

Abstract
AIM: We have previously reported that inducible over-expression of Bak may prolong cell cycle in G1 phase and lead to apoptosis in HCC-9204 cells. This study is to investigate whether p27KIP1 plays an important role in this process.
METHODS: In order to elucidate the exact function of p27KIP1 in this process, a zinc inducible p27KIP1 stable transfectant and transie nt p27KIP1-GFP fusion transfectant were constructed. The effects of inducible p27KIP1 on cell growth, cell cycle arrest and apoptosis were examined in the mock, control pMD vector, and pMD-KIP1 transfected HCC-9204 cells.
RESULTS: This p27KIP1-GFP transfectant may transiently express the fusion gene. The cell growth was reduced by 35% at 48 h of p27KIP1 induction with zinc treatment as determined by trypan blue exclusion assay. These differences remained the same after 72 h of p27KIP1 expression. p27KIP1 caused cell cycle arrest after 24 h of induction, with 40% inc rease in G1 population. Prolonged p27KIP1 expression in this cell line induced apoptotic cell death reflected by TUNEL assay. Fourty-eighth and 72 h of p27KIP1 expression showed a characteristic DNA ladder on agarose gelelec trophoresis.
CONCLUSION: Bak may induce cell cycle arrest in G1 phase through upregul ating expression of p27KIP1 and subsequently lead to apoptosis in HCC-9204 cells. The p27KIP1-GFP fusion protein can be transiently expr essed in HCC-9204 cells. The inducible p27KIP1 expressing cell line provides a model to assess p27KIP1 function.
Keywords: p27KIP1; apoptosis; cell cycle; inducible expression system; carcinoma , hepatocellular; liver neoplasms
INTRODUCTION
Cell cycle regulatory proteins play a critical role in both normal cell growth and tumorigenesis. The cyclin dependent kinases (CDKs) are the major regulators o f cell cycle progression, and thus are important candidates for therapeutic tumo r suppression[1]. The CDKs, which are responsible for the phosphorylati on of the retinoblastoma protein (pRb) and pRb related proteins, are in turn regulated by changes in cyclin levels, phoshphorylation and the presence of cyclin kinase inhibitors (CKIs)[2-3]. Two classes of CKIs are present within mammalian cells. One class, the INK4 (inhibitors of CDK4) family, is a group of ankyrin repeat protein whose members p16INK4A, p15INK4b, p18INK4c and p19INK4d are specific inhibitors of cyclin D1/CDK4 or CDK6 complexes[4-6]. The second class of CKIs, the p21 family, c onsists of p21CIP/WAF1, p27KIP1 and p57KIP2 which are general inhibitors of the G1/S CDKs[7-9]. Homology between the f amily members is limited to a conserved amino-terminal 60-residue domain respo nsible for kinase binding and inhibition[10].
p27KIP1 regulates cell cycle progression by interacting with,and the reby inhibiting,various cyclin-CDK complexes. Physiologically, p27KIP1 is believed to act primarily to regulate progression of cells from late G1 into S through its interaction with cyclin E-CDK2 complexes[11]. p27KIP1 has been implicated as a mediator of growth arrest due to TGF-β, cAMP, and other extracellular factors [12]. Moreover, elevated expression of p27KIP1 leads to G1 arrest in many cell types and promotes neuronal differentiation in mouse neuroblastoma cells, while inhibition of p27KIP1 expression through use of antisense technology prevents G1 arrest and/or suppresses entry of fibroblasts into a state of quiescence in resp onse to mitogen depletion[13-14]. The phenotype of mice null for the p27KIP1 gene includes increased body size, female sterility and a high incidence of spontaneous pituitary tumors[15].
We have previously shown that overexpression of Bak might prolong cell cycle in G1 phase though this new property of Bak had never been reported before. We provided a hypothesis that Bak’s translocated expression from the cytoplasm to the nuclei of HCC-9204 cells may trigger the expression of certain cell cycle regulators such as cyclins, CDKs, or CDK inhibitors through DNA-protein or prote in-protein interaction. Since p27KIP1 is one of major cyclin-CDK reg ulators in G1 phase, we currently executed an investigation of whether overexpression of Bak in HCC-9204 cells upregulates the expression of p27KIP1 and whether p27KIP1 itself may induce cell cycle arrest in G1 pha se and even apoptosis in HCC-9204 cells.
MATERIALS AND METHODS
Cell line and cell culture
HCC-9204 cell line derived from a human hepatocellular carcinoma and established in our laboratory was cultured in Eagle’s medium containing phenol red, with 50 mL/L fetal bovine serum (FBS) (HyClone Laboratories, USA) in a humidified at mosphere of 950 mL/L air, 50 mL/L CO2 at 37 °C[16]. All media were s upplemented with 2 mmol/L-L-glutamine, 100 mg/L penicillin and 100 kU/L str epto mycin. Culture medium and supplements were obtained from GIBCO BRL. Of the HCC- 9204 cell line, p53 was mutated. Medium was changed every 3 days. Cells were removed from culture flasks for passage by washing once with Hank’s balanced salt solution, followed by a 5 min incubation with 0.5 mmol/L EDTA and 0.5 g/L trypsin at pH 7.4, RT.
RT-PCR analysis
Total RNA was extracted from HCC-9204 cells using an RNA extraction reagent, TR IZOL (Life Technologies, USA), according to standard acid-guanidium-phenol-ch loroform method[17]. About 5 μg of total RNA were revers etra nscribed at 42¡æ for 60 min in a total 20 μL reaction volume using a first-Strand cDNA synthesis kit (Boehringer Mannheim, Germany). cDNA was inc ubated at 95 °C for 5 min to inactivate the reverse transcriptase, and was serve d as template DNA for 32 rounds of amplification using the GeneAmp PCR System 2400 (Perkin-Elmer Applied Biosystems, CA, USA). PCR was performed in a standard 50 μL reaction mixture consisting of 10 mmol/L Tris-HCl, 50 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride (pH 8.3), 0.2 mmol/L dNTPs, 50 pmol of each sense and antisense primer and 2.5 U of Taq DNA polymerase (M BI, Canada). Amplification was performed for 30 s at 94 °C, 45 s at 58 °C and 45 s at 72 °C after heat-start for 5 min. Finally, an additional extension step wa s carried out for 7 min. As negative control, the DNA template was omitted in th e reaction. The amplification products were separated on 12 g/L agarose gels and visualized by ethidium bromide staining. PCR primers for p27KIP1 were as follows: forward primer, 5’-ggggtaccatgtcaaacgtgcgagt-3’; reverse primer , 5’-gcgtcgacacgtttgacgtcttc-3’: according to the p27KIP1 gene structure in Genebank. PCR product of 597 bp was obtained. Kpn-I and Sal-I sites were designed in the 5’and 3’end of cDNA encoding p27KIP1 and the terminal code for p27KIP1 was mutated so as to construct a fu sion protein.
