2; rash: n = 2; chest pain: n = 1; lightheadedness: n = 1; tingling and stinging sensation in mouth and throat: n = 1). Other adverse reactions occurred in 3 of 14 children, including infections in 2 (skin infection: n = 1; mastoiditis: n = 1) and immunoglobulin deficiency without infectious complications in 1 (this patient received 4 rituximab courses of 750 mg/m2 × 2).
Rituximab efficacy.
Six patients were relapse-free during rituximab treatment (patients 9, 10, 11, 13, 14, and 15) (table 2; figure 1). In these 6 relapse-free patients, the rate of use of other immune therapies during rituximab (corticosteroids: n = 4; IVIg: n = 2; plasma exchange: n = 0; second-line immune therapies: n = 0) was similar to the rate in the other 10 patients (corticosteroids: n = 9; IVIg: n = 4; plasma exchange: n = 4; mycophenolate mofetil + cyclophosphamide: n = 1; azathioprine: n = 1). In the 10 relapsing patients, a total of 21 events occurred during rituximab treatment (mean 2.1, median 1.5, range 1–5) (table 2). Relapses occurred a mean of 9.1 months (median 8.1, range 1.2–27.8) after the last rituximab course (figure 2A). There was a statistically significant reduction between pre- and post-rituximab ARR when first events were included (p = 0.003) or excluded (p = 0.014) (table 2). There was also a significant reduction in the ARR in the year after rituximab initiation compared to the year before (p = 0.002).
Table 2.
Duration of disease, number of events, and ARR pre- and post-rituximab
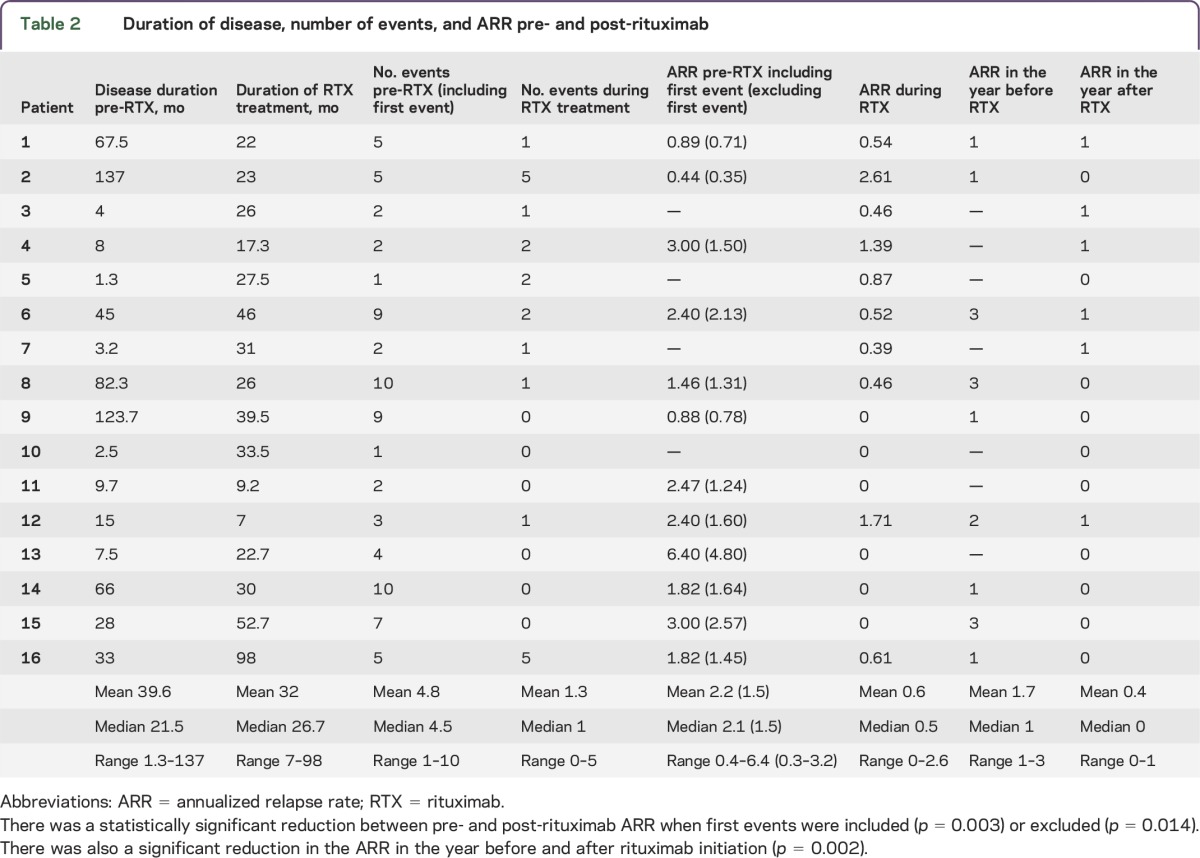
Figure 2. Time to relapse and time to B cell repopulation after rituximab.

