

Abstract
We report a PS1 gene mutation (Val 97Leu) in a Chinese familial Alzheimer’s disease (FAD) pedigree and a cell model of FAD built by transfecting PS1 v97L mutants into human neuroblastoma SH-SY5Y cells. To test our hypothesis that the PS1 v97L mutation is pathogenic, we investigated possible alterations in transport regulation and intracellular Ca2+ homeostasis in endoplasmic reticulum (ER). Grp78 is an ER-resident chaperone mediating the unfolded protein response (UPR) and is a key regulator of ER stress transducers. KDEL is a 4-amino-acid retention sequence made of Lys-Asp-Glu-Leu-COO. KDEL is a “resident” sequence as protein residence in ER is consistently associated with KDEL at the C-extremity. Our group used KDEL recognizing anti-Grp78 monoclonal antibody to detect the level of Grp78. We found increased KDEL level in all the transfected cells including cells transfected with PS1 V97L genes, wild-type and the mock. However cells with PS1 V97L mutation expressed a relatively lower KDEL compared with the wild-type and the mock, and a significantly lower Grp78 level compared with the wild-type, the mock and control. These results suggest that PS1 V97L mutation impedes intracellular transport regulation in ER. PS1 V97L mutation mediates increased ER Ca2+ content in human neuroblastoma SH-SY5Y cells. The increased intracellular Ca2+ release is due to depleted Ca2+ storing content of ER but not due to extracellular environment as capacitative Ca2+ entry (CCE) is invariant. PS1 V97L mutation interferes with intracellular Ca2+ homeostasis. Abnormal transport regulation and Ca2+ homeostasis attributed to PS1 V97L mutation may be associated with the pathology of Chinese familial FAD.
Keywords: Familial alzheimer’s disease (FAD), presenilin1 (PS1) genes, gene mutation; ER stress, Ca2+ homeostasis
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder which is characterized by β-amyloid (Aβ) peptides deposition and formation of neurofibrillary tangles in the brain [1]. Familial AD (FAD) tends to be early-onset with a relatively adverse prognosis. Presenilin-1 (PS1) genes have been found in FAD [2]. However, the mechanisms underlying the pathogenic mutations are still unclear, especially in Chinese. At the cellular level, different PS1 mutations have been consistently found to selectively increase the production and deposition of longer and more fibrillogenic form of the Aβ42 peptide [3-5]. Our group had described a novel PS1 gene mutation (Val 97 Leu) associated with early-onset FAD in a Chinese family [6]. This mutation is located in the first transmembrane (TM1) domain of PS1, which was considered as one of the critical functional or conformational domains in PS1 according to the model of three-dimensional structure of the PS1 protein [6]. PS1 v97L mutation may alter the preferred cleavage site for γ-secretase to generate more Aβ42, which is a putative neurotoxin precipitating AD [7]. PS1 v97L mutation may play an important role in impeding the transport regulation in ER and lead to Aβ42 deposition. KDEL is a 4-amino-acid sequence made of Lys-Asp-Glu-Leu-COO. KDEL is a “resident” sequence as protein residence in ER is consistently found to have KDEL at the C-extremity. Grp78 is one of ER-resident chaperones that facilitate protein folding [8]. Grp78 binding assists in the folding of newly synthesized proteins and prevents protein aggregation and maintains the proteins in an inactive state. To test our hypothesis that the PS1 v97L mutation resulted in FAD, we constructed a cell model of FAD by transfecting PS1 v97L mutants into human neuroblastoma SH-SY5Y cells and investigated possible alterations in KDEL and Grp78 and intracellular Ca2+ homeostasis in ER.
Materials and methods
The Chinese pedigree of FAD
We have reported a Chinese pedigree of FAD-associated PS1 V97L mutation [6,7]. The family consisted of 23 members in three successive generations (13 men, 10 women). The family members were all of Han nationality aged 36.5 ± 15.9 years. Three of them were diagnosed with Alzheimer’s disease. One suspected woman (I1) died of pneumonia 12 years ago. The proband (II1) was a 68-year-old woman who first had memory and counting difficulties, with a noticed irritability, at the age of 58. Patient 2 (III3) was a 51-year-old man whose memory disturbance, dyscalculia, apathy and personality change were first noticed at age 47. Patient 3 (III5) was a 49-year-old woman who had similar symptom onset at age 46. The three patients had an abnormal mobility pattern of the PS1 gene exon 4 in PCR-SSCP analysis. Consistent with the PCR-SSCP results, DHPLC analysis of exon 4 showed dual peaks in these three AD patients, with DNA sequencing revealing a missense mutation (G→T) at position 289 in exon 4 of the PS1 gene, leading to a Val 97 Leu substitution, while no mutations were found in any other family members and controls.
