

Abstract
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disorder. Inflammatory responses and autophagy have been implicated in the amyloid-β (Aβ) aggregation in Alzheimer’s disease. Although major evidence indicates that macro autophagy is involved in the pathogenesis of AD, its exact role is still unclear. β-asarone, a major component of Acorus tatarinowii Schott, has various neuroprotective effects. However, little is known about the protection of β-asarone against inflammation response and autophagy. In the present study, we investigated the neuroprotective effects of β-asarone on Aβ25-35 induced inflammatory responses and autophagy, and the possible mechanism. Our results showed that β-asarone attenuated inflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6 production. Meanwhile, β-asarone could significantly reduce Beclin-1, LC3B and increase anti-apoptotic protein Bcl-2 level. These results showed that β-asarone protected cells from Aβ25-35 induced inflammation and attenuated autophagy via Bcl-2/Beclin-1 pathway. Our findings suggested that β-asarone might be a potential preventive drug for AD.
Keywords: Alzheimer’s disease, β-asarone, inflammation response, autophagy
Introduction
Alzheimer’s disease (AD) is an age-related and progressive neurodegenerative disease. AD patients suffer from severe progressive cognitive dysfunction, memory impairment, behavioral symptoms and loss of independence [1]. The pathological hallmarks of AD are extracellular amyloid plaques, intra-neuronal neurofibrillary tangles (NFTs), and cerebral atrophy [2,3]. Actually, abundant Aβ generation and clearance dysfunction contribute to the amyloid plaques [4]. Aβ25-35 is a synthetic peptide of 11 amino acids that corresponds to a fragment of Aβ1-40 and Aβ1-42, which is widely used for the establishment of in vitro cell models of AD [5].
Autophagy, a ubiquitous lysosomal degradative pathway, which is essential for cell growth, survival, differentiation, development as well as protein homeostasis, and eliminates abnormal protein aggregates in neuronal cells, has been implicated in the inflammatory responses of many neurodegenerative diseases including AD [6,7]. Inflammatory responses induce autophagy and increase autophagic vacuoles [8]. Furthermore, inflammatory cytokines can up-regulate Bcl-2 expression and inhibit the level of Beclin-1 [9].
β-asarone, the major component of Acorus tatarinowii Schott, can easily pass through the blood brain barrier [10], and shows various neuroprotective effects [11]. Previous studies have indicated β-asarone protected cells from Aβ1-42 induced cytotoxicity and attenuated autophagy via activation of Akt-mTOR signaling pathway, which could be involved in neuroprotection of β-asarone against Aβ toxicity [12]. β-asarone exhibits anti-inflammatory effects by suppressing the production of pro-inflammatory mediators through NF-κB signaling and the JNK pathways in activated microglial cells [13]. However, β-asarone activities on preventing Aβ-induced inflammatory and autophagy are still not fully understood.
In this study, we treated the human neuroblastoma cell line, the SH-SY5Y cell line, with Aβ25-35 to observe the neurotoxicity and analyze the role of autophagy. The aim of this study was to investigate whether β-asarone could ameliorate Aβ25-35-induced inflammatory responses and autophagy, and was also to explore the possible mechanism on regulating the Bcl-2/Beclin-1 pathway.
Materials and methods
Reagents, chemicals and the preparation of drugs
β-asarone (cis forms of 2,4,5-trimethoxy-1-propenylbenzene) (purity ≥ 99.55%) was dissolved in dimethylsulfoxide (DMSO) to make a stock solution of 100 mM. Amyloid β-Protein Fragment 25-35 was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Aβ25-35 was dissolved in double-distilled water to 1 mM. Before use, the Aβ25-35 solution was incubated at 37°C for 7-10 days to form aggregated diffusible oligomers, and then diluted in medium. Sodium fluoride (NaF), phenylmethylsulfonyl fluoride (PMSF), protease and phosphatase inhibitor cocktails, sterile filtered dimethyl sulfoxide (DMSO) Hybri-Max, and Triton X-100, 3-MA were purchased from Sigma-Aldrich. Dulbecco’s modified Eagle’s medium (DMEM), FBS certified, 0.05% Trypsin-EDTA (1X), Phenol Red and Penicillin-Streptomycin were purchased from GIBCO. Anti-Beclin-1, anti-LC3B, and anti-Bcl-2 were all supplied from Cell signaling and secondary anti-rabbit mouse/IgG from Sigma-Aldrich.