(A) Relapses during rituximab (RTX) treatment according to the time from last rituximab course (total 21 relapses in 10 patients). (B) Inter- and intraindividual variability in the time to B cell repopulation after rituximab in 9 patients. To assess the variability in the intraindividual time to repopulation, these 9 patients were selected based on the availability of at least 2 rituximab courses with evidence of a repopulated CD19 count after demonstrated depletion and the fact that the same dose regimen was administered (rituximab regimen specified for each patient next to the bar). The horizontal bars represent the range of intraindividual variability in the time to repopulation, and the dots represent the actual measurements. There is significant variability between patients, although the intrapatient variability appears to be less.
CD19 count monitoring and repopulation on rituximab.
During the total 42.6 years of cohort rituximab treatment, a total of 196 CD19 counts were measured (mean 12.2 per patient, median 9, range 1–36). All patients had documented B cell depletion after at least 1 rituximab course. In the 31 rituximab courses (in 13 patients) with documented B cell depletion followed by repopulation, the mean time from rituximab administration to the last demonstrated depleted CD19 count was 4.5 months (median 5.1, range 0.9–8.7), and the mean time to the first demonstrated repopulated CD19 count was 6.8 months (median 6.7, range 2.7–12.2). The 2 shortest times to repopulation occurred 2.7 and 2.9 months after rituximab (375 mg/m2 × 1 and 375 mg/m2 × 2, respectively) in 2 different patients. We observed notable interpatient variability in the time to B cell repopulation and some intrapatient variability (figure 2B). The mean time to repopulation in the first rituximab courses (mean 7.4 months, median 7.2, range 3.6–12.2; calculated in 8 courses) was similar to that in subsequent courses (mean 6.7 months, median 6.8, range 2.7–11.2; calculated in 23 courses). Time to B cell repopulation was faster in the younger patients (18 courses in 5 patients with adequate data; age range 8.2–11.7 years at rituximab initiation) than in the older patients (12 courses in 7 patients; age range 13.3–15.9 years at rituximab initiation) (mean 5.9 vs 8.1 months, median 5.6 vs 8.5 months). Where adequate data were available (n = 10 courses), once B cells repopulated over the threshold of 10 × 106 cells/L, B cell counts never redepleted spontaneously. In contrast, according to available data (n = 9 courses), only 22% of CD19 counts 1–9 × 106 cells/L were followed by repopulated CD19 values ≥10 × 106 cells/L within 1 month.
CD19 count and relationship to relapses.
The 21 relapses that occurred in 10 children during rituximab treatment are detailed in table e-1 at Neurology.org/nn (adequate CD19 data in 20 of 21 relapses). Most of the events (13 of 20) occurred with B cell repopulation and are defined as “repopulation” relapses. In these 13 “repopulation” relapses, the mean CD19 value at relapse was 192.3 × 106 cells/L (median 130, range 10–449), and in 4 of these 13 events the CD19 count was 10–50 × 106 cells/L (10, 16, 37, and 40 × 106 cells/L). Of the 13 “repopulation” relapses, there was a lack of monitoring (defined as ≤1 CD19 count in the 4 months preceding the relapse) in 10, a delay in redosing (defined as ≥10 days between detection of repopulation and rituximab redosing) during which the relapse occurred in 5, and there was no inadequate monitoring or delayed redosing in 2.
The remaining 7 clinical events occurred in 2 patients: 3 relapses in patient 2 (rituximab 750 mg/m2 × 2 infusions 2 weeks apart) occurred despite B cell depletion and were defined as “depletion” relapses, whereas the 4 relapses in patient 16 (rituximab 1,000 mg × 2 infusions 4 weeks apart) occurred with documented persistent nondepleted CD19 counts (mean 6.2 CD19 counts/relapse, median 6, range 2–11) and were defined as “depletion failure” relapses. Examples of relapse freedom after treatment and of “repopulation,” “depletion,” and “depletion failure” relapses are presented in figures 3 and e-1.