Cell culture
Human neuroblastoma SH-SY5Y cells were cultured in a mixture of Eagle’s minimum essential medium (GibcoBRL, USA) with non-essential amino acids and Ham’s F12 medium (GibcoBRL, USA) (1:1mixture), supplemented with 10% (v/v) heat-inactivated fetal bovine serum (GibcoBRL, USA). Cells were incubated at 37°C in a humidified incubator gassed with 95% air and 5% CO2, passaged every 7 days and used for up to 20 passages.
Plasmid preparation and transfection
pcDNA3.1/PS1wt (a full length human PS1wt cDNA had been subcloned into the EcoRI and XhoI sites of pcDNA3.1) was a generous gift of the Department of Neurology, Harvard Medical School. Mutant human PS1 (PS1 V97L) cDNA was generated using site-directed mutagenesis as described [8] and sequenced using T7 and BGH general primers subsequently to confirm cDNA integration. Plasmid DNA was isolated from transformed E.coli DH5α cells using QIAGEN columns (QIAGen, Germany). Plasmid DNA was diluted to the final concentration of 2 µg/µl. Vectors containing the cDNA encoding either human PS1 wt or PS1 V97L mutation and pcDNA void vector were successfully introduced into SH-SY5Y cells using lipofectamineTM 2000 (Invitrogen, USA) according to the manufacturer’s procedures. Stably transfected SH-SY5Y cells, expressing the different PS1 constructs, were maintained with the antibiotic Zeocin at a final concentration of 400 μg/mL. The PS1 expression in these cell lines was confirmed by sequencing and checked by immunoblotting using a PS1 N-terminal antibody. Sequencing of the RT-PCR products revealed a missense mutation (G→T) at position 289 in exon 4 in cells transfected with PS1 V97L, proving that the mt PS1 gene transfection was successful at the mRNA level.
RT-PCR
Total RNA from cultures was isolated using the Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions from different cell lines, respectively. First strand cDNA was made using SuperScriptTM First-Strand synthesis kit (Promega, USA) according to the manufacturer’s procedures. Aliquots of 1.5 µl of the final reaction product were used as templates in a standard PCR reaction of 35 cycles (30 s at 94°C, 50 s at 49°C, 60 s at 72°C) with the primer pairs 5’-ACA GTT GCT CCA ATG ACA GA-3’ and 5’-CCC CAA GTA AAT GAA TGA AA-3’ located upstream and downstream of the exon4 and exon5 of PS1, respectively, including the mutant site. The PCR products were analyzed on a 1.5% agarose gel and DNA sequenced (performed by Bioengineering Co, Shanghai, China) to identify the integration of mutant PS1 gene into the genome. To test for differences in PS1 mRNA expression among samples, mRNA level of β-actin as the endo-control was also determined by RT-PCR. The β-actin primers were: 5’CATCTCTTGCTCGAAGTCCA3’ and 5’ATCATGTTTGAGACCTTCAACA3’.
Immunofluorescence
Cells were fixed with 0.1% Triton X-100 for 10 min, washed three times using phosphate-bufered saline (PBS) and sealed with serum (1:10) for 30 min. The cells were incubated with mouse anti-KDEL monoclonal antibody (StressGen Biotechnologies, Canada 1:40) for 6 h at 4°C, and washed with PBS. Cells were stained with FITC-conjugated anti-mouse IgG (Zhongshan Golden Bridge Bio, China 1:64) for 40 min at 25°C. The stained cells were observed using a confocal microscope.