Cell culture
Human neuroblastoma cell SH-SY5Y cells were maintained in DMEM supplemented with 10% FBS and 100 U/mL PS at 37°C in 5% CO2 humidified atmosphere. At 85% confluence, the cells were used for further experiments. Approximately 24 hours later, cells were differentiated by treatment with complete DMEM supplemented.
MTT assay
The cells were cultured at an initial density of 4 × 104 cells/ml in 96-well plates for 24 h. Cells were pre-incubated with or without β-asarone (10, 50, or 100 nM) for 2 h, following incubation with aggregated Aβ25-35 for 24 h. 10 μL/well of MTT solution (5 mg/ml) was added and cells were incubated at 37°C for 4 h. After discarding the medium, 100 μL DMSO was added to dissolve the formazan crystals for 10 min. The number of viable cells was determined at 570 nm on a microplate reader.
RNA isolation and real-time polymerase chain reaction (PCR)
Total RNA was isolated from the cells using TRIzol reagent (CWBIO, Beijing, China). First-strand cDNA was synthesized using SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Real-time PCR were performed by using SYBR®Premix Ex Taq™ kit (Takara Biotechnology, Dalian, China) in an ABI7300 real-time PCR system (Applied Biosystems, Foster City, CA, USA). Primers used were as follows: GAPDH sense, 5’-AAATGAGCCCCGCCTTCT-3’; GAPDH antisense, 5’-AGGATGTCAGCGGGAGCCGG-3’; IL-6sense, 5’-CCTTCGGTCCAGTTGCCTTCT-3’; IL-6antisense, 5’-CCAGTGCCTCTTTGCTGCTTTC-3’; IL-1βsense, 5’-GGGATGATGATGATAACCTG-3’; IL-1βantisense, 5’-TGGTCGTTGCTTGGTTCTCCT-3’; TNF-sense, 5’-GCTCTTCTGCCTGCTGCACTTTGG-3’; TNF-anti-sense, GTTGACCTTGGTCTGGTAGGAGACGG-3’. Ct (threshold cycle) data were collected using the Sequence Detection Software version. The relative quantification of gene expression was analyzed by the 2-ΔΔCt method [14]. Fold change = 2-ΔΔCt, where ΔΔCt = (Cttarget gene -CtGAPDH)-(Ctcontrol-CtGAPDH).
Western blot analysis
The cells were harvested and washed twice with ice-cold PBS buffer in 1.5 ml EP tube, centrifugedat 3000 g for 5 min. The supernatant was removed and RIPA buffer with protein inhibitor was added. The mixture was placed in ice for 30 min. Lysates were centrifuged at 12,000 g for 30 min, and the protein in the supernatant was denatured with lysis buffer for 5 min at 100°C and then subjected to SDS-PAGE gels. The gels were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, CA); the membranes were blocked with 5% nonfat milk in TBST for 1 h and incubated with a primary antibody overnight at 4°C. The primary antibodies used were rabbit anti-Beclin-1 (1:1000, CST, D40C5), rabbit anti-LC3 B (1:1000, CST, 12513S), rabbit anti-bcl-2 (1:1000, 2870 s) and mouse anti-β actin (1:5000, sigma). Bound secondary antibodies were visualized using an enhanced chemiluminescence (ECL) kit (Pierce Biotechnology) with ChemDoc XRS with Quantity One software (BioRad, Hercules, CA).
Statistical analysis
The data were analyzed using the SPSS 19.0 program software and expressed by means ± SEM. The one-way ANOVA analysis of variance was used to compare the scores of different groups. “P<0.05” was considered to indicate a statistically significant difference.