DNA sequencing
The PCR product was purified through Wizard Plus Minipreps DNA Purification System (Promega, USA). The pGEM-T Easy Vector Systems (Promega, USA) was applied for the cloning of the PCR product. Then the recombinant plasmid DNA was trans formed into E.coli and was sequenced by dideoxy-mediated chain-termination method, using ABI PRISM-TM Dye Terminator Cycle Sequencing Ready Reaction Kit (Perkin Elmer, USA). This kit was developed specifically for the preparation of samples for sequence analysis on the ABI PRISM 377 DNA Sequencer. Cycle sequ encing was performed on the GeneAmp PCR systems 2400 (Perkin Elmer, USA) according to the instructions.
Plasmid construction
Plasmids were constructed using standard molecular biology techniques[18]. In order to construct a hybrid DNA encoding a chimerical p27KIP1-GFP fusion protein, pGEM-T Easy KIP1 was digested with Kpn-I and Sal-Ito yield a 0.6 kb fragment. The resulting fragment then ligated to Kpn-I and Sal-I digested pGREEN LANTERN-1 (Clontech, USA) containing the humanized Thr65 GFP cDNA under the control of CMV enhancer/promoter and the SV40 poly adenylation signal. Correct reading framework of the fusion gene was confirmed by DNA sequencing. Human p27KIP1 (pBluescript SK-p27) was a gift from Dr. Polyak[19-20]. PCR primers for p27KIP1 were as foll ows: forward primer, 5’-ggggtaccatgtcaaacgtgcgagt-3’; reverse primer, T7 primer. PCR product of 1.0 kb was obtained and subsequently ligated into pGEM-T Easy Vector System. To construct pMD-KIP1, pGEM-T Easy KIP1 was digested with EcoR-I and Xho-I to yield a 1.0 kb fragment. T he resulting fragment then ligated to EcoR-I and Xho-I digested pMD-neo. Plasmids for transfection were purified using a kit (Quiagen Inc., USA).
Transient and stable transfection
pKIP1-GFP and pMD-KIP1 were used for transient and stable transfections respec tively. Clonfectin (Clontech, USA) was used to transfect plasmids into HCC-9204 cells. According to the instructions, Clonfectin transfection reagent was an effective liposome transfection reagent for many mammalian cell types with high tr ansfection efficiencies and required only 1-4 hour incubation for optimal resu lts, especially effective transfections in serum-containing media. Green fluore scence was detected 4, 24 and 48 h after transfection. For stable transfection, the transfected HCC-9204 cells were grown for 2 weeks in a medium containing 0. 5 g/L G418 (Life Technologies, Inc., USA), after which the G418 concentration wa s reduced to 0.2 g/L. Stable cell line of HCC-9204 tranfected with pMD-KIP1 vector was treated by continuous exposure to 100 µmol/L of ZnSO4.
Cell viability
Cells were seeded at 105 per dish. HCC-9204 cells viability was determined by Nikon Eclipse TE200 inverted microscopic examination of cells stained by 1 g/L trypan blue (trypan blue exclusion), counting cells on a hemocytometer at the ti me point indicated[21].
Apoptosis analyzed by TUNEL assay
Cells were harvested for TUNEL staining. The proportion of cells showing DNA fra gmentation was measured by incorporation of fluorescein isothiocyanate (FITC)-1 2-dUTP into DNA by using terminal deoxynucleotidyltransferase (TdT)[22]. Briefly, a kit from Boehringer Mannheim (In Situ Cell Death Detection Kit, F ITC) was used. After a 30 min (RT) incubation with 30 g/L BSA, 200 mL/L normal bovine serum in PBS, pH 7.4, slides were covered with the TUNEL mix (calf thy mus TdT, FITC-12-dUTP and cobalt chloride in 1 × reaction buffer) for 1.2 h at 37 °C. The morphologic features were visualized by fluorescence microscopy. Routine HE staining was also conducted. Negative control was performed by omitting TdT. We used paraffin-embedded sections of HCC as a positive control[23].
DNA laddering
Laddering of DNA in extracts from untransfected and transfected cells was carried out. DNA was extracted from 5 × 106 cells 24 h post-induction. DNA fragmenta tion was assayed as described previously[24]. Briefly, 24 h following ad dition of 100 µmol/L ZnSO4, 5 × 10 6 cells were lysed in buffer c ontaining 5 mmol/L Tris (pH 8.0), 20 mmol/L EDTA and 5 g/L Triton X-100 onice for 30 min. High molecular weight DNA was removed by centrifugation at 14000 r/min for 15 min at 4 °C, and the supernatant was sequentially extracted with phenol: chlorofo rm, and chloroform. The low molecular weight DNA was recovered by ethanol precip itation, resuspended in 30 µL of TE buffer, and treated with RNase A for 3 h at 37 °C prior to electrophoresis on 12 g/L agarose gels.
Western blot analysis of transgene expression
Monolayers were rinsed with PBS and lysed with SDS-PAGE loading buffer (50 mmol /L Tris HCl pH 6.8, 100 mmol/L dithiothreitol, 20 g/L SDS). Mock and pMD-neo tr ansfected cells served as a control. Samples were analyzed by SDS-PAGE and tran sferred into Hybond-C super membranes (Amersham, UK). The membranes were blocke d with 50 mL/L skim milk, 1 g/L Tween-20 and then probed with primary antibody overnight according to manufacturer’s instructions, washed in PBS, 2 g/L Tween-20 and then incubated with the appropriate horseradish- peroxidase (HRP) conju gated secondary antibody. After washing, the membranes were developed by DAB det ection reagents according to manufacturer’s guide (Dako Co. USA). p27K IP1, rabbit polyclonal antibody (Santa Cruz Biotechnology, USA) was detected f ollowed by HRP-conjugated anti-rabbit IgG (Fc) was used. The level of β-actin was used as a control for equal loading of protein.