Immunoblot analysis
The Grp78 expression was analyzed by Western blotting. To obtain cell extracts, cells were washed twice with ice-cold phosphate-buffered saline (PBS), harvested with a cell-scraper, and collected by 3000 rpm centrifugation for 3 min. Pellets were resuspended in 100 μL of RIPA lysis buffer (10 mmol Tris-HCl pH 8.0 10 mmol NaCl 0.1% SDS, 0.5% NP-40, 1 mmol PMSF, 10 mg/mL aprotinin 1 mmol EDTA, and 0.1% sodium deoxycholate, 0.1% SDS). After lysis on the ice for 30 min, the cell lysates were cleared by centrifugation at 14,000 rpm for 30 min at 4°C. Samples were stored at -20°C until use. Protein amounts in cell extracts were quantified using the protein assay kit (Pierce, Rockford, USA). Proteins were loaded 25 µg per lane onto a 10% Tris-glycine SDS-PAGE gel, transferred onto PVDF membranes (Millipore, USA), and incubated with presenilin-1 monoclonal antibody B-6 (SANTA CRUZ, USA) (1:500), mouse anti-Grp78 antibody (SPA-827, StressGen Biotechnologies, Canada) (1:1000) and mouse anti-actin monoclonal antibody C-2 (SANTA CRUZ, USA) (1:500), followed by incubation with horseradish peroxidase (HRP)-coupled goat anti-mouse IgG (Zhongshan Golden Bridge Bio, China) (1:2000). Antibody binding was visualized using the ECL Western Blotting detection reagent (Amersham Biosciences Ltd, Hongkong, China), captured on film and analyzed by Glyko bandscan 4.30 software (Microsoftware Inc. USA).
Intracellular calcium measurements
Four groups were measured respectively (Control: untransfected cells; Mock: cells transfected with void vector; WT: cells transfected with PS1 wt; MT: cells transfected with PS1 V97L). Cytosolic Ca2+ concentration ([Ca2+] i) was monitored with the Ca2+ indicator Fluo-3. Maintaining the cells in a Ca2+-free medium, the Ca2+ store depletion was first induced by adding the SERCA inhibitor thapsigargin (Tg, 1.0 μM)). Capacitative Ca2+ entry (CCE) was subsequently monitored upon CaCl2 addition (Ca2+, 1 mM). The SERCA inhibitor induced the passive release of Ca2+ from intracellular stores, leading to a transient increase in [Ca2+] i. After 5-7 min, addition of CaCl2, in the continuous presence of Tg, induced a second large [Ca2+] i peak followed by a sustained [Ca2+] i plateau due to Ca2+ influx across the plasma membrane. Cells were loaded with serum-free medium containing 5 μM Fluo-3 AM ester, 0.5% (v/v) PluronicF-127 at room temperature for about 30 min. Basal [Ca2+] I was measured by removing the medium and adding Ca2+-/Mg2+-free PBS. Basal [Ca2+] i was measured repeatedly for 10 min at 37°C until a steady state of [Ca2+] i was reached. The sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor thapsigargin (Tg, 1.0 μM) was added to PBS at the indicated concentrations at 37°C for 10 min when basal [Ca2+] i was measured. Fluorescence was measured using laser confocal microscopy (Olympus FV500, Japan), at the excitation and emission wavelengths of 485 and 530 nm, respectively, using slit widths of 20 nm. Apparent [Ca2+] i corresponding to fluorescent value F was calculated using the formula [Ca2+] i = Kd (F-Fmin)/(Fmax-F), where Kd for Fluo-3-Ca2+ was taken as 390 nm, as indicated in the protocol in the manufacturer’s manual (Molecular Probes, USA). Fmin was determined by measuring the signal of the unloaded cells, and was defined by addition of a 100 mM CaCl2 buffer containing 1% (v/v) Triton X-100 to the cells.
Statistical analysis
Analyses of inter-group differences were carried out by one-way analysis of variance (ANOVA), followed by Tukey-Kramer multiple comparison procedure. A value of P < 0.05 was considered statistically significant.
Results
PS1 levels in PS1wt and PS1 V97L cells
High levels of PS1 expression were detected in PS1wt and PS1 V97L stably transfected SH-SY5Y neuroblastoma cell lines. The PS1 expression in these cell lines was confirmed by sequencing and checked by immunoblotting using a PS1 N-terminal antibody. Sequencing of the RT-PCR products revealed a missense mutation (G→T) at position 289 in exon 4 in cells transfected with PS1 V97L, proving that the mt PS1 gene transfection was successful at the mRNA level. Untransfected cells and cells transfected with void vector were found to express very low levels of PS1 holoprotein, most of PS1 identified as a 28 kDa band corresponding to endogenous PS1 N-terminal fragment (NTF). PS1-WT cells showed accumulation of full-length PS1 (48 kDa) and as well as of NTF. Both WT and mutant clones showed similar PS1 levels following transfection.