Figure 3. Summary figure exemplifying 4 different types of response to rituximab treatment observed in our patients.

Relapse freedom with rituximab (RTX) (A, B), occurrence of relapses with a repopulated B cell count (“repopulation” relapses; C, D), occurrence of relapses with a depleted B cell count (“depletion” relapses; E), occurrence of relapses in association with failure to reach B cell depletion (“depletion failure” relapses; F). (A) Relapse freedom (no relapses during rituximab): rituximab redosing after B cell repopulation (patient 14). (B) Relapse freedom (no relapses during rituximab): rituximab redosing before B cell repopulation (patient 10). (C) “Repopulation” relapses (relapses with B cell repopulation): repopulation was detected only at the time of the relapse (third and fourth relapses); subsequent rituximab courses were administered after the relapse (second and third rituximab courses) (patient 4). (D) “Repopulation” relapses (relapses with B cell repopulation): repopulation was noticed at CD19 count monitoring and rituximab was administered, but clinical relapse occurred a few days after rituximab, before depletion was achieved (second and third relapses) (patient 5). (E) “Depletion” relapses (relapses despite B cell depletion in the last 3 relapses) (patient 2). (F). “Depletion failure” relapses (relapses associated with failure to reach B cell depletion in the first, second, and third rituximab courses). In this patient, B cell depletion was achieved in subsequent rituximab courses (total 9 courses; same rituximab regimen used in all the courses, 1,000 mg × 2). The figure shows only the first 5 courses; no relapses occurred subsequently.
Outcome.
At a mean follow-up of 6.1 years from disease onset (median 5.1 years, range 1.6–13.6), mean EDSS score was 2.4 (median 2.5, range 0–6.5), and no ongoing problems (EDSS 0) were reported in 5 patients. There was a trend of worse EDSS scores at follow-up in the patients who had more relapses during the disease course, but this was not statistically significant (r = 0.49, p = 0.051). The most common neurologic problem at follow-up was reduced visual acuity, reported in 10 of 16 cases; in 6 of these 10 patients visual acuity was severely reduced (worse eye with maximal visual acuity corrected less than 20/200). Pyramidal signs in the lower limbs were reported in 2 of 16 cases, upper limb involvement in 0 of 16, and bowel or bladder impairment in 1 of 16.
DISCUSSION
We retrospectively studied 16 children with NMO treated with ≥2 rituximab courses with the aim of optimizing rituximab monitoring and redosing to prevent relapses. This represents the largest reported therapeutic study of pediatric NMO.
Confirming published literature, there was a female predominance in our cohort, and most of the clinical events were monosymptomatic TM, ON, and brainstem events. Our patients had a high relapse rate and a long disease course before rituximab initiation. Fifteen of the 16 patients were NMO-IgG positive, although the single MOG-IgG-positive patient had 2 events in the 3 months preceding rituximab initiation and 1 relapse during rituximab, suggesting a highly relapsing disease. Although long-term data regarding MOG-IgG-associated disease are lacking, early reports suggest that MOG-IgG-associated NMOSD is more reversible and less severe and carries less risk of permanent disability compared to NMO-IgG-associated NMOSD.19,22
All patients received immunosuppressive therapies before rituximab: all received IV methylprednisolone and oral steroids, half received IVIg, half received plasma exchange, and 62.5% were given other second-line immune therapies before rituximab. Rituximab was generally initiated after ≥2 events, although it was started after the first event in 3 cases. Overall, our cohort likely represents a more severe end of the pediatric NMO spectrum.
In our cohort, the protocols of rituximab induction, redosing, and monitoring were heterogeneous, reflecting the multicenter nature of our cohort and the lack of guidelines and consensus opinion. Rituximab was redosed at a mean of 7.9 months, although with considerable variability (range 2.2–28.3 months). Redosing occurred for different reasons, including occurrence of relapses, detection of B cell repopulation, or, more rarely, planned redosing (figures 3 and e-1).
Rituximab treatment was relatively well tolerated with no major complications in 42.6-year cohort treatment. There was evidence of efficacy in our cohort; 6 of the patients were relapse-free during treatment (figures 3, A and B and e-1). There was a significant reduction in ARR using all measures, although it is important to note that the ARR may decline during the course of disease regardless of treatment.23 The use of other immune therapies was similar in the 6 relapse-free patients compared to the other patients, suggesting that the lack of relapses was not due to other concomitant therapies administered with rituximab.