Immunohistochemistry analysis
HCC-9204 cells were harvested at different times of induction with ZnSO4. Cel l preparations were fixed with 700 mL/L ethanol for two hours, washed in PBS, an d incubated with p27KIP1 antibody diluted 1 × 100 in PBS containing 10 mL/L bovine serum albumin (BSA, Sigma, USA). Immunostaining was performed using LSAB1 kit (Dako, Peroxidase, USA), according to the instructions of the manufac turer.
Fluorescence microscopy and laser confocal microscope
Nikon Eclipse TE200 inverted fluorescence microscope was used to observe and pho tograph. Images were collected on a laser confocal microscope, model MRC-1024 ( BioRad, UK) with a Plan-Ne-ofluar 40 × 1.3 NA Apochromat objective (Zeiss, Ger many). The 488 nm line of a krypton/argon laser was used for fluorescence excita tion of FITC. Images were processed using Adobe Photoshop 5.0 software (Adobe Sy stems Inc., USA)
RESULTS
RT-PCR for p27KIP1 in Bak transfected HCC 9204 cells
Bak transfected HCC-9204 cells were collected 0, 24 and 48 h post-induction as the previous study mentioned and were used to extract total RNA by TRIZOL reagent. The findings as shown in Figure 1 suggested that overexpression of Bak const antly induced expression of CDK inhibitor, p27KIP1, at mRNA level in H CC-9204 cell line.
Figure 1.
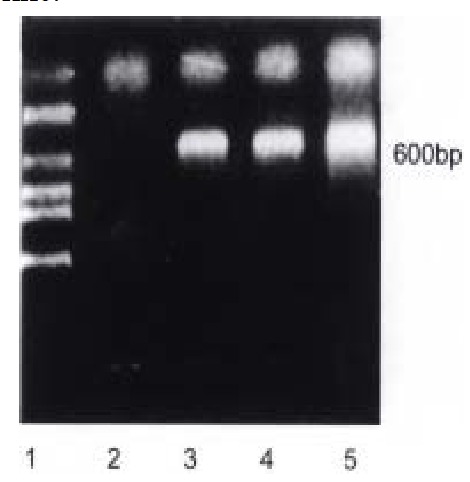
RT-PCR results of human p27KIP1 RNA in Bak transfected HCC-9204 cell line. Predicted PCR product size is about 600 bp. lane 1: Molecular we ight Marker (Takara, DL 2000); lane 2: Bak (0 h); lane 3: Bak (24 h); lane 4: Bak (48 h)
Sequencing of p27KIP1 and p27KIP1-GFP fusion genes
The purified p27KIP1-PCR product was cloned into pGEM T-Easy vector and sequenced. Comparison of the sequences with published data in Genebank indica ted only one base pair of disparity while the amino acid residue encoded was the same as what the Genebank indicated (Figure 2). pGEM-T Easy KIP1 was digested with Kpn-I and Sal-I to yield a 0.6 kb fragment. The resulting fragment then ligated to Kpn-I and Sal-I digested pGREE N LANTERN-under the control of CMV enhancer/promoter and the SV40 polyadenylation signal. Correct reading framework of the fusion gene was confirmed by restriction enzyme analysis or DN A sequencing.
Figure 2.

The nucleotide sequences of p27KIP1. Only one nucleotide is changed from c→t at site No 524, while the sequences of amino acids are the same
Transient expression of p27KIP1-GFP fusion proteins in HCC-9204 cell line
reen fluorescence was visible 8 h after transient transfection with pKIP1-GFP fusion constructs. The p27KIP1-GFP fusion proteins localized primarily to cell nuclei, with some GFP presented in the cytoplasm (Figure 3) . However, 24 h post transfection, the green fluorescence disappeared. Some cells exhibited typical active cell death including condensed and rounded cells, cell detachment under Nikon Eclipse TE200 inverted microscope as shown in Figure 4.
Figure 3.
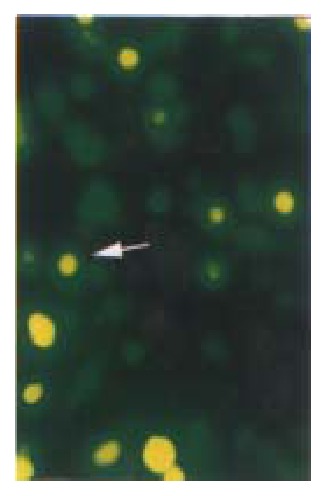
Cell slides under Nikon Eclipse TE200 fluorescence microscope using excitation light at either 488 nm to detect GFP (emission filter 522 nm). Distribution of GFP p27KIP1 fusion protein expressed in living HCC-9204 cells 8 after 100 μmol/L ZnSO4 treatment, indicated by an arrow. × 200
Figure 4.
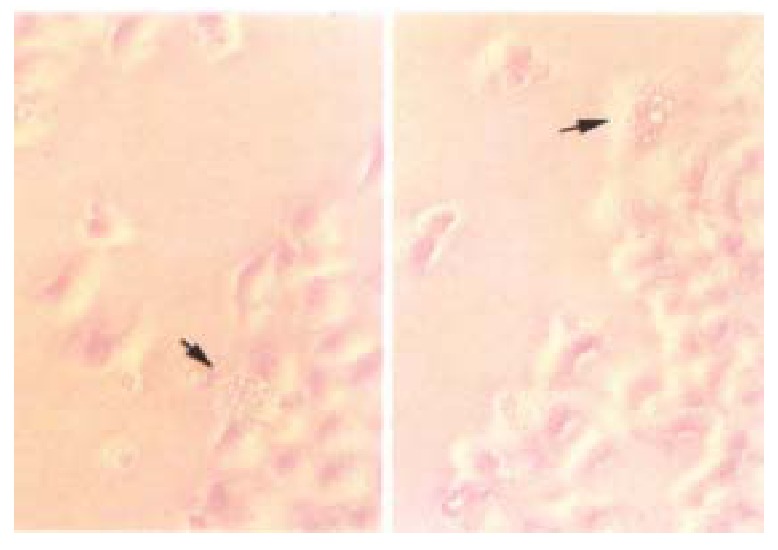
Transient transfected HCC-9204 cells exhibiting typical active cell death such as cell blebbing, condensed and rounded cells as seen under, Nikon Eclipse TE200 phase contrast microscope indicated by an arrow. × 200
pMD vectors direct the inducible expression of p27KIP1
Inducible pMD-KIP1 vectors were constructed to express human cyclin kinase inhi bitors, p27KIP1, as shown in Materials and Methods. Transgene expression was under the control of the metallothionein-II (MT-II) promoter and the BGH polyadenylation signal. Previously it was found that MT-II promoter gave higher expression levels in HCC-9204 cells as compared with those obtained with the HCMV promoter. To study the effects of pMD-vector driven expression of p27KIP1, HCC-9204 cells were left untre ated (mock), or were transfected with control pMD-vector or pMD-KIP1, at 4, 24 and 48 h, respectively after addition of 100 μmol/L ZnSO4. Western blot analysis showed a strong signal of Mr 27 000 protein appeared in H CC-9204 24 h post induction. However no band was detected in normal HCC-9204 cells as shown in Figure 5. Immunohistochemistry of p27KIP1 as shown in Figure 6 was consistent with Western blot analysis. Nuclear staining signals were observed in p27KIP1 stable transfectant 24 h post addition of ZnSO4.