KDEL in transfected cells Figure 1 illustrates the immunofluorescence of KDEL in the four groups. According to the immunofluorescence values, the level of KDEL increased in all the transfected groups. However, cells with PS1 V97L mutants (MT) expressed a relatively lower KDEL compared with the wild-type (WT) and the mock while WT was not statistically different compared with the control (Figure 1). That was detected 24 h after transfection. (*P < 0.001 Mock vs. Control, MT vs. Control, WT vs Control, and MT vs. WT).
Figure 1.
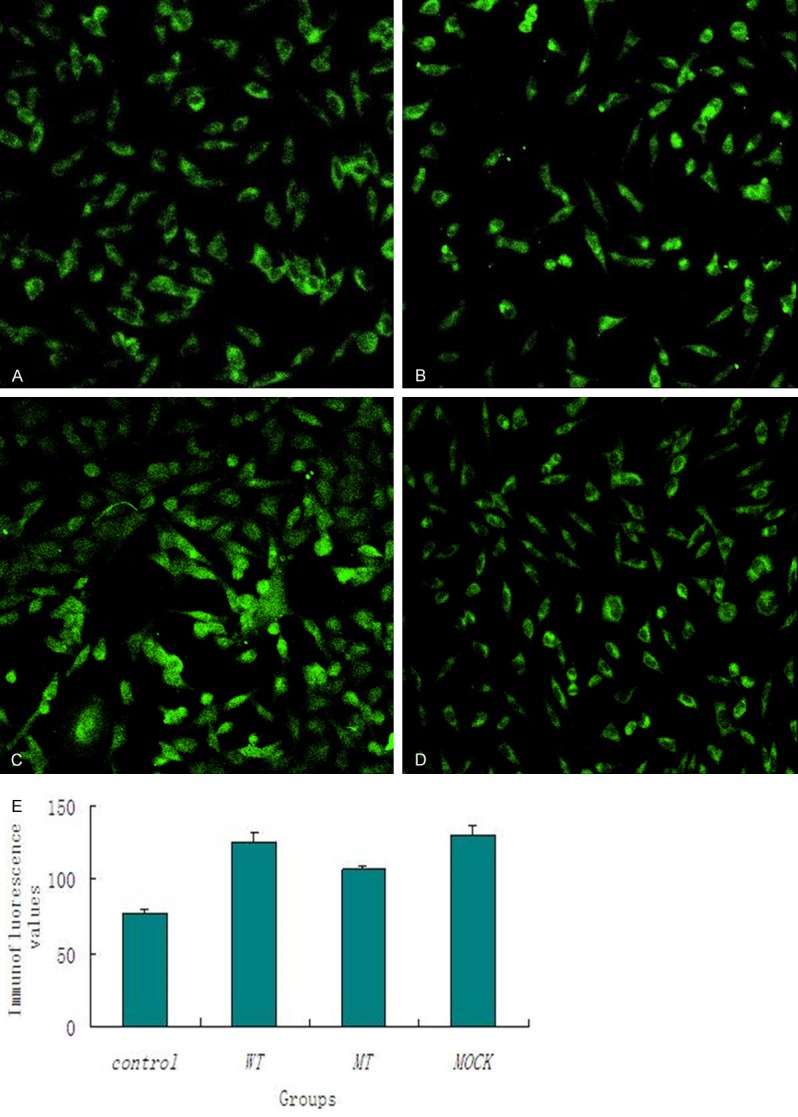
Immunofluorescence analysis of KDEL in MT (A), control (B), WT (C) and Mock (D) (200 ×). KDEL in SH-SY5Y cells according to the immunofluorescence values (E). (*P < 0.001 Mock vs. Control, MT vs. Control, WT vs. Control, and MT vs. WT).