Results
Aβ25-35 inhibits the growth of SH-SY5Y cells
More evidence suggests that Aβ has neurotoxic effects both in vitro and in vivo. To determine the cytotoxicity in SH-SY5Y cells, we pretreated Aβ25-35 and examined the effect on the viability of SH-SY5Y cells by MTT assay. Cell viability was significantly decreased after exposure to 5, 10, 20 and 30 μM Aβ25-35 for 24 h, and founded IC50 was 20 μM. Next we used Aβ25-35 (20 μM) after treatment for 6, 12, 24 and 36 h (Figure 1A and 1B). Therefore, Aβ25-35 could inhibit cell growth with dose and time-dependently.
Figure 1.
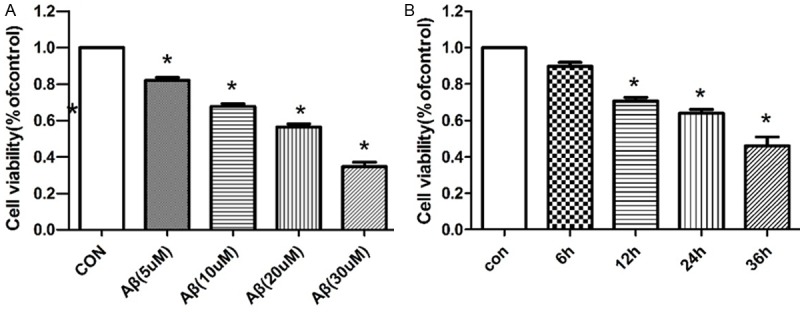
Inhibiton of Aβ25-35 against the growth of SH-SY5Y cells. Cell viability was examined by MTT assay in SH-SY5Y cells. Cells were treated with various doses of Aβ25-35 for 24 h (A) and 20 mM Aβ25-35 for various times (B) (*P<0.05).
β-asarone alleviated the toxic effect of Aβ25-35 in SH-SY5Y cells
Cell viability was increased by β-asarone, there was a significant difference between the β-asarone treated groups and Aβ treated group (P<0.05, Figure 2). Among three doses of β-asarone (10, 50, 100 μM), β-asarone concentration of 50 μM increased cell viability greatly.
Figure 2.

Increase of β-asarone on cell viability. SH-SY5Y cells were pretreated with β-asarone (10, 50, 100 μM respectively) for 2 h before treatment with Aβ25-35 for 24 h (*P<0.05).
The effects of β-asarone on expressions of LC3, Beclin-1 and Bcl-2
To explore the autophagy mechanism of β-asarone in Aβ25-35 induced SH-SY5Ycells, the cells were treated as described above. The levels of Beclin-1, LC3 and Bcl-2 in cells were determined by western blot. As shown in Figure 3, Aβ25-35 could significantly increase the levels of Beclin-1 and LC3B whereas decrease the level of antiapoptotic protein Bcl-2. Interestingly, both β-asarone (50 μM) and autophagy inhibitor 3-MA (5 mM) reversed these expressions. Pretreatment with β-asarone and autophagy inhibitor 3-MA significantly decreased Beclin-1 and LC3B expression but increased Bcl-2 expression compared with the Aβ treated group. These results demonstrated that β-asarone protected Aβ25-35 induced autophagy via Beclin-1/Bcl-2 pathway.
Figure 3.
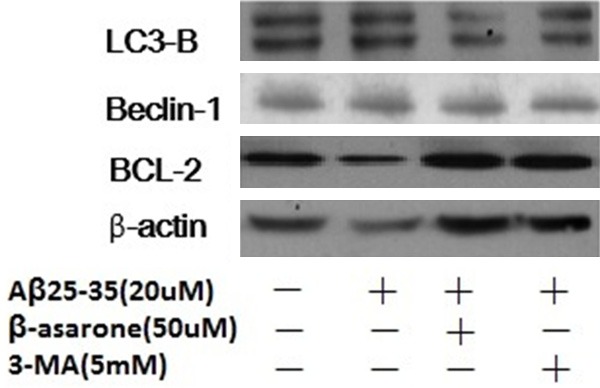
Effects of β-asarone on Aβ25-35-induced autophagy and Beclin-1, LC3 and Bcl-2 protein expressions. SH-SY5Y cells were pretreated with β-asarone (50 μM) and 3-MA (5 mM) respectively for 2 h before treatment with Aβ25-35 for 24 h. After exposure to Aβ25-35, the level of Beclin-1, LC3 and Bcl-2 were evaluated by western blot analysis.
