

Abstract
Dyskeratosis congenita is a multisystem genetic disorder. Although hepatic involvement is reported in about 7% of patients with dyskeratosis congenita, it is not well characterized and often attributed to hemochromatosis from frequent blood transfusions. A few case reports describe cirrhosis and hepatic cell necrosis in affected individuals in autosomal dominant pedigrees. Bone marrow failure and malignancies are the principal causes of death in dyskeratosis congenita. We describe the first case of living donor liver transplantation, in dyskeratosis congenita for decompensated cirrhosis with portal hypertension. The patient also had associated severe hepatopulmonary syndrome, interstitial lung disease, bilateral hip replacement for avascular necrosis of the head of femur, and a past history of bone marrow transplantation for bone marrow failure.
Abbreviations: BMF, bone marrow failure; DKC, dyskeratosis congenita; HPS, hepatopulmonary syndrome; LDLT, living donor liver transplantation; MAA, microaggregated albumin scan; PHT, portal hypertension; SCE, saline contrast echocardiography
Keywords: chronic liver disease, dyskeratosis congenita, hepatopulmonary syndrome, liver transplantation, microaggregated albumin scan
Dyskeratosis congenita (DKC), also known as a Zinsser Engman Cole syndrome, is a rare genetically heterogenous multisystem disorder characterized by the triad of abnormal skin pigmentation, nail dystrophy, and mucosal leukoplakia.1 Bone marrow failure, which occurs in approximately 50% of patients, and the predisposition to malignancy are the principal causes of early mortality.2 X-linked recessive, autosomal recessive, and autosomal dominant forms of the disease are recognized,3 along with genitourinary, pulmonary, skeletal, neurological, ophthalmic, dental, and gastrointestinal abnormalities.2 Gastrointestinal findings, such as esophageal strictures, hepatomegaly or cirrhosis, are seen in 10% of patients.4 A possible association with noncirrhotic portal hypertension has also been suggested.5 Considerable diverse presentation and disease severity may be present within the same family in DKC.
Here, we report a case of an adult male patient of DKC who underwent living donor liver transplantation (LDLT) for end stage liver disease (ESLD) with severe hepatopulmonary syndrome (HPS).
Case Report
A 34-year-old male with no addiction was diagnosed to have DKC at the age of 24 years when he presented with bone marrow failure (BMF) requiring multiple blood transfusions and associated skin hyperpigmentation, nail dystrophy, and tongue leukoplakia. He received allogenic bone marrow transplantation (BMT) at the age of 25 years, for BMF. He was on cyclosporine and steroid for 3 years after which immunosuppression was stopped in view of bilateral hip pathology. He eventually underwent bilateral hip replacement for avascular necrosis of the head of femur at the age of 29 years. He developed hemetemesis at the age of 31 years for which endoscopic variceal ligation was done and on evaluation was found to have chronic liver disease (CLD). During the current admission, the patient presented with complaints of gradually progressive dyspnea on exertion, easy fatigability, and generalized weakness of 3 years duration. He also had decompensation in the form of ascites, spontaneous bacterial peritonitis (SBP), and jaundice.
At admission, physical examination showed severe malnutrition (BMI 16.8), pallor, icterus, clubbing, pedal edema, and ascites. Dermatologic findings included reticulated skin pigmentation, dystrophic nail changes with longitudinal striations, and deformities (Figure 1). Leucoplakia was present on the tongue (Figure 2).
Figure 1.
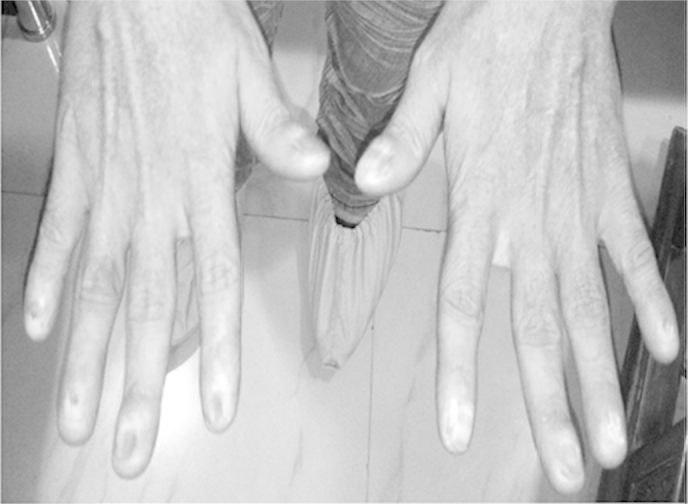
Nail dystrophy of finger nails.