Grp78 expression
Cells with PS1 mutants (MT) expressed a significantly lower Grp78 level compared with the wild-type, the mock and control. Figure 2 illustrates the Western blot of Grp78 expression. (Mean gray value ± S.D. 82.5281 ± 1.615 vs. 214.3156 ± 1.477, P < 0.001, n = 3 MT vs. WT; 82.5281 ± 1.615 vs. 216.8571 ± 0.5791, P < 0.001, n = 3 MT vs. mock; 82.5281 ± 1.615 vs. 208.5176 ± 1.317, P < 0.001, n = 3 MT vs. control). However, WT and Mock showed increased expression of Grp78 compared with control. (Mean gray value ± S.D. 214.3156 ± 1.477 vs. 208.5176 ± 1.317, P < 0.01, n = 3 WT vs. control; 216.8571 ± 0.5791 vs. 208.5176 ± 1.317, P < 0.001, n = 3 mock vs. control).
Figure 2.
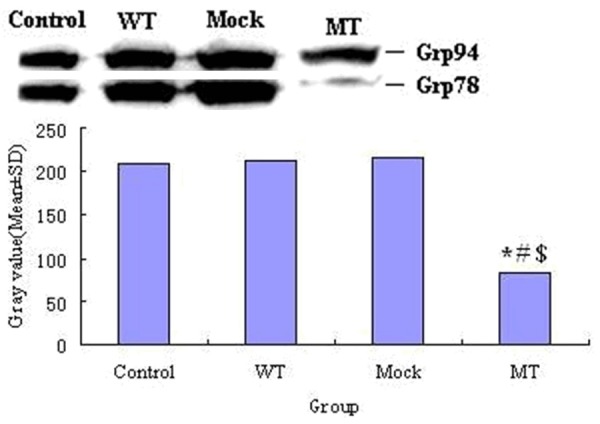
Western blot of Grp78. MT expressed a significantly lower Grp78 level compared with WT, Mock and control.
Increased Ca2+ release from intracellular stores and normal CCE in SH-SY5Y cells expressing PS1 V97L mutation
The first large [Ca2+] i peak appeared when Tg was used in a Ca2+-free medium, a fast rise in [Ca2+] i, due to prompt and complete discharge of intracellular stores, followed by a rapid return to the pre-stimulatory level. In the continuous presence of Tg, activation of CCE was detected as a second large [Ca2+] i peak, upon CaCl2 addition (Ca2+, 1 mM). Intracellular Ca2+ release was increased in MT groups, compared with WT, mock and control. The average peak rise in amplitude for intracellular Ca2+ release as well as discharge of intracellular stores in MT was much higher than the other three groups, as shown in Figure 3. ([Ca2+] i peak rise amplitudes (n M), MT, 30.09 ± 1.11; WT, 23.73 ± 1.32; Mock, 21.88 ± 0.92; control, 21.82 ± 0.58; n = 20, MT vs. WT, Mock, control $*#P < 0.001). CCE of each group was not statistically different.
Figure 3.
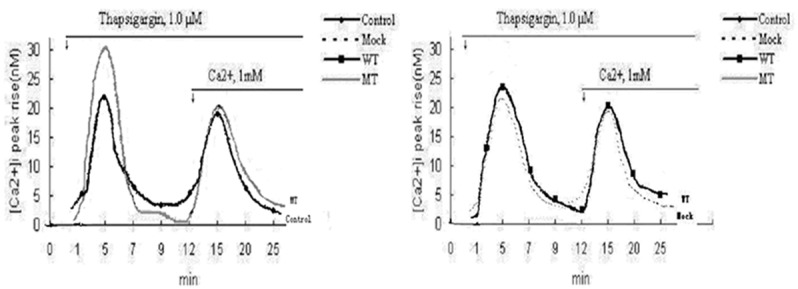
Average peak amplitudes of intracellular Ca2+ release in MT higher than in other three groups. (Control: untransfected cells; Mock: cells transfected with void vector; WT: cells transfected with PS1 wt; MT: cells transfected with PS1 V97L).