Effect of β-asarone on production of pro-inflammatory cytokines in SH-SY5Y cells
β-asarone attenuated pro-inflammatory cytokines (IL-6, IL-1β and TNF-α) in Aβ25-35, stimulated SH-SY5Y cells as shown in Figure 4. To explore whether β-asarone represses the production of pro-inflammatory cytokines (IL-6, IL-1β and TNF-α), SH-SY5Y cells were induced with Aβ25-35 (20 μM) in the presence or absence of β-asarone at the dose of 50 μM. RT-PCR analysis showed that the mRNA levels of these cytokines were prevented by β-asarone. These results indicated that β-asarone inhibited the production of pro-inflammatory cytokines, especially IL-1β and TNF-α (*P<0.05).
Figure 4.
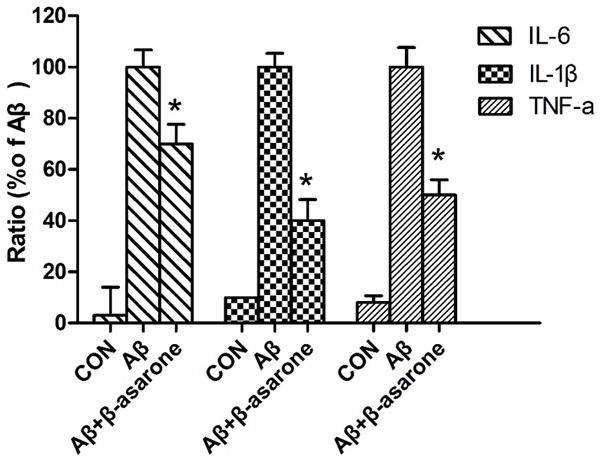
Inhibitory effect of β-asarone on the production of pro-inflammatory cytokines. The cells were treated as described above. After 24 h, the levels of TNF-α, IL-1β and IL-6 mRNA levels were determined by RT-PCR. The results are expressed as the ratio of IL-6/GAPDH, IL-1β/GAPDH and TNF-α/GAPDH for three independent experiments. *P<0.05 compared with the Aβ-treated group by One-way ANOVA.
Discussion
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disorder. Although multiple drugs have now been approved, the effective means for its treatment are lack. β-asarone is the most important and efficient component of A. tatarinowii Schott [15], which can pass through the BBB quickly and distribute in the brain stem, hippocampus, cortex and cerebellum widely [10,16]. It can inhibit neuronal apoptosis via the CaMKII/CREB/Bcl-2 signaling pathway in in vitro models and in APP/PS1 mice [17]. More evidence suggests that β-asarone protects cells from Aβ1-42 induced cytotoxicity and attenuats autophagy via activation of Akt-mTOR signaling pathway [12], and protects against beta-amyloid-induced neurotoxicity in PC12 cells via JNK signaling and modulation of Bcl-2 family proteins [18]. Previous studies have shown that β-asarone protects PC12 cells against OGD/R-induced injury partly due to attenuating Beclin-1-dependent autophagy caused by decreasing (Ca2+) and increasing MMP [19]. β-asarone treatment may be a promising therapeutic tool for AD and these results also suggest that β-asarone could attenuate autophagy.
Autophagy is a lysosome degradation process that turns over cytoplasmic materials and helps the cell to maintain homeostasis. It maintained at low levels under normal conditions but can be augmented rapidly as a cytoprotective response when cells suffer oxidative stress, infection, or protein aggregate accumulation [20]. Autophagosomes accumulate abnormally in the brain of AD. Several neurodegenerative disorders including AD inflammation response and autophagy have been found to play an important role in recycling cellular waste and eliminating potentially toxic damaged organelles and protein aggregates [21]. Alterations of autophagy and excessive inflammatory response are two hallmarks to AD [22]. Evidence shows that inflammation-stimulated autophagy or autophagy-triggered inflammatory responses lead to neurodegenerative disorders, such as Aβ accumulation. Inflammatory response is critical for immune system responding to stimuli by producing a variety of pro-inflammatory cytokines, including TNF-α, IL-1β and IL-6 [23,24]. In this study, β-asarone significantly inhibited Aβ induced production of TNF-α, IL-6 and IL-1β in SH-SY5Y cells, indicating that β-asarone might be beneficial for delaying the progression of inflammatory conditions.