Figure 2.
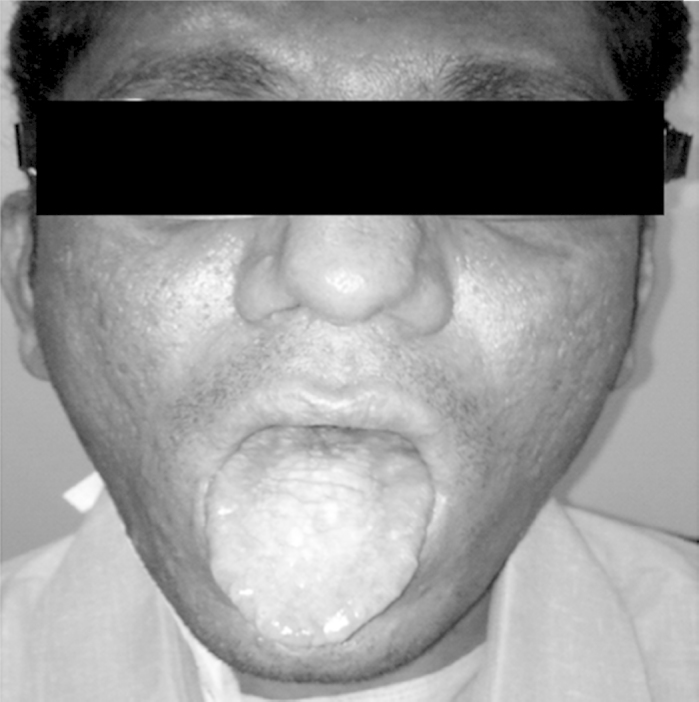
Leukoplakia of tongue.
Complete hemogram revealed normocytic normochromic to macrocytic cells with hemoglobin 13.9 g/dl, total leucocytes count 4600/mm3, and platelet count 64,000/mm3. Prothombin time (PT) was 16.5 s and International normalized ratio (INR) was 1.7 at the time of admission. Liver function test (LFT) revealed serum bilirubin 1.58 mg/dl, AST 61 IU/L (ref. range = 5–40), ALT 50 IU/L (ref. range = 10–40), SAP 140 IU/L (ref. range = 32–92), and serum albumin 2.6 g/dl (ref. range = 3.5–5.2) and was chronically deranged since last 3 years. Reticulocyte count was 2.24% (ref. range = 0.2–2) which was an indirect evidence that bone marrow function was satisfactory.
Abdominal ultrasound revealed CLD with cirrhotic changes, portal hypertension (PHT), gross ascites, and splenomegaly. Ascitic fluid analysis at the time of admission at our hospital was turbid in appearance with 100/mm3 white blood cells (predominantly neutrophils). Serum ascites albumin gradient was 2.04 g/dl (ascitic fluid albumin = 0.56 g/dl and Serum albumin 2.6 g/dl). Upper gastrointestinal endoscopy showed grade II–III esophageal varices and severe portal hypertensive gastropathy.
Etiological workup for all possible causes of liver cirrhosis (viral, metabolic, and autoimmune) was negative. Transjugular liver biopsy confirmed liver cirrhosis with Child–Pugh score of 10/15 and MELD of 16. Further evaluation was done to establish the diagnosis and severity of HPS. In view of low oxygen saturation on room air (), Blood gas exchange (ABG) in the sitting position was obtained that indicated severe hypoxemia (; ) with a very high alveolar-arterial oxygen gradient [D(A − aO2) = 67]. Repeat ABG obtained with 100% oxygen showed of 158 mmHg. The chest radiograph and pulmonary function test was normal. High-resolution computed tomography (HRCT) of chest showed interstitial lung disease (ILD) involving bilateral lower lobes and apices.
The severe hypoxemia with absent major findings on CT scan raised the suspicion of an intra-cardiac or pulmonary right to left shunt. An echocardiogram ruled out intra-cardiac shunt. There were no signs of pulmonary hypertension or left ventricular dysfunction. Agitated Saline Contrast echocardiography (SCE) showed severe shunting. An albumin lung perfusion scan (Tc99mMAA) confirmed an intra-pulmonary arteriovenous shunt equal to 21% (Figure 3).