Figure 5.
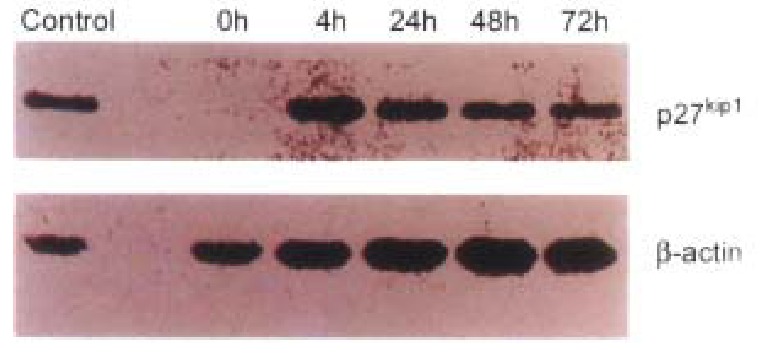
The results of Western blots of lysates from transfected HCC-9204 cells, probed with antibodies against human p27KIP1. Excellent p27KIP1 expression was observed in HCC-9204 cells even 4 h after induction with zinc.
Figure 6.
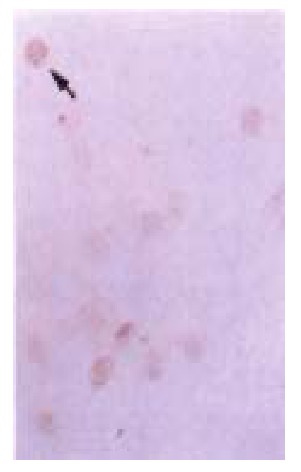
p27KIP1 detected by DAKO LABC kit in transfected HCC-9204 cells after 24 h of addition of ZnSO4. p27KIP1 dark-brown granules are present in the nuclei of p27KIP1 transfected HCC-9204 cells, as in dicated by an arrow. × 200
Inhibition of tumor cell growth by exogenous p27KIP1
p27KIP1 gave a very pronounced inhibition of growth. HCC-9204 cells viability was determined by microscopic examination of cells stained by 1 g/L try pan blue (trypan blue exclusion) (Figure 7). Time course effect of p27KIP1 on HCC-9204 cells growth was observed under Nikon Eclipse TE200 inverted mi croscopic (Figure 8). Within one day post-induction, a block in cell growth was observed in HCC-9204 cells with a few cells staining positive with Trypan blue, indicating that the exogenous p27KIP1 was having a cytostatic effect on growth. More dramatic effects were observed 24 h after addition of zinc. Moc k and control pMD vector transfected cells grew until confluent (day 3) and then entered a growth arrest following which significant numbers of trypan blue-sta ining cells were observed, indicative of cell death, accounting for the decline in the cell number, which was especially apparent in confluent HCC-9204 cells. By 3 days post-induction, cells staining positive with trypan blue-staining cells were observed in the cultures transfected with p27KIP1 even though the monolayers were not confluent, and this cell death accounted for the over- all reduction in cell number that was observed during the remainder of the exper iment. Cell death was greatest in HCC9204 cells transfected with pMD-KIP1.
Figure 7.
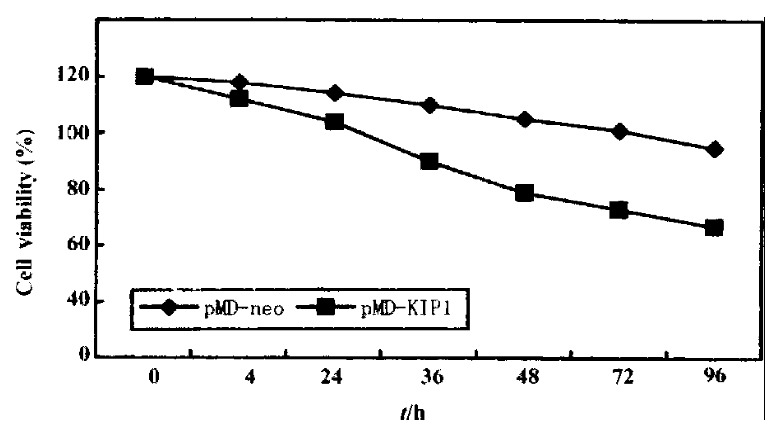
Time course of growth of cells transfected with pMD-KIP1 expressing p27KIP1. HCC-9204 cells were induced by 100 μmol/L ZnSO4. HCC-9204 cells viability was determined by microscopic examination of cells stained by 1 g/L trypan blue, counting cells on a hemocytometer at the time point indicated. Each treatment was triplicate.
Figure 8.