To investigate the possible mechanism by β-asarone pretreatment that alleviated the cytotoxity of Aβ25-35 in SH-SY5Y cells, we explored the effect of β-asarone on Aβ-induced autophagy. Aβ25-35 induced the expression of LC3B and beclin-1 levels, which suggested that β-asarone could inhibit Ab-induced autophagy in SH-SY5Y cells. It has been shown that Bcl-2 can regulate autophagy by binding to Beclin-1 and thereby can inhibit the association between Beclin-1 and hVps34, which leads to autophagy and inflammatory responses [9]. Beclin-1 is essential for autophagy, which indirectly implicates an important role in numerous biological cellular processes, including aging and cell death [25]. In our previous study, downregulation of Beclin-1, LC3B and up regulation of BCL-2 by β-asarone might inhibit Aβ induced autophagy and contribute to its protective effect against Aβ related cytotoxicity.
In conclusion, we showed that Aβ25-35 inhibited SH-SY5Y cell growth and induced autophagy. Furthermore, β-asarone could provide neuroprotection against Aβ, which might be related to its inhibition autophagy. In conclusion, we found that β-asarone could inhibit Beclin-1 and LC3B expressions; meanwhile, it also could enhance Bcl-2 expression. Collectively, these findings indicated that the β-asarone treatment could attenuate the neurons damage induced by Aβ via suppressing the activity of autophagy and it might be explored as a potential therapeutic agent in AD treatment.
Disclosure of conflict of interest
None.
References
- 1.Forstl H, Kurz A. Clinical features of Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 1999;249:288–90. doi: 10.1007/s004060050101. [DOI] [PubMed] [Google Scholar]
- 2.Giaccone G, Pedrotti B, Migheli A, Verga L, Perez J, Racagni G, Smith MA, Perry G, De Gioia L, Selvaggini C, Salmona M, Ghiso J, Frangione B, Islam K, Bugiani O, Tagliavini F. Beta PP and Tau interaction. A possible link between amyloid and neurofibrillary tangles in Alzheimer’s disease. Am J Pathol. 1996;148:79–87. [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M, Sisodia SS, Price DL. Neurofibrillary tangles and beta-amyloid deposits in Alzheimer’s disease. Curr Opin Neurobiol. 1991;1:441–7. doi: 10.1016/0959-4388(91)90067-h. [DOI] [PubMed] [Google Scholar]
- 4.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaminsky YG, Marlatt MW, Smith MA, Kosenko EA. Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: evidence for Abeta (25-35) Exp Neurol. 2010;221:26–37. doi: 10.1016/j.expneurol.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013;36:7–16. doi: 10.1007/s10059-013-0140-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol. 2012;226:255–73. doi: 10.1002/path.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen GY, Yang HJ, Lu CH, Chao YC, Hwang SM, Chen CL, Lo KW, Sung LY, Luo WY, Tuan HY, Hu YC. Simultaneous induction of autophagy and toll-like receptor signaling pathways by graphene oxide. Biomaterials. 2012;33:6559–69. doi: 10.1016/j.biomaterials.2012.05.064. [DOI] [PubMed] [Google Scholar]
- 9.Zhaorigetu S, Yang Z, Toma I, McCaffrey TA, Hu CA. Apolipoprotein L6, induced in atherosclerotic lesions, promotes apoptosis and blocks Beclin 1-dependent autophagy in atherosclerotic cells. J Bio Chem. 2011;286:27389–98. doi: 10.1074/jbc.M110.210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang YQ, Shi C, Liu L, Fang RM. Pharmacokinetics of beta-asarone in rabbit blood, hippocampus, cortex, brain stem, thalamus and cerebellum. Die Pharmazie. 2012;67:120–3. [PubMed] [Google Scholar]