Discussion
Endoplasmic reticulum(ER) regulates protein synthesis, folding and trafficking, cellular responses to stress, and intracellular Ca2+ levels [9]. Mutant protein expression, abnormal Ca2+ homeostasis, and other cellular stresses cause accumulation of unfolded or misfolded proteins in ER lumen, resulting in ER stress [9]. ER stress can occur under various physiological settings that have significant implications for health and disease, including AD [10]. Normal cells respond to ER stress by increasing transcription of genes encoding ER-resident chaperones such as Grp78, Grp94 and protein disulphide isomerase (PDI) to facilitate protein folding [11]. ER stress elicits an adaptive response triggering a signaling cascade called unfolded protein response (UPR), which alleviates stress by induction of ER-localized chaperones, initiation of degradation and attenuation of protein synthesis. A master regulator (Grp78) and three classes of UPR transducers (IRE1, PERK and ATF6) have been identified in this process [12]. Grp78 is the ER-located Hsp70 analogue. It binds transiently to a variety of newly synthesized proteins and more persistently to some misfolded proteins. Further, Grp78 is known to assist folding and assembly of newly synthesized proteins by recognition and binding of hydrophobic stretches of unfolded proteins. Binding of Grp78 prevents protein aggregation and maintains the proteins in a folded and oligomerization-competent state [12]. In non-stressed cells, Grp78 binds to all three transducers mentioned above, which are maintained in an inactive state. Under ER stress, all three sensors are released from Grp78 and activated. Thus, Grp78 is a key regulator of ER stress transducers, which are released during ER stress. Therefore, Grp78 induction is a general indicator of ER stress and initiation of UPR [11,12].
KDEL is a four-amino acid peptide sequence (Lys-Asp-Glu-Leu-COO) occurring at the carboxyl terminus of the protein. KDEL plays an important role in processing precursor protein and intracellular membrane transport. Protein residence in ER is consistently found to have KDEL at the C-terminus, which recognizes and binds to KDEL-receptor in the endomembrane of ER and Golgi complex [13]. Protein without KDEL turns into secretory protein, which is transported through the membrane of organelle. KDEL-containing protein is resident, and contributes to the accumulation of unfolded or misfolded proteins in the ER lumen, resulting in ER stress. KDEL plays an important role in intracellular protein transport regulation and is a retention indicator of UPR signaling pathway [14]. Thus, using anti-Grp78 monoclonal antibody against KDEL to detect Grp78 levels, the UPR signaling pathway and the protein transport regulation under ER stress can be monitored.
Based on the FAD-linked PS1 gene mutation in a Chinese FAD pedigree, we developed a cell model of FAD by transfecting PS1 v97L mutants into human neuroblastoma SH-SY5Y cells in vitro. Our group had reported that PS1 v97L mutation may generate Aβ42, which is a putative neurotoxin resulting in AD [7]. We investigated alterations in KDEL and Grp78 level in the FAD cell model to investigated FAD under ER stress.
The results showed that Grp78 was significantly reduced compared with WT, mock and control. Therefore, Grp78 expression in WT and Mock increased compared with control. On that we can say ER stress was successfully induced by the transfection as Grp78 was an important indicator of ER. However, ER stress was significantly impeded in cells expressing PS1 V97L (MT). One of the mechanisms may be associated with blocking the protein transport in UPR signaling pathway. Immunofluorescence data suggest increased KDEL levels in all the transfected groups. However, cells with PS1 V97L mutation (MT) expressed a relatively lower KDEL compared with the wild-type (WT) and the mock while WT was not statistically different compared with the control. As a retention indicator of UPR, increasing KDEL suggests abnormal protein processing. Our findings indicated that PS1 V97L mutation may perturb the ER stress response, and the decreased induction of Grp78 in PS1 mutant cells resulted from attenuation of the UPR signaling pathway.
Our results suggests that PS1 mutation plays a role in signaling of UPR, while wild-type PS1 does not, suggesting that PS1 mutations perturb the ER stress response through a gain-of-function. Perturbed ER stress response may be mediated by the inhibition of activation of ER stress master regulator Grp78 and interfering with the protein transport in UPR signaling pathway. Interfering with the protective systems to ER stress in PS1 mutation-bearing cells may lead to susceptibility to various cellular stresses [15].
Ca2+ homeostasis plays an important role in ER function and membrane transport, as well as the balance of internal environment. The intracellular Ca2+ level is mediated by the release of Ca2+ stores in ER and CCE through the cell membrane. Abnormal intracellular Ca2+ transference under various physiological settings may have significant implications in health and disease, including AD [10]. However, Ca2+ storage capacity in ER in different cell models may be increased [16,17], decreased [18,19], or almost normal [20], as reported. The intracellular Ca2+ storing in ER significantly increased in rats transfected with PS1 A246E and PS1 M146V while neurons were more susceptible to stress [21]. Neurons of rats transfected with PS1 A246E showed increased CCE while fibroblast with PS1 M146V showed stable CCE [22]. Nearly all reports suggest that PS1 mutation results in increased neuronal apoptosis and vulnerable to excitotoxicity.