The clinical events during rituximab occurred a mean of 9.1 months after the last rituximab course, although the timing of relapses was very widely distributed.
We chose a CD19 count of 10 × 106 cells/L as a threshold for B cell repopulation, as previously used,16,20 partly because absolute values (rather than percentages) would allow adequate comparison across centers. The observation that 4 of the 13 “repopulation” relapses in our cohort occurred during early repopulation (10–50 × 106 cells/L) confirms the clinical validity of 10 × 106 cells/L as a threshold. We also observed that once B cells repopulated beyond 10 × 106 cells/L, CD19 counts continued to rise and there was no spontaneous return to B cell redepletion.
We confirmed a relationship between B cell repopulation and relapses. Most relapses occurred with CD19 repopulation, and only 1 patient had relapses with depleted CD19 counts, confirming that depletion appears to be protective in most patients. In most of the 13 “repopulation” relapses, CD19 monitoring was inadequate and B cell repopulation went unnoticed until subsequent clinical relapse. In 5 of the “repopulation” relapses, there was delayed rituximab redosing after detection of B cell repopulation and relapses occurred while waiting to admit the patient for redosing. Furthermore, B cell depletion can take up to 1 month after rituximab administration,21 allowing for a “window of vulnerability” for relapses (as shown in figure 3D). The remaining 7 relapses occurred in 2 patients, defined as “depletion” relapse in one patient (figure 3E) and “depletion failure” relapse in the other (figure 3F). Therefore, only 1 of the 16 patients (patient 2) had relapses despite adequate monitoring and documented B cell depletion and can therefore be defined as having true rituximab failure. The reason for the different response to rituximab in these 2 patients with “depletion” and “depletion failure” relapses is not clear, although some investigators have suggested that B cell activating factor of the tumor necrosis factor family may be relevant.20,24–26 Some studies in adult patients have shown a relationship of anti-AQP4 antibodies with B cell status and clinical relapses,11,27,28 although others have not found a convincing relationship.20 Unfortunately, longitudinal anti-AQP4 antibodies were not available in our cohort.
In light of the above considerations on the relationship between B cell repopulation and relapses, understanding the timing of repopulation after rituximab is critical for preventing relapses. The mean time for B cell repopulation in our cohort was between 4.5 (mean time of last depletion) and 6.8 months (mean time of first repopulation) after the last rituximab course. However, repopulation as early as 2.7 months post-rituximab and persistent B cell depletion up to 8.7 months were observed, suggesting large interpatient variability in CD19 count effects (figure 2B), similar to other published data.29 In contrast, the intrapatient variability appeared to be smaller, implying a relative predictability of B cell repopulation in individuals, which will help monitoring. We noted that the shortest time to repopulation occurred with the lowest rituximab doses and the longest with the highest doses, as previously observed,29 although other variables likely play a role in B cell repopulation. We also observed that younger patients repopulated faster than older patients.
In our study, we observed that genuine treatment failure with rituximab occurred in only 1 patient, whereas relapses were otherwise attributable to the challenges associated with monitoring and redosing. We confirm that rituximab efficacy is associated with CD19 depletion, and our data suggest that the detection of repopulation over 10 × 106 cells/L should alert the clinician to the likely possibility of further B cell rise and relapse risk. Considering the significant variability observed in the time to B cell repopulation, further efforts should be made to optimize rituximab monitoring. A possible individualized strategy to minimize relapses involves rigorous CD19 monitoring (i.e., monthly, especially after the third month), particularly during first courses, and rapid redosing on repopulation detection. Given the latency of B cell depletion after rituximab infusion, there is a risk of relapse during this repopulated period, especially when there is delay in redosing. In view of this, an alternative strategy involves planned rituximab redosing at regular intervals, before B cell repopulation occurs, as previously described9,10,20 and shown in figure 3B, which may reduce the relapse risk but will result in increased therapy cost. As shown in figure 2B, there is significant variability in B cell repopulation between patients, including early repopulation (3–6 months) even for higher dose regimens (375 mg/m2 × 4 and 1,000 mg/m2 × 2). Therefore, planned redosing would need to be at short intervals (3–4 months) to minimize the chance of B cell repopulation.