Figure 3.

Technetium-99m labeled macroaggregated albumin lung perfusion scan showing 21% uptake in the brain.
CT pulmonary angiography showed no direct arteriovenous communication and no vascular dilation. The severe hypoxemia and positive T99mMAA results in the context of CLD were consistent with very severe HPS.
It was felt that at the time of presentation to us, his decompensated CLD with severe HPS was more likely to shorten his survival than any other complication of DKC. After a multidisciplinary discussion and optimization of clinical condition, LDLT was done on 29 August 2013 after consensus that the predominant pulmonary pathology was HPS due to CLD and not ILD. He received a left hemiliver graft from his brother. Intra-operatively, 500 mg of methylprednisolone was given in the anhepatic phase; it was tapered to 100 mg, 80 mg, 60 mg, and 40 mg on days 1, 2, 3, and 4 respectively. On day 5, the patient was started on tacrolimus, mycophenolate mofetil (MMF), and prednisolone 20 mg which were titrated as per trough levels, clinical and laboratory parameters. On follow-up, his immunosuppression has stabilized at tacrolimus 0.5 mg BD and MMF 1 g BD. Prednisolone was tapered and stopped by 3 months.
He was extubated on post-operative day 2. There were features of type 1 respiratory failure on initial blood gases after extubation, for which intermittent noninvasive ventilation (NIV) and high flow oxygen therapy were given. He was off NIV from day 10 and maintained oxygen saturation (, ) on room air from day 20. The patient was discharged on day 28. Explant histopathology was suggestive of cirrhosis with no evidence of veno-occlusive disease, iron overload, drug-induced liver injury, nodular regenerative hyperplasia (NRH), or any other specific etiology.
HPS has since improved dramatically. One year post transplantation (hospital follow-up in September 2014), the patient was doing well, maintaining oxygen saturation ( = 98%, = 92 mmHg) with room air. Agitated SCE showed mild shunting. He had gained weight (BMI 24) and was gainfully employed. On last hospital follow-up in July 2015 (22 month post transplantation), the patient had no complaints and had a normal PFT. HRCT chest showed ILD involving bilateral lower lobes and apices, similar to previous scan prior to transplantation. Complete hemogram and LFT were normal.
Discussion
To the best of our knowledge, this is the first ever case of LDLT in DKC for decompensated cirrhosis with PHT, complicated by severe HPS described in literature. The liver disease was secondary to DKC, as PHT with or without cirrhosis has been reported to be associated with DKC.2, 5 Many systems were affected in this patient, including the skin, bone marrow, skeletal system, liver, and lung (severe HPS as well as ILD).
The main causes of mortality in DKC mentioned in the literature are BMF (60–70%), pulmonary disease (10–15%), and malignancy (10%). Our patient was unusual and his main life-threatening problem at the time of presentation was decompensated CLD with severe HPS. As the patient's life expectancy without liver transplant was limited due to severe HPS, the family opted for this option. A multidisciplinary meeting was held in many sessions to ensure that the pulmonary morbidity was potentially reversible after liver transplantation and not solely due to restrictive lung disease or DKC per se. There was no malignancy or other medical conditions that could have resulted in short-term mortality and thus not justifying a major undertaking such as liver transplantation.
HPS prevalence varies between 5% to 32% in patients with cirrhosis or severe PHT.6 HPS is characterized by an intra-pulmonary right to left shunt in the setting of CLD.7 The diagnostic criteria are hypoxemia (PaO2 < 80 mmHg or D (A − aO2) ≥ 15 mmHg), pulmonary vascular dilatation (confirmed by positive findings on agitated SCE or abnormal uptake in the brain (>6%) with radioactive lung-perfusion scanning), and association with CLD (PHT with or without cirrhosis). The most sensitive and least invasive diagnostic method is agitated SCE.7 Currently, no effective medical therapies for HPS exist; LT is the only successful treatment. The severity of HPS is, however, a significant determinant of post-transplant survival, with the presence of an arterial oxygen concentration of ≤50 mmHg, or a shunt fraction >20% strongly predictive of a high postoperative mortality.8 In the largest single center series, patients with HPS had a 5-year survival rate of 76% after LT, a rate comparable to patients without HPS.9
Potentially, LDLT could be a sensible strategy for patients with HPS due to the rapid deterioration of lung function while awaiting transplant. Only a few case series exist of LDLT in this setting and the largest describes 14 patients with excellent results.10 They found that the overall time to extubation was 2 (1–32 days) and hospital stay was 20 (17–46 days). However, the length of time of normalization of arterial hypoxemia post LT is highly variable. One of the retrospective studies showed complete resolution of their pretransplant oxygen dependence in an average of 53.5 ± 35.7 days in severe HPS.11
Pulmonary complications are reported in 20% of patients with DKC.12 ILD has also been described with DKC after BMT.13 Our patient had minimal bilateral ILD prior to LT which was most probably post BMT lung changes. Even after 2 years post LT, the skin changes and pulmonary changes remain stable.