To investigate the Ca2+ content of intracellular stores, cells were challenged with Tg (1 μM). In a Ca2+-free medium, Tg induced a rapid rise in [Ca2+] i, due to prompt and complete discharge of intracellular stores in ER, followed by a rapid return to the pre-stimulatory level. The rapid Ca2+ response is typical of agonists coupled to phospholipase C activation and has been described in a large variety of cell types [23,24]. In the continuous presence of Tg, activation of CCE was detected as a second large [Ca2+] i peak, upon CaCl2 addition (Ca2+, 1 mM). Depletion of intracellular stores increased the [Ca2+] i level in the MT groups, which was significantly higher than in the other three groups. Therefore, the PS1 V97L mutation appears to be linked to an increased Ca2+ content of intracellular stores. However, no difference was found in the WT, mock and control. This phenomenon excludes possible unspecific effects of exogenous PS1 expression on this Ca2+ pathway. No significant difference was found in CCE among the four SH-SY5Y clones, suggesting that the increased intracellular Ca2+ release was due to depletion in ER Ca2+ storing capacity and not the extracellular environment as CCE was normal. PS1 V97L mutation mediates the increase in ER Ca2+ content in human neuroblastoma SH-SY5Y cells. In our experiment, both wide-type and mutant clones showed similar PS1 levels following transfection, indicating that PS1 V97L mutation but not PS1 over-expression affected Ca2+ homeostasis of transfected SH-SY5Y cells. The result was consistent with recent studies reporting that abnormal cellular Ca2+ signaling is common in FAD pathogenesis [25].
Abnormal Ca2+ homeostasis could be mediated by the following two pathways. First, the PS1 V97L mutation affected or altered the protein structure of Ca2+ channel in the ER membrane, leading to altered intracellular Ca2+ homeostasis. Second, Aβ has also been shown to trigger Ca2+ release from the ER and induce ER stress and neurotoxicity [26].
Our results suggest that the mutant PS1 V97L but not wild-type played a role in altered Ca2+ homeostasis, suggesting that PS1 V97L mutation altered susceptibility to various cellular stresses. The PS1 V97L mutation affects the intracellular Ca2+ content as well as the ER function but not by extracellular Ca2+ influx. In conclusion, our findings suggest that interference with intracellular transport regulation and abnormal Ca2+ homeostasis due to PS1 V97L mutation may result in FAD. PS1 V97L mutation exerts a primary effect on ER function. The direct role of PS1 and other proteins in ER stress and Ca2+ regulation remains to be established and requires further investigation.
Acknowledgements
We acknowledge the support and help from the Department of Neurology, Harvard Medical School.
Disclosure of conflict of interest
None.
References
- 1.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–70. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 2.Yasuda Y, Kudo T, Katayama T, Imaizumi K, Yatera M, Okochi M, Yamamori H, Matsumoto N, Kida T, Fukumori A, Okumura M, Tohyama M, Takeda M. FAD-linked presenilin-1 mutants impede translation regulation under ER stress. Biochem Biophys Res Commun. 2002;296:313–8. doi: 10.1016/s0006-291x(02)00859-8. [DOI] [PubMed] [Google Scholar]
- 3.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–13. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 4.Citron M, Westaway D, Xia W, Carlson G, Diehl T. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 5.Lleo A, Berezovska O, Growdon JH, Hyman BT. Clinical, pathological, and biochemical spectrum of Alzheimer disease associated with PS-1 mutations. Am J Geriatr Psychiatry. 2004;12:146–56. doi: 10.1097/00019442-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Jia JP, Xu E, Shao YK, Jia JM, Sun YX. One novel presenilin-1 gene mutation in a Chinese pedigree of familial Alzheimer’s disease. J Alzheimers Dis. 2005:119–24. doi: 10.3233/jad-2005-7204. [DOI] [PubMed] [Google Scholar]