Time course effect of p27KIP1-on HCC-9204 cells growth. Cells were seeded at 10\+5 per dish. HCC-9204 cells were induced by 100 μmol/L ZnSO4 at the time indicated. Cells morphology was observed under Nikon Eclipse TE 200 phase-contrast microscopic at 0, 24 and 48 h post addition of zinc. “Uppe r-row” represents pMD-neo transfected cells; “Lower-row” represents pMD- Bak transfected cells. × 200
From Figure 7, we could obviously figure out that the cell growth was reduced by 35% after 48 h of p27KIP1 induction with zinc treatment as determined by trypan blue exclusion assay. These differences remained the same at 72 h , and even at 96 h post addition of zinc.
pMD vector expression of p27KIP1 alters cell cycle distribution profiles
To see if the p27KIP1 had an ability to induce G1 arrest, cell cycle analysis was performed on transfected HCC-9204 cells. At 24 h post-induction, HCC-9204 cells expressing exogenous p27KIP1 showed an accumulation of cells in G1 compared to mock and control pMD transfected cells, consistent with p27KIP1 inhibiting tumor cell growth. (Figure 9)
Figure 9.
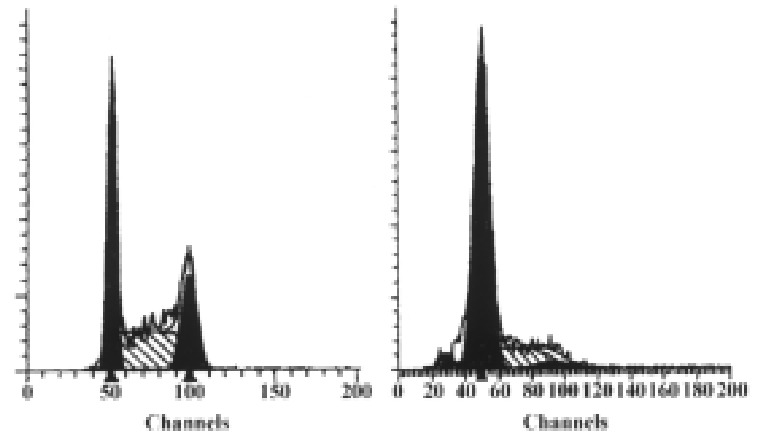
Result of flow cytometric analysis. Cells were stably transfectd with p27KIP1 and induced by 100 μmol/L ZnSO4. The percentage of each phase for each group (before or 24 h after addition of zinc) is indicated in each panel.
FACS analysis showed that overexpression of p27KIP1 resulted in an i ncrease of G1 population in HCC-9204 cells from 35.21% to 76.31%. Together, these data indicated that overexpression of p27KIP1 alone is sufficie nt to suppress the proliferation of human tumor cells. We are currently investig ating the role of p27KIP1 in mediating cellular responses to a variety of external signals and the mechanism of inhibition.
Induction of apoptosis in cells transfected with vectors expressing p27KIP1
Since by day 3, a significant proportion of HCC-9204 cells transfected with the vectors expressing p27KIP1 stained positive with trypan blue, indicat ive of cell death, we attempted to ascertain whether p27KIP1 was induc ing apoptosis. To confirm that over-expression of p27KIP1 induced cel l death was apoptotic, a variety of apoptosis associated detection assays were a pplied. In stable transfectants after addition of 100 µmol/L ZnSO4, cell growth was significantly inhibited according to FACS analysis as shown in Figure 9. TUNEL analysis (Figure 10) and DNA laddering (Figure 11) confirmed that part of cells underwent apoptosis 48 h or 72 h after addition of 100 µmol/L ZnSO4.
Figure 10.
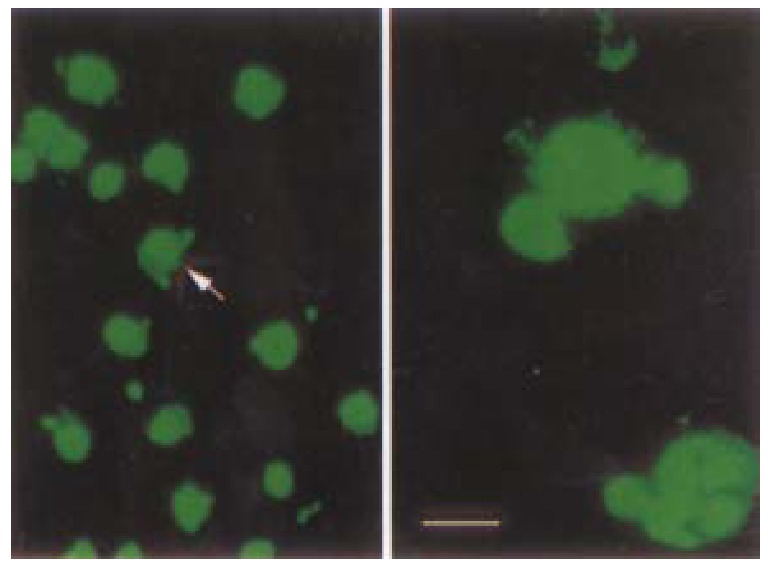
TUNEL assay demonstrating obvious apoptotic changes in HCC-9204/p27KIP1 24 h after addition of zinc under laser confocal microscope as in dicated by an arrow. (Bar represents 10 μm)
Figure 11.

Analysis of DNA ladder formation at 0 (lane 1), 4 h (lane2), 24 h (lane 3), 48 h (lane 4), 72 h (lane 5) post addition of zinc in HCC-9204 cell line. Eac h lane was loaded with 10 μg DNA.
Taken together, p27KIP1 overexpression induced rapid morphological changes in HCC-9204 cells. p27KIP1 overexpression inhibited prolifera tion of HCC-9204 cells and subsequently induced apoptosis.
DISCUSSION
Since its introduction into cell biological research, the green fluorescent protein (GFP) of the jellyfish-Aequorea victoria has become a versatile tool for the analysis of protein function and dynamics at the cellular level[25] . GFP, which consists of 238 amino acids, has been used as a tag for localizatio n of a broad range of proteins in a wide variety of eukaryotic cells[26]. A mutant of GFP, S65T with an excitation peak of 489 nm and an emission peak of 511 nm, emits fourto six times more fluorescence energy compared with wild-type GFP. GFP may be fused generically to a target protein, and the fluorophore of GFP forms spontaneously in the presence of oxygen, thus rendering it an ideal probe for in vivo applications. The in vivo expression of GFP obviates the fixation and permeablization of cells for immunofluorescence or the microinj ection of labeled proteins[27].