The retrospective design, the lack of standardization, and the relatively small number of patients are the main limitations of our study. However, we have confirmed rituximab efficacy, demonstrated challenges in monitoring, and provided data on B cell repopulation. This will improve redosing of rituximab in children with NMO and other serious autoimmune disorders of the CNS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families. M. Nosadini has appreciated funding from Petre Foundation (Australia) and the University of Padua (Italy).
GLOSSARY
- AQP4
aquaporin-4
- ARR
annualized relapse rate
- EDSS
Expanded Disability Status Scale
- IVIg
IV immunoglobulin
- MS
multiple sclerosis
- MOG
myelin oligodendrocyte glycoprotein
- NMO
neuromyelitis optica
- NMOSD
NMO spectrum disorders
- ON
optic neuritis
- TM
transverse myelitis
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
Dr. Margherita Nosadini: data analysis and first draft, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Gulay Alper: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Catherine J. Riney: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Leslie A. Benson: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Shekeeb S. Mohammad: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Sudarshini Ramanathan: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Melinda Nolan: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Richard Appleton: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Richard J. Leventer: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Kumaran Deiva: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Fabienne Brilot: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Mark P. Gorman: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Amy T. Waldman: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Brenda Banwell: acquisition of data, critical revision of the manuscript for important intellectual content, editing and approval of final draft. Dr. Russell C. Dale: project conception, design, and modification; data analysis and first draft; critical revision of the manuscript for important intellectual content; study supervision.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
M. Nosadini and G. Alper report no disclosures. C.J. Riney received speaker honoraria and/or travel funding from Biogen Idec and UCB Australia Pty Ltd and received research support from Novartis and UCB. L.A. Benson received research support from Boston Children's Hospital. S.S. Mohammad received travel funding from the Movement Disorders Society and received research support from NHMRC. S. Ramanathan received research support from NHMRC. M. Nolan reports no disclosures. R. Appleton served on the scientific advisory board for SHIRE, received speaker honoraria from EISAI, and received publishing royalties from Oxford University Press and Cambridge University Press. R.J. Leventer received travel funding and speaker honoraria from Asia Oceania Congress of Child Neurology, received research support from NHMRC and Campbell Edwards Trust Research. K. Deiva received travel funding from Biogen Idec, Merck Serono, and Genzyme. F. Brilot is an associate editor for Journal of Visualized Experiments and received research support from NHMRC, The Star Scientific Foundation Australia, The Trish Multiple Sclerosis Foundation Australia, Multiple Sclerosis Research Australia, Multiple Sclerosis Angels Melbourne Australia, and Petre Foundation Australia. M.P. Gorman received research support from NIH, United States Department of Defense, and Multiple Sclerosis Society. A.T. Waldman is on the scientific advisory board for Johns Hopkins University and Children's Hospital of Philadelphia; received travel funding from Novartis; received publishing royalties from UpToDate; has consulted for OptumInsight Life Sciences Inc; participated in NIH Loan Repayment Program; received research support from Biogen Idec, NIH/NINDS, and Children's Hospital of Philadelphia; and holds stock or stock options in Pfizer and Spark Therapeutics. B. Banwell served on the scientific advisory board for Biogen Idec, Sanofi, Eli Lilly, and Novartis; received travel funding and/or speaker honoraria from Biogen Idec, Merck Serono, Teva Neuroscience, and Bayer; is on the editorial board for Neurology and Multiple Sclerosis and Related Disorders; has consulted for Biogen Idec, Eli Lilly, and Sanofi; has spoken at an event supported by the Consortium of MS Centers; received research support from Canadian Institute of Health Research, Multiple Sclerosis Society of Canada, Multiple Sclerosis Scientific Research Foundation, and National Multiple Sclerosis Society. R.C. Dale served on the scientific advisory board for Queensland Children's Medical Institute, received speaker honoraria from Biogen Idec and Bristol-Myers-Squibb, is an editorial advisory board member for Multiple Sclerosis and Related Disorders and an editorial board member for Neurology: Neuroimmunology & Neuroinflammation and European Journal of Paediatric Neurology, received publishing royalties from Biogen and Bristol-Myers-Squibb, and received research support from NHMRC and Multiple Sclerosis Research Australia. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Wingerchuk DM, Weinshenker BG. Neuromyelitis optica (Devic's syndrome). Handb Clin Neurol 2014;122:581–599. [DOI] [PubMed] [Google Scholar]