- 11.Fang YQ, Fang RM, Fang GL, Jiang Y, Fu SY. Effects of beta-asarone on expression of c-fos in kindling epilepsy rat brain. Zhongguo Zhong Yao Za Zhi. 2008;33:534–6. [PubMed] [Google Scholar]
- 12.Xue Z, Guo Y, Zhang S, Huang L, He Y, Fang R, Fang Y. Beta-asarone attenuates amyloid betainduced autophagy via Akt/mTOR pathway in PC12 cells. Eur J Pharmacol. 2014;741:195–204. doi: 10.1016/j.ejphar.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Lim HW, Kumar H, Kim BW, More SV, Kim IW, Park JI, Park SY, Kim SK, Choi DK. β-Asarone (cis-2,4,5-trimethoxy-1-allyl phenyl), attenuates pro-inflammatory mediators by inhibiting NF-kappaB signaling and the JNK pathway in LPS activated BV-2 microglia cells. Food Chem Toxicol. 2014;72:265–72. doi: 10.1016/j.fct.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Wang JH, Wu QD, Bouchier-Hayes D, Redmond HP. Hypoxia upregulates Bcl-2 expression and suppresses interferon-gamma induced antiangiogenic activity in human tumor derived endothelial cells. Cancer. 2002;94:2745–55. doi: 10.1002/cncr.10520. [DOI] [PubMed] [Google Scholar]
- 16.Fang YQ, Shi C, Liu L, Fang RM. Analysis of transformation and excretion of beta-asarone in rabbits with GC-MS. Eur J Drug Metab Pharmacokinet. 2012;37:187–90. doi: 10.1007/s13318-012-0083-z. [DOI] [PubMed] [Google Scholar]
- 17.Wei G, Chen YB, Chen DF, Lai XP, Liu DH, Deng RD, Zhou JH, Zhang SX, Li YW, Lii H, Liu LF, Wang Q, Nie H. β-Asarone inhibits neuronal apoptosis via the CaMKII/CREB/Bcl-2 signaling pathway in an in vitro model and AbetaPP/PS1 mice. J Alzheimers Dis. 2013;33:863–80. doi: 10.3233/JAD-2012-120865. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Xing G, Dong M, Zhou L, Li J, Wang G, Zou D, Wang R, Liu J, Niu Y. Beta-asarone protection against beta-amyloid-induced neurotoxicity in PC12 cells via JNK signaling and modulation of Bcl-2 family proteins. Eur J Pharmacol. 2010;635:96–102. doi: 10.1016/j.ejphar.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Mo ZT, Fang YQ, He YP, Zhang S. β-Asarone protects PC12 cells against OGD/R-induced injury via attenuating Beclin-1-dependent autophagy. Acta Pharmacol Sin. 2012;33:737–42. doi: 10.1038/aps.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glick D, Barth S, Macleod KF. Macleod, Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Orenstein RA. Nixon, Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37:1949–61. doi: 10.1111/ejn.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC, Tan L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging. 2014;35:941–57. doi: 10.1016/j.neurobiolaging.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Kavanaugh A. Interleukin-6 inhibition and clinical efficacy in rheumatoid arthritis treatmentdata from randomized clinical trials. Bull NYU Hosp Jt Dis. 2007;65(Suppl 1):S16–20. [PubMed] [Google Scholar]
- 24.Lee C, Lim HK, Sakong J, Lee YS, Kim JR, Baek SH. Janus kinase-signal transducer and activator of transcription mediates phosphatidic acid-induced interleukin (IL)-1 beta and IL-6 production. Mol Pharmacol. 2006;69:1041–7. doi: 10.1124/mol.105.018481. [DOI] [PubMed] [Google Scholar]
- 25.Huang L, Deng M, He Y, Fang Y. β-asarone and levodopa coadministration protects against 6-OHDA-induced damage in parkinsonian rat mesencephalon by regulting autophagy: downexpression Beclin-1 and LC3B and upexpression p62. Clin Exp Pharmacol Physiol. 2015;42:269–77. doi: 10.1111/1440-1681.12344. [DOI] [PubMed] [Google Scholar]