GFP fusion proteins constitute a major advance in the study of the dynamics of intracellular processes in living cells[28-32]. A major concern in the application of GFP as a fluorescent tag relates to whether the distribution of GFP fluorescence is identical to that of the protein to which it is fused. We have constructed GFP p27KIP1 fusion gene and transfected into HCC-9204 cells. The results indicated that the fusion protein was still express in the nuclei of HCC-9204 cells.
Previous studies indicated that cell survival following transfection with pGRE EN LANTERN-1 was similar to transfection with pCMV·SPORT-β gal. No trypan blue-stained cells were seen to fluoresce green, and the fluorescence intensity of green fluorescent cells treated with trypan blue remained the same when judged by microscopic observation indicating that fluorescent cells were viable cells[33]. A comparative study has been performed to determine wheth er apoptotic cells still can express GFP. A GFP positive clone was chosen and co unterstained with PI. The cells which had a property of apoptosis were stained with PI and no longer expressed GFP. We used PI counter-staining to show that the cells appearing as red fluorescence were not viable and only viable cells had a potential to express GFP. The fluorescence green vanished as soon as the cells become unviable (Figure 12). Therefore, transient expression of KIP1-GFP fusion protein was disrupted when these cells underwent apoptosis.
Figure 12.
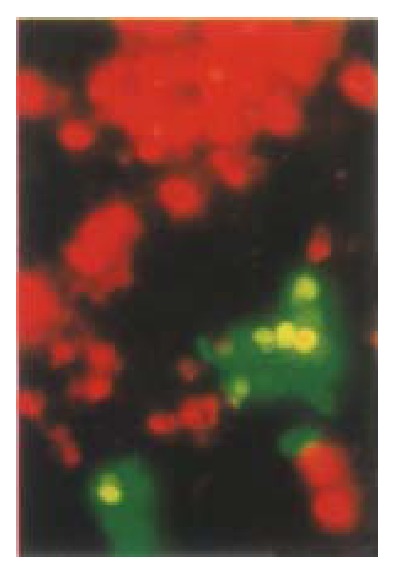
Comparative study of whether apoptotic cells can still express GFP. A GFP positive clone was chosen and counterstained with PI. Apoptotic cells were stained with PI and no longer expressed GFP. HCC-9204 cells were observed under Nikon Eclipse TE200 fluorescent phase-contrast microscopic. × 200
The cell cycle progression is controlled by a family of cyclins and their spec ific catalytic partners, cyclin-dependent kinases (CDKs). D-type cyclins g over n the G1/S transition in association with their proper physiological partners, CDK4 or CDK6, and CDK2,B respectively[34-35]. For cells to complete the G1/S transition, the activities of both D-type cyclins and cyclin E are essential. CDK inhibitors stop the cell cycle progression by negatively and stoichi ometrically regulating cyclin/CDK activities and are the targets of various extr acellular growth-modifiers. These inhibitors have similar N-terminal regio ns which are responsible for the blockade of CDK kinases. They can inhibit any G1 cyclin/CDK activity. The CDK inhibitors also served as the common mediators of various biological processes including DNA-damages, cell differentiation, cell to cell contact inhibition and cell senescence[36]. Recent studies indicate that oncogene signals converge to cyclin/CDK and CDK inhibitors[37].We have reported that overexpression of Bak may prolong cell cycle in G1 phase in HCC-9204 cells. We postulated that translocated expression of Bak in this process transferred signals to the cell cycle regulators in the nulcei, and thus induced cell cycle arrest in G1 phase.
p27KIP1, as a member of CDKIs, is constitutively expressed in many ce lls, and the level of this protein is elevated in G0 phase and declines as cells in culture enter the cell cycle[38-40]. p27KIP1 may play an important role in governing the growth factor restriction point by ensuring that CDK activity is suppressed during G0 and early G1 phase[41]. Int erestingly, in many types of tumors including breast, colon, prostate and ovaria n carcinomas, the expression of p27KIP1 gene was down-regulated[42-43]. From the sequencing analysis, we could figure out that in some types of tumor, p27KIP1 gene was null or mutated, while in other types it was still intact. HCC-9204 cells, as a target cell line, has intact p27KIP1 which was confirmed by fluorescence in situ hybridization (FISH) as shown in Figure 13 and DNA sequencing. Researchers are attempting to discover the mech anism of controlling the expression of p27KIP1 and cyclin-CDK activat ion in these malignant cells.
Figure 13.
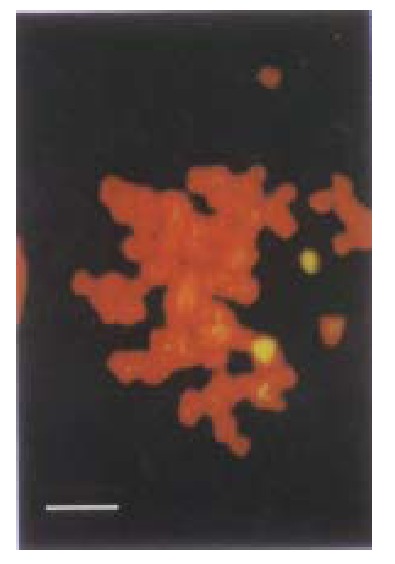
In situ fluorescence hybridization with FITC labelled p27KIP1 probe. Two signals were observed in the chromosome under laser confocal micro scopy. (Bar represents 5 μm)
The correct timing of cyclin-CDK activation is regulated at several levels, in cluding the control of cyclin-encoding gene expression, cyclin stability and cellular localization, as well as CDK subunit phosphorylation and dephosphorylation[44-46]. Another level of regulation is provided by the CDKIs which consist of two families. The potential role of p27KIP1 in control of the hepatocyte cell cycle is suggested by the implication of mitogens and anti-mit ogens known to control p27KIP1 levels in the regulation of liver cell proliferation[47]. The discovery of p27KIP1 has facilitated the elucidation of these mechanisms. In addition, in all organs including the liver, p27/, but not p21/mice show an increased cell density withou t any obvious alteration in liver cells differentiation or structure[48].