Conclusion
DKC can present with end stage liver disease and HPS. We suggest that LT be considered for carefully selected patients with DKC in whom liver disease is the prime driver for mortality as it has the potential to dramatically improve their quality of life.
Conflicts of Interest
The authors have none to declare.
References
- 1.Drachtman R.A., Alter B.P. Dyskeratosis congenital: clinical and genetic heterogeneity: report of a new case and review of the literature. Am J Pediatr Hematol Oncol. 1992;14:297–304. [PubMed] [Google Scholar]
- 2.Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000;110:768–779. doi: 10.1046/j.1365-2141.2000.02109.x. [DOI] [PubMed] [Google Scholar]
- 3.Knight S., Vulliamy T., Copplestone A., Gluckman E., Mason P., Dokal I. Dyskeratosis congenita (DC) registry: identification of new features of DC. Br J Haematol. 1998;103:990–996. doi: 10.1046/j.1365-2141.1998.01103.x. [DOI] [PubMed] [Google Scholar]
- 4.Brown K.E., Kelly T.E., Myers B.M. Gastrointestinal involvement in a woman with dyskeratosis congenita. Dig Dis Sci. 1993;38:181–184. doi: 10.1007/BF01296794. [DOI] [PubMed] [Google Scholar]
- 5.Yazgan Y., Demirtuk L., Ozel M., Basekim C. A case of dyskeratosis congenita with portal hypertension associated with jugular venous anomaly. Turk J Gastroenterol. 2006;17:66–69. [PubMed] [Google Scholar]
- 6.Schenk P., Schöniger-Hekele M., Fuhrmann V., Madl C., Silberhumer G., Müller C. Prognostic significance of the hepatopulmonary syndrome in patients with cirrhosis. Gastroenterology. 2003;125:1042–1052. doi: 10.1016/s0016-5085(03)01207-1. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez-Roisin R., Krowka M.J. Hepatopulmonary syndrome—a liver-induced lung vascular disorder. N Engl J Med. 2008;358:2378–2387. doi: 10.1056/NEJMra0707185. [DOI] [PubMed] [Google Scholar]
- 8.Arguedas M.R., Abrams G.A., Krowka M.J., Fallon M.B. Prospective evaluation of outcomes and predictors of mortality in patients with hepatopulmonary syndrome undergoing liver transplantation. Hepatology. 2003;37:192–197. doi: 10.1053/jhep.2003.50023. [DOI] [PubMed] [Google Scholar]
- 9.Swanson K.L., Wiesner R.H., Krowka M.J. Natural history of hepatopulmonary syndrome: impact of liver transplantation. Hepatology. 2005;41:1122–1129. doi: 10.1002/hep.20658. [DOI] [PubMed] [Google Scholar]
- 10.Saigal S., Choudhary N., Saraf N. Excellent outcome of living donor liver transplantation in patients with hepatopulmonary syndrome: a single centre experience. Clin Transpl. 2013;27:530–534. doi: 10.1111/ctr.12126. [DOI] [PubMed] [Google Scholar]
- 11.Collisson E.A., Nourmand H., Fraiman M.H. Retrospective analysis of the results of liver transplantation for adults with severe hepatopulmonary syndrome. Liver Transpl. 2002;8:925–931. doi: 10.1053/jlts.2002.35544. [DOI] [PubMed] [Google Scholar]
- 12.Alder J.K., Chen J.J., Lancaster L. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yabe M., Yabe H., Hattori K. Fatal interstitial pulmonary disease in a patient with dyskeratosis congenita after allogenic bone marrow transplantation. Bone Marrow Transplant. 1997;19:389–392. doi: 10.1038/sj.bmt.1700674. [DOI] [PubMed] [Google Scholar]