- 7.Fang B, Jia L, Jia J. Presenilin-1 V97L mutation enhanced intracellular and extracellular Aβ42 level in SH-SY5Y neuroblastoma cells. Neurosci Lett. 2006;406:33–7. doi: 10.1016/j.neulet.2006.06.072. [DOI] [PubMed] [Google Scholar]
- 8.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–92. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–80. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 10.Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer’s disease. J Chem Neuroanat. 2004;28:67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Sidrauski C, Chapman R, Walter P. The unfolded protein response: an intracellular signaling pathway with many surprising features. Trends Cell Biol. 1998;8:245–9. doi: 10.1016/s0962-8924(98)01267-7. [DOI] [PubMed] [Google Scholar]
- 12.Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005:373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Capitani M, Sallese M. The KDEL receptor: new functions for an old protein. FEBS Lett. 2009;583:3863–71. doi: 10.1016/j.febslet.2009.10.053. [DOI] [PubMed] [Google Scholar]
- 14.Young CL, Raden DL, Robinson AS. Analysis of ER resident proteins in Saccharomyces cerevisiae: implementation of H/KDEL retrieval sequences. Traffic. 2013;14:365–81. doi: 10.1111/tra.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, Mori K, Rozmahel R, Fraser P, George-Hyslop PS, Tohyama M. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s diseaselinked presenilin-1 mutations. J Biol Chem. 2001;276:43446–54. doi: 10.1074/jbc.M104096200. [DOI] [PubMed] [Google Scholar]
- 16.Hirashima N, Etcheberrigaray R, Bergamaschi S, Racchi M, Battaini F, Binetti G, Govoni S, Alkon DL. Calcium responses in human fibroblasts: a diagnostic molecular profile for Alzheimer’s disease. Neurobiol Aging. 1996;17:549–55. doi: 10.1016/0197-4580(96)00074-7. [DOI] [PubMed] [Google Scholar]
- 17.Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:534–8. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grossmann A, Kukull WA, Jinneman JC, Bird TD, Villacres EC, Larson EB, Rabinovitch PS. Intracellular calcium response is reduced in CD4+ lymphocytes in Alzheimer’s disease and in older persons with Down’s syndrome. Neurobiol Aging. 1993;14:177–85. doi: 10.1016/0197-4580(93)90094-r. [DOI] [PubMed] [Google Scholar]
- 19.Peterson C, Ratan RR, Shelanski ML, Goldman JE. Altered response of fibroblasts from aged and Alzheimer donors to drugs that elevate cytosolic free calcium. Neurobiol Aging. 1988;9:261–266. doi: 10.1016/s0197-4580(88)80063-0. [DOI] [PubMed] [Google Scholar]
- 20.Gibson GE, Vestling M, Zhang H, Szolosi S, Alkon D, Lannfelt L, Gandy S, Cowburn RF. Abnormalities in Alzheimer’s disease fibroblasts bearing the APP670/671 mutation. Neurobiol Aging. 1997;18:573–80. doi: 10.1016/s0197-4580(97)00149-8. [DOI] [PubMed] [Google Scholar]
- 21.Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195–200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- 22.Schneider I, Reverse D, Dewachter I, Ris L, Caluwaerts N, Kuiperi C, Gilis M, Geerts H, Kretzschmar H, Godaux E, Moechars D, Van Leuven F, Herms J. Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J Biol Chem. 2001;276:11539–44. doi: 10.1074/jbc.M010977200. [DOI] [PubMed] [Google Scholar]
- 23.Freichel M, Suh SH, Pfeifer A, Schweig U, Trost C, Weissgerber P, Biel M, Philipp S, Freise D, Droogmans G, Hofmann F, Flockerzi V, Nilius B. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat Cell Biol. 2001;3:121–7. doi: 10.1038/35055019. [DOI] [PubMed] [Google Scholar]
- 24.Pizzo P, Fasolato C, Pozzan T. Dynamic properties of an inositol 1, 4, 5-trisphosphate- and thapsigargin-insensitive calcium pool in mammalian cell lines. J Cell Biol. 1997;136:355–66. doi: 10.1083/jcb.136.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 26.Suen KC, Lin KF, Elyaman W, So KF, Chang RC, Hugon J. Reduction of calcium release from the endoplasmic reticulum could only provide partial neuroprotection against beta-amyloid peptide toxicity. J Neurochem. 2003;87:1413–26. doi: 10.1111/j.1471-4159.2003.02259.x. [DOI] [PubMed] [Google Scholar]