Most studies examining the function of p27KIP1 had been performed in tissue culture systems using transformed cell lines or embryonic cells. Previous studies suggested that p27KIP1 overexpression causes cell cycle arrest in G1 phase and/or apoptotic death of mammalian cells and supported the potential utility of gene therapeutic approaches aimed at elevating p27KIP1 expression for treatment of human cancers and inhibition of tumorigenicity[49]. Recent evidences demonstrated that high levels of p27KIP1 protein, induced by adenovirus vector, led to growth arrest as well as enhancement of apoptosis in several cell lines from different species and tissues of various origins[50]. However, as far as we know, no study has been conducted previously to examine the expression of p27KIP1 in human hepatocelluar carcinoma tissue in relation to apoptosis. We obtained p27KIP1 gene fragment in Bak transfected HCC-9204 cells and constructed a transient or stable transfectant. The purpose of the study was to examine the potential role of p27KIP1 during Bak overexpression in HCC-9204 cells. The results suggest that overexpression of Bak may induce expression of p27KIP1 at least in mRNA level. p27KIP1 may exert its effect on G1 phase elongatio n induced by Bak and p27KIP1 itself may play an important role in the regulation of cell cycle during this process.To our knowledge, this is the firs t time that a significant correlation between p27KIP1 expression and cell cycle arrest and apoptosis has been demonstrated in HCC-9204 cell line.
However, the mechanism of tumor growth suppression appears to include not only p27KIP1 but also various other proteins, such as p53 etc. As weknow, p53 is mutated in HCC-9204 cell line and is often null in other cell lines[51-53]. Therefore, how these cell types undergo cell death is of co ncern since the p53-dependent apoptosis pathway has been abolished. p53 serves as a major G1 checkpoint regulator of the induction of p21-CIP1 but not of p27KIP1 and p57KIP2, while it induces apoptosis[54]. The p53-mediated CDK inhibitor p21CIP1 does not lead to significant apoptosis, though both p21CIP1 and p27KIP1 induce G1 arrest through their potential abilities to inhibit CDK activity[55-56]. These findings suggest that the inhibition of CDK activity may not be sufficient to induce apoptotic cell death. A possible explanation of these differences between p27KIP1 and p53-mediated p21CIP1ªª functions is that p27KIP1 may have other functions in addition to its notable function as a CDK inhibitor. It can also be speculated that p27KIP1, which has the potential to induce growth arrest and apoptosis, may play a more important role in tumor growth suppression than p53-mediated p21CIP1.
ACKNOWLEDGEMENTS
Special thanks are extended to Mr. Dong Bo for technical assistance with f luorescence-activated cell sorter analysis; Ms. Zhang Fan for technical assista nce with laser confocal microscope; Dr. Polyak SS for pBluescript SK-p27; Dr. Zhang Jie for pMD-neo plasmid and technical support; Ms. An Jianhua for photo processing; Dr. Zheng Jian-Yong for other cooperation.
Footnotes
Edited by Zhou XH proofread by Mittra S
References
- 1.Favrot M, Coll JL, Louis N, Negoescu A. Cell death and cancer: replacement of apoptotic genes and inactivation of death suppressor genes in therapy. Gene Ther. 1998;5:728–739. doi: 10.1038/sj.gt.3300661. [DOI] [PubMed] [Google Scholar]
- 2.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7:331–342. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Q, Zou JX, Chen YL, Yu HZ, Wang LD, Li YX, Guo HQ, Gao SS, Qiu SL. Alteration of tumor suppressor gene p16 and Rb in gastric cancinogenesis. China Natl J New Gastroenterl. 1997;3:262. doi: 10.3748/wjg.v3.i4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 5.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 6.Guan KL, Jenkins CW, Li Y, Nichols MA, Wu X, O'Keefe CL, Matera AG, Xiong Y. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 1994;8:2939–2952. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 7.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 8.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 9.Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci USA. 1994;91:5291–5295. doi: 10.1073/pnas.91.12.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 11.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 12.Kato JY, Matsuoka M, Polyak K, Massagué J, Sherr CJ. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell. 1994;79:487–496. doi: 10.1016/0092-8674(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 13.Winston J, Dong F, Pledger WJ. Differential modulation of G1 cyclins and the Cdk inhibitor p27kip1 by platelet-derived growth factor and plasma factors in density-arrested fibroblasts. J Biol Chem. 1996;271:11253–11260. doi: 10.1074/jbc.271.19.11253. [DOI] [PubMed] [Google Scholar]
- 14.Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger WJ. Repression of p27kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol Cell Biol. 1996;16:4327–4336. doi: 10.1128/mcb.16.8.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 16.Hu CM, Liu YF, Sui YF, Xu LQ, Liu CG. Establishment of a human hepatocel lular carcinoma cell line HCC-9204 and its characteristics. J Fourth Milit Me dUniv. 1995;16:92–95. [Google Scholar]
- 17.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning, a labora-tory m anual, 2nd ed. Cold Spring Harbor Laboratory Press. 1989 [Google Scholar]
- 19.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 20.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 21.Begg AC, editor Principles and practice of the tumor growth delay assay. Oxford, England: Pergamon Press. 1987:125–127. [Google Scholar]
- 22.Li J, Wang WL, Liu B, Wang BY. Apoptosis in human hepatocel-lular carcino ma by terminal deoxynucleotidyl transferase medi-ated dUTP-FITC nick end labeli ng. Shijie Huaren Xiaohua Zazhi. 1998;6:491–494. [Google Scholar]
- 23.Labat-Moleur F, Guillermet C, Lorimier P, Robert C, Lantuejoul S, Brambilla E, Negoescu A. TUNEL apoptotic cell detection in tissue sections: critical evaluation and improvement. J Histochem Cytochem. 1998;46:327–334. doi: 10.1177/002215549804600306. [DOI] [PubMed] [Google Scholar]