- 2.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 3.Wegner C. Recent insights into the pathology of multiple sclerosis and neuromyelitis optica. Clin Neurol Neurosurg 2013;115:S38–S41. [DOI] [PubMed] [Google Scholar]
- 4.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler 2012;18:1480. [DOI] [PubMed] [Google Scholar]
- 5.Kleiter I, Hellwig K, Berthele A, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 2012;69:239–245. [DOI] [PubMed] [Google Scholar]
- 6.Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler 2012;18:113–115. [DOI] [PubMed] [Google Scholar]
- 7.Trebst C, Jarius S, Berthele A, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 2014;261:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dale RC, Brilot F, Duffy LV, et al. Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology 2014;83:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacob A, Weinshenker BG, Violich I, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 2008;65:1443–1448. [DOI] [PubMed] [Google Scholar]
- 10.Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler 2011;17:1225–1230. [DOI] [PubMed] [Google Scholar]
- 11.Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol 2013;70:1110–1117. [DOI] [PubMed] [Google Scholar]
- 12.Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol 2014;71:324–330. [DOI] [PubMed] [Google Scholar]
- 13.Lotze TE, Northrop JL, Hutton GJ, Ross B, Schiffman JS, Hunter JV. Spectrum of pediatric neuromyelitis optica. Pediatrics 2008;122:e1039–e1047. [DOI] [PubMed] [Google Scholar]
- 14.McKeon A, Lennon VA, Lotze T, et al. CNS aquaporin-4 autoimmunity in children. Neurology 2008;71:93–100. [DOI] [PubMed] [Google Scholar]
- 15.Beres SJ, Graves J, Waubant E. Rituximab use in pediatric central demyelinating disease. Pediatr Neurol 2014;51:114–118. [DOI] [PubMed] [Google Scholar]
- 16.Longoni G, Banwell B, Filippi M, Yeh EA. Rituximab as a first-line preventive treatment in pediatric NMOSDs: preliminary results in 5 children. Neurol Neuroimmunol Neuroinflamm 2014;1:e46. 10.1212/NXI.0000000000000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 18.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815. [DOI] [PubMed] [Google Scholar]
- 19.Dale RC, Tantsis EM, Merheb V, et al. Antibodies to MOG have a demyelination phenotype and affect oligodendrocyte cytoskeleton. Neurol Neuroimmunol Neuroinflamm 2014;1:e12. 10.1212/NXI.0000000000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pellkofer HL, Krumbholz M, Berthele A, et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 2011;76:1310–1315. [DOI] [PubMed] [Google Scholar]
- 21.Kosmidis ML, Dalakas MC. Practical considerations on the use of rituximab in autoimmune neurological disorders. Ther Adv Neurol Disord 2010;3:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79:1273–1277. [DOI] [PubMed] [Google Scholar]
- 23.Kim SM, Park J, Kim SH, et al. Factors associated with the time to next attack in neuromyelitis optica: accelerated failure time models with random effects. PLoS One 2013;8:e82325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Wang K, Zhong X, et al. Cerebrospinal fluid BAFF and APRIL levels in neuromyelitis optica and multiple sclerosis patients during relapse. J Clin Immunol 2012;32:1007–1011. [DOI] [PubMed] [Google Scholar]
- 25.Nakashima I, Takahashi T, Cree BA, et al. Transient increases in anti-aquaporin-4 antibody titers following rituximab treatment in neuromyelitis optica, in association with elevated serum BAFF levels. J Clin Neurosci 2011;18:997–998. [DOI] [PubMed] [Google Scholar]
- 26.Gredler V, Mader S, Schanda K, et al. Clinical and immunological follow-up of B-cell depleting therapy in CNS demyelinating diseases. J Neurol Sci 2013;328:77–82. [DOI] [PubMed] [Google Scholar]
- 27.Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008;131:3072–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He D, Yu Y, Yan W, Dai Q, Xu Z, Chu L. Individualized rituximab treatment for relapsing neuromyelitis optica: a pediatric case report. Pediatr Neurol 2014;51:255–258. [DOI] [PubMed] [Google Scholar]
- 29.Greenberg BM, Graves D, Remington G, et al. Rituximab dosing and monitoring strategies in neuromyelitis optica patients: creating strategies for therapeutic success. Mult Scler 2012;18:1022–1026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.