- 24.Kouchi Z, Saido TC, Ohyama H, Maruta H, Suzuki K, Tanuma S. The restrictive proteolysis of alpha-fodrin to a 120 kDa fragment is not catalyzed by calpains during thymic apoptosis. Apoptosis. 1997;2:84–90. doi: 10.1023/a:1026443926962. [DOI] [PubMed] [Google Scholar]
- 25.Yang F, Moss LG, Phillips GN. The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 26.Yokoe H, Meyer T. Spatial dynamics of GFP-tagged proteins investigated by local fluorescence enhancement. Nat Biotechnol. 1996;14:1252–1256. doi: 10.1038/nbt1096-1252. [DOI] [PubMed] [Google Scholar]
- 27.Chalfie M, Kain S. Edited green fluorescent protein: properties, applications and protocols. Wiley Liss, 1998. ISBN 0 471 17839X [Google Scholar]
- 28.Lewis PJ, Marston AL. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene. 1999;227:101–110. doi: 10.1016/s0378-1119(98)00580-0. [DOI] [PubMed] [Google Scholar]
- 29.Imreh G, Beckman M, Iverfeldt K, Hallberg E. Noninvasive monitoring of apoptosis versus necrosis in a neuroblastoma cell line expressing a nuclear pore protein tagged with the green fluorescent protein. Exp Cell Res. 1998;238:371–376. doi: 10.1006/excr.1997.3846. [DOI] [PubMed] [Google Scholar]
- 30.Mahajan NP, Linder K, Berry G, Gordon GW, Heim R, Herman B. Bcl-2 and Bax interactions in mitochondria probed with green fluorescent protein and fluorescence resonance energy transfer. Nat Biotechnol. 1998;16:547–552. doi: 10.1038/nbt0698-547. [DOI] [PubMed] [Google Scholar]
- 31.Wiemer EA, Wenzel T, Deerinck TJ, Ellisman MH, Subramani S. Visualization of the peroxisomal compartment in living mammalian cells: dynamic behavior and association with microtubules. J Cell Biol. 1997;136:71–80. doi: 10.1083/jcb.136.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans K, Nelson PH, Schifferli K, Jessee J. pGREEN LANTERN 1, a sup e rior green fluorescent protein mammalian cell transfection reporter. Focus. 1996;15:40–43. [Google Scholar]
- 34.Kato JY, Sherr CJ. Inhibition of granulocyte differentiation by G1 cyclins D2 and D3 but not D1. Proc Natl Acad Sci USA. 1993;90:11513–11517. doi: 10.1073/pnas.90.24.11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993;73:487–497. doi: 10.1016/0092-8674(93)90136-e. [DOI] [PubMed] [Google Scholar]
- 36.Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 37.Schreiber M, Muller WJ, Singh G, Graham FL. Comparison of the effectiveness of adenovirus vectors expressing cyclin kinase inhibitors p16INK4A, p18INK4C, p19INK4D, p21(WAF1/CIP1) and p27KIP1 in inducing cell cycle arrest, apoptosis and inhibition of tumorigenicity. Oncogene. 1999;18:1663–1676. doi: 10.1038/sj.onc.1202466. [DOI] [PubMed] [Google Scholar]
- 38.Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 39.Reed SI, Bailly E, Dulic V, Hengst L, Resnitzky D, Slingerland J. G1 control in mammalian cells. J Cell Sci Suppl. 1994;18:69–73. doi: 10.1242/jcs.1994.supplement_18.10. [DOI] [PubMed] [Google Scholar]
- 40.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 41.McIntyre M, Desdouets C, Sénamaud-Beaufort C, Laurent-Winter C, Lamas E, Bréchot C. Differential expression of the cyclin-dependent kinase inhibitor P27 in primary hepatocytes in early-mid G1 and G1/S transitions. Oncogene. 1999;18:4577–4585. doi: 10.1038/sj.onc.1202815. [DOI] [PubMed] [Google Scholar]
- 42.Singh SP, Lipman J, Goldman H, Ellis FH, Aizenman L, Cangi MG, Signoretti S, Chiaur DS, Pagano M, Loda M. Loss or altered subcellular localization of p27 in Barrett's associated adenocarcinoma. Cancer Res. 1998;58:1730–1735. [PubMed] [Google Scholar]
- 43.Tsihlias J, Kapusta LR, DeBoer G, Morava-Protzner I, Zbieranowski I, Bhattacharya N, Catzavelos GC, Klotz LH, Slingerland JM. Loss of cyclin-dependent kinase inhibitor p27Kip1 is a novel prognostic factor in localized human prostate adenocarcinoma. Cancer Res. 1998;58:542–548. [PubMed] [Google Scholar]
- 44.Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14:2077–2086. doi: 10.1128/mcb.14.3.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, Sherr CJ. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- 46.Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Albrecht JH, Poon RY, Ahonen CL, Rieland BM, Deng C, Crary GS. Involvement of p21 and p27 in the regulation of CDK activity and cell cycle progression in the regenerating liver. Oncogene. 1998;16:2141–2150. doi: 10.1038/sj.onc.1201728. [DOI] [PubMed] [Google Scholar]
- 48.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Gorospe M, Huang Y, Holbrook NJ. p27Kip1 overexpression causes apoptotic death of mammalian cells. Oncogene. 1997;15:2991–2997. doi: 10.1038/sj.onc.1201450. [DOI] [PubMed] [Google Scholar]
- 50.Kwon TK, Nordin AA. Overexpression of cyclin E and cyclin-dependent kinase inhibitor (p27Kip1): effect on cell cycle regulation in HeLa cells. Biochem Biophys Res Commun. 1997;238:534–538. doi: 10.1006/bbrc.1997.7335. [DOI] [PubMed] [Google Scholar]
- 51.Yang SM, Zhou H, Chen RC, Wang YF, Chen F, Zhang CG, Zhen Y, Yan JH, Su JH. Sequencing of p53 mutation in established human hepatocellular carcinoma cell line of HHC4 and HHC15 in nude mice. World J Gastroenterol. 1998;4:506–510. doi: 10.3748/wjg.v4.i6.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deng ZL, Ma Y. Aflatoxin sufferer and p53 gene mutation in hepatocellular carcinoma. World J Gastroenterol. 1998;4:28–29. doi: 10.3748/wjg.v4.i1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng XM, Peng WW, Yao JL. Codon 249 mutations of p53 gene in development of hepatocellular carcinoma. World J Gastroenterol. 1998;4:125–127. doi: 10.3748/wjg.v4.i2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirama T, Koeffler HP. Role of the cyclin-dependent kinase inhibitors in the development of cancer. Blood. 1995;86:841–854. [PubMed] [Google Scholar]
- 55.Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA. p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene. 1999;18:2789–2797. doi: 10.1038/sj.onc.1202615. [DOI] [PubMed] [Google Scholar]
- 56.Wang LD, Yang WC, Zhou Q, Xing Y, Jia YY, Zhao X. Changes of p53 and Wafl p21 and cell proliferation in esophageal carcinogenesis. China Nat l J New Gastroenterol. 1997;3:87. doi: 10.3748/wjg.v3.